
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynchrotron radiation X-ray diffraction computed tomography (XRDCT): a new tool in cultural heritage and stone conservation for 3D non-destructive probing and phase analysis of inorganic re-treatments†
Elena
Possenti
 *a,
Nicoletta
Marinoni
b,
Claudia
Conti
a,
Marco
Realini
a,
Gavin B. M.
Vaughan
c and
Chiara
Colombo
a
*a,
Nicoletta
Marinoni
b,
Claudia
Conti
a,
Marco
Realini
a,
Gavin B. M.
Vaughan
c and
Chiara
Colombo
a
aIstituto di Scienze del Patrimonio Culturale (ISPC), Consiglio Nazionale delle Ricerche (CNR), Via R. Cozzi 53, 20125 Milano, Italy. E-mail: elena.possenti@cnr.it; claudia.conti@cnr.it; marco.realini@cnr.it; chiara.colombo@cnr.it; Tel: +39 02 66173392
bDipartimento di Scienze della Terra, Università degli Studi di Milano, Via S. Botticelli 23, 20133 Milano, Italy. E-mail: nicoletta.marinoni@unimi.it
cEuropean Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: vaughan@esrf.fr
First published on 27th February 2024
Abstract
The issue of preserving carbonatic stones of cultural heritage (CH) restored in the past that have undergone new decay phenomena is strongly emerging and conservation science has not yet found a reliable solution. In this paper, we propose the application of synchrotron radiation X-ray diffraction computed tomography (XRDCT) to explore the effects of using inorganic-mineral products (ammonium oxalate; ammonium phosphate) in sequence as a novel, compatible and effective re-treatment approach to consolidate decayed carbonatic stones already treated with inorganic-mineral treatments. High-quality XRDCT datasets were used to qualitatively/quantitatively investigate and 3D localize the complex mixture of crystalline phases formed after the conservation re-treatments within a porous carbonatic stone substrate. The XRDCT reconstruction images and the structural refinements of XRD patterns with the Rietveld methods showed that the phase composition of reaction products, their volume distribution, and weight fraction vary as a function of the treatment sequence and penetration depth. The high potential of XRDCT allows (i) assessment of peculiar trends of each treatment/treatment sequence; (ii) exploration of the reaction steps of the sequential treatments and (iii) demonstration of the consolidating effect of inorganic re-treatments, non-destructively and at the micron scale. Above all, our study (i) provides new analytical tools to support the conservation choices, (ii) showcases new analytical possibilities for XRDCT in conservation science, including in investigations of CH materials and decay processes, and (iii) opens up new perspectives in analytical chemistry and material characterisation for the non-destructive and non-invasive analysis of reactions within heterogeneous polycrystalline systems.
Introduction
Carbonatic stone materials of cultural heritage (CH) are prone to decay, resulting in compositional variations, microstructural decohesion, and material losses, even if, in many cases, they have been restored in the past with the application of conservation treatments.1–3 By settling inside the porous matrices, the conservation products give rise to novel multi-phase systems where the decayed stone substrate and the conservation product coexist.4–7 Over time, the unavoidable interactions of these “systems” with the surrounding environment promote further decay patterns on the already decayed material as well as on the conservation product. This process implies that more and more CH masterpieces will show decayed treated stone materials which need a further conservation procedure.Heritage science is now facing a new frontier of conservation, that is to say, the pressing need to find effective, sustainable, and compatible technological solutions to preserve the material integrity of “decayed treated systems” that are progressively more complex and more fragile.
To date, a wide gap exists in knowledge of the interactions occurring between the new conservation products and (i) the remains of the past decayed conservation products; and (ii) the decay products of the decayed stone matrix.
Moreover, a wide range of possible conservation treatments is available for the consolidation and protection of carbonatic stone materials (both organic-polymeric products and inorganic-mineral ones) and almost all these products may have been used in the past or can be applied nowadays. This gives rise to a very broad scenario of possible one-to-one combinations, whose effects are, for the most part, unexplored. The need for studies on this topic is also demonstrated by the lack of specific terms to label the application of conservation treatments to decayed, already treated, stone matrixes. Therefore, this conservation procedure is labelled in the following as “re-treatment”.
Here, the focus of the study is to explore the effects induced by conservation re-treatments carried out with inorganic-mineral products of a porous carbonatic stone material already treated with an inorganic-mineral treatment. Within this context, inorganic products based on ammonium oxalate (AmOx, (NH4)2C2O4·H2O) and ammonium phosphate (DAP, (NH4)2HPO4)8 are here proposed as a valid option to restore the microstructural cohesion of carbonatic matrixes already treated with AmOx or DAP inorganic treatment, being an unexplored and compatible alternative to re-treatments with organic products.
The core idea of inorganic treatments is that they penetrate within the porous matrix of stone materials, react with the substrates, and partially transform the minerals of the matrix (in general, Ca and Mg carbonatic minerals) in newly formed crystalline phases through a dissolution – recrystallization process.8–11 These new phases (having natural counterparts, such as hydroxyapatite and whewellite) nucleate on calcite grains creating a 3D crystalline network that re-binds detached stone grains, improving their mechanical properties. By nucleating within the stone materials, AmOx and DAP induce variations in the crystallochemical composition and in 3D microstructural features of the stone materials from the micro to the macro scale.6,12,13
AmOx and DAP treatments have peculiar hallmarks, which include the kinetics of the reaction, the formation of specific phase assemblages (oxalates and phosphates, respectively), and a diffusion/penetration depth within the treated materials.14–21 The different crystallization depths of reaction products formed by AmOX and DAP are the reason why the former is generally used for the protection of stone materials while the latter is employed mainly for consolidation.
Recent studies have investigated the crystal-chemistry and 2D distribution of newly formed calcium oxalates and calcium phosphates formed by AmOx and DAP with lab-scale instruments and large-scale sources13,15,17,20,22,23,48 and have shown that a lot of variables (e.g., the minerals, microstructure and grain size of the substrate; pH; free calcium ion availability, etc.) affect the reaction of inorganic treatments with stone materials.
3D tomographic investigations have been carried out as well on AmOx and DAP treatments, with a focus on the 3D microstructural variations of the porous network6,24 and on the volume distribution of generic “H-containing reaction products”.21,25,26
The crystallisation induced by inorganic re-treatments on stone material already treated with inorganic products can be completely different from the crystallisation path occurring on untreated substrates but a few similarities are seen with regard to: (i) the 3D distribution of calcium oxalates formed by AmOx in porous stone materials as well as their spatial arrangement on the surface of calcite grains of the lithotype; and (ii) the interaction of inorganic re-treatments with the crystalline phases formed by previous inorganic treatments.
As proof of the need for new insights into this topic, some pioneering studies have been carried out on double inorganic treatments.27,28 However, many of them have been performed on compact carbonatic stone materials and/or have been carried out using 1D or 2D analytical techniques, while a 3D spatially resolved analytical approach is also required to explore the volume and crystallochemical variations induced by sequential inorganic re-treatments on porous substrates. Moreover, many of these studies were not able to detect minor phases, whose identification and localisation are highly important as, in many cases, they are also metastable phases prone to phase transformations (dissolution, re-crystallisation) over time.
This lack of knowledge also faces an analytical challenge, namely the need to simultaneously and non-invasively couple the crystallochemical characterization of reaction phases with the 3D tomographic investigation of their distribution within selected regions of the treated lithotypes, allowing the correlation of the new crystalline phases to their specific positions inside the treated volume, which, at the moment, is one of the most demanding open issues in the field of inorganic re-treatments.
Synchrotron radiation (SR) X-ray diffraction computed tomography (XRDCT) is a recently developed analytical technique of high potential as it allows the phase composition and the spatial distribution of crystalline phases (and potentially amorphous ones) in heterogeneous and multi-phase samples to be studied simultaneously, with high spatial resolution, high crystallochemical selectivity and 3D outcomes.29–31 This makes of diffraction imaging a groundbreaking and powerful approach in those research fields where material characterisation combining space-resolved and non-destructive structural information of phases in complex mixtures simultaneously is required. This is the case for cultural heritage and conservation science, as XRDCT can perform non-destructive, and potentially non-invasive, structural analysis of fragments or minute objects from CH that cannot be ground or micro-sampled.32–34
Recently, the ability of XRDCT to simultaneously provide XRD and tomographic data was used in stone conservation to 3D map the crystallization of calcium phosphates formed by DAP treatments.33 However, to date, no XRDCT data are available on crystalline phases formed within porous stone substrates treated by AmOx as well as on the 3D and compositional interaction of AmOx and DAP inorganic treatments applied in sequence on the same stone substrate.
Starting from here, we carry out a step forward. This study demonstrates the feasibility and high potential of XRDCT to 3D identify, quantify and spatially localise at a micrometric voxel size resolution the stable and metastable crystalline phases formed in the complex mixture after the reaction of inorganic re-treatments with porous carbonatic stone materials. The study is carried out on the highly porous matrix of Noto limestone, a calcarenite widely used in the important Baroque built heritage located in UNESCO sites in south-eastern Sicily (Italy),35 treated by DAP, by AmOx, and by a sequential combination of these treatments (AmOx followed DAP, AmOx → DAP; DAP followed AmOx, DAP → AmOx). Qualitative analysis, quantitative analysis and image analysis of XRDCT datasets collected on the samples treated with a single inorganic treatment (DAP, AmOx) are used as the XRDCT reference benchmark to:
(i) investigate the crystallochemical composition and volume distribution of major, minor and trace phases formed by the inorganic re-treatments;
(ii) quantify via the structural refinement of XRD patterns with the Rietveld method the weight fraction of newly formed crystalline phases as a function of the specific depth of penetration;
(iii) explore possible phase transformations between single and sequential inorganic re-treatments, in order to understand whether and how the treatment sequence influences the composition, localization, abundance and penetration depth of stable and metastable newly formed phases; and
(iv) correlate the abundance/localisation of phases to the intrinsic microstructural heterogeneity of the stone material.
