
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Post column infusion of an internal standard into LC-FT-ICR MS enables semi-quantitative comparison of dissolved organic matter in original samples†
Rebecca
Rodrigues Matos
 a,
Elaine K.
Jennings
a,
Jan
Kaesler
a,
Thorsten
Reemtsma
ab,
Boris P.
Koch
cd and
Oliver J.
Lechtenfeld
*ae
a,
Elaine K.
Jennings
a,
Jan
Kaesler
a,
Thorsten
Reemtsma
ab,
Boris P.
Koch
cd and
Oliver J.
Lechtenfeld
*ae
aDepartment of Environmental Analytical Chemistry, Helmholtz Centre for Environmental Research – UFZ, Permoserstr. 15, 04318 Leipzig, Germany. E-mail: oliver.lechtenfeld@ufz.de
bInstitute of Analytical Chemistry, University of Leipzig, 04103 Leipzig, Germany
cAlfred-Wegener-Institute Helmholtz Centre for Polar and Marine Research, Ecological Chemistry, Am Handelshafen 12, 27570 Bremerhaven, Germany
dHochschule Bremerhaven, University of Applied Sciences, An der Karlstadt 8, 27568 Bremerhaven, Germany
eProVIS–Centre for Chemical Microscopy, Helmholtz Centre for Environmental Research – UFZ, Permoserstr. 15, 04318 Leipzig, Germany
First published on 14th May 2024
Abstract
Ultrahigh resolution mass spectrometry hyphenated with liquid chromatography (LC) is an emerging tool to explore the isomeric composition of dissolved organic matter (DOM). However, matrix effects limit the potential for semi-quantitative comparison of DOM molecule abundances across samples. We introduce a post-column infused internal standard (PCI-IS) for reversed-phase LC-FT-ICR MS measurements of DOM and systematically evaluate matrix effects, detector linearity and the precision of mass peak intensities. Matrix effects for model compounds spiked into freshwater DOM samples ranging from a headwater stream to a major river were reduced by 5–10% for PCI-IS corrected mass peak intensities as compared to raw (i.e., untransformed) intensities. A linear regression of PCI-IS corrected DOM mass peak intensities across a typical DOM concentration range (2–15 mg dissolved organic carbon L−1) in original, non-extracted freshwater samples demonstrates excellent linearity of the detector response (r2 > 0.9 for 98% of detected molecular formulas across retention times). Importantly, PCI-IS could compensate for 80% of matrix effects across an environmental gradient of DOM composition from groundwater to surface water. This enabled studying the ionization efficiency of DOM isomers and linking the observed differences to the biogeochemical sources. With PCI-IS original, non-extracted DOM samples can be analysed by LC-FT-ICR MS without carbon load adjustment, and mass peak intensities can be reliably used to semi-quantitatively compare isomer abundances between compositionally similar DOM samples.
Introduction
Even though dissolved organic matter (DOM) is the major form of organic carbon in oceans and freshwater, its individual molecular constituents are still incompletely identified and characterized.1–3 The extreme complexity of this mixture and the low concentration of its individual constituents are the main obstacles in the complete decoding of DOM molecular and structural composition.4 The state-of-the-art technique to analyse DOM composition is ultrahigh resolution mass spectrometry, such as Fourier transform ion cyclotron resonance (FT-ICR) or Orbitrap mass spectrometry, routinely detecting more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular formulas (MF) in a single sample.5,6
000 molecular formulas (MF) in a single sample.5,6
The commonly used workflow for non-targeted DOM analysis is pre-concentration and desalting of samples using solid phase extraction (SPE)7 followed by direct infusion mass spectrometry (DI-MS). While DI-MS allows for simultaneous and sensitive detection of many DOM molecules, this technique is prone to matrix effects,8–11 caused by the competitive ionization and/or transfer from the liquid to the gas phase of co-eluting analytes and sample constituents.8 In addition, the structural information obtained from a DI mass spectrum is limited since it is not possible to differentiate between isomers and every MF in DOM is likely to represent a large number of different structures.3,12 Thus, the magnitude of a single mass peak in a DI mass spectrum of highly complex mixtures such as DOM corresponds to the concentration-weighted average of response factors of isomers with yet unknown proportions.10,13 As a consequence, the relative intensities (e.g. normalized to the base peak or the sum of all assigned peaks) are extensively used to evaluate compositional differences between DOM components.14,15 However, this approach lacks a quantitative dimension to extract predominant environmental processes as large (relative) intensity differences may also be caused by variable ionization (efficiency), extraction biases or matrix effects.11,16
Liquid chromatography (LC) coupled with FT-ICR or Orbitrap MS is increasingly applied to study DOM because it increases the amount of molecular-level information, particularly for heteroatom-containing molecules and those of very high or low polarity, and improves the sensitivity to disentangle complex environmental processes.6,12,17,18 In addition, the isomeric composition, along with the physico-chemical properties of the respective DOM isomers, can be explored simultaneously.12,17,19 An additional benefit for LC-MS approaches in general is that chromatographic separation reduces matrix effects caused by competitive ionization of co-eluting analytes and sample constituents.8,9,20 Matrix effects decrease the accuracy of a measurement and potentially decouple signal intensity from concentration.9,20,21 Organic trace analysis and many “omics” fields commonly utilize LC-MS to reduce matrix effects.22,23 However, for DOM research, the benefit of matrix effect reduction by LC-MS has not been systematically explored.
At the compound level, the response of a given analyte obtained by MS detection is a combination of random and systematic errors, matrix effects, as well as ionization efficiency and concentration of the analyte.9,24 When chromatography is applied, the above-mentioned factors may thus all influence the intensity and shape of the chromatographic peak, impacting the accuracy and precision of the analytical results. To study and compensate for matrix effects, as well as the instrument and sample preparation variability in LC-MS, an internal standard (IS) – a target analyte's structural analogue not expected to be present in the analysed sample – is usually spiked to the sample.25 Typically, the standard addition along with the standard curve allows for absolute quantification of the target compound.26 However, since DOM is an extremely complex mixture of potentially millions of structurally unknown components, analyte-matched internal standardization is still not feasible.
As an alternative approach, an IS can be continuously infused after the chromatography column (here referred to as the post-column infused internal standard, PCI-IS) and before electrospray ionization (ESI).27–29 This ensures volumetric mixing and that both – IS and analyte – ionize at the same flow rate and solvent composition.30 In addition, the IS signal can be tracked throughout an entire chromatographic run, and matrix effects can be simultaneously compensated for various analytes. It has been demonstrated that matrix effects in electrospray ionization are comparable even for analytes of different physicochemical properties that elute at the same retention time.31 Accordingly, matrix effects may be considered in part to be retention time dependent rather than solely analyte dependent. As a result, one IS may compensate for matrix effects even of structurally different analytes.32 In particular, for non-targeted metabolomics where severe matrix effects are common (e.g. urine and blood fluids), PCI-IS improves the reliability of analyses and quantitative comparisons, although individual standards are not available.29,33 This fact is especially attractive to DOM research since the chemical structure of individual DOM molecules and isomers is yet unknown. While this approach may not be as accurate and robust as analyte-matched internal standardization, we expect that PCI-IS will improve the robustness of analysis (e.g. during multi-day sequences) and will enable semi-quantification (i.e. the evaluation of concentration differences) of DOM molecules across samples.
