
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcoustic detection of a mutation-specific Ligase Chain Reaction based on liposome amplification†
Nikoletta
Naoumi
ab,
Monica
Araya-Farias
cd,
Maria
Megariti
b,
Lucile
Alexandre
cd,
George
Papadakis
b,
Stephanie
Descroix
 cd and
Electra
Gizeli
*ab
cd and
Electra
Gizeli
*ab
aDepartment of Biology, University of Crete, Vassilika Vouton, Heraklion, 70013, Greece
bInstitute of Molecular Biology and Biotechnology-FORTH, 100 N. Plastira Str., Heraklion 70013, Greece. E-mail: gizeli@imbb.forth.gr
cLaboratoire Physico-Chimie Curie, CNRS UMR 168, Institut Curie, PSL Research University, Paris, France
dInstitut Pierre-Gilles de Gennes for Microfluidic (IPGG), Paris, France
First published on 17th May 2024
Abstract
Single nucleotide variants (SNVs) play a crucial role in understanding genetic diseases, cancer development, and personalized medicine. However, existing ligase-based amplification and detection techniques, such as Rolling Circle Amplification and Ligase Detection Reaction, suffer from low efficiency and difficulties in product detection. To address these limitations, we propose a novel approach that combines Ligase Chain Reaction (LCR) with acoustic detection using highly dissipative liposomes. In our study, we are using LCR combined with biotin- and cholesterol-tagged primers to produce amplicons also modified at each end with a biotin and cholesterol molecule. We then apply the LCR mix without any purification directly on a neutravidin modified QCM device Au-surface, where the produced amplicons can bind specifically through the biotin end. To improve sensitivity, we finally introduce liposomes as signal enhancers. For demonstration, we used the detection of the BRAF V600E point mutation versus the wild-type allele, achieving an impressive detection limit of 220 aM of the mutant target in the presence of the same amount of the wild type. Finally, we combined the assay with a microfluidic fluidized bed DNA extraction technology, offering the potential for semi-automated detection of SNVs in patients’ crude samples. Overall, our LCR/acoustic method outperforms other LCR-based approaches and surface ligation biosensing techniques in terms of detection efficiency and time. It effectively overcomes challenges related to DNA detection, making it applicable in diverse fields, including genetic disease and pathogen detection.
Introduction
The detection of single nucleotide variants (SNVs), commonly known as point mutations, plays a pivotal role in advancing our understanding of genetic diseases, cancer development, evolutionary biology, and personalized medicine.1 Ligase-based amplification and detection techniques have been extensively utilized for SNVs discrimination in pathogen detection2 genetic diseases2,3 and cancer diagnosis.4–7 In these techniques, a DNA ligase is employed to favor the amplification of one DNA target versus the target which carries a mismatch at the ligation site.1 Ligation-based amplification techniques can be roughly divided into two main categories: those that use a ligase for the mismatch discrimination and a polymerase with strong strand displacement activity for the amplification of the “ligated” target like the Rolling Circle Amplification (RCA), the Strand Displacement Amplification (SDA)2 and the past few years the ligase-based isothermally exponential amplification (LIEXA)8 and Loop-mediated isothermal amplification (LAMP)9 reactions. The other category includes methods mainly based on the ligation for the detection such as the Oligonucleotide Detection Assay (OLA) and the Ligase Detection Reaction (LDR), the Ligase Chain Reaction (LCR) and LCR variants (Gap-LCR, Nested-LCR). The OLA reaction is based on the covalent joining of two adjacent oligonucleotides by a DNA ligase when they are hybridized with full complementarity to the DNA target usually combined with a PCR pre-amplification step.10 On the other hand, the ligase detection reaction (LDR) achieves linear target amplification by repeated ligation of one pair of complementary to the target probes through a cycling protocol of denaturation, annealing and ligation. Finally, LCR leads to target amplification through a cycling protocol as well, however, in this case two pair of probes are added to the reaction leading to exponential amplification.2During LCR, a thermostable DNA-ligase is mixed with 2 pairs of oligonucleotide probes, each pair complementary to the corresponding strand of the target DNA. Upon hybridization of the probes with the target, the ligase connects the two adjacent oligos only when perfect complementation occurs. This procedure requires the cycling of two temperatures for denaturation and annealing/ligation and leads to the formation of double stranded DNA (dsDNA) products. The formed ligated products serve as templates in subsequent cycles as in PCR leading to exponential amplification.2 Reports on LCR efficiency indicate that this is high only if the number of cycles employed during the amplification process is kept relatively low.11 Compared to techniques based on primer extension, such as PCR,12 LCR has been reported to exhibit a greater ability to discriminate mismatches12. Moreover, in contrast with PCR, LCR does not introduce any uncontrolled amplification bias, i.e., the preferential amplification of some DNA templates over the others within the same reaction, and errors.13–15