Experimental
Materials
Noto yellowish limestone is a calcarenite outcropping in the Iblean Plateau, south-eastern Sicily, and it has been extensively used as a construction and ornamental stone material for the elaborate UNESCO Baroque monuments of the Val di Noto.35–39 This biomicrite,40 primarily composed of calcite, along with subordinate clay minerals, quartz, and iron oxides, exhibits high porosity (25–37%) within the micro-pore range.6,39 Inorganic treatments were carried out on freshly quarried prismatic specimens (50 × 50 × 20 mm) of Noto Yellowish limestone with a poultice containing 0.76 M DAP aqueous solution (10% w/w; DAP CAS number 7783-28-0, assay ≥99.0%, reagent grade) or 0.35 M AmOx aqueous solution (5% w/w; AmOx CAS number 6009-70-7, assay ≥98.0%, reagent grade). The poultice (ratio ∼5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DAP solution:cellulose pulp; dry cellulose pulp MH 300 Phase, Italy; approximately 1–1.5 cm thick) was applied to the specimen surface, and a layer of Japanese paper was inserted between the paper poultice and the stone surface to prevent damage from the poultice sticking after drying. Throughout the treatment, the specimens were sealed in plastic film to avoid the drying of the solutions. After 2 hours, the plastic film was removed, and the specimens were left to dry at room temperature for 24 hours with the poultice intact. Subsequently, the poultice was removed, and all specimens underwent three rounds of rinsing by immersion in Milli-Q water before being dried again. For sequential inorganic re-treatments, the DAP and AmOx treated samples were re-treated with AmOx and DAP solutions with the same procedure as described above.
:1 DAP solution:cellulose pulp; dry cellulose pulp MH 300 Phase, Italy; approximately 1–1.5 cm thick) was applied to the specimen surface, and a layer of Japanese paper was inserted between the paper poultice and the stone surface to prevent damage from the poultice sticking after drying. Throughout the treatment, the specimens were sealed in plastic film to avoid the drying of the solutions. After 2 hours, the plastic film was removed, and the specimens were left to dry at room temperature for 24 hours with the poultice intact. Subsequently, the poultice was removed, and all specimens underwent three rounds of rinsing by immersion in Milli-Q water before being dried again. For sequential inorganic re-treatments, the DAP and AmOx treated samples were re-treated with AmOx and DAP solutions with the same procedure as described above.
Hereafter, the labelling of the samples/treatments will be “DAP” for the Noto limestone treated only with DAP solution; “AmOx” for specimens treated only with AmOx solution; “DAP → AmOx” in the case of the Noto limestone initially treated with DAP and then re-treated with AmOx; and “AmOx → DAP” in the case of the Noto limestone initially treated with AmOx and then re-treated with DAP.
Suitable fragments for XRDCT (cylinders with a diameter 1.5–1.0 mm) were sampled from the treated 50 × 50 × 20 mm specimens using a core driller, as shown in the scheme in Fig. S1 in the ESI.† The cylinders were drilled starting from the treated surface down to the inner portion of the Noto limestone (height ∼5–6 mm); the drilling regions were selected far from the external boundaries of the 50 × 50 × 20 mm specimens to avoid possible boundary effects. After drilling, the cylinders were washed twice by immersion in Milli-Q water to remove any powder formed by the drilling procedure and then they were left to dry at room temperature for 24 hours.
Methods
The XRDCT investigations were conducted at the ID15a beamline30 of the ESRF (European Synchrotron Radiation Facility; Grenoble, France) during the allocated beamtime for proposal number 80828. Cylindrical samples were affixed to a sample stage with capabilities for translational and rotational movements. The monochromatic beam's energy was adjusted to 42 keV using a double-bounce bent Laue monochromator and focused down to the micrometer scale (35 μm (horizontal) × 15 μm (vertical)) through a compound refractive lens transfocator. The photon flux during the experiment was 1013 ph s−1. The XRDCT scans along the y-axis were acquired through a 180-degree rotation (ω).41 The slice thickness matched the vertical beam size (15 μm). The spatial step size of the XRDCT measurements was equivalent to the voxel size (35 μm horizontal and 15 μm vertical). The angular step of the experiment, set at 0.0054 Å−1 (0.014° of 2Theta), was determined by the size of the detector pixel (172 μm), by the sample-detector distance, and by the oversampling of the data during integration and then rebinning to a constant step. Each XRDCT dataset required a total measurement time of ∼8 hours.A Pilatus 3 × 2 M CdTe area detector was utilized to capture the diffractograms in this study. The detector distance and other geometric parameters were calibrated based on diffractograms from a capillary sample of NIST SRM674b CeO2 using PyFAI. A locally modified version of PyFAI42,43 was employed to azimuthally integrate the projections, with outlier rejection to mitigate potential skewing effects caused by the presence of large crystals. Projections were organized into sinograms, which were then centred and corrected for background scattering. The reconstruction was done point by point in the diffraction patterns, via filtered back projection.44–46 Beamline scripts facilitated tomographic reconstructions of the integrated data. With this experimental setup, the field of view (FOV) measured and thus the size of the analysed volume was 1.8 × 1.8 × 1.5 mm, and the resulting reconstructed images (tomograms) had a final voxel size of 35 × 35 × 15 μm. Samples were positioned within the FOV to focus on either the treated surface or the inner regions.
The RGB reconstruction images of specific crystalline phases were extracted from the XRDCT tomographic datasets by plotting the integrated area of marker XRD peaks against all pixels of the array. These reconstruction images, illustrating the 3D spatial distribution of specific XRD peaks, are presented in a false-colour map ranging from red to green–blue hues, corresponding to regions with high or low integrated areas, respectively. Each slice's size, and thus each XRDCT reconstruction image, obtained with this setup, is 1800 × 1800 × 15 μm.
The XRDCT reconstruction images and patterns were obtained from the XRDCT tomographic datasets using the software/Python scripts available at the ID15a beamline. During this data processing, the scripts perform image interpolation to fill in and smooth the existing data in XRDCT maps. This digital process increases the number of pixels in the false-colour maps extracted as a TIFF file from the XRDCT datasets, enhancing the graphical output of the RGB maps. XRD patterns, extracted from individual voxels or whole slices of the 3D phase maps, underwent no smoothing or post-processing. The qualitative analysis of the XRD patterns was conducted with the X'Pert HighScore 2.2.0 software and database (PANalytical). Visualization and RGB splitting of the XRDCT reconstructed slices utilized the open-source Fiji software,47 while 3D image renderings were generated using the commercial software VGStudio 2.0 (Volume Graphics, Germany).
Results and discussion
Qualitative analysis of newly formed crystalline phases formed by inorganic re-treatments
Fig. 1 shows the reconstructed XRD patterns of a representative slice extracted using XRDCT datasets of the Noto limestone treated by DAP and by AmOx as well as by their sequential treatments. For clarity, the XRD pattern of the untreated Noto limestone is shown as well.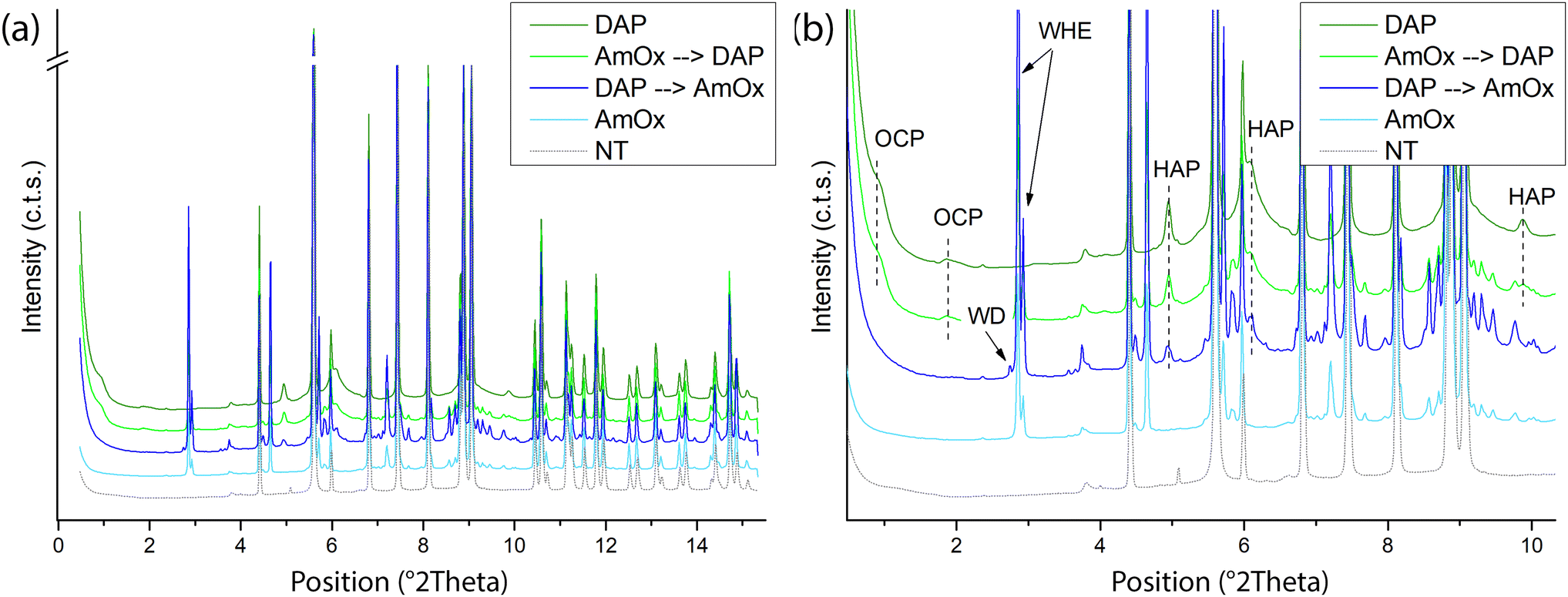 | ||
| Fig. 1 (a) XRD patterns extracted from a representative XRDCT slice of the untreated (NT) and of the Noto limestone treated with DAP, AmOx, DAP → AmOx and AmOx → DAP. (b) A zoom of the 2θ range between ∼1 and 10 Å−1 to highlight the marker peaks of OCP, HAP, WHE and WD. | ||
The qualitative phase analysis of XRD patterns shows that calcite (CaCO3) is the dominant crystalline phase in all the treated lithotypes (Fig. 1).