Here, we report on the first application of a PCI-IS combined with LC-FT-ICR MS with the aim to enable semi-quantitative non-targeted analysis of original DOM samples (i.e., non-extracted and not concentration adjusted). As PCI-IS we use stable isotope labelled Naproxen reflecting structural motifs in terrestrial DOM. First, we tested the compensation of matrix effects and inter-sample variability by PCI-IS and evaluated the accuracy (based on chromatographic peak areas) of a set of model compounds (MCs) in various DOM matrices. Second, we tested the compensation of instrument variability and evaluated the repeatability for PCI-IS transformed DOM mass peak intensities from repeat injections of a DOM reference sample. Third, we tested the ability to use variable sample concentration with a concentration series of a DOM reference sample and evaluated the linearity of the detector response. Fourth, we tested the potential for semi-quantification of single DOM compounds with two non-extracted freshwater samples from a forest stream and a large river, representing a variable sample matrix and structurally diverse compounds, and a mixture of both and evaluated the compensation of matrix effects by comparing theoretical and measured detector response factors. Our approach can be readily extended to other non-targeted analyses of complex mixtures, such as metabolomics and petroleomics, where molecule abundance estimates rely on robust peak intensity determination.
Materials and methods
Chemicals
Ultrapure water (MQW, 18.2 MΩ, <5 ppb TOC; Milli-Q system, Merck, Darmstadt, Germany) was used to dissolve Suwannee River fulvic acid (SRFA, International Humic Substances Society, SRFA II; 2S101H) for a stock solution of 1 mg mL−1. MQW and methanol (HPLC grade; Lab-Scan) were used as solvents for LC-FT-ICR MS both with 0.1% formic acid (Sigma-Aldrich). Model compounds (MCs) – originally introduced by Patriarca et al.12 for QC – D-glucuronic acid, 2-(4-(2,2-dicarboxy-ethyl)-2,5-dimethoxy-benzyl)-malonic acid, fraxin, isoferulic acid 3-O-β-D-glucuronide, leu-enkephalin, and vanillic acid were analytical grade (Table SI 1.1†). The MCs 2-(4-(2,2-dicarboxy-ethyl)-2,5-dimethoxy-benzyl)-malonic acid, fraxin, and isoferulic acid 3-O-β-D-glucuronide are isomers and are therefore important to demonstrate suitability of the PCI-IS approach. Naproxen-D3 (Sigma-Aldrich) was diluted to a final concentration of 50 ng mL−1. Naproxen-D3 was selected as IS because its structural motifs – an aromatic ring, methoxy group and carboxylic acid – are common among DOM and the mass of the tri-deuterated variant is not expected to be detectable in DOM.Samples
The SRFA stock solution (1 mg C L−1 in MQW) was diluted to the following final dissolved organic carbon (DOC) concentrations: 2.0, 2.5, 5.0, 10, 15, and 20 mg C L−1, covering a typical DOC concentration range in terrestrial surface waters.34 Samples were collected from the Ströbecker Flieβ (“Strob.”, 2.40 mg C L−1), a small creek draining agricultural fields, the spring of the river Holtemme (6.39 mg C L−1) in the forested catchment of mid-range mountains and the river Elbe (5.27 mg C L−1) (details of samples and sampling sites can be found in ESI SI 1.1 and Fig. SI 1.1†). Samples were collected in pre-combusted glass bottles (400 °C, 4 h), transferred to the lab and filtered through glass fiber filters (GF/F, 0.7 μm, Whatman) within 24 h after sampling. DOC concentrations were determined with the high temperature combustion (HTCO) method (DIMATOC 2100, Dimatec Analysentechnik, Essen, Germany) following DIN EN 12260 (total organic carbon, TOC). The three freshwater samples were used at their original concentration without extraction.Furthermore, the samples from Elbe, Holtemme, and a 1:1 mixture of Elbe and Holtemme (Elbe:Holtemme) were prepared as dilution series with the following concentrations: Elbe: 5.27, 4.27, 3.27, 2.77, and 2.27 mg C L−1; Holtemme: 6.39, 5.39, 4.39, 3.39, and 2.39 mg C L−1; and Elbe:Holtemme: 5.83, 4.83, 3.83, 3.33, and 2.83 mg C L−1.
All MCs were spiked in MQW, SRFA 10 mg C L−1, Elbe, and Strob. at the following concentrations: D-glucuronic acid: 12.5, 25, 37.5, 50, and 75 ng mL−1; 2-(4-(2,2-dicarboxy-ethyl)-2,5-dimethoxy-benzyl)malonic acid: 2, 4, 6, 8, 10, and 12 ng mL−1; fraxin: 4, 8, 12, 16 and 24 ng mL−1; isoferulic acid 3-O-β-D-glucuronide: 2, 4, 6, 8, 10, and 12 ng mL−1; leu-enkephalin: 2.5, 5, 7.5, 10, and 12.5 ng mL−1; vanillic acid: 25, 50, 75, 100, and 150 ng mL−1. Each sample was injected in triplicate (n = 3) and samples were analysed from low to high concentrations.
PCI-IS-LC-FT-ICR MS
A reversed phase liquid chromatography method developed in our lab was adapted to implement the PCI-IS.19 Briefly, an ultrahigh performance liquid chromatography system (UHPLC; UltiMate 3000RS, Thermo Fisher Scientific, Waltham, MA, U.S.A.) equipped with a reversed-phase polar end-capped C18 column (ACQUITY HSS T3, 1.8 μm, 100 Å, 150 × 3 mm, Waters, Milford, U.S.A.) and a guard column (ACQUITY HSS T3 VanGuard, 1.8 μm, 100 Å, 2.1 × 5 mm, Waters) was used to separate the DOM. The flow of the main system was combined after the RP column with the flow from a second HPLC pump (LPG-3400SD) to add a counter gradient that mirrors the main gradient yielding a constant MQW/MeOH ratio at the ESI source. The PCI-IS (naproxen-D3) was added to the solvent bottles of the second pump solvents (A: MQW, and B: MeOH) at a concentration of 50 ng mL−1 (Fig. 1). The sample injection volume was 100 μL. | ||
| Fig. 1 Instrumental setup of the LC-FT-ICR MS with counter gradient and PCI-IS introduced by the second (2nd) pump. The flow of the 2nd pump (0.2 mL min−1) containing naproxen-D3 (50 ng mL−1) is mixed into the main pump flow (0.2 mL min−1) after the chromatography column. As the solvent gradient of the 2nd pump mirrors the gradient of the first pump (including a delay time to compensate for the different void and LC column volumes) resulting in a 1:1 MQW:MeOH ratio after the T-joint. The summed flow is then split, and 0.3 mL min−1 is directed to the DAD detector while 0.1 mL min−1 is transferred to the ESI source of the FT-ICR MS. | ||
The flow from the LC was continuously introduced into an FT-ICR mass spectrometer equipped with a dynamically harmonized analyzer cell (solariX XR, Bruker Daltonics, Billerica, MA, U.S.A.), a 12 T refrigerated actively shielded superconducting magnet (Bruker Biospin, Wissembourg, France), and an electrospray ionization source (ESI, Apollo II, Bruker Daltonics; capillary voltage: 4.3 kV, nebulizer gas pressure: 1.0 bar, dry gas temperature: 250 °C, dry gas flow rate: 8.0 L min−1). All samples were analyzed in negative ionization mode, with an ion accumulation time of 200 ms, a data size of 2 MWord (∼0.84 s transients, Fig. SI 1.2†), and broadband detection in magnitude mode (m/z 150 to 1000).
Data processing
Full profile LC-MS chromatograms were split into one minute segments between 11 min and 18 min retention time (RT) for SRFA samples and between 11 min and 17 min for Elbe, Holtemme and Elbe:Holtemme samples. These narrow RT ranges as compared to previous work were chosen because, at the lowest prepared concentrations, the number of shared molecular formulas in earlier (<11 min) and later segments (>17/18 min) was small (n < 120) and thus omitted for statistical analysis. All scans in each minute (64 scans) were averaged using DataAnalysis (version 5.0, Bruker Daltonics) and treated as a single spectrum resulting in six – seven mass spectra per sample. The peak picking signal-to-noise (S/N) threshold was set to 4, and each spectrum was calibrated with masses commonly found in NOM (n = 232, Table SI 1.2†). The resulting root-mean-squared mass error was less than 0.20 ppm (n = 120).