Although LCR can exponentially amplify the target, the overall amplification efficiency remains low. Combination of LCR with other amplification techniques such as PCR2,4 or RCA7,11 has been extensively applied for more efficient SNV and mutation detection. Although these approaches improve the sensitivity and specificity of the assay, detecting the products is still difficult. Separating them from the reaction mixture and eliminating interference from remaining probes, primers, and reaction components is challenging. Detection usually involves electrophoretic-based methods5 or real-time fluorescence coupled with prior treatment,16,17 which, along with the extra amplification step, greatly increase the complexity of the assay.7 DNA biosensors are an alternative means that can contribute to the development of a simpler detection technique. There are several publications reporting the detection of SNVs through on-sensor ligation; in these assays, a DNA target hybridizes to a surface-immobilized capture probe followed by the loading of a 2nd probe which is hybridized to the captured target. A DNA ligase connects the two adjacent oligos only upon perfect complementation. Then a thermal18–20 or chemical treatment21,22 step is included to dehybridize and wash away the non-ligated probe followed by a signal amplification step through the binding of nanoparticles,21 enzymatic catalysis18,20,22 and molecular beacons.19 However, publications reporting biosensing detection of LCR are minimal, and all of them concern electrochemical sensors.3,23,24
In this work, instead of combining LCR with another enzymatic amplification technique and sophisticated detection analysis, we enhanced the LCR signal during its detection utilizing an acoustic biosensor. Regarding acoustic biosensor operation, the presence of an analyte on the sensor surface affects the propagation characteristics of the acoustic wave, i.e., its velocity and energy, which are expressed as changes in frequency (ΔF) and dissipation (ΔD). ΔF correlates with the amount of the deposited mass on the sensor;25 ΔD and (ΔD/ΔF) correlate, among other things, with the viscoelastic properties of the surface-attached layer and hydrodynamic properties26–28 or conformation29–32 of discretely-bound molecules. We designed an acoustic detection assay during which firstly the amplified dsDNA products were directly immobilized on the device surface and then were detected through the binding of highly dissipative liposomes of a diameter of 200 nm. Liposomes acted as acoustic signal enhancers,33 significantly improving the sensitivity of the LCR compared to the standard gel electrophoresis analysis. The approach was applied for the detection of the BRAF V600E point mutation versus the wild type (wt) allele achieving a detection limit of 3300 copies or 5.5 zmol or 220 aM of mutant (mt) target.
Experimental section
Acoustic experiments
The 5 MHz QCM sensors (AWS, S.L. Paterna, Spain) were monitored at the 7th overtone (35 MHz) using the quartz crystal microbalance with dissipation monitoring (QCM-D) (Q-Sense E4, Biolin Scientific, Sweden) platform (see also S1†). The dissipation (D) and frequency (F) signals were recorded in real time; F results are reported here as raw data, i.e., without dividing with the overtone no.Acoustic detection of b-BSA & NAv
200 μL of Neutravidin (NAv-Invitrogen) (0.2 mg mL−1) or 250 μL biotinylated-BSA (b-BSA) (0.2 mg mL−1) diluted in PBS pH = 7.4 (Sigma-Aldrich) were applied on the device surface; 200 μL of 0.05 mg mL−1 of NAv was further applied on the b-BSA layer. b-BSA was prepared as described in S2.†Liposome preparation
POPC liposomes (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) from Avanti Polar Lipids (Alabaster, AL, USA.) were prepared as described before.34 Briefly, an initial solution of 2 mg lipid per mL was prepared in PBS buffer and extruded through a polycarbonate filter; the filtered solution was stored (up to three days) and was used at a dilution of ten times for the sensing experiments. The extrusion process results in rather narrow size distributions with average diameters near those of the employed filter pore (here 200 nm).35–37 Liposome polydispersity effects on the recorded QCM signal are of minor importance.38 Nevertheless, care was taken to use, as much as possible, the same particle batch (preparation).Acoustic analysis of LCR
A sample of 20 μL BRAF LCR reaction, diluted in a total volume of 125 μL, was applied on the QCM sensor (flow rate: 25 μL min−1), which was pre-coated with b-BSA/NAv. In the case of the 99 cycles LCR, a suspension of POPC liposomes was added at a volume 500 μL. Regarding the analysis of the 30 cycles LCR, after LCR injection, 200 μL × 500 nM 20 nt-DNA-chol were loaded followed by 500 μL liposome addition.Phosphorylation of the LCR probes
