
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic isolation and microfluidic electrophoresis analysis of residual dsRNA impurities in mRNA vaccines and therapeutics†
Adriana
Coll De Peña
 a,
Matei
Vaduva
b,
Nina S.
Li
a,
Shreyas
Shah
c,
Menel
Ben Frej
c and
Anubhav
Tripathi
*a
a,
Matei
Vaduva
b,
Nina S.
Li
a,
Shreyas
Shah
c,
Menel
Ben Frej
c and
Anubhav
Tripathi
*a
aCenter for Biomedical Engineering, School of Engineering, Brown University, Providence, RI, USA. E-mail: anubhav_tripathi@brown.edu
bDepartment of Molecular Biology, Cell Biology, and Biochemistry, Division of Biology and Medicine, Brown University, Providence, RI, USA
cApplied Genomics, Revvity, Hopkinton, MA, USA
First published on 23rd January 2024
Abstract
The versatility, rapid development, and ease of production scalability of mRNA therapeutics have placed them at the forefront of biopharmaceutical research. However, despite their vast potential to treat diseases, their novelty comes with unsolved analytical challenges. A key challenge in ensuring sample purity has been monitoring residual, immunostimulatory dsRNA impurities generated during the in vitro transcription of mRNA. Here, we present a method that combines an enzyme, S1 nuclease, to identify and isolate dsRNA from an mRNA sample with a microfluidic electrophoresis analytical platform to characterize the impurity. After the method was developed and optimized, it was tested with clinically relevant, pseudouridine-modified 700 and 1800 bp dsRNA and 818–4451 nt mRNA samples. While the treatment impacted the magnitude of the fluorescent signal used to analyze the samples due to the interference of the buffer with the labeling of the sample, this signal loss was mitigated by 8.8× via treatment optimization. In addition, despite the mRNA concentration being up to 400× greater than that of the dsRNA, under every condition, there was a complete disappearance of the main mRNA peak. While the mRNA peak was digested, the dsRNA fragments remained physically unaffected by the treatment, with no change to their migration time. Using these samples, we detected 0.25% dsRNA impurities in mRNA samples using 15 μL with an analytical runtime of 1 min per sample after digestion and were able to predict their size within 8% of the expected length. The short runtime, sample consumption, and high throughput compatibility make it suitable to support the purity assessment of mRNA during purification and downstream.
Introduction
During the COVID-19 pandemic, the importance of mRNA vaccines was highlighted due to their low production cost, fast manufacturing, and effectiveness, among other advantages.1–6 As the first vaccine of its kind to be approved by the FDA, it has accelerated the research interest in mRNA-based vaccines and therapies, showing potential in treating genetic, infectious, cancer, and other diseases and preventing them.1,6–8 Despite the immense potential of this technology to combat diseases, manufacturing challenges remain that allow for residual impurities in the final product.9–11 Numerous filtration and chromatographic purification steps are integrated into the manufacturing of mRNA to remove the numerous nucleic acid by-products and impurities generated during the process. However, double-stranded RNA (dsRNA), which can be a by-product of the in vitro transcription (IVT) of mRNA, is a notable challenge for manufacturers, as it behaves similarly to mRNA in current quality control assays and is notoriously difficult remove during purification.12–14 To address this, improvements have been made in terms of enzyme technology and purification methods,15 however, residual dsRNA at trace concentrations can remain in the final product.16 dsRNA is a molecule of interest due to its ability to significantly increase the immune response to mRNA vaccines,17 leading to both local and systemic inflammation in treated individuals.18,19 Further, due to the strong immune response to dsRNA,20 it has also been found to decrease the effectiveness of the treatment due to the inhibition of translation.16 The current analytical standard for dsRNA monitoring is dot blot, which is capable of detecting low concentrations of dsRNA contaminants in mRNA samples. Despite its widespread use, dot blot only provides information regarding the concentration and not the length of the dsRNA impurities. Given the potential implications for the efficacy of the therapeutics and the length-dependent adverse effects dsRNA can cause,20 it is essential to monitor both the concentration and the length of these impurities.Microfluidic electrophoresis has been used to analyze mRNA molecules21–25 and can detect dsRNA molecules.26 Its rapid turnaround time, high throughput compatibility, and low sample consumption27,28 make it particularly suited to support the development of biopharmaceutical products. However, in addition to the main mRNA peak and the residual dsRNA of interest, other by-products, such as truncated or degraded mRNA fragments, may still be present,12 making it so that mobility differences alone between mRNA and dsRNA cannot be used for their characterization.26 Therefore, it is crucial to integrate a method to identify the peak-producing molecules before assessing their size and concentration.
We previously presented an identification method that differentiated dsRNA from mRNA via their dynamic staining response.26 While it presented an interesting identification method, in practice, the significantly larger mRNA peak tended to mask the significantly smaller dsRNA peak, and the need to use two different gel dyes caused minor shifts in the electropherogram that made it difficult to match the peaks. Instead, this study introduces an analytical approach that utilizes an enzyme, S1 nuclease, to digest all single-stranded fragments in the sample, enabling the characterization of the remaining dsRNA products (Fig. 1). Post digestion, the samples are analyzed using a custom microfluidic method we developed that enables the detection of low concentrations of dsRNA and mRNA but using a single gel dye mixture.26 By integrating the isolating effect of S1 nuclease digestion into a microfluidic detection regiment, this method provides a rapid, reproducible, and efficient means for analyzing dsRNA impurities in mRNA formulations.