In-house software was used to assign molecular formulas (MF) to the mass peaks using the following configuration: m/z range between 150–1000, relative mass error ± 0.5 ppm, elemental ranges C: 1–60, 13C: 0–1, H: 1–122, N: 0–2, O: 0–40, S: 0–1, 34S: 0–1, and elemental ratios 0.3 < H/C < 3, 0 < O/C < 1.2, 0 < N/C < 1.5, 0 < DBE (double bond equivalent) < 25, and −10 < DBE-O (double bond equivalent minus oxygen) < 10.35 An MF was excluded if it was found in the blank with higher peak magnitude than the same MF in the sample (at the respective RT).
The chromatographic peaks of the MCs’ m/z were manually extracted and their intensity averaged using DataAnalysis. The MC baseline was corrected based on the intensity of the respective m/z in a non-spiked sample.
PCI-IS method evaluation
Throughout this work, we use the term (mass peak) magnitude for the raw mass peak magnitudes (abbreviated RAW). Naproxen-D3 (m/z 232.1056) was used to transform the DOM RAW values by dividing with the naproxen-D3 peak magnitude to obtain an internal standard normalized peak intensity (ISN). Since the PCI-IS was infused after the chromatography column, naproxen-D3 could be detected across all RTs and the calculation of the ISN was done for each segment individually. We evaluated the applicability of the method in the following ways: | (1) |
Matrix effects were calculated both for RAW and ISN values.
Furthermore, matrix effects were also assessed as the difference of the slopes of the analytical curve calculated as linear regression of the (transformed) intensity of a MC against its concentration between a sample matrix and in MQW. Using the slopes instead of individual concentrations allowed studying the concentration dependence of matrix effects as an important prerequisite for semi-quantitative comparisons of mass peak intensities without adjusting the DOC concentration to same values before injection into the LC.
The linearity for non-extracted freshwater samples (Elbe, Holtemme, Elbe:Holtemme) was evaluated as above using all concentration levels (Fig. SI 1.4†). Due to the expected compositional differences between Elbe and Holtemme, the expected slope of the Elbe:Holtemme sample was calculated according to (eqn (2)) considering only the MFs shared between the three samples.
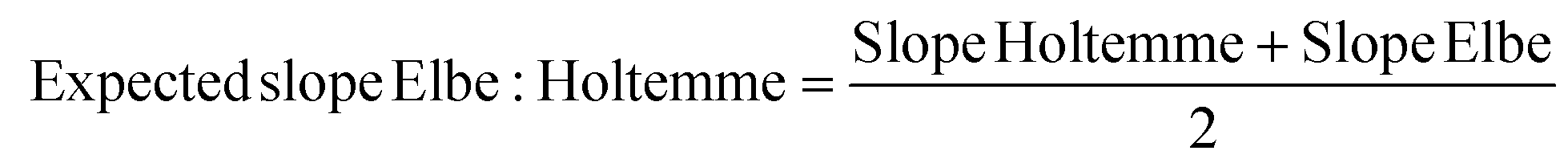 | (2) |
The remaining matrix effects were evaluated by plotting the expected slope against the experimentally determined slope from the measurement of the concentration series of the Elbe:Holtemme mixture. A deviation from the 1:1 line then indicates uncompensated matrix effects.
Results and discussion
Performance of the PCI-IS for variable sample concentrations
Even with the use of separation techniques like LC, matrix effects and instrument variability impact the MS analysis of complex mixtures like DOM and should be compensated for. For this purpose, an IS was continuously infused after the chromatographic separation. During the main DOM elution (10–18 min), the total ion chromatogram (TIC) magnitude of SRFA increased with the injected amount of carbon (Fig. 2A). More importantly, the sum of intensity of all assigned peaks detected over all studied RT segments increased linearly with the SRFA concentration (Fig. 2C). This linear increase indicated that for the applied measurement conditions and selected carbon concentration range (i.e., 2.0–15 mg C L−1), ion detection was within the linear dynamic range of the ICR cell. Notably, if higher amounts of carbon are injected, the ICR cell might suffer from overloading effects and a deviation from the linear relationship can be observed, eventually requiring adaptation of the ion accumulation time or dilution of the samples (Fig. SI 2.1†).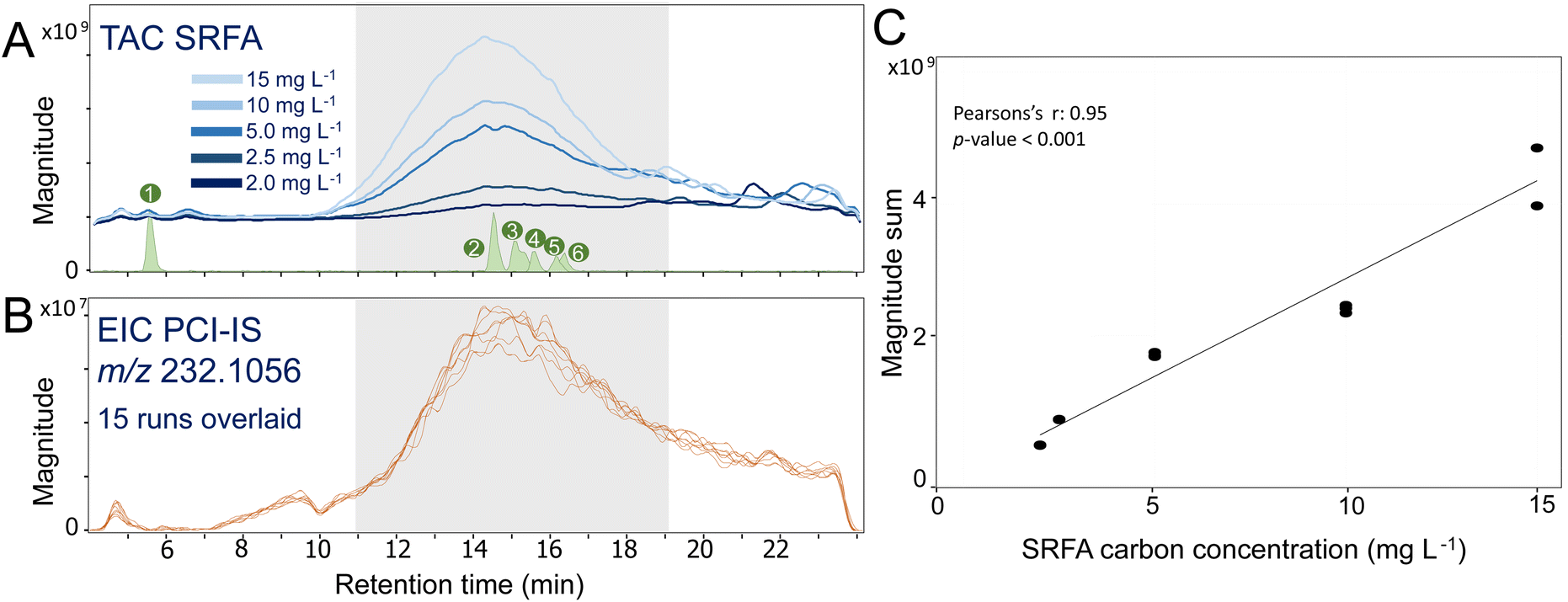 | ||
| Fig. 2 (A) Total assigned chromatogram (TAC) of SRFA (raw peak magnitudes, RAW) at different concentrations (2.0, 2.5, 5.0, 10, and 15 mg C L−1) and model compounds (MCs, green): 1: D-glucuronic acid; 2: 2-(4-(2,2-dicarboxy-ethyl)-2,5-dimethoxy-benzyl)malonic acid; 3: fraxin, 4: isoferulic acid 3-O-β-D-glucuronide; 5: leu-enkephalin; 6: vanillic acid, (B) extracted ion chromatogram (EIC) of naproxen-D3 (PCI-IS) from 15 consecutive injections using the SRFA concentration in (A), and (C) linear regression of sum of all assigned peak magnitudes versus SRFA carbon concentration. The grey area in (A) and (B) represents the retention time range considered in this work. | ||
Despite variable SRFA concentrations, the extracted ion chromatogram (EIC) of the PCI-IS naproxen-D3 was highly reproducible with a largest CV of its intensity of 14% at 11 min (Table SI 2.1†). However, we observed a variable naproxen-D3 intensity across the run (Fig. 2B) also previously observed38 that may be attributed to variations in system pressure during gradient elution. However, the use of naproxen-D3 as PCI-IS has little impact on the ionization of DOM (average ratio between DOM RAW intensity with and without naproxen-D3 ranges from 0.8 to 1.1; Fig. SI 2.2†), pointing to moderate co-ionization effects with DOM.