Hydroxyapatite (HAP, Ca5(PO4)3OH) and octacalcium phosphate (OCP, Ca8(HPO4)2(PO4)4·5H2O) are the crystalline phases newly formed by the DAP treatment while whewellite crystals (WH, CaC2O4·H2O) are formed after AmOx treatment. These findings are in line with the typical phase composition of the reaction products formed by DAP and AmOx treatments on stone substrates composed of micritic calcite.27,33,48,49
Novel findings concern the phase composition of reaction products formed when DAP and/or AmOx treatments are sequentially applied on the same stone substrate.
In the case of the Noto limestone initially treated with DAP and then treated with AmOx (DAP → AmOx), a mixture of hydroxyapatite, whewellite and weddellite (WD, CaC2O4·(2 + x)H2O) is identified within the stone matrix.
In contrast, whewellite, hydroxyapatite and octacalcium phosphate are formed when the AmOx treatment is followed by DAP application (AmOx → DAP).
During the reaction, the DAP and AmOx solutions form thermodynamically stable phases (HAP and WHE, respectively) and metastable phases (OCP and WD).48,50 The crystallisation of the latter (OCP and WD), which are formed during the second treatment, depends on particular reaction conditions (pH, calcium availability). OCP and WD are precursor phases and they tend to transform to HAP and WHE,27,50,51 respectively. This parent-to-product phase transformation may take different times depending on the surrounding environmental conditions14,48,52 and studies on their stability showed a relationship with humidity.48,53–55
In previous studies,14,48,52 OCP crystals formed by DAP treatments appeared quite stable even after years while WD crystals formed by AmOx treatments were detected in the initial steps of the AmOx reaction with calcite, and then they were completely transformed to WHE after a few seconds or tens of minutes during the reaction. Here, the novelty is that (i) the OCP crystallites are stable when only DAP treatments are carried out or when DAP treatments are applied to AmOx substrates (AmOx → DAP); (ii) WD crystals are stable for up to several months only in DAP → AmOxtreatments. This suggests that the coexistence with the crystalline phases formed by the first treatment may have created the conditions to make this phase more stable, slowing or hindering the complete WD-to-WHE phase transformation. This speculation is also in line with literature data showing that metastable WD crystals became more stable in historical calcium oxalate films in the presence of other hydrated crystalline phases, and in particular of gypsum and apatite.18,56
Peak profile analysis was carried out with the Rietveld method and it highlighted that both the newly formed calcium oxalate (CaOx) phases have well-defined and sharp XRD peaks (Fig. 1). This finding suggests that CaOx phases are quite crystalline, as also demonstrated by the structural refinement of the full pattern profile carried out with the Rietveld method (i.e., estimated crystal size and microstrain of WHE of ∼0.49 μm and ∼3000, respectively). In contrast, calcium phosphate (CaP) phases have broad and weak XRD peaks which could be attributed to a defective structure, the presence of ionic substitutions and/or the nucleation of sub-micrometric crystallites (i.e., the estimated crystal size and microstrain of HAP and OCP as obtained by the structural refinement are ∼0.01–∼0.05 μm and ∼3000–∼4000, respectively). The formation of poorly crystalline non-stoichiometric calcium phosphates is a recurring feature of DAP treatments and it is due to the variation of the pH and ionic strength (i.e. free Ca2+ availability, presence of CO32−) during the reaction.12,15,23,48,57 Interestingly, our XRDCT results show that the crystallinity of CaOx, and especially of CaP phases, does not change among single and sequential re-treatments, as well as by varying the sequence of the treatments.
2D/3D distribution of re-treatment newly formed products: spatial localisation
The XRDCT tomographic reconstruction images of Fig. 2, obtained by subtracting a local linear background and by plotting the integrated area of the Bragg marker peaks in each voxel of the same slice, show the spatial distribution of calcite, HAP, OCP, WHE and WD in four representative slices of the Noto limestone treated by DAP, by AmOx, by DAP → AmOx and by AmOx → DAP as well as the Miller index used to map that phase. The XRDCT phase maps display the semi quantitative distribution of specific crystalline phases in a false colour thermal map, spanning from blue (absence/low intensity of the crystalline phase) to red (high intensity of that phase).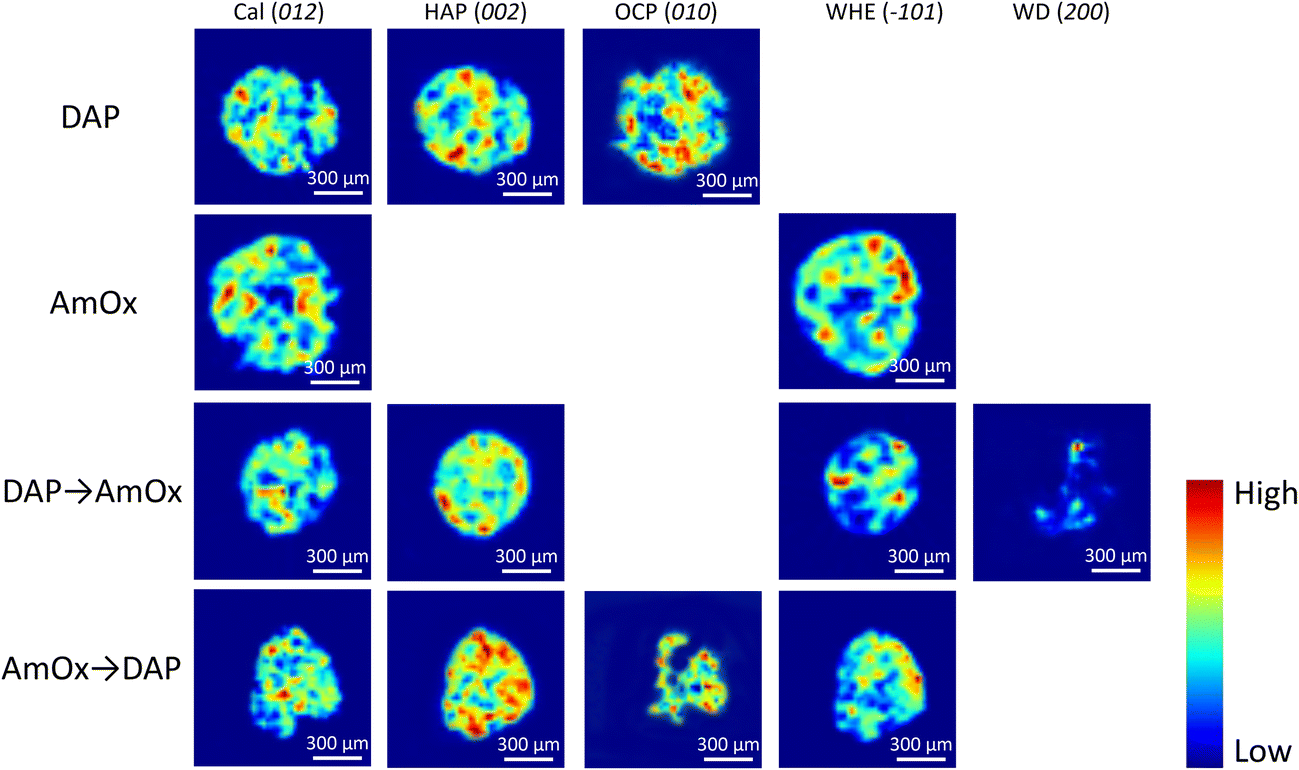 | ||
| Fig. 2 XRDCT reconstruction images showing the spatial distribution of calcite (Cal), hydroxyapatite (HAP), octacalcium phosphate (OCP), whewellite (WHE) and weddellite (WD) formed after the DAP, AmOx, DAP → AmOx and AmOx → DAP conservation treatments. The indexing of the selected Bragg peaks of each crystalline phase is shown in the round brackets. | ||
The XRD marker peaks used in this study to map the localisation of specific crystalline phases (integrated Q-range, interplanar distance and indexing of the selected Bragg peaks) are listed in Table 1.