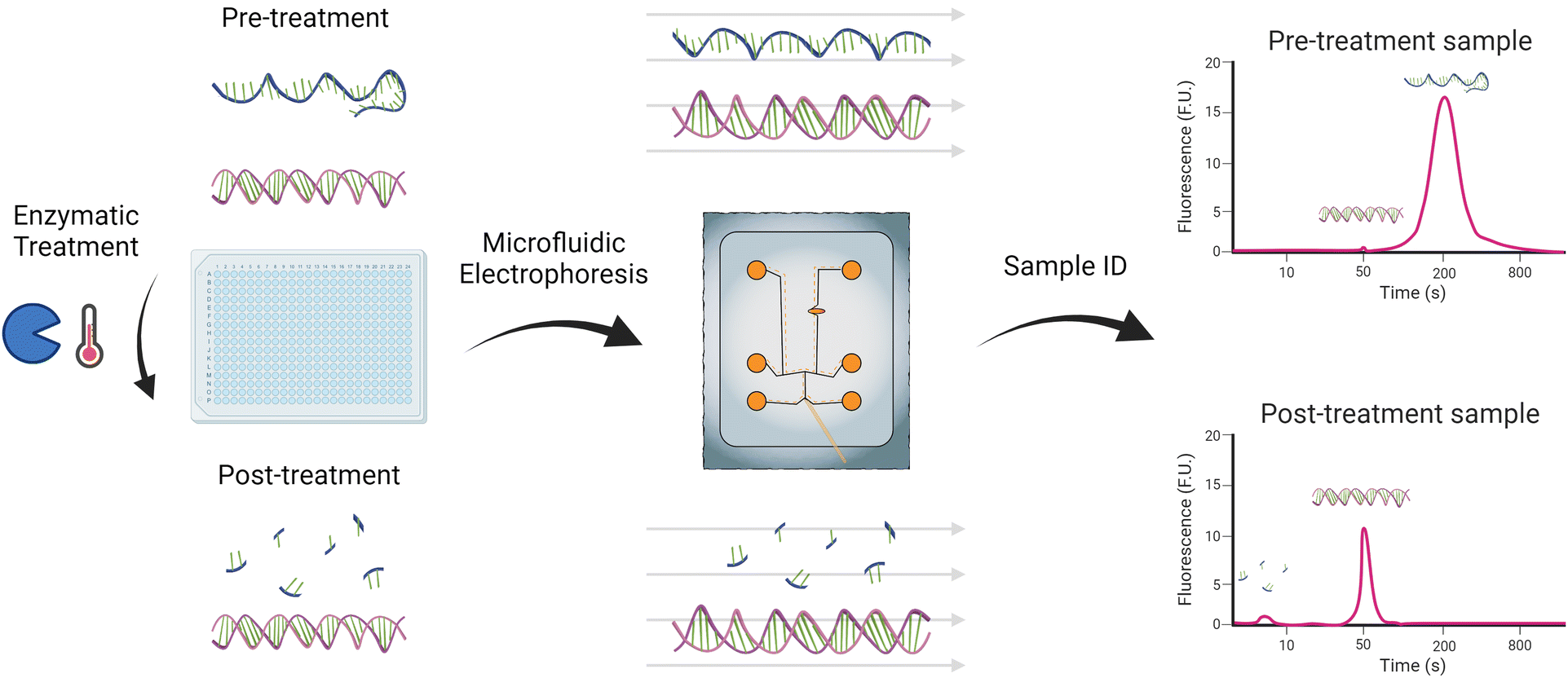 | ||
| Fig. 1 Enzymatic identification and microfluidic electrophoresis characterization workflow of dsRNA contaminants and impurities in mRNA samples. Figure created using BioRender.com and Adobe Illustrator. | ||
This study aims to assess the efficacy and specificity of the S1 nuclease-assisted microfluidic approach for detecting dsRNA impurities in mRNA formulations. While the manufacturer-recommended S1 nuclease treatment showed great initial promise, it was initially developed for single-stranded DNA digestion (i.e., overhangs) and had to be optimized to be more fit for purpose. The primary considerations during its optimization were maximizing its compatibility with the microfluidic analytical platform by mitigating its impact on the dsRNA signal and adjusting the reaction buffer to enzyme ratios to ensure the method was robust and would perform well against different lengths and concentrations of mRNA. The optimization of the nuclease treatment enabled an 8.8-fold increase in the dsRNA limit of detection post-digestion from 2.83 ng μL−1 using the standard protocol to 0.32 ng μL−1 after optimization. Capable of digesting mRNA concentrations of at least 200 ng μL−1, this suggests the method has a theoretical dsRNA contaminant sensitivity of 0.16% of the total mRNA, and the lowest percentage tested was 0.25%, which yielded reproducible results. However, the actual percentage is likely even lower, as the maximum loading of the system was not tested beyond 200 ng μL−1, based partly on sample availability.