Compensation of matrix effects by PCI-IS
The potential to compensate matrix effects by PCI-IS was assessed based on six MCs spiked into SRFA of varying carbon concentrations and into two non-extracted freshwater samples (Elbe and Strob.). Using RAW intensities, a signal suppression in SRFA as compared to MQW was observed for five out of six MCs, with increasing suppression for increasing carbon concentrations (Fig. 3). Vanillic acid showed a signal enhancement (despite baseline correction due to co-eluting DOM with the same m/z value) also observed for other acidic compounds in SRFA.11 The use of ISN reduced matrix effects by 5 to 10% – resulting in matrix effects closer to 100% for all MCs. This indicates that a PCI-IS can effectively diminish matrix effects caused by variable DOM carbon concentrations.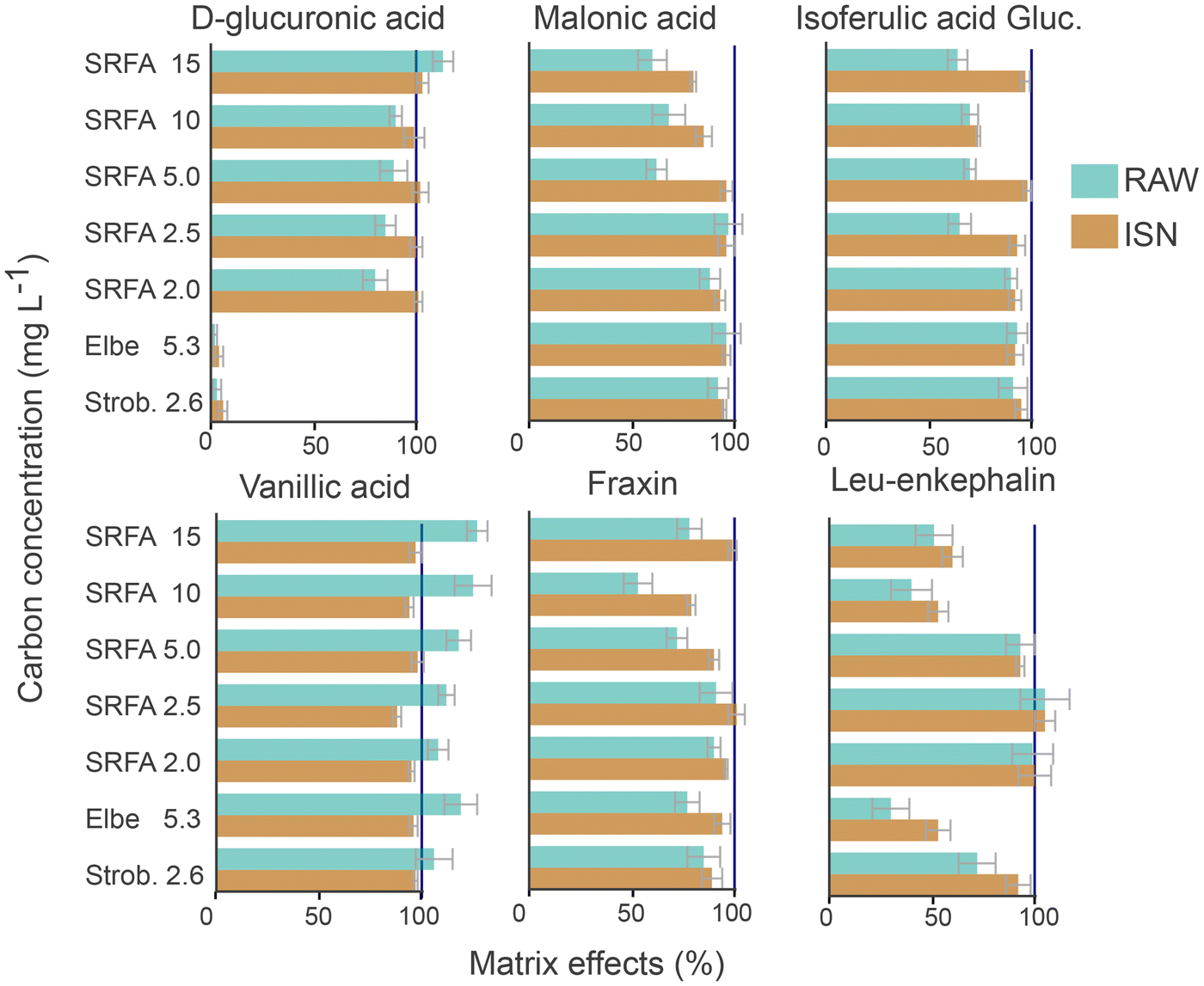 | ||
| Fig. 3 Matrix effects calculated for internal standard normalized peak intensities (ISN, brown) and raw peak magnitude (RAW, blue) for the model compounds (MC) spiked in SRFA with varying carbon concentration (from 2 to 15 mg C L−1), Ströbecker Fließ (Strob., 2.6 mg C L−1), and Elbe (5.3 mg C L−1). The dark blue vertical line at 100% represents the expected values (as obtained from the MCs in MQW). All MCs were baseline corrected. Note that due to the elution of D-glucuronic acid close to the void volume (tR = 4.8 min), this peak is affected by co-eluting inorganic constituents of the non-extracted samples Strob. and Elbe (Fig. SI 3.2†). | ||
Also, for the two non-extracted samples, the matrix effects were smaller using ISN (median = 93%) as compared to RAW (median = 88%, n = 12) for all MCs (Fig. 3). Notably, D-glucuronic acid (co-elution with salt close to the void volume) and leu-enkephalin (last eluting MC) were suppressed by the sample matrix and this effect could not be fully compensated by the PCI-IS. This indicates that different DOM compositions – represented by the non-extracted samples – lead to more pronounced matrix effects at the beginning and end of the chromatography run, potentially exaggerated by the lower response of the naproxen-D3 in this RT range. For the three isomers of m/z 369 (2-(4-(2,2-dicarboxy-ethyl)-2,5-dimethoxy-benzyl)-malonic acid, fraxin, isoferulic acid 3-O-β-D-glucuronide) and vanillic acid, matrix effects for ISN transformed intensities were smaller (>95%) as compared to RAW intensity (<95%). Considering that these four MCs elute in the same RT range as the majority of DOM (i.e., between 14 and 16.5 min), the PCI-IS can likely compensate the matrix effects for a large range of DOM constituents.
Overall these results suggest that we can consider two types of matrix effects in LC-FT-ICR MS analyses of DOM. Firstly, for the same sample (e.g. SRFA) where the chemical composition is unaffected with changing carbon concentration an increasing suppression or enhancement of individual analytes related to an increasing number of DOM ions eluting at the same time is observed (“ME type I”). Secondly, changes in the chemical composition (e.g. comparison of Elbe and Strob.) of DOM impact the MC intensity differently (“ME type II”). In both cases, matrix effects can be effectively controlled by ISN. However, our results also indicate that an accurate assessment of matrix effects needs to consider both, the concentration-dependent ME type I (in the case where samples are injected at variable carbon concentrations) and the composition-dependent ME type II (in the case where the sample matrix changes, which is likely for environmental gradients), in order to accurately represent individual molecule concentrations (differences) by their mass peak intensities.