| Crystalline phase | Acronym | Chemical formula | Integrated Q-range (Å−1) | Integrated 2θ range (°) | Interplanar distance (Å) | Indexing of selected Bragg peak |
|---|---|---|---|---|---|---|
| Calcite | Cal | CaCO3 | 1.61–1.66 | 4.34–4.47 | 3.85 | 012 |
| Hydroxyapatite | HAP | Ca5(PO4)3OH | 1.80–1.87 | 4.85–5.04 | 3.44 | 002 |
| Octacalcium phosphate | OCP | Ca8(HPO4)2(PO4)4·5H2O | 0.30–0.42 | 8.08–1.13 | 18.70 | 010 |
| Whewellite | WHE | CaC2O4·H2O | 1.04–1.07 | 2.80–2.88 | 5.93 | −101 |
| Weddellite | WD | CaC2O4·(2 + x)H2O | 1.00–1.04 | 2.69–2.80 | 6.18 | 200 |
RGB colour imaging of XRDCT thermal maps can be used to obtain further semi-quantitative and spatially resolved information on the volume distribution of specific crystalline phases. The procedure, described in ref. 33, implies the splitting of the XRDCT thermal image of a phase into its red, green, and blue component and generates three distinct maps illustrating the spatial arrangement of each channel. These three new false colour maps range from black (indicating absence or low intensity) to red (or green or blue, depending on the channel, signifying high intensity of that specific channel). The red channel highlights areas where the diffraction peak is most intense, providing a visual representation of the primary regions within the sample volume where the crystallization of that phase predominantly occurs. Conversely, the distribution map of the blue channel indicates the lack of that integrated peak, corresponding to voids within the sample and/or regions occupied by other crystalline phases. Lastly, the green channel's distribution map represents the volume where the integrated peak area is less intense but still observable. When exploring the effects of inorganic treatments on carbonate stone matrixes by XRDCT, the green channel proves to be the richest source of information for unravelling the binding network of new crystalline phases within such complex multiphase 3D systems, while the red channel emerges as the most interesting channel to explore the crystallisation sequence from a single “grain” of calcite. Therefore, in order to focus on the crystallisation sequence from calcite grains, in the following we exclusively used the red channel for all the crystalline phases and we applied the RGB correlation to emphasize their mutual spatial distribution. This leads to the RGB correlation of distribution maps of Fig. 3, illustrating the spatial distribution of the red channels for all phases under consideration. I.e., in Fig. 3a, the red channel of calcite is represented in blue, the red channel of HAP is represented in red and the red channel of OCP is represented in green. Their RGB correlation shows that, in the case of DAP treatment, HAP and OCP crystals nucleate from calcite grains of the substrate and they bind together the grains of calcite forming the consolidating shell (pink, yellow and light blue regions in Fig. 3a). At the same time, there are more regions of overlap between Cal and HAP (in pink in Fig. 3a), than between Cal and OCP (in light blue in Fig. 3a), showing that mainly HAP nucleates on the surface of calcite grains. The predominant presence of overlapping zones between Cal and HAP compared to Cal and OCP can be schematized in a phase stratigraphy as shown in the scheme (not to scale) of Fig. 4a.
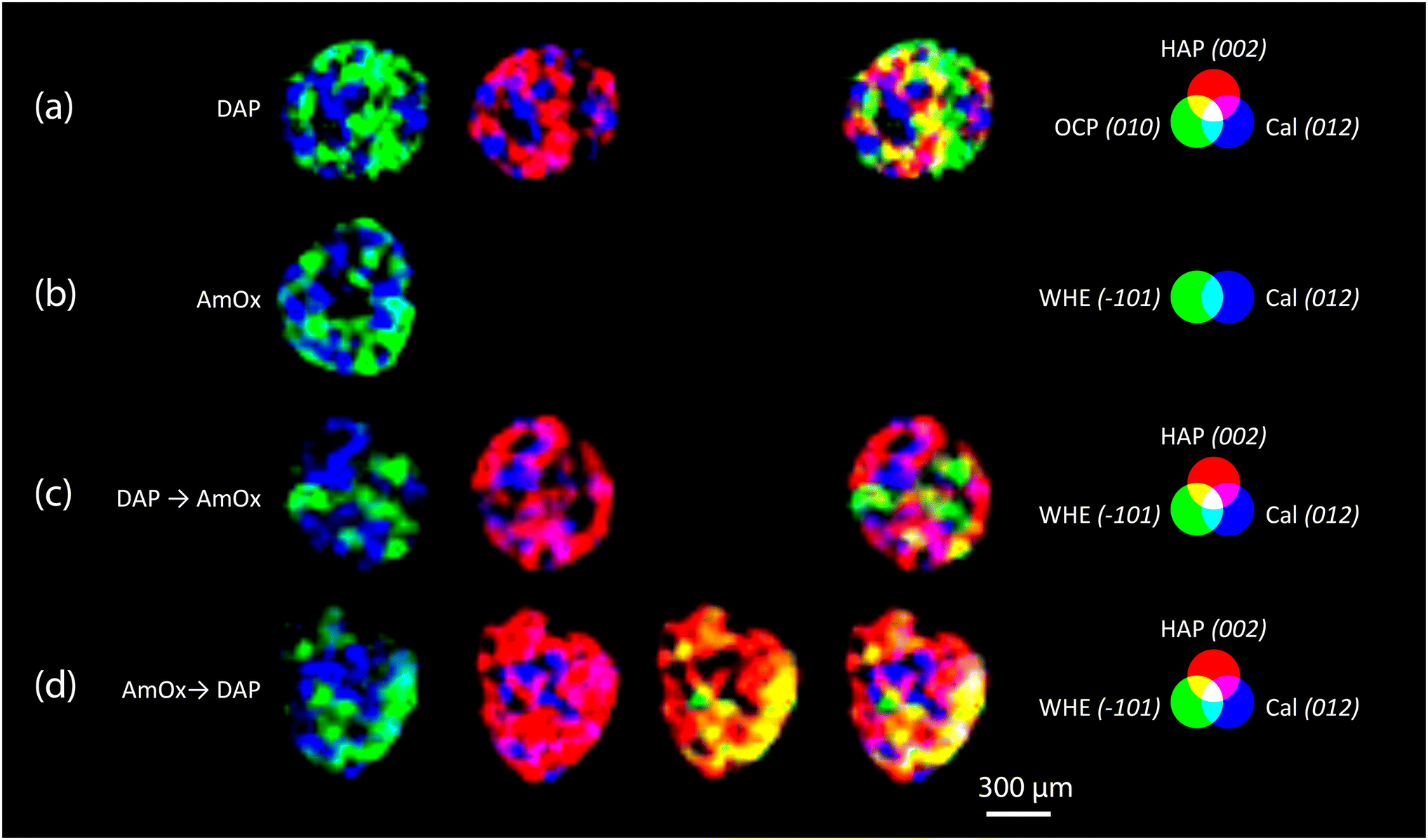 | ||
| Fig. 3 XRDCT image reconstruction and RGB correlation (red channels,33) of the Noto limestone treated with DAP (a), AmOx (b), DAP → AmOx (c) and AmOx → DAP (d). For each typology of treatment, the circles of the additive synthesis show the false colour attributed to the red channel of specific crystalline phases (HAP, OCP, WHE and Cal). The indexing of the selected Bragg peaks are reported in round brackets after the acronym of the crystalline phases. Red, blue and green primary colours indicate the regions where the phase is alone. The light blue, pink, yellow and white regions indicate the spatial overlapping of two or three phases characterised by the primary colours. | ||
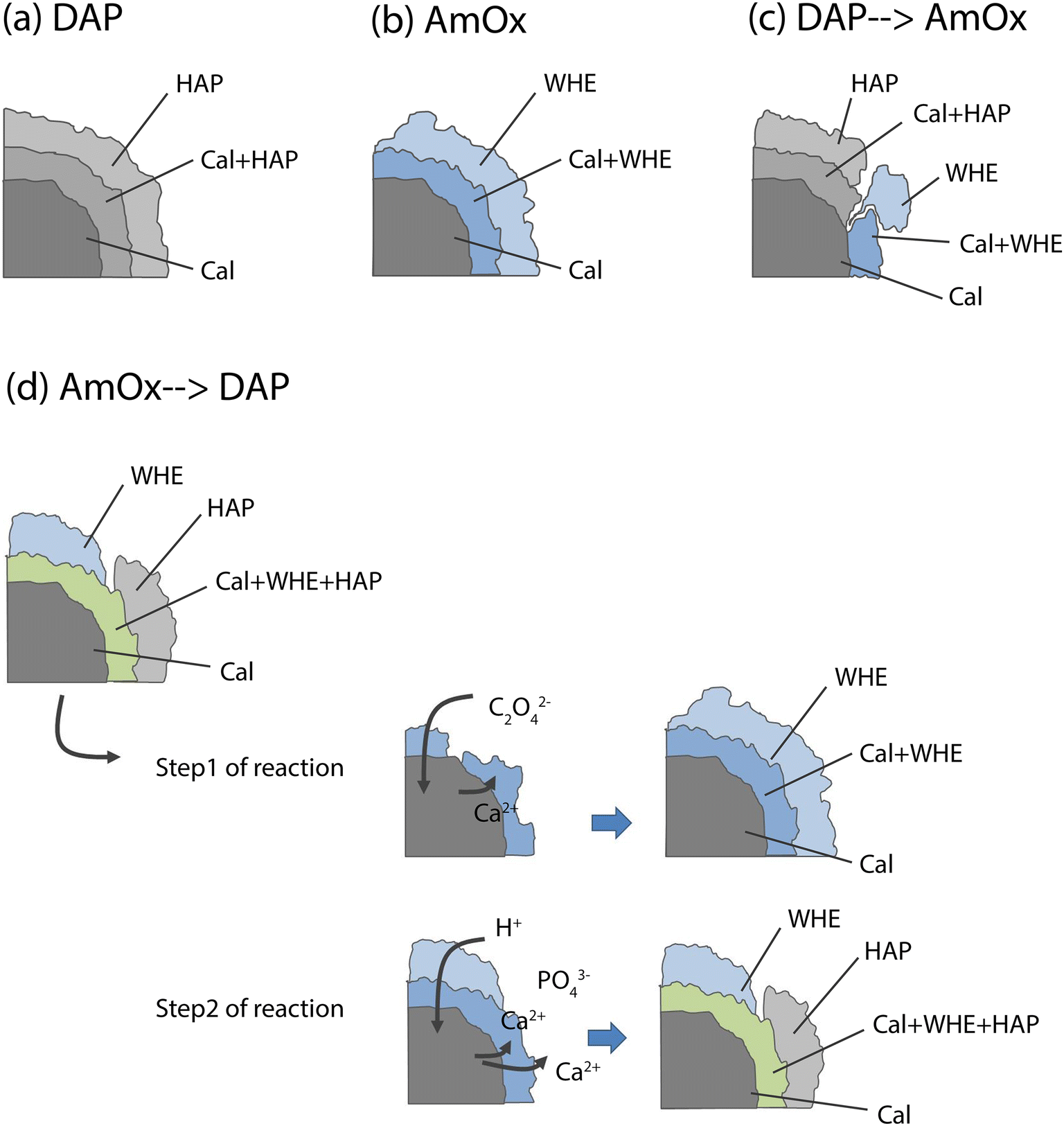 | ||
| Fig. 4 Scheme (not to scale) of the newly formed HAP and WHE phases crystallised after the DAP (a), AmOx (b), DAP → AmOx (c) and AmOx → DAP (d) treatments showing the phase sequence nucleated from calcite (Cal) grains. For clarity (i) the spatial localisation OCP and WD (which are minor phases that nucleate only close to the treated surface and their crystallisation is affected by the sequence of the re-treatments) are not reported in this scheme; (ii) in this scheme, only calcite is highlighted as a source of Ca2+ ions. | ||
In the AmOx treatment (Fig. 3b), the RGB correlation of XRDCT reconstruction images of Cal and WHE shows the growth of a highly interconnected crystal network formed by WHE. For the first time, we can assert the 3D consolidating action of AmOx treatments on calcite grains of porous Noto limestone. The phase sequence observed by XRDCT on calcite grains treated by AmOx is schematised in Fig. 4b.