Materials and methods
Materials
The mRNA and dsRNA samples used in this study were pseudouridine chemically modified since this chemical modification has been shown to increase the stability of the molecule and is of great clinical relevance.29–31 The pseudouridine chemically modified mRNA (818, 1198, 1913, 3406, and 4451 nt) and dsRNA (700 and 1800 bp) fragments were purchased from CATUG Biotechnology (CatPure™; Cambridge, MA) at respective concentrations of 2000 ng μL−1 and 200 ng μL−1. The chemically modified samples used in this study are clinically relevant as they were synthesized following the same method (in vitro transcription) and chemical modification (pseudouridine) as the Moderna Therapeutics and Pfizer-BioNTech COVID-19 vaccines.30 The dsRNA ladder used to assess the length of the impurities was composed of a dsRNA ladder 21–500 bp (New England Biolabs, Ipswich, MA) and a 4001 bp custom synthesized dsRNA construct (Azenta, Burlington, MA). Before use, the samples were diluted to the specified concentrations in 1× TE buffer, pH 8.0. The S1 nuclease (100 U μL−1) and its 5× reaction buffer were purchased from ThermoFisher Scientific (Franklin, MA) for the enzymatic treatment. They were diluted in nuclease-free water to the specified concentration. S1 nuclease was selected for this study because it is expected to degrade single-stranded nucleic acids (ThermoFisher Scientific), potentially enabling the differentiation between mRNA and dsRNA. For the microfluidic analysis, a custom glass nucleic acid microfluidic chip with a metal sipper and custom chip reagents compatible with the LabChip GXII Touch platform were obtained from Revvity (Waltham, MA).Enzymatic treatment
Per the manufacturer, ThermoFisher Scientific, the standard operating procedure (SOP) treatment protocol for the use of S1 Nuclease recommends the use of 10 U (0.1 μL) of enzyme for every 1 μg of nucleic acid and 6 μL of 5× reaction buffer per 10 U of enzyme, in a total volume of 30 μL. Once mixed, it is incubated for 30 minutes at 25 °C, then 2 μL of 0.5 M EDTA are added, and the mixture is heated at 70 °C for 10 minutes for enzyme inactivation. Due to the lower concentration at which mRNA samples tend to be available, we adjusted the initial protocol to a total volume of 15 μL and 0.20 μg of nucleic acid (0.150 μg of mRNA and 0.075 μg of dsRNA). As such, the SOP became 2.25 U of the enzyme (2.25 μL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution) and 1.35 μL of 5× reaction buffer for 10 ng μL−1 of mRNA and 5 ng μL−1 of dsRNA, mixed with nuclease-free water to a final volume of 15 μL, and then incubated at 25 °C for 30 min. When specified, 1 μL of 0.5 M EDTA was added before the heat inactivation of the sample at 70 °C for 10 min.
:100 dilution) and 1.35 μL of 5× reaction buffer for 10 ng μL−1 of mRNA and 5 ng μL−1 of dsRNA, mixed with nuclease-free water to a final volume of 15 μL, and then incubated at 25 °C for 30 min. When specified, 1 μL of 0.5 M EDTA was added before the heat inactivation of the sample at 70 °C for 10 min.
During the study, adjustments to the treatment components will be reported as changes relative to the latter protocol described above. For instance, if 5 U of the enzyme is used instead of the standard 2.25 U, it will be reported as 2× enzyme. Regardless of the adjustments made to the components, the treatment volume was kept constant across the study. In the case of changes to the nucleic acid concentration, it will refer solely to changes in the mRNA concentration. In contrast, the dsRNA concentration will be kept constant across the individual experiments unless specified for the limit of detection (LOD) assessment.
Microfluidic measurements and analysis