Matrix effects of the set of MCs were further quantified with SRFA and the two non-extracted samples (Strob. and Elbe) as matrices by comparing the slopes obtained from the detector response across the MC concentration range (Table SI 3.1†). Compared to MCs in MQW we observed smaller slopes for 5 out of 6 MCs. The lower response is in line with the results from a single concentration level (cf.Fig. 3). Across the three different DOM types, the mean slope for all MCs in the two non-extracted samples was 78 ± 30 (72 ± 39) % for ISN (RAW) as compared to the respective slopes of MCs (except for D-glucuronic acid) in MQW (considered 100%, Table SI 3.1†). The lower mean value and larger spread of slopes for the RAW as compared to ISN points to a strong effect of co-eluting/co-ionizing sample matrix on the sensitivity of the measurement (ME type II). It is noted that such sample dependent matrix effects have also shown to impact DI-MS measurements after SPE.10 The relative intensity of MC spiked into various samples was found to vary across different types of matrices and carbon loads in DI-MS analyses; e.g. it decreased more than 15% with increasing SRFA carbon concentrations (0–40 mg C L−1), suggesting that the peak intensities do not correlate with compound concentration. Our results with LC-FT-ICR MS, on the other hand, indicate that the combination of matrix and DOM separation via RP-LC with counter gradient and PCI-IS can reduce these two types of matrix effects, similar to other LC-ESI-MS approaches.28,39 This enables semi-quantitative comparisons of the same molecular features (m/z × RT) across samples, even though individual DOM molecule standards are not available for absolute quantification.
Repeatability and effect of PCI-IS on DOM MFs
The variability of peak intensities beyond MCs was assessed using the set of shared MF between triplicate measurements of SRFA. The mean and maximum CV of peak intensities were 13% (13%) and 52% (54%) for ISN (RAW), respectively (Fig. 4, Table SI 4.1†). The variability of low intensity peaks was inherently higher as compared to higher intensity peaks, as previously observed.40 This pattern was observed regardless of the SRFA concentration and RT (Table SI 4.1†). Also, a good agreement between the CV values using ISN and RAW was found (Fig. 4) indicating that for extracted samples, the concentration related matrix effects are only minor in LC-FT-ICR MS. The results also indicate that while the inclusion of a counter gradient stabilizes the ESI spray and helps to improve sample-to-sample repeatability, the major source of intensity variability is directly related to the measured mass peak intensities or S/N ratios (and hence independent of the LC).40 Nevertheless, PCI-IS will aid in compensating instrument variability and increasing the accuracy and comparability of data acquired over a longer period of time.41 | ||
| Fig. 4 Coefficient of variation (CV) for individual molecular formulas (MF, n = 162925) in triplicate measurements using RAW and ISN for SRFA 10 mg C L−1. The grey lines represent the mean of CV for both treatments. The black line indicates the 1:1 ratio. MFs are colored by the intensity percentiles (25th, 50th, 75th, 100th). All segments (n = 8, RT: 11 – 18 min) were combined. The mean CV of testable MFs is available in Table SI 4.1.† | ||
Linearity of DOM MF peak intensities
The linear correlation between the sum of the total assigned intensity and carbon concentration indicated a linear detector behaviour within the considered concentration range (Fig. 2). However, as ion suppression and space charge effects in the analyser cell are a known phenomenon in ICR, the linear relationship between individual peak intensities and carbon concentration needed to be tested.Within the considered range of SRFA carbon concentrations (2–15 mg C L−1), the linear regression of ISN values versus sample carbon concentration was significant (with r2 > 0.9, α = 0.005) for 98% of testable MFs (Fig. 5) and slightly better as compared to RAW (96% of testable MFs, Fig. SI 5.1†). Similar to the instrument variability, the fit of the regression decreased with decreasing peak intensity (Fig. 5) but even for low intensities (25th percentile) the r2 values of the linear regression were significant and above 0.8 (p < 0.005).
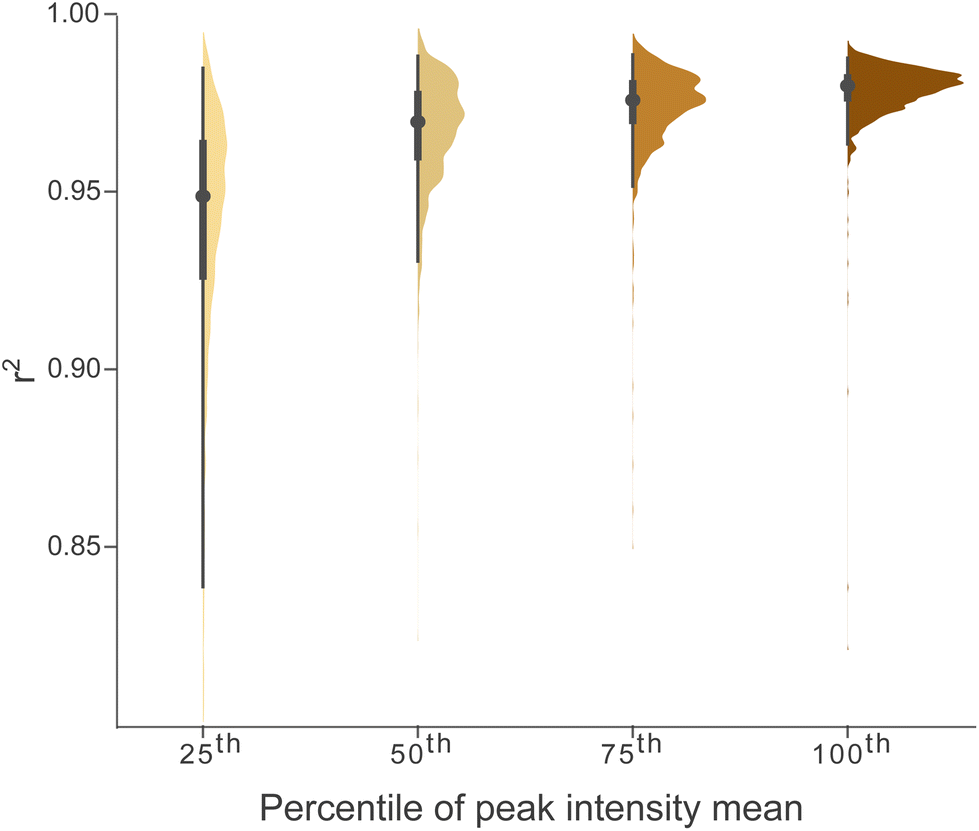 | ||
| Fig. 5 Distribution of squared linear correlation coefficient (r2) values for testable MFs, grouped and colored by percentiles (25th, 50th, 75th, 100th) of the mean ISN values (across carbon concentration range 2–15 mg L−1, n = 81751, Table SI 1.3†). All segments (RT: 11–18 min) were combined. | ||
Likewise, for the non-extracted samples (Elbe, Holtemme, Elbe:Holtemme), the linear regression of ISN values versus sample carbon concentration was significant (with r2 > 0.9, α = 0.005) for 80% of testable MFs and better as compared to RAW (75% of the testable MFs; Fig. SI 5.2†) and no trend was observed for different RTs (11–18 min; Fig. SI 5.3†). Overall, these results indicate that for LC-FT-ICR MS measurement of DOM, the observed peak intensity of a particular MF correlates well with the sample's carbon concentration as has been observed with DI (cf. ESI Fig. 3 in Osterholz et al. (2015)49). However, the non-extracted samples revealed a pronounced non-linear pattern when comparing the correlation coefficients calculated for ISN and RAW (Fig. SI 5.2†), pointing to an impact of the sample matrix on the measured peak intensities (here RAW). Since, in case of a concentration series, the matrix is the same this effect can be attributed to concentration differences (ME type I) and the general improvement of the correlation coefficients when using ISN confirms that this type of matrix effect can be efficiently compensated by PCI-IS. Further matrix effect related to different sample composition (ME type II) will be discussed in the next section.