As for the Noto limestone treated by DAP → AmOx (Fig. 3c), a shell of HAP is formed on the surface of calcite grains and reconnects them similarly to what is observed in the case of DAP treatments (cfr with Fig. 3a). Notwithstanding the abundant crystallisation of HAP, the stone substrate remains reactive toward the subsequent AmOx re-treatment and WHE crystals are formed as well. Interestingly, WHE crystals appear to grow mainly on HAP crystals, rather than Cal ones, as demonstrated by the yellow regions in Fig. 3c (which are RGB plots of the overlap of volume regions where WHE and HAP coexist) and by the low amount of light blue regions (coexistence of WHE and Cal). However, it is worth noting that a minor part of WHE crystals is observed also directly on Cal crystals (Fig. 3c). This complex crystalline sequence gives rise to a sort of phase stratigraphy composed of Cal–HAP–WHE, as shown in the scheme of Fig. 4c.
The volume distribution of the phases in the AmOx → DAP treatment is even more interesting (Fig. 3d). Indeed, we can clearly see the crystallisation of WHE crystals, as well as the coexistence of WHE and Cal on the external portion of calcite grains (Fig. 3d) as shown in step 1 of the reaction in Fig. 4d. At the same time, the RGB correlation imaging also shows that HAP, formed by the subsequent DAP treatment, is abundantly overlapped with calcite grains. This means that the WHE + Cal system remains highly permeable, and reactive, toward the subsequent DAP re-treatment (step 2 of the reaction), which is able to pass through the WHE shell, and reach the calcite grains to form calcium phosphates. These CaPs (HAP + OCP) crystallise on the surface of calcite grains, most likely growing between WHE crystals. Such crystallisation gives rise to a different phase sequence as shown in the scheme given in Fig. 4d.
Having stated the above, the phase sequences of Fig. 4 can be considered a sort of reference stratigraphy of calcium oxalate and phosphate phases crystallised on the Noto limestone grains as a function of the treatment and re-treatment sequence. For these reasons, the spatial localisations of OCP and WD are not reported in the scheme of Fig. 4 as they are minor and metastable phases, nucleate only close to the treated surface and are affected by the sequence of the treatments.
3D distribution of re-treatment newly formed products: depth of penetration and phase quantification
The newly formed crystalline phases are more abundant close to the treated surface of the specimens and undergo a decrease of their relative abundance toward the bulk of the samples. This trend is confirmed by the volume renderings of Fig. 5 as well as by the sequential refinement with the Rietveld method of all the extracted XRD patterns.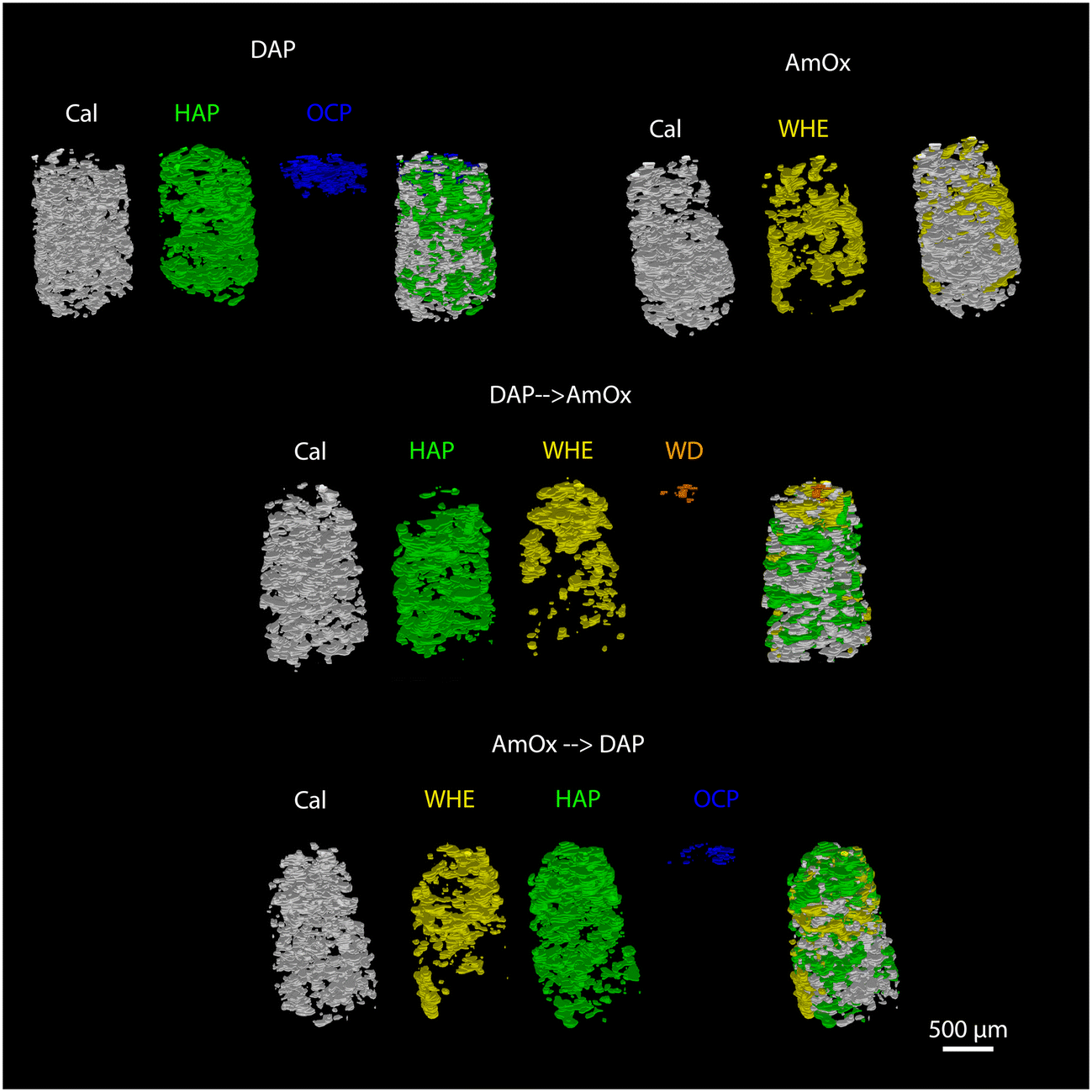 | ||
| Fig. 5 Volume renderings of the Noto limestone treated with DAP, AmOx, DAP → AmOx and AmOx → DAP showing the 3D spatial distribution of calcite (Cal; in grey), hydroxyapatite (HAP; in green), octacalcium phosphate (OCP; in blue), whewellite (WHE; in yellow), and weddellite (WD; in orange). The spatial distributions of specific phases are superimposed to show their relative spatial localization. | ||
This trend is in common to all the newly formed phases, while differences among the different treatments can be appreciated in terms of the localization, penetration depth and quantity of reaction products.
As for the depth of penetration, HAP and WHE are present in the entire volume investigated by XRDCT which, in our measurements, corresponded to a ∼1.5 mm depth from the treated surface. A depth of penetration in the range of ∼1.5 mm from the treated surface is particularly remarkable in the case of AmOx treatments, as the reaction products formed by this treatment are generally confined to the most superficial hundreds of micrometers from the treated surface of the porous matrixes (i.e. plasters and frescoes).13,16 Here, such a significant penetration depth can be directly ascribed to the highly interconnected porous structure of the Noto limestone, which promotes the diffusion and crystallization of new phases down to the inner portions of the stone materials and notwithstanding the high reactivity of this reagent. Secondly, it is remarkable that the crystallisation of WHE occurs so deep even after the application of the DAP treatment.
OCP and WD are localized only close to the most superficial regions of the treated specimens. More precisely, OCP is detected down to ∼250–300 μm while WD can be detected down to ∼180 μm (volume renderings of Fig. 5 and quantitative analysis of Fig. 6,7). The depth of penetration of OCP is more or less constant in DAP and AmOx → DAP treatments. The same cannot be stated for WD, as this phase is detected only in the case of DAP → AmOx treatments.