Once the samples were loaded onto a 96- or 384-well plate, which could be done before or after digestion, the plate and the microfluidic chip were transferred onto a LabChip GXII Touch platform (Revvity). The pressure, electric fields, and robotic motion to load and analyze the sample onto the chip were controlled using the LabChip platform. We describe this system, the setup, and the electrokinetic script used here in a previous study.26The electropherograms yielded by the LabChip GXII Touch platform were analyzed using the LabChip GX Reviewer software, version 5.11 (Revvity). The data extracted from the LabChip GX Reviewer software was then input into GraphPad Prism 10.1 for statistical analysis and visualization. The statistical significances reported through the study were obtained from a Tukey post hoc test with a confidence interval of 95%, where *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. In addition, JMP Pro 17 was used for the factorial design and analysis described in the study, which helped determine the main effects and all two-factor interactions of the enzyme, buffer, and mRNA concentrations using three values for each.
To determine the sensitivity of the method, the limit of detection, LOD, of the platform was calculated using the peak area as follows:
 | (1) |
 | (2) |
Results and discussion
S1 nuclease treatment baseline
mRNA and dsRNA, particularly longer fragments, have significantly different electrophoretic mobilities.26 Therefore, to characterize contaminant or residual dsRNA in mRNA samples, it is essential to first identify the peak-producing molecule before using a ladder to determine its length. S1 nuclease is an enzyme that degrades single-stranded nucleic acids (Fig. 2a) and presents a promising method for identifying dsRNA fragments in mRNA samples prior to microfluidic characterization. To assess the potential of the method, we compared an untreated sample to a sample treated following the standard operating procedure (SOP), which contains EDTA (Fig. 2b and c). While EDTA is commonly used due to its ability to chelate magnesium ions required by nuclease enzymes, protecting nucleic acids from further degradation, in excess, it can affect the performance of microfluidic analyses. Therefore, we also investigated the need for EDTA in the treatment (Fig. 2d). When the untreated sample was compared to the treated samples, a complete disappearance, or digestion, of the mRNA peak (∼37.7 s), but retention, albeit at a significantly lower magnitude, of the dsRNA peak (∼31.4 s), as highlighted in Fig. 2. Interestingly, EDTA did not have a measurable impact on the dsRNA peak area or migration time, and neither did the treatment, regardless of the use of EDTA, compared to the untreated sample. Since the goal is to develop a streamlined and robust protocol for detecting residual dsRNA in mRNA samples, and EDTA did not appear to impact the digestion performance, it was removed from the workflow.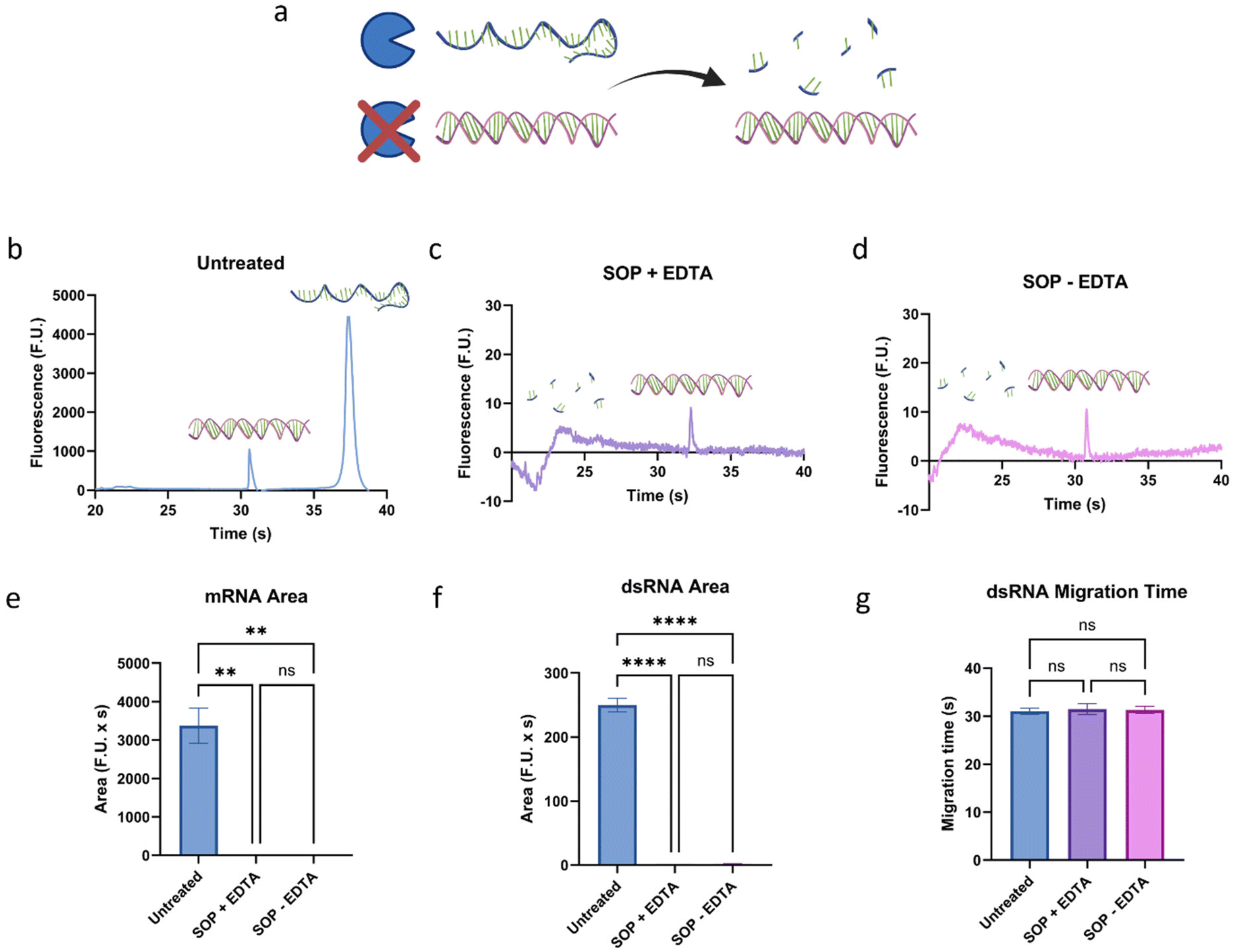 | ||
| Fig. 2 Assessment of the impact of the (a) S1 nuclease treatment on mRNA and dsRNA. Electropherograms of 5 ng μL−1 of a 700 bp dsRNA fragment and 10 ng μL−1 of a 1198 nt mRNA fragment (b) untreated, (c) treated following the SOP, which contains EDTA, and (d) treated following the SOP without EDTA. When present, the dsRNA peak has an average migration time of 31.4 s while the mRNA peak has an average migration time of 37.7 s; between 20 and 25 s, a small peak can usually be observed representing the lower marker. Summarized (e) mRNA peak area, (f) dsRNA peak area, and (g) dsRNA peak migration time under the three conditions. “*” indicates the significance level in the difference (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001), while “ns” indicates no statistical difference. This figure was created using BioRender.com and GraphPad Prism. | ||
Ensuring that dsRNA remains intact, confirmed by the lack of change in migration time (Fig. 2g), is crucial for the robustness of the method as it will allow correct length determination downstream. However, while the S1 nuclease treatment continues to show great promise, there was a significant decrease of over two orders of magnitude in the dsRNA peak area, going from 249.8 to an average of 2.0 F.U. × s from untreated to treated. This suggests that the limit of detection (LOD) of the method, at this stage, was 2.83 ng μL−1, calculated using the average yielded by eqn (1) and (2). Considering the target is to be able to detect ∼1% dsRNA by concentration in mRNA samples, and these samples can be present as smears, this would require the loading of over 300 ng μL−1 of the mRNA sample. Therefore, the next goal of the study is to mitigate the signal decrease of dsRNA caused by the treatment to increase the sensitivity of the method.