Matrix effects in original, non-extracted freshwater samples
To test the matrix effect compensation by PCI-IS caused by different sample matrices and compositions (ME type II), the dilution series of two non-extracted samples Elbe, Holtemme, and Elbe:Holtemme was used (Fig. SI 1.3†). The slopes (linear regression of ISN versus sample's carbon concentration) of MF shared between the two contrasting freshwater samples were 0.8 ± 0.5 times larger for Elbe than for the Holtemme sample (Fig. 6A). This is expected due to the differing DOM sources and hence DOM composition in both samples, resulting in different mixtures of DOM chemical structures and accordingly, different ionization efficiencies (see ‘Response factors of individual DOM MF and its relationship with molecule polarity’).
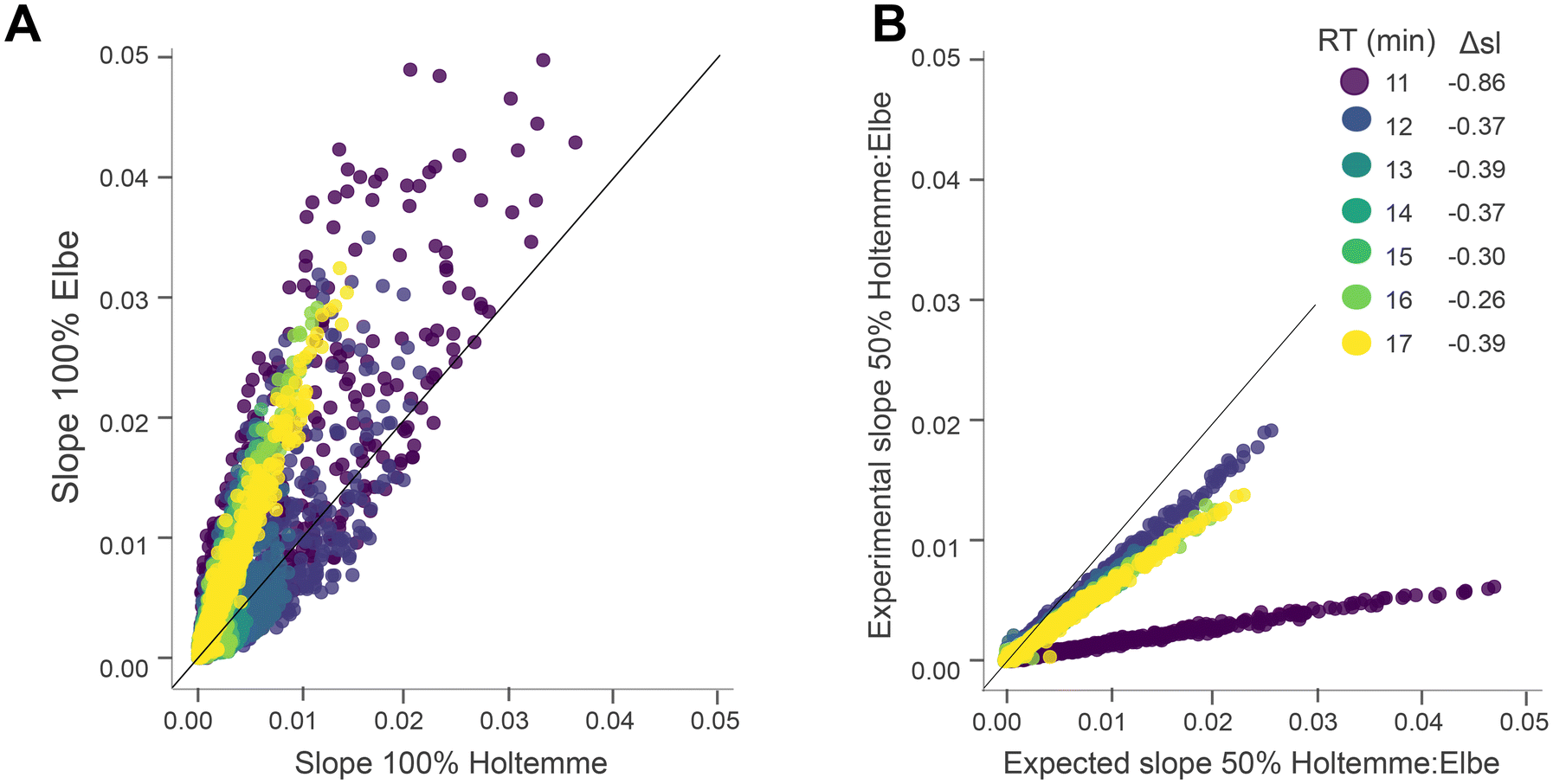 | ||
| Fig. 6 Response factors for DOM compounds. Slopes were calculated from the linear regression analysis of ISN peak intensity against sample carbon concentration. (A) Slopes of individual molecular formulas (MF) in 100% Elbe plotted against their slope in 100% Holtemme. (B) Expected slope of 50% Holtemme:Elbe (calculated as the average of the slope from 100% Holtemme and Elbe, cf. Fig. SI 1.3†) plotted against the experimental slope of 50% Holtemme:Elbe and the deviation of the slope (Δsl) from ideal conformity (1:1 ratio, black line). MF are colored by the retention time (RT). Note: the chromatography peaks at RT 11 min (Fig. SI 1.2†) in the 1:1 mixture might impact the accuracy of the slopes for this segment. | ||
Along environmental gradients (e.g. surface water flow along increasing catchment sizes or baseflow–event flow gradients),42,43 the DOM composition and the matrix changes, potentially affecting analysis of (non-extracted) samples spanning large gradients. To evaluate the impact of variable sample matrices on peak intensities (ME type II), the expected slopes of Elbe:Holtemme (calculated from the individual slopes in Elbe and Holtemme) were plotted against the experimental slopes (Fig. 6B). Between 12 and 17 min, sample-derived matrix effects within the compositional gradient between Holtemme and Elbe were well compensated, with Δsl ranging from −0.26 (16 min) to −0.39 (13 min). Of note, the strong Δsl of −0.86 at 11 min was possibly caused by the sample handling (Fig. SI 1.3b:† chromatographic peaks at 11 min), highlighting that the most polar segment might be more prone to strong matrix effects due to the co-eluting (in-)organic matrix as commonly observed for LC-MS.17,19 Our results indicate that matrix effects in DOM can be substantially improved using PCI-IS.
Overall, PCI-IS transformed peak intensities (ISN) for non-extracted samples can be used as a representative measure of the (relative) concentration of the underlying DOM compounds. Furthermore, a comparison of ISN derived intensities allows semi-quantitative assessment of concentration differences between samples, provided that the compositional and matrix gradients are not too large. This can be readily tested by the presented approach of end-member mixing and regression slope analysis. Further assessment of matrix effects and their compensation by PCI-IS might be required for other sets of samples to confirm the semi-quantitative aspect along (larger) environmental gradients (e.g. across the land–ocean aquatic continuum).