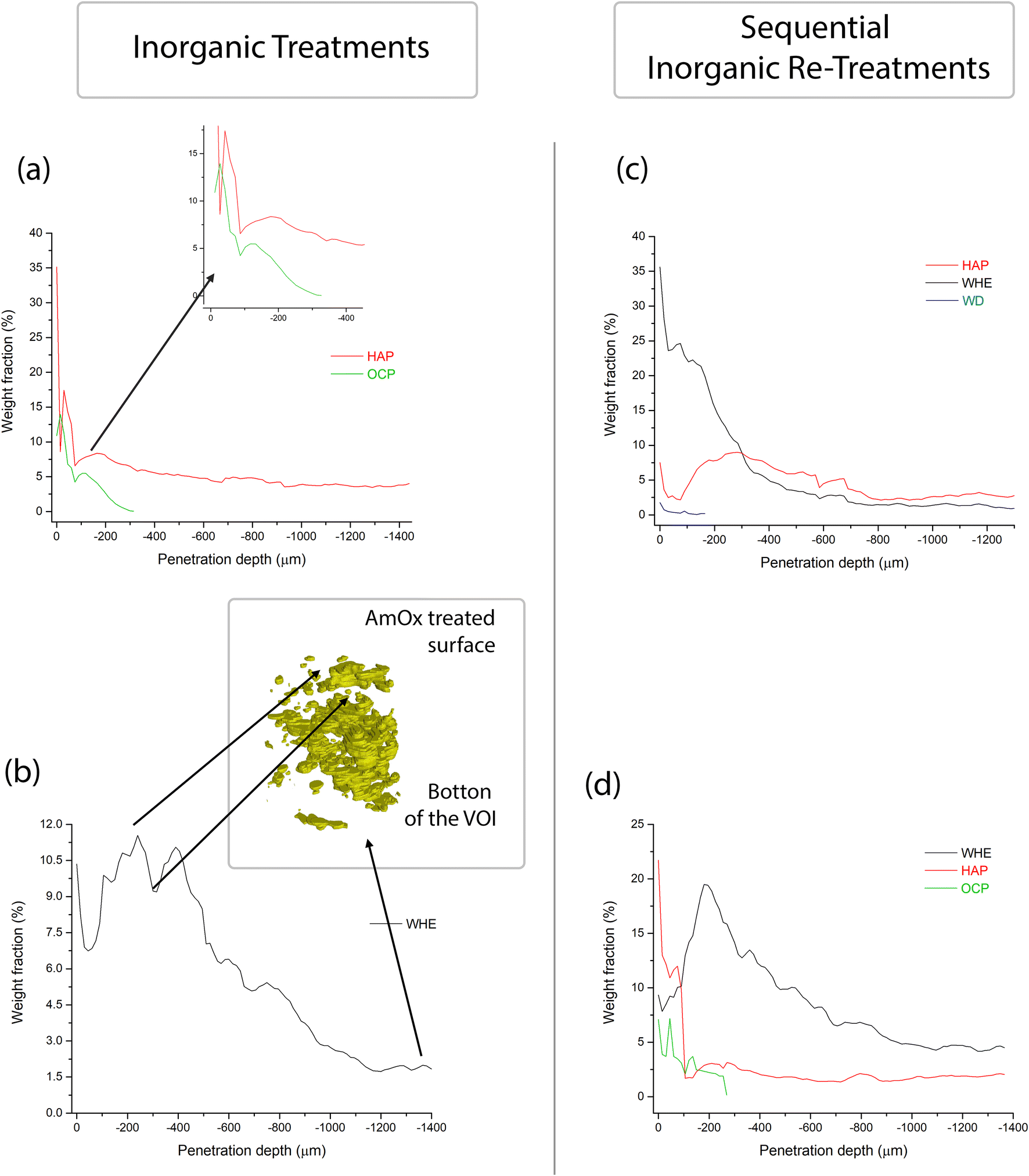 | ||
| Fig. 6 Weight fractions calculated with the full profile fit with the Rietveld method as a function of the penetration depth (0 μm corresponds to the treated surface) of hydroxyapatite (HAP), octacalcium phosphate (OCP), whewellite (WHE) and weddellite (WD) crystallised within the Noto limestone treated with DAP (a), AmOx (b), DAP → AmOx (c) and AmOx → DAP (d). The volume rendering in (b) shows the 3D distribution of WHE in the Noto limestone treated with AmOx and points out the correlation between the weight fraction of WHE at a certain depth from the treated surface in relation to its spatial distribution and volume hindrance. | ||
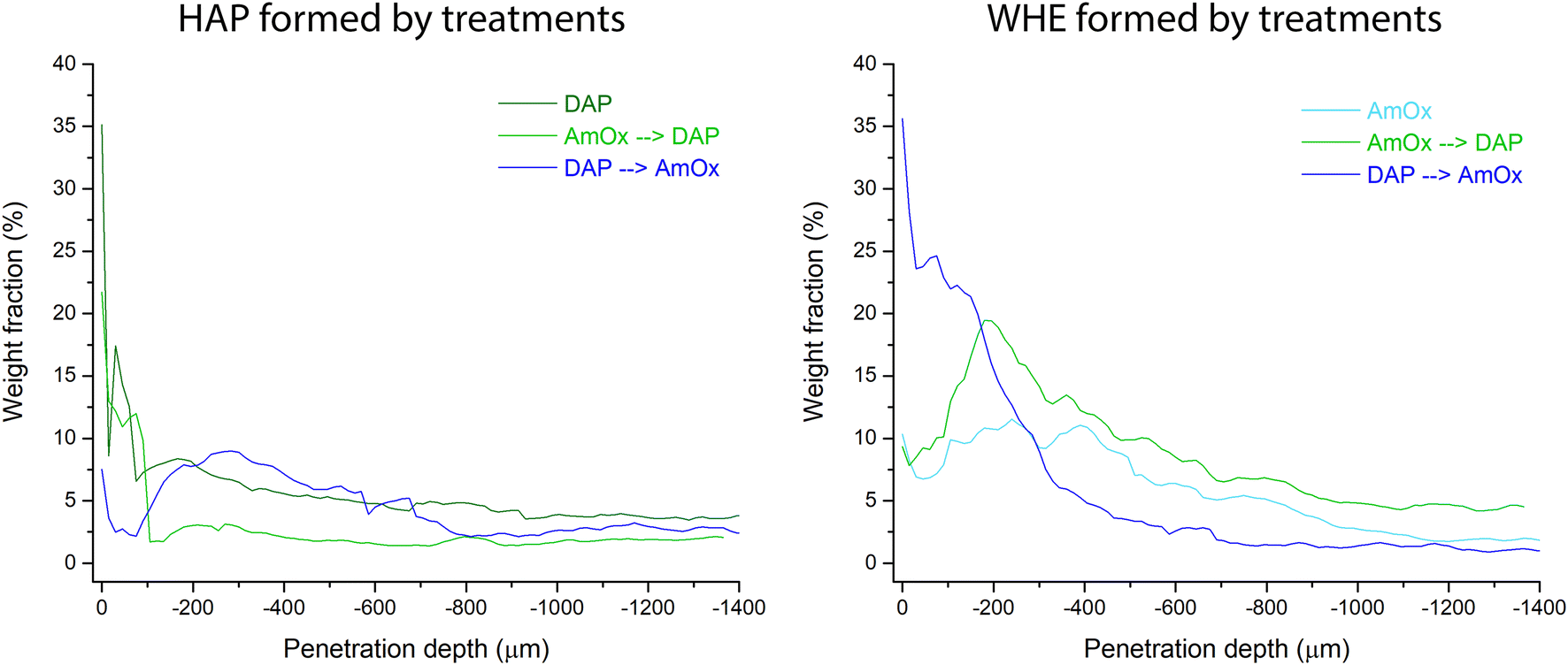 | ||
| Fig. 7 Comparison of the weight fractions calculated with the full profile fit with the Rietveld method of hydroxyapatite (HAP) and whewellite (WHE) formed by DAP, AmOx, DAP → AmOx and AmOx → DAP treatments. | ||
The full profile fit of the XRD patterns carried out with the Rietveld refinement allowed quantification of the weight fraction of the newly formed phases as a function of the penetration depth from the treated surface (the quantitative data and the refined XRD patterns are shown in Fig. S2–S5 and in Tables ST1–ST4 in the ESI†). The quantitative analysis using the full profile fit was conducted on the reconstructed diffractogram of each entire slice, and this process was sequentially performed for every slice within the investigated volume. The trend of the weight fraction of each newly formed crystalline phase vs. the penetration depth is shown in Fig. 6.
The weight fraction of HAP shows a bimodal trend with two maxima, one localized very close to the surface treated by DAP (0–100 μm from the treated surface) and the other at a penetration depth of between 100 and 400 μm. This occurs either when the lithotype is treated only with DAP or in the case of combined treatments (DAP → AmOX; AmOx → DAP). The main difference between the three treatment modalities lies in the amount of HAP formed. More specifically, in the case of Noto limestone treated only with DAP, HAP is 8–18% of the total weight fraction in the first 200 μm from the treated surface. The weight fraction of HAP decreases from 8% to about 4% at a penetration depth from 200 to 1000 μm. This penetration depth corresponds to a plateau and the weight fraction of HAP remains stable at ∼4% down to the bottom of the investigated VOI.
As for DAP → AmOx treatments, the weight fraction of HAP has a first maximum of 8–9% close to the surface. The weight fraction has a second maximum at 8–9% at about 300 μm from the treated surface and then it is followed by a decrease until a plateau of 2.5–3 wt% is achieved at below ∼800 μm from the treated surface. It is worth highlighting that the weight fraction of HAP in the most superficial 200 μm shows some fluctuations. The different values (i.e. 8–18% in DAP vs. 8–9% in DAP → AmOx) can be ascribed to the heterogeneity of the stone materials while the bimodal trend and the decrease of the weight fraction with depth are key features of the crystallisation promoted by the DAP solution.
In AmOx → DAP treatments, the trend of the amount of HAP is quite similar to that of previous treatments but the weight fraction is significantly lower (bimodal trend, 2–13% in the first peak in the first ∼50 μm, ∼3% in the second peak at −200 μm and then a progressive decrease until the plateau of 1.5–2% from −400 μm to the bottom of the specimen). This is most likely due to the fact that when DAP is applied after AmOx, the substrate is already partially passivated and it is less reactive, therefore a lower amount of HAP is formed with respect to DAP treatments carried out on untreated stone substrates.
The weight fraction of OCP has a very similar trend to that of HAP treatment. It measures between 14% and 4% close to surface, then it decreases dramatically from 4% to 0% between 200 and 300 μm from the treated surface. This trend is observed both in the case of individual DAP treatments and in situations involving AmOx → DAP re-treatment.
WHE formed by AmOx shows some fluctuations as well. In the case of AmOx treatments, the weight fraction of WHE is 10% close to the surface. It decreases to ∼7% at 60 μm depth, increases again to 10–11% at 250–400 μm depth (with a new minimum of 9% at ∼320 μm) and then progressively diminishes until it is 2% at the bottom of the VOI.