Optimization of the buffer to enzyme ratio of the digestion
The relationship between buffer, enzyme, and mRNA concentrations was evaluated to increase the sensitivity of the method. First, some initial experiments not included in this study were conducted to narrow down the optimal concentrations of each parameter. During these experiments, it was observed that these parameters can interact with each other, so we designed a factorial experiment to test three levels of each of these parameters. We tested buffer concentrations of 1/16, 1/8, and 1/4× SOP concentration, enzyme concentrations of 2, 10, and 50× SOP concentration, and mRNA concentrations of 1/2, 1, and 2 the initial concentration of 10 ng μL−1 (Table S1†). As can be observed in Fig. 2, and as we confirmed during initial tests, under different conditions, a smear can appear next to the lower marker. While no experiments were conducted to characterize the nature of the smear further, it is believed it is primarily composed of fragments of mRNA that were not fully digested.Therefore, to determine the performance of each condition, the dsRNA peak area and height and the undesired peak area were analyzed and compared (Fig. S1†), and the most promising (high dsRNA peak area and height and low undesired peak area) were chosen for further investigation, as highlighted in Fig. 3a and b. A factorial analysis (Fig. S2†) was used to determine the optimal formulation to maximize the dsRNA peak and minimize the undesired peak. Based on these parameters, the optimal condition was determined to be condition 13, with a buffer concentration of 1/16× SOP, an enzyme concentration of 10× SOP, and 1× the mRNA concentration (Fig. 3c). Overall, it was observed that inadequate buffer to enzyme concentrations for a given mRNA concentration can lead to insufficient digestion of the mRNA. Considering that using this condition, the LOD is ∼0.26 ng μL−1, which would require a concentration of over 30 ng μL−1 of mRNA to detect the impurities effectively, it was decided, based on a similar performance, to test further condition 14 (Fig. 3d), which only differed in buffer concentration (1/8 instead of 1/16×) from condition 13, against different mRNA concentrations to ensure the robustness of the method.
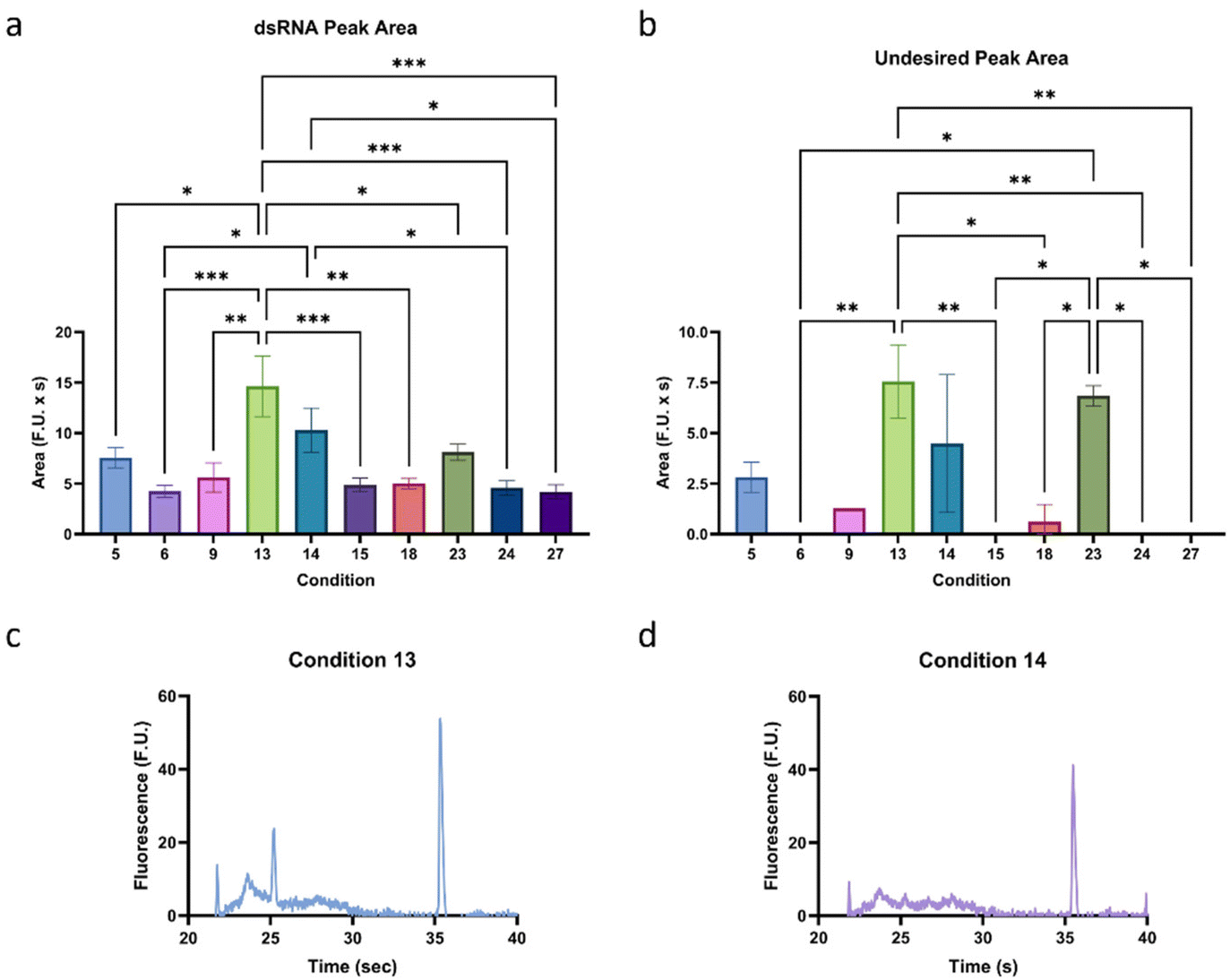 | ||
| Fig. 3 Assessment of the optimal buffer to enzyme and mRNA concentrations (Table S1†). Summarized (a) dsRNA peak area and (b) undesired peak area of the top performing conditions. Electropherograms of conditions (c) 13 and (d) 14, which were selected for further analysis. “*” indicates the significance level in the difference (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001), while “ns” indicates no statistical difference. This figure was created using GraphPad Prism. | ||
Since condition 13 (1/16× buffer, 10× enzyme) was the preferred condition based on the factorial analysis and condition 14 (1/8× buffer, 10× enzyme) was semi-qualitatively our preferred condition based on overall peak magnitudes and shapes, we conducted an experiment where mRNA concentrations of 10, 100, and 200 ng μL−1 were used to test the robustness of each method. We found that the undesired peak area is positively correlated with mRNA concentration for both conditions, and the dsRNA peak area is independent of changes in mRNA concentration (Fig. 4a and b). Furthermore, the distance between the undesirable and dsRNA peaks in both conditions was sufficient to avoid peak overlap, highlighting the robustness of the method. As predicted, condition 13 had significantly higher undesirable peak areas than condition 14 at higher concentrations (Fig. 4c–f). Despite this, the dsRNA peak area was similar for the two conditions, which means that condition 14 has a higher dsRNA peak area to undesirable peak area ratio. As a result, Condition 14 was chosen as the optimal treatment formulation.