Response factors of individual DOM MF and their relationship with molecule polarity
Next to the quantitative aspect, the slopes of the linear regression analysis of carbon concentration against ISN can provide additional information on the ionization efficiency and structure of underlying DOM compounds (here for Holtemme and Elbe). Generally, for a given sample and MF, the slope obtained from the linear regression of the sample between the concentration and RAW represents the analytes’ response factor. However, if ISN is used (and matrix effects are thus minimized) the slopes are a proxy of the ionization efficiency of the underlying compounds. In addition, the combination of PCI-IS with the constant solvent composition provided by the counter gradient also allows comparison of the response factors of the same MF observed across different datasets and even investigation of the ionization behaviour of DOM homologous series within one RT segment.Overall, slopes of the Elbe river sample were larger than those of the Holtemme sample – reaching a magnitude of 0.03 and 0.01 for Elbe and Holtemme at RT 15 min, respectively (Fig. 6 and 7). This indicates that the underlying DOM compounds in the Holtemme sample have lower ionization efficiency compared to DOM compounds in the Elbe sample. The river Elbe drains a large range of land use types: forested, agricultural, and mixed catchments, thus integrating different DOM sources (with additional input from photoautotrophic DOM).44 This translates into an increased percentage of heteroatoms (up to 40% of CHNO in early RTs and 25% in late RT) and a higher number of MFs with relatively low masses across the polarity range (Fig. SI 6.1a and b†). The Holtemme spring, in contrast, represents groundwater originating from a mountain range with mostly coniferous vegetation45 contributing DOM characterized by higher aromaticity (larger aromaticity index) and unsaturation (lower H/C ratios) (Fig. SI 6.1d and e†). The steeper slopes obtained for the Elbe river agrees with a study of the ionization efficiency of negatively charged compounds where less aromatic and low molecular weight DOM compounds show greater ionization efficiency.24
 | ||
| Fig. 7 Ionization efficiency of DOM compounds. (A) and (B) Kendrick mass defect plot (CO2 base mass) coloured by the slope of the analytical curve for the 16 min section. The upper insets highlight changes in slopes of carbon concentration against ISN intensity. Note that the different absolute values may be caused by the different sample compositions, (cf.Fig. 6). (C) and (E) O/C plotted against m/z. (D) and (F) H/C plotted against O/C. | ||
A detailed inspection of the slopes within a KMD series revealed that the slopes were dependent on the number of CO2 units (Fig. 7). For a particular KMD series, the slope first increased and then decreased, generally corresponding to the intensity pattern across a KMD series and potentially related to the lower ionization efficiency of larger, less polar molecules.11 This indicates that not all oxygens atoms in a DOM MF contribute to the ionization process (since the most oxygen-rich molecules do not have the largest slopes), and that the observed intensity pattern across KMD series or nominal masses may be a representation of ionization efficiencies and not solely of relative molecule abundance.46–48 This is further confirmed by the fact that even hydrophilic fractions contain MF with high O/C ratios (Fig. SI 6.1†), pointing to a potential masking of oxygen in non-carboxylic functional groups.
Conclusion
The use of PCI-IS for LC-FT-ICR MS analysis of DOM results in an improved comparability of mass peak intensities enabling semi-quantitative analysis of samples with variable carbon concentrations – here ranging from 2 to 15 mg C L−1 as usually found in terrestrial waters – without adjusting instrumental settings or carbon concentration/injection volumes. Matrix effects caused by different sample compositions can be compensated by using PCI-IS transformed intensities and the regression analysis performed on the non-extracted samples showed good linearity for most of the MFs. Overall, LC-FT-ICR MS with PCI-IS opens the possibility to move beyond compositional comparison of DOM samples and instead apply semi-quantification of single DOM molecules, since matrix effects are largely reduced (which would otherwise also contribute to intensity changes). This may be useful in extracting key features of DOM that indicate quantitatively most important compositional changes, e.g. during incubation or manipulation experiments (sorption, UV degradation).Analysing the slopes of DOM molecules (by using concentration series) allows the study of their ionization behaviour and gaining further insights into DOM functional groups across samples and polarity ranges. This approach can easily be adapted for other non-targeted analyses of complex mixtures (e.g. petroleomics, metabolomics) where authentic standards are not available for quantification. Since the use of PCI-IS helps in reducing instrument variability (caused by different tunings) and compensates for sensitivity changes during longer measurement sequences, it may also help to improve inter-lab comparability. In addition, a PCI-IS can also be used as an internal calibrant when lock masses are not found in a given RT. For this purpose, the performance of other internal standards with different masses and structures can be tested to widen the range of internal calibrants and to extend the compensation for matrix effects of different compound classes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Norbert Kamjunke and the captain and crew of the RV Albis for sampling the Elbe river water and Ronald Krieg for providing samples from the Ströbecker Fließ and Holtemme. We are grateful for using the analytical facilities of ProVIS Centre for Chemical Microscopy within the Helmholtz Centre for Environmental Research Leipzig, that is supported by European Regional Development Funds (EFRE – Europe funds Saxony) and the Helmholtz Association. We also thank Johann Wurz for software development. R. R. M. received funding provided by the German Research Foundation, DFG, project number 45025664 and the Helmholtz Association (HiDA, project number 14256) that allowed this research to be conducted. Finally, we thank the editor and two anonymous reviewers for their constructive comments that improved the manuscript.References
- N. Hertkorn, M. Harir, B. P. Koch and B. Michalke, Biogeosciences, 2013, 10, 1583–1624 CrossRef CAS.
- M. Witt, J. Fuchser and B. P. Koch, Anal. Chem., 2009, 81, 2688–2694 CrossRef CAS PubMed.
- M. Zark and T. Dittmar, Nat. Commun., 2018, 1–8 CAS.
- N. Hertkorn, M. Frommberger, M. Witt, B. P. Koch, P. Schmitt-kopplin and E. M. Perdue, Anal. Chem., 2008, 80, 8908–8919 CrossRef CAS PubMed.
- Y. Qi, Q. Xie, J. J. Wang, D. He, H. Bao, Q. L. Fu, S. Su, M. Sheng, S. L. Li, D. A. Volmer, F. Wu, G. Jiang, C. Q. Liu and P. Fu, Carbon Res., 2022, 1–22 Search PubMed.
- Y. Qi, C. Ma, S. Chen, J. Ge, Q. Hu, S. L. Li, D. A. Volmer and P. Fu, ACS ES&T Water, 2021, 1, 1975–1982 Search PubMed.
- T. Dittmar, B. Koch, N. Hertkorn and G. Kattner, Limnol. Oceanogr.: Methods, 2008, 6, 230–235 CrossRef CAS.
- A. De Nicolò, M. Cantù and A. D'Avolio, Bioanalysis, 2017, 9, 1093–1105 CrossRef PubMed.
- H.-C. Liu, D.-L. Lin and H. H. McCurdy, Forensic Sci. Rev., 2013, 25, 65–78 Search PubMed.
- W. Kew, A. Myers-Pigg, C. Chang, S. Colby, J. Eder, M. Tfaily, J. Hawkes, R. Chu and J. Stegen, EGUsphere, 2022, 2022, 1–26 Search PubMed.
- C. Patriarca, A. Balderrama, M. Može, P. J. R. Sjöberg, J. Bergquist, L. J. Tranvik and J. A. Hawkes, Anal. Chem., 2020, 92, 14210–14218 CrossRef CAS PubMed.
- J. A. Hawkes, C. Patriarca, P. J. R. Sjöberg, L. J. Tranvik and J. Bergquist, Limnol. Oceanogr. Lett., 2018, 3, 21–30 CrossRef CAS.
- J. A. Hawkes, J. D'Andrilli, J. N. Agar, M. P. Barrow, S. M. Berg, N. Catalán, H. Chen, R. K. Chu, R. B. Cole, T. Dittmar, R. Gavard, G. Gleixner, P. G. Hatcher, C. He, N. J. Hess, R. H. S. Hutchins, A. Ijaz, H. E. Jones, W. Kew, M. Khaksari, D. C. Palacio Lozano, J. Lv, L. R. Mazzoleni, B. E. Noriega-Ortega, H. Osterholz, N. Radoman, C. K. Remucal, N. D. Schmitt, S. K. Schum, Q. Shi, C. Simon, G. Singer, R. L. Sleighter, A. Stubbins, M. J. Thomas, N. Tolic, S. Zhang, P. Zito and D. C. Podgorski, Limnol. Oceanogr.: Methods, 2020, 18, 235–258 CrossRef CAS.