The trend of the weight fraction of WHE in the AmOx → DAP treatment is comparable with that of the AmOx treatment, with an initial weight fraction value of 9.5%, a decrease to 7.5% at a depth of 30 μm, an increase up to 20% at a depth of ∼200 μm, and a progressive decrease all through the VOI until 4% at the bottom.
In the DAP → AmOx sample, the weight fraction of WHE shows very high values (35.5%) close to the treated surface. It decreases progressively with depth and reaches a plateau of 1.0–1.5% below a depth of 700 μm. The trend of this treatment sequence seems to be in contrast with that of the previous ones. However, it should be considered that the very high amount of WHE in the most external region is coupled with the quite low amount of HAP formed in the same region. On the other hand, where the crystallization of HAP in the DAP → AmOx sample has values found also in the single DAP treatment (i.e. from 300 μm of depth, weight fraction 8–9%; then a decrease to a plateau of HAP at 2.5–3%) the crystallization of WHE records the lowest values of the three treatments, showing that the surface of pore walls of the Noto limestone is quite passivated by the HAP crystallization. A comparison of the amounts of WHE and HAP formed from the different treatments and treatment sequences is shown in Fig. 7. The trends and wt% amounts of HAP formed by DAP and by AmOx → DAP are quite similar (higher abundance of the phase close to the treated surface; progressive wt% decrease with the penetration depth). The same trend can be found for WHE crystals formed by AmOX and by AmOx → DAP.
Spot fluctuations of the weight fraction of a particular crystalline phase for the phase trend are also correlated with the particular microstructure of the Noto limestone in the corresponding volume. For example, the abrupt drop in WHE wt% in Fig. 6 can be directly correlated with a volume region where WHE is less present (Fig. 5 and 6), which in turn is due to it being a more compact and less reactive region of the Noto limestone.
DAP → AmOx treatment shows a contrasting trend. Here, HAP is mainly localised at the depth from ∼200 to 700 μm while abundant crystallisation of WHE occurs from the surface to a depth of ∼400 μm.
Overall discussion
The different crystallisation trend detected in the DAP → AmOx treatment with respect to the AmOx → DAP is due to the different kinetics of the AmOx and DAP inorganic products. Indeed, DAP slowly dissolves Ca2+ ions from the surface and subsurface calcite grains. These Ca2+ ions are mainly spread within the bulk of the stone by capillarity with the DAP solution, where they form CaP phases; crystallisation of CaP phases (especially metastable) also occurs close to the surface as observed for only DAP treatments. This slow process takes place several times during the treatment, which promotes the diffusion of the CaP phases and their high penetration depth. At the same time, this process of repeated dissolution of calcite crystals close to the surface leaves them with a high specific surface area and a very rough surface. This is the reason why, when the subsequent AmOx treatment is applied, the oxalate ions form a highly reactive substrate with a very high specific surface area, which promotes a quick crystallisation of CaOx phases mainly close to the treated surface.So far, the high quality of XRDCT datasets allows demonstration that the application of a second inorganic treatment partially changes the composition of the crystalline phases promoted by the first treatment. It transforms the metastable phases formed by the first treatment into more stable ones but does not change or improve the crystallinity of poorly ordered phases. In the light of the crystalline phases detected in this study, the following considerations can be stated.
The presence of reaction products formed by the first treatment influences the composition of the crystalline phases formed during the second one. Most likely, the second treatment acts on a substrate which is partially “passivated” because the surface of calcite crystals is covered by the consolidating shell of the first consolidating treatment. By being surrounded by the shell, these calcite grains are less reachable from the DAP or AmOx solution of the second treatment, and the metastable phases formed by the first treatment act as a further Ca2+ source to form new phases during the second inorganic treatment. At the same time, the treated stone substrates remain reactive toward the second treatment as (i) calcite grains which experienced the first inorganic treatment present a high specific surface area; (ii) notwithstanding the presence of a calcium phosphate or oxalate shell on pore walls of the stone substrate, the newly formed shell does not hinder the ion exchange with inorganic treatments; and (iii) the metastable phases, formed by the first single treatment and mainly localised close to the surface, act as further Ca2+ source for the crystallisation of new phases during the subsequent inorganic re-treatment. This condition stands for both the treatment sequences (DAP → AmOx, AmOx → DAP). This is a key achievement as it broadens our knowledge of the reaction of inorganic treatments and integrates previous literature data on the reactivity of already-treated stone materials, especially in the case of AmOx → DAP treatments where the main part of the Ca2+ ions of calcite grains was found to be not reactive anymore to further reaction with DAP.27 These findings support the possibility of re-applying inorganic treatments on decayed stone substrates which had been treated in the past, and it points out that the application of such treatments now will not hamper the possibility of re-treating the same stone substrates in the future.
Moreover, our XRDCT image reconstructions exhibit the volume localisation of the newly formed phases (both CaOx and CaP) that nucleate from calcite grains of the substrate and show that there are regions where calcite and new phases coexist. By doing this, the new phases give rise to a 3D network of crystals (a crystalline shell) able to bind together grains of calcite and, at the same time, preserve the voids within the stone material. Additionally, our findings clearly reveal the consolidating action of calcium oxalates formed by the AmOx treatments (both single and sequential).
The collected data and the quantitative analysis show how the sequence of the treatments strongly influences the amount of newly formed crystalline phases as a function of the specific depth of penetration. In particular, they show that the first 200–300 μm of depth from the treated surface is a complex region where the crystallization and amount of phases show fluctuations due to complex reaction processes. Both the treatments and their couplings tend to reach a plateau of the weight fraction of the newly formed phases. This plateau is reached at a depth between 600 and 800 μm. In the case of double treatments, the weight fraction of reaction products formed with the second treatment is lower (∼half) than that of the phases formed on untreated substrates. It is worth pointing out that these values are representative of the particular system investigated in this study. These results arise from selected factors/reaction conditions (lithotype, molarity, treatment duration). The same parameters (weight fraction at a particular penetration depth, crystallochemical composition, crystallinity, domain size, etc.) are expected to vary by varying some of these factors (i.e. different stone substrate). Therefore, the main outcome of this study is not, or not only, an investigation of the particular features (and fluctuations) of a selected treatment (or treatment sequence) on a particular stone system but also pointing out the high potential of XRDCT to extrapolate specific trends which, irrespective of all these variables, can be considered peculiar to the treatments in their application to different stone materials.
Conclusions
This paper shows the application and high analytical potential of SR XRDCT to non-destructively investigate the effects induced by AmOx and DAP inorganic mineral re-treatments of sedimentary stones of cultural heritage previously treated with inorganic-mineral products. XRDCT measurements allowed the performance of phase characterization and 3D localization of calcium phosphates and calcium oxalates having very similar crystal structures and chemical composition.By carrying out qualitative/quantitative analysis of the XRD patterns extracted from the XRDCT dataset and image analysis, volume renderings and RGB correlation of 3D datasets, it was possible to quantify and localise the newly formed phases as a function of the penetration depth from the treated surface. This approach allowed us to explore the crystallinity, phase stability, space orientation and volume distribution of trace phases in a complex mixture.
As a major outcome of the study, it was possible to (i) assess trends (composition of phases, weight fraction, spatial localisation) peculiar to each treatment/re-treatment sequence and (ii) explore the reaction steps of sequential re-treatments, and hence to (iii) demonstrate the possibility of re-treating with inorganic treatments stone materials already treated with inorganic treatments.
Looking at the broader picture, the strength of this study lies in its ability to generate innovative analytical perspectives. By showcasing the potential of XRDCT to investigate the effects induced by conservation products in CH stone materials, our study (i) provides new analytical tools to support the choices of conservation methods for decayed treated artefacts of CH, (ii) unveils new analytical possibilities for XRDCT in conservation science, including investigations into the nature and volume distribution of CH materials, along with exploring manufacturing techniques, decay processes and conservation procedures within complex multi-phase systems, and (iii) opens up new horizons in analytical chemistry and material characterisation, including the possibility of performing qualitative and quantitative phase characterisation, non-destructive and non-invasive analysis, and in situ and ex situ studies of heterogeneous polycrystalline systems.
Author contributions
Conceptualization, E.P. and Ch.C. (Chiara Colombo); methodology, E.P. and N.M.; investigation, E.P., Cl.C. (Claudia Conti), N.M., and G.B.M.V.; formal analysis and data curation, E.P.; writing – original draft, E.P.; supervision, E.P., M.R., and N.M.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the European Synchrotron Radiation Facility for the allocation of beamtime and use of synchrotron radiation facilities at beamline ID15a (proposal number 80828). We also acknowledge Dr Stefano Gandelli for contributing to data processing during his MSc research thesis, Dr Marco Cantaluppi for contributing to GSAS data processing, Dr Valentina Barberini for supplying the core driller and Dr Roberta Possenti for her support in the image graphic design.References
-
E. Doehne and C. A. Price, Stone Conservation. An overview of current research, the Getty Conservation Institute, Los Angeles, 2nd edn, 2010 Search PubMed
.