 | ||
| Fig. 4 Comparison of conditions 13 and 14 at 1 M (10 ng μL−1), 10 M (100 ng μL−1), and 20 M (200 ng μL−1) of the 1198 nt sample with the 700 bp dsRNA concentration kept constant at 5 ng μL−1. Summarized areas of the undesired and dsRNA peaks using (a) condition 13 and (b) condition 14. Electropherograms of (c) condition 13 and (d) condition 14 at 20M. Summarized areas of the (e) undesired peak and the (f) dsRNA peak for conditions 13 and 14. “*” indicates the significance level in the difference (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001), while “ns” indicates no statistical difference. This figure was created using GraphPad Prism. | ||
Validation of the method
Once the treatment conditions were optimized (1/8× buffer, 10× enzyme), its performance was validated in terms of sensitivity (Fig. S3†) and length dependency (Fig. 5). To assess the LOD, a 700 bp dsRNA sample was analyzed at 1.0, 2.5, and 5.0 ng μL−1, and the peak area and height of each sample were used to determine the LOD using eqn (1) and (2) (Fig. S3†). A strong correlation can be observed between peak area and concentration (Fig. S3a†). No observable difference was observed between the area- and height-based estimations of the LOD, suggesting strong confidence in the LOD of the method estimated to be 0.32 ± 0.07 ng μL−1 of treated dsRNA. Next, to assess the length-dependency of the assay, it was tested with different dsRNA and mRNA lengths, specifically dsRNA of lengths 700 bp and 1800 bp and mRNA of lengths 818 nt, 1198 nt, 1913 nt, 3406 nt, and 4451 nt (Fig. 5). More specifically, Sample 1 contained 700 bp dsRNA and 818 nt mRNA, Sample 2 contained 700 bp dsRNA and 1198 nt mRNA, Sample 3 contained 700 bp dsRNA and 1913 nt mRNA, Sample 4 contained 700 bp and 1800 bp dsRNA and 1913 nt mRNA, Sample 5 contained 700 bp and 1800 bp dsRNA and 3406 nt mRNA, and Sample 6 contained 700 bp and 1800 bp dsRNA and 4451 nt mRNA. The concentration of dsRNA and mRNA in all samples was 1 ng μL−1 and 200 ng μL−1, respectively. For all samples tested, there was a complete digestion of the mRNA peak, and, as can be highlighted in Fig. 5, the dsRNA migration time was preserved for both the 700 and 1800 bp fragments. Notably, this method can identify different dsRNA impurities in the same sample without interference. | ||
| Fig. 5 Assessing the robustness of the method against different mRNA lengths at a concentration of 200 ng μL−1 spiked with a 700 bp dsRNA or a 700 bp and a 1800 bp dsRNA each at a concentration of 1 ng μL−1. Since all the mRNA peaks were fully digested by the enzyme, the dsRNA fragments were analyzed to determine whether the treatment affects the migration and integrity of the fragments that are not targeted by the enzyme. Migration time of (a) the 700 bp and (b) the 1800 bp dsRNA for the samples containing each fragment. Peak area for (c) the 700 bp and (d) the 1800 bp dsRNA for the samples containing each fragment. Representative electropherograms of (e) the untreated 1913 nt mRNA, (f) the treated 1913 nt mRNA and 700 bp dsRNA, and (g) the treated 1913 nt mRNA, 700 bp dsRNA, and 1800 bp dsRNA. Panels f and g highlight the complete disappearance through digestion of the 1913 nt mRNA peak. “*” indicates the significance level in the difference (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001), while “ns” indicates no statistical difference. This figure was created using GraphPad Prism. | ||
Once it was confirmed that the integrity of the dsRNA fragments was not affected, we tested the ability of the method to estimate the length of dsRNA impurities. To this end, once again a 1913 nt fragment was spiked with two dsRNA fragments to simulate contaminants, however, this time the dsRNA fragments were added at a concentration of 0.25% relative to the mRNA concentration (Fig. 6). Once the dsRNA fragments were identified, a ladder combined in-house was used to determine the length of the dsRNA impurities with an average error of 8%, ranging from 4–11%, with no statistical difference from the expected value. It must be noted that while the error is within our desired threshold, the ladder used to generate the curve that was used to estimate the lengths was composed of non-chemically modified dsRNA, while the samples were spiked with pseudouridine chemically modified dsRNA. This suggests that while their mobilities may differ slightly, a non-modified ladder can be used to estimate the length of pseudouridine modified dsRNA, and potentially other chemical modifications.