- P. Herzsprung, N. Kamjunke, C. Wilske, K. Friese, B. Boehrer, K. Rinke, O. J. Lechtenfeld and W. von Tümpling, Water Res., 2023, 232, 119672 CrossRef CAS PubMed.
- N. Kamjunke, W. von Tümpling, N. Hertkorn, M. Harir, P. Schmitt-Kopplin, H. Norf, M. Weitere and P. Herzsprung, Water Res., 2017, 123, 513–523 CrossRef CAS PubMed.
- J. D’Andrilli, S. J. Fischer and F. L. Rosario-Ortiz, Environ. Sci. Technol., 2020, 54(19), 11654–11656 CrossRef PubMed.
- E. Jennings, A. Kremser, L. Han, T. Reemtsma and O. J. Lechtenfeld, Environ. Sci. Technol., 2022, 56, 1894–1904 CrossRef CAS PubMed.
- E. K. Jennings, M. Sierra Olea, J. M. Kaesler, U. Hübner, T. Reemtsma and O. J. Lechtenfeld, Water Res., 2023, 229, 119477 CrossRef CAS PubMed.
- L. Han, J. Kaesler, C. Peng, T. Reemtsma and O. J. Lechtenfeld, Anal. Chem., 2021, 93, 1740–1748 CrossRef CAS PubMed.
- S. Kittlaus, J. Schimanke, G. Kempe and K. Speer, J. Chromatogr. A, 2012, 1218, 8399–8410 CrossRef PubMed.
- W. M. A. Niessen, P. Manini and R. Andreoli, Mass Spectrom. Rev., 2006, 25, 881–899 CrossRef CAS PubMed.
- G. A. Evans, Nat. Biotechnol., 2000, 18, 2000 CrossRef PubMed.
- M. Raida, P. Schulz-Knappe, G. Heine and W. G. Forssmann, J. Am. Soc. Mass Spectrom., 1999, 10, 45–54 CrossRef CAS PubMed.
- M. Oss, S. Tshepelevitsh, A. Kruve, P. Liigand, J. Liigand, R. Rebane, S. Selberg, K. Ets, K. Herodes and I. Leito, Rapid Commun. Mass Spectrom., 2021, 35, 1–21 CrossRef PubMed.
- H. Liao, G. Chen, I. Tsai and C. Kuo, J. Chromatogr. A, 2014, 1327, 97–104 CrossRef CAS PubMed.
- B. Yao, D. Sun, Y. Ren and M. Wang, J. Chem. Educ., 2022, 99, 603–611 CrossRef CAS.
- D. Chepyala, H. Kuo, K. Su, H. Liao, S. Wang, S. R. Chepyala, L. Chang and C. Kuo, Anal. Chem., 2019, 91, 10702–10712 CrossRef CAS PubMed.
- H. W. Liao, C. H. Kuo, H. C. Chao and G. Y. Chen, J. Pharm. Biomed. Anal., 2020, 178, 112956 CrossRef CAS PubMed.
- J. Rossmann, L. D. Renner, R. Oertel and A. El-Armouche, J. Chromatogr. A, 2018, 1535, 80–87 CrossRef CAS PubMed.
- M. Huang, H.-Y. Li, H.-W. Liao, C.-H. Lin, C.-Y. Wang, W.-H. Kuo and C.-H. Kuo, Rapid Commun. Mass Spectrom., 2020, 34, e8581 CrossRef CAS PubMed.
- H. Stahnke, T. Reemtsma and L. Alder, Anal. Chem., 2009, 81, 2185–2192 CrossRef CAS PubMed.
- B. K. Choi, A. I. Gusev and D. M. Hercules, Anal. Chem., 1999, 71, 4107–4110 CrossRef CAS.
- D. Chepyala, H. C. Kuo, K. Y. Su, H. W. Liao, S. Y. Wang, S. R. Chepyala, L. C. Chang and C. H. Kuo, Anal. Chem., 2019, 91, 10702–10712 CrossRef CAS PubMed.
- M. P. da Silva, K. Blaurock, B. Beudert, J. H. Fleckenstein, L. Hopp, S. Peiffer, T. Reemtsma and O. J. Lechtenfeld, J. Geophys. Res.: Biogeosci., 2021, 126, 1–18 Search PubMed.
- P. Herzsprung, N. Hertkorn, W. Von Tumpling, M. Harir, K. Friese and P. Schmitt-Kopplin, Anal. Bioanal. Chem., 2014, 406, 7977–7987 CrossRef CAS PubMed.
- E. Kendrick, Anal. Chem., 1963, 35, 2146–2154 CrossRef CAS.
- C. A. Hughey, C. L. Hendrickson, R. P. Rodgers, A. G. Marshall and K. Qian, Anal. Chem., 2001, 73, 4676–4681 CrossRef CAS PubMed.
- H. W. Liao, I. L. Tsai, G. Y. Chen, Y. S. Lu, C. H. Lin and C. H. Kuo, J. Chromatogr. A, 2014, 1358, 85–92 CrossRef CAS PubMed.
- H. Liao, G. Chen, M. Wu, W. Liao, C. Lin and C. Kuo, J. Proteome Res., 2017, 16, 1097–1104 CrossRef CAS PubMed.
- R. L. Sleighter, H. Chen, A. S. Wozniak, A. S. Willoughby, P. Caricasole and P. G. Hatcher, Anal. Chem., 2012, 84, 9184–9191 CrossRef CAS PubMed.
- C. Dusny, M. Lohse, T. Reemtsma, A. Schmid and O. J. Lechtenfeld, Anal. Chem., 2019, 91, 7012–7018 CrossRef CAS PubMed.
- S. Wagner, J. H. Fair, S. Matt, J. D. Hosen, P. Raymond, J. Saiers, J. B. Shanley, T. Dittmar and A. Stubbins, J. Geophys. Res.: Biogeosci., 2019, 124, 759–774 CrossRef CAS.
- H. Peter, G. Singer, A. J. Ulseth, T. Dittmar, Y. T. Prairie and T. J. Battin, J. Geophys. Res.: Biogeosci., 2020, 125, 1–17 Search PubMed.
- N. Kamjunke, L. M. Beckers, P. Herzsprung, W. von Tümpling, O. Lechtenfeld, J. Tittel, U. Risse-Buhl, M. Rode, A. Wachholz, R. Kallies, T. Schulze, M. Krauss, W. Brack, S. Comero, B. M. Gawlik, H. Skejo, S. Tavazzi, G. Mariani, D. Borchardt and M. Weitere, Sci. Total Environ., 2022, 828, 154243 CrossRef CAS PubMed.
- N. Kamjunke, N. Hertkorn, M. Harir, P. Schmitt-Kopplin, C. Griebler, M. Brauns, W. von Tümpling, M. Weitere and P. Herzsprung, Water Res., 2019, 164, 114919 CrossRef CAS PubMed.
- L. J. Tranvik, J. Bergquist, J. A. Hawkes, C. Patriarca and P. J. R. Sj, Limnol. Oceanogr. Lett., 2018, 21–30 Search PubMed.
- N. Hertkorn, R. Benner, M. Frommberger, P. Schmitt-kopplin, M. Witt, K. Kaiser, A. Kettrup and J. I. Hedges, Geochim. Cosmochim. Acta, 2006, 70, 2990–3010 CrossRef CAS.
- A. Zherebker, E. Shirshin, A. Rubekina, O. Kharybin, A. Kononikhin, N. A. Kulikova, K. V. Zaitsev, V. A. Roznyatovsky, Y. K. Grishin, I. V. Perminova and E. N. Nikolaev, Environ. Sci. Technol., 2020, 54, 2667–2677 CrossRef CAS PubMed.
- H. Osterholz, J. Niggemann, H.-A. Giebel, M. Simon and T. Dittmar, Nat. Commun., 2015, 6, 7422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00119b |
| This journal is © The Royal Society of Chemistry 2024 |