-
Stone in Architecture, ed. S. Siegesmund and R. Snethlage, Springer, Berlin, Heidelberg, 4th edn, 2011 Search PubMed
-
A. Henry, Stone Conservation: Principles and Practice, Routledge, 2006 Search PubMed
- S. Bugani, M. Camaiti, L. Morselli, E. Van de Casteele and K. Janssens, Anal. Bioanal. Chem., 2008, 391, 1343–1350 CrossRef CAS PubMed
- V. Cnudde, A. Cwirzen, B. Masschaele and P. J. S. Jacobs, Eng. Geol., 2009, 103, 76–83 CrossRef
- E. Possenti, C. Colombo, C. Conti, N. Marinoni, M. Merlini, R. Negrotti, M. Realini and G. D. Gatta, Mater. Charact., 2019, 154, 315–324 CrossRef CAS
- M. Bertasa, E. Possenti, A. Botteon, C. Conti, A. Sansonetti, R. Fontana, J. Striova and D. Sali, Analyst, 2017, 142, 4801–4811 RSC
- M. Matteini, S. Rescic, F. Fratini and G. Botticelli, Int. J. Archit. Herit. Conserv. Anal. Restor., 2011, 5, 717–736 CrossRef
- E. Hansen, E. Doehne, J. Fidler, J. Larson, B. Martin, M. Matteini, C. Rodriguez-Navarro, E. S. Pardo, C. Price, A. de Tagle, J. M. Teutonico and N. Weiss, Stud. Conserv., 2003, 48, 13–25 CrossRef
- M. Matteini, Conserv. Sci. Cult. Herit., 2008, 8, 13–27 Search PubMed
- E. Sassoni, S. Naidu and G. W. Scherer, J. Cult. Herit., 2011, 12, 346–355 CrossRef
- E. Possenti, C. Colombo, C. Conti, L. Gigli, M. Merlini, J. R. Plaisier, M. Realini, D. Sali and G. D. Gatta, Constr. Build. Mater., 2019, 195, 557–563 CrossRef CAS
- M. Cotte, V. Gonzalez, F. Vanmeert, L. Monico, C. Dejoie, M. Burghammer, L. Huder, W. de Nolf, S. Fisher, I. Fazlic, C. Chauffeton, G. Wallez, N. Jiménez, F. Albert-Tortosa, N. Salvadó, E. Possenti, C. Colombo, M. Ghirardello, D. Comelli, E. Avranovich Clerici, R. Vivani, A. Romani, C. Costantino, K. Janssens, Y. Taniguchi, J. McCarthy, H. Reichert and J. Susini, Molecules, 2022, 27, 1997 CrossRef CAS PubMed
- C. Conti, M. Casati, C. Colombo, M. Realini, L. Brambilla and G. Zerbi, Spectrochim. Acta, Part A, 2014, 128, 413–419 CrossRef CAS PubMed
- E. Possenti, C. Colombo, M. Realini, C. L. Song and S. G. Kazarian, Anal. Chem., 2021, 93, 14635–14642 CrossRef CAS PubMed
- C. Conti, C. Colombo, D. Dellasega, M. Matteini, M. Realini and G. Zerbi, J. Cult. Herit., 2011, 12, 372–379 CrossRef
- N. Calore, A. Botteon, C. Colombo, A. Comunian, E. Possenti, M. Realini, D. Sali and C. Conti, Vib. Spectrosc., 2018, 98, 105–110 CrossRef CAS
- C. Conti, I. Aliatis, C. Colombo, M. Greco, E. Possenti, M. Realini, C. Castiglioni and G. Zerbi, J. Raman Spectrosc., 2012, 43, 1604–1611 CrossRef CAS
- G. Graziani, E. Sassoni, G. W. Scherer and E. Franzoni, Constr. Build. Mater., 2017, 148, 571–578 CrossRef CAS
- E. Possenti, C. Conti, G. D. Gatta, M. Merlini, M. Realini and C. Colombo, Anal. Methods, 2020, 12, 1587–1594 RSC
- G. Graziani, C. Colombo, C. Conti, E. Possenti, E. Perelli Cippo, M. Realini and E. Sassoni, Constr. Build. Mater., 2018, 187, 238–247 CrossRef CAS
- C. Conti, L. Cutard, A. Botteon, L. Brambilla, N. Marinoni, M. Realini, M. Catrambone, E. Possenti and C. Colombo, Crystals, 2023, 13, 1212 CrossRef CAS
- E. Possenti, C. Colombo, C. Conti, L. Gigli, M. Merlini, J. R. Plaisier, M. Realini and G. D. Gatta, Appl. Phys. A, 2018, 124, 383 CrossRef
- G. Massinelli, E. Possenti, C. Colombo, G. D. Gatta, M. Realini and N. Marinoni, Constr. Build. Mater., 2023, 397, 132348 CrossRef CAS
- M. Realini, C. Colombo, C. Conti, F. Grazzi, E. Perelli Cippo and J. Hovind, Anal. Bioanal. Chem., 2017, 409, 6133–6139 CrossRef CAS PubMed
- C. Conti, C. Colombo, G. Festa, J. Hovind, E. P. Cippo, E. Possenti and M. Realini, J. Cult. Herit., 2016, 19, 463–466 CrossRef
- I. Osticioli, G. Botticelli, P. Matteini, S. Siano, R. Pini and M. Matteini, J. Raman Spectrosc., 2017, 48, 966–971 CrossRef CAS
- M. Matteini, F. Fratini, S. Rescic, M. Baldan, L. Campana and O. A. Cuzman, Int. J. Conserv. Sci., 2020, 11, 405–424 CAS
- M. Voltolini, M. C. Dalconi, G. Artioli, M. Parisatto, L. Valentini, V. Russo, A. Bonnin and R. Tucoulou, J. Appl. Crystallogr., 2013, 46, 142–152 CrossRef CAS
- G. B. M. Vaughan, R. Baker, R. Barret, J. Bonnefoy, T. Buslaps, S. Checchia, D. Duran, F. Fihman, P. Got, J. Kieffer, S. A. J. Kimber, K. Martel, C. Morawe, D. Mottin, E. Papillon, S. Petitdemange, A. Vamvakeros, J. P. Vieux and M. Di Michiel, J. Synchrotron Radiat., 2020, 27, 515–528 CrossRef CAS PubMed
- G. Artioli, T. Cerulli, G. Cruciani, M. C. Dalconi, G. Ferrari, M. Parisatto, A. Rack and R. Tucoulou, Anal. Bioanal. Chem., 2010, 397, 2131–2136 CrossRef CAS PubMed
- F. Vanmeert, G. Van der Snickt and K. Janssens, Angew. Chem., Int. Ed., 2015, 54, 3607–3610 CrossRef CAS PubMed
- E. Possenti, C. Conti, G. D. Gatta, N. Marinoni, M. Merlini, M. Realini, G. B. M. Vaughan and C. Colombo, iScience, 2022, 25, 105112 CrossRef CAS PubMed
- P. Bordet, F. Kergourlay, A. Pinto, N. Blanc and P. Martinetto, J. Anal. At. Spectrom., 2021, 36, 1724–1734 RSC
- L. Anania, A. Badalà, G. Barone, C. M. Belfiore, C. Calabrò, M. F. La Russa, P. Mazzoleni and A. Pezzino, Constr. Build. Mater., 2012, 33, 122–132 CrossRef
- M. F. La Russa, G. Barone, C. M. Belfiore, P. Mazzoleni and A. Pezzino, Environ. Earth Sci., 2011, 62, 1263–1272 CrossRef
- G. Alessandrini, A. Bocci, R. Bugini, D. Emmi, R. Peruzzi and M. Realini, in 7th Internationa congress on deterioration and conservation of stone, 1993, pp. 11–20.
- C. Urzì and M. Realini, Int. Biodeterior. Biodegrad., 1998, 42, 45–54 CrossRef
- G. Barbera, G. Barone, V. Crupi, F. Longo, G. Maisano, D. Majolino, P. Mazzoleni, S. Raneri, J. Teixeira and V. Venuti, Eur. J. Mineral., 2014, 26, 189–198 CrossRef CAS
- G. Barone, V. Crupi, F. Longo, D. Majolino, P. Mazzoleni, S. Raneri, J. Teixeira and V. Venuti, J. Instrum., 2014, 9, 1–9 Search PubMed
- A. Vamvakeros, S. D. M. Jacques, M. Di Michiel, P. Senecal, V. Middelkoop, R. J. Cernik and A. M. Beale, J. Appl. Crystallogr., 2016, 49, 485–496 CrossRef CAS PubMed
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed
- J. Kieffer, S. Petitdemange and T. Vincent, J. Synchrotron Radiat., 2018, 25, 612–617 CrossRef CAS PubMed
- S. D. M. Jacques, M. Di Michiel, S. A. J. Kimber, X. Yang, R. J. Cernik, A. M. Beale and S. J. L. Billinge, Nat. Commun., 2013, 4, 2536 CrossRef PubMed
- S. D. M. Jacques, M. Di Michiel, A. M. Beale, T. Sochi, M. G. O'Brien, L. Espinosa-Alonso, B. M. Weckhuysen and P. Barnes, Angew. Chem., Int. Ed., 2011, 50, 10148–10152 CrossRef CAS PubMed
- M. G. O'Brien, S. D. M. Jacques, M. Di Michiel, P. Barnes, B. M. Weckhuysen and A. M. Beale, Chem. Sci., 2012, 3, 509–523 RSC
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed
- E. Possenti, C. Colombo, D. Bersani, M. Bertasa, A. Botteon, C. Conti, P. P. Lottici and M. Realini, Microchem. J., 2016, 127, 79–86 CrossRef CAS
- E. Sassoni, RILEM Tech. Lett., 2017, 2, 14–19 CrossRef
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS PubMed
- C. Drouet, BioMed Res. Int., 2013, 2013, 1–12 CrossRef PubMed
- E. Possenti .
- C. Conti, C. Colombo, M. Realini, G. Zerbi and P. Matousek, Appl. Spectrosc., 2014, 68, 686–691 CrossRef CAS PubMed
- C. Conti, M. Casati, C. Colombo, E. Possenti, M. Realini, G. D. Gatta, M. Merlini, L. Brambilla and G. Zerbi, Spectrochim. Acta, Part A, 2015, 150, 721–730 CrossRef CAS PubMed
- C. Conti, L. Brambilla, C. Colombo, D. Dellasega, G. D. Gatta, M. Realini and G. Zerbi, Phys. Chem. Chem. Phys., 2010, 12, 14560–14566 RSC
- T. Dreyfuss, J. Cult. Herit., 2020, 42, 56–63 CrossRef
- E. Possenti, C. Conti, G. D. Gatta, M. Realini and C. Colombo, Coatings, 2019, 9, 169 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an02208k |
| This journal is © The Royal Society of Chemistry 2024 |