 | ||
| Fig. 6 Assessment of the ability to estimate the dsRNA impurity fragment length from a sample containing a 700 bp and a 1800 bp dsRNA fragment at a concentration of 0.5 ng μL−1 each and a 1913 nt mRNA fragment at a concentration of 10 ng μL−1 (untreated) and 200 ng μL−1 (treated). The untreated electropherogram is for illustrative purposes with the same dsRNA concentration as the treated but an mRNA concentration 20× lower due to the undigested mRNA concentration maximum loading capacity of the chip, which prevents us from loading more than 13 ng μL−1 (ref. 26) of untreated mRNA. Electropherograms of the (a) untreated and (b) treated samples. (c) Plot highlighting the logarithmic fit for the migration of the ladder peaks (80, 150, 300, 500, 4001 bp), y = 9.82log(x) + 7.04 with an R2 = 0.99. (d) Comparison of the expected (black) and estimated fragment length of the two dsRNA peaks using the dsRNA ladder fit, showing no statistical difference between the expected and estimated values for both peaks. Figure created using GraphPad Prism. | ||
Conclusion
This study presents a novel method for characterizing dsRNA impurities in mRNA formulations. Microfluidic electrophoresis offers a rapid, high-throughput analytical tool for the fragment-based analysis of both dsRNA and mRNA fragments, enabling the determination of their lengths through ladders. However, as discussed in our previous study,26 long dsRNA and mRNA molecules can differ significantly in electrophoretic mobility, requiring nucleic acid-specific ladders for size determination. Therefore, since, in addition to the residual dsRNA, truncated or degraded mRNA fragments may also be present in the sample,11 it is crucial to determine the identity of the dsRNA peaks before assessing their size.To this end, we developed an enzymatic identification method compatible with our microfluidic system that uses S1 nuclease to digest all single-stranded fragments in the sample, only leaving behind the dsRNA fragments. While the standard protocol for the enzyme treatment yielded acceptable results, we optimized the concentrations of buffer and enzyme used in the protocol to increase the sensitivity and robustness of accepting a wide range of mRNA concentrations. Upon optimizing these conditions, the LOD of the method for dsRNA post-treatment was calculated to be 0.32 ± 0.07 ng μL−1, 8.8-fold greater than using the SOP, and the maximum loading concentration of mRNA tested in this study was 200 ng μL−1. Therefore, the lowest percentage of dsRNA contaminant experimentally tested in an mRNA sample here was 0.25%; however, based on the LOD, we expect the system to be capable of detecting percentages as low as 0.16% when 200 ng μL−1 of mRNA are loaded. It should be noted that it is also possible that higher concentrations of mRNA may be analyzed using the proposed method, further lowering the percentage of impurities that can be detected, but it was not directly assessed during this study.
The focus of this paper was primarily the development of an enzymatic identification method for residual dsRNA in mRNA samples. Once the peak was identified, we were able to use the line of best fit yielded by a ladder we combined in-house to estimate the length of the impurities within 8% of the expected length. While in this study, a ladder had to be run on the same plate as the sample and the migration over length of the ladder peaks had to be fit to generate the curve used to estimate the sizes, we are currently working on developing a more streamlined sizing method. Using experimental data, we are developing a physics-informed neural network model capable of predicting dsRNA and mRNA electrophoretic mobility under different gel concentrations using our microfluidic platform.
Requiring only 10–15 μL of sample, the proposed method has an analytical run time of 1 min per sample post enzyme treatment. Since the analytical platform is compatible with 96- and 384-well plates, and the enzyme treatment can be conducted directly on the well plate, this method is considered high throughput, requiring human intervention during the microfluidic analysis. The low sample requirements and high throughput nature of the method make it highly suitable for the batch-to-batch monitoring of residual dsRNA and for the mRNA process development optimization in the biopharmaceutical industry.
Author contributions
Conceptualization, A.C.D.P.; data curation, A.C.D.P., M.V., and N.L.; formal analysis, A.C.D.P., and M.V.; funding acquisition, A.T.; investigation, A.C.D.P., N.L., S.S., and M.B.F.; methodology, A.C.D.P., and M.V.; project administration, A.C.D.P., and A.T.; resources, A.T.; supervision, A.C.D.P., and A.T.; validation, A.C.D.P., M.V., N.L., S.S., and M.B.F.; visualization, A.C.D.P., M.V., and N.L.; writing – original draft, A.C.D.P., and M.V.; writing – reviewing and editing, A.C.D.P., M.V., N.L., S.S., M.B.F., and A.T.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was in part supported by Revvity's research grant to Brown University. A. T. is a paid scientific advisor/consultant and lecturer for Revvity.References
- S. Qin, X. Tang, Y. Chen, K. Chen, N. Fan, W. Xiao, Q. Zheng, G. Li, Y. Teng, M. Wu and X. Song, Signal Transduction Targeted Ther., 2022, 7(1), 1–35 CrossRef PubMed.
- K. K. W. To and W. C. S. Cho, Expert Opin. Drug Discovery, 2021, 16, 1307–1317 CrossRef CAS PubMed.
- G. Maruggi, C. Zhang, J. Li, J. B. Ulmer and D. Yu, Mol. Ther., 2019, 27(4), 757–772 CrossRef CAS PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17(4), 261–279 CrossRef CAS PubMed.
- R. Feng, S. Patil, X. Zhao, Z. Miao and A. Qian, Front. Mol. Biosci., 2021, 8, 710738 CrossRef CAS PubMed.
- A. J. Barbier, A. Y. Jiang, P. Zhang, R. Wooster and D. G. Anderson, Nat. Biotechnol., 2022, 40(6), 840–854 CrossRef CAS PubMed.
- G. Zhang, T. Tang, Y. Chen, X. Huang and T. Liang, Signal Transduction Targeted Ther., 2023, 8(1), 1–30 CrossRef PubMed.
- H. H. Wei, L. Zheng and Z. Wang, Fundam. Res., 2023, 3, 749–759 CrossRef CAS.
- N. Pardi, M. J. Hogan and D. Weissman, Curr. Opin. Immunol., 2020, 65, 14–20 CrossRef CAS PubMed.
- F. J. Triana-Alonso, M. Dabrowski, J. Wadzack and K. H. Nierhaus, J. Biol. Chem., 1995, 270, 6298–6307 CrossRef CAS PubMed.
- S. S. Rosa, D. M. F. Prazeres, A. M. Azevedo and M. P. C. Marques, Vaccine, 2021, 39, 2190 CrossRef CAS PubMed.
- M. Baiersdörfer, G. Boros, H. Muramatsu, A. Mahiny, I. Vlatkovic, U. Sahin and K. Karikó, Mol. Ther. – Nucleic Acids, 2019, 15, 26 CrossRef PubMed.
- J. Martínez, V. Lampaya, A. Larraga, H. Magallón and D. Casabona, Front. Mol. Biosci., 2023, 10, 1248511 CrossRef PubMed.
- J. Zhang, Y. Liu, C. Li, Q. Xiao, D. Zhang, Y. Chen, J. Rosenecker, X. Ding and S. Guan, Pharmaceutics, 2023, 15, 2182 CrossRef CAS PubMed.
- X. Piao, V. Yadav, E. Wang, W. Chang, L. Tau, B. E. Lindenmuth and S. X. Wang, Mol. Ther. – Nucleic Acids, 2022, 29, 618–624 CrossRef CAS PubMed.
- K. Karikó, H. Muramatsu, J. Ludwig and D. Weissman, Nucleic Acids Res., 2011, 39, e142 CrossRef PubMed.
- X. Mu and S. Hur, Acc. Chem. Res., 2021, 54, 4012 CrossRef CAS PubMed.
- L. Alexopoulou, A. C. Holt, R. Medzhitov and R. A. Flavell, Nature, 2001, 413, 732–738 CrossRef CAS PubMed.
- H. Mitoma, S. Hanabuchi, T. Kim, M. Bao, Z. Zhang, N. Sugimoto and Y. J. Liu, Immunity, 2013, 39, 123 CrossRef CAS PubMed.
- N. McGarry, C. L. Murray, S. Garvey, A. Wilkinson, L. Tortorelli, L. Ryan, L. Hayden, D. Healy, E. W. Griffin, E. Hennessy, M. Arumugam, D. T. Skelly, K. J. Mitchell and C. Cunningham, Brain, Behav., Immun., 2021, 95, 413–428 CrossRef CAS PubMed.
- J. Raffaele, J. W. Loughney and R. R. Rustandi, Electrophoresis, 2022, 43, 1101–1106 CrossRef CAS PubMed.
- Y. Sun, Z. X. Lu, M. Miller, T. Perroud and Y. Tong, NAR: Genomics Bioinf., 2023, 5(1), lqad011 Search PubMed.
- R. Radpour, M. Sikora, T. Grussenmeyer, C. Kohler, Z. Barekati, W. Holzgreve, I. Lefkovits and Y. Z. Xiao, J. Proteome Res., 2009, 8, 5264–5274 CrossRef CAS PubMed.
- J. G. Slagter-Jäger, C. A. Nicolette and I. Y. Tcherepanova, J. Pharm. Biomed. Anal., 2012, 70, 657–663 CrossRef PubMed.
- N. Verma, S. Walia and A. Pandya, Prog. Mol. Biol. Transl. Sci., 2022, 186, 85–107 CAS.
- A. C. De Peña, N. Li, M. Vaduva, L. Bwanali and A. Tripathi, Analyst, 2023, 148(16), 3758–3767 RSC.
- G. M. Whitesides, Nature, 2006, 442(7101), 368–373 CrossRef CAS PubMed.
- E. K. Sackmann, A. L. Fulton and D. J. Beebe, Nature, 2014, 507(7491), 181–189 CrossRef CAS PubMed.
- E. Dolgin, Nature, 2021, 597, 318–324 CrossRef CAS PubMed.
- P. Morais, H. Adachi and Y.-T. Yu, Front. Cell Dev. Biol, 2021, 9, 789427 CrossRef PubMed.
- K. Karikó, H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato, S. Akira and D. Weissman, Mol. Ther., 2008, 16, 1833–1840 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Conditions tested during the factorial analysis. Fig. S1: Comparison of the dsRNA and undesired peak areas yielded by the factorial analysis. Fig. S2: Factorial analysis of the buffer, enzyme, and mRNA concentrations experiment. Fig. S3: Assessment of the dsRNA limit of detection. See DOI: https://doi.org/10.1039/d3an02157b |
| This journal is © The Royal Society of Chemistry 2024 |