300 pmol of the BRAF-cp1, BRAF-p2(-chol), BRAF-cp1.2 or BRAF-p2.2 were mixed with 1× T4 PNK reaction buffer (NEB), 1 mM ATP (NEB), 0.1 μg μL−1 BSA (NEB) and 10 U T4 PNK Enzyme in a total reaction volume of 50 μL. The mixture was incubated at 37 °C for 60 min followed by heat inactivation at 65 °C for 20 min. The reactions were stored at −20 °C.LCR protocols
In total, two LCR protocols were developed; an initial protocol consisting of 99 cycles and a second optimized protocol consisting of 30 cycles. About the 99 cycles LCR, reactions were performed in the thermal cycler peqSTAR 2× Gradient at the final volume of 25 μL containing 5 pmol of each probe (BRAF-p1(-b), BRAF-p2(-chol), BRAF-cp1, BRAF-cp2), 1× AmpLigase Reaction buffer, 0.1 μg μL−1 BSA, 2.5U of the thermostable AmpLigase DNA Ligase enzyme (Lucigen) and the appropriate amount of BRAF V600E 50% or BRAF wt PCR-derived target (see S3†). Each reaction was subjected to 99 cycles of denaturation at 92 °C for 5 s and annealing/ligation at 75 °C for 10 s. For the 30 cycles LCR, the reactions were run in the thermal cycler FastGene® Ultra Cycler Gradient (NIPPON Genetics Europe) at a final volume of 25 μL. Each reaction contained 2 pmol of each probe (BRAF-p1(-b), BRAF-p2.2, BRAF-cp1.2, BRAF-cp2.2), 1× AmpLigase Reaction buffer, 0.1 μg μL−1 BSA, 1U of the thermostable AmpLigase DNA Ligase enzyme and the desired amount of the 277 bp BRAF purified PCR target (see S3†) or 15 μL of the beads/released target. Reactions were heated at 92 °C for 5 s and 65 °C for 5 s for 30 cycles. For both protocols, a no template control (NTC) was included in every run (see also Table S1†). More information about both LCR protocols’ optimization you can find in S4.†Agarose gel electrophoresis
The LCR products were analyzed with 2% (w/v) agarose gel electrophoresis (AGE) stained with GelRed Nucleic Acid Gel Stain (Biotium) in 1 × TBE buffer (4.5 mM Tris–HCl, pH 7.9, 4.5 mM boric acid, 0.2 mM ethylene diamine tetraacetic acid) at 120 V for 35 min at room temperature. The results were visualized under UV light (imaging system).Capture of ctDNA from human plasma using the FB automated platform
Plasma samples (300 μL) pretreated with Proteinase K (2.5 mg L−1) (Invitrogen) for 2 h at 37 °C were mixed with the BRAF-mt-277 bp or BRAF-wt-277 bp targets at various concentrations ranging from 10 pM to 10 fM and with 0.4 μM of each biotinylated capture-probe BRAF-fw-80 bp and BRAF-rv-80 bp. The sample was heated at 95 °C to denature the dsDNA followed by cooling down allowing the capture probes to anneal at each strand of the DNA. Then, 150 μL of the treated plasma sample was injected in a PDMS chip filled with 500 μg of streptavidin (SAv)-coated magnetic beads Dynabeads MyOne Streptavidin T1 (1 μm) (Invitrogen) & the Dynabeads M-270 Carboxylic Acid (2.8 μm) (Invitrogen) mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (250 μg each) at the flow rate of 5 μL min−1. The capturing of the dsDNA on the beads was evaluated by LCR and acoustic analysis (see also S5 & Table S1†).
:1 ratio (250 μg each) at the flow rate of 5 μL min−1. The capturing of the dsDNA on the beads was evaluated by LCR and acoustic analysis (see also S5 & Table S1†).
Results and discussion
Principle of acoustic-based detection of LCR products through liposomes
The main goal of this work was to design a methodology for the detection of low quantities of SNVs obviating the occasionally biased PCR and instead employing a simple LCR assay coupled with acoustic detection. Although, LCR theoretically amplifies exponentially the target of interest, when we started with ultralow amounts of target, even upon enzymatic amplification, the resulted product remained low. For this reason, we employed a second amplification step during the acoustic detection based on the use of highly dissipative liposomes.The principle of SNV detection using the LCR followed by acoustic analysis is illustrated in Fig. 1. To develop the assay, we use as a model target a 277 bp DNA corresponding to the BRAF gene, with (mt) or without (wt) the V600E point mutation, i.e., a T-A transition. As shown in Fig. 1, LCR is performed by two pairs of probes -p2/p1 & cp2/cp1–50 nt each. The 1st pair of probes -p2/p1- is complementary to the upper strand of the BRAF gene and the 2nd pair -cp2/cp1- to the lower strand, while the two pairs are complementary to each other, too. The 3′-end of the p1 and the 5′-end of the cp1 probes are complementary to the T-A SNV, corresponding to the BRAF V600E point mutation. Regarding the 1st pair of probes, the p1 is biotinylated at the 5′-end, while the p2 probe is modified by a cholesterol at the 3′-end. During LCR of several repeats, DNA fragments of 100 bp employing both a biotin and a cholesterol molecule are exponentially produced only in the case of the mt target where fully complementarity occurs. In contrast, in the case of wt target no amplified products are generated due to the presence of the mismatch on the ligation site. Ligation products are then loaded directly on a NAv-modified sensor, without prior purification or sample heating. The presence of NAv allows the capturing of the biotin/cholesterol-modified amplicons in the case of the mt target and the unused biotinylated probe with its complementary strand in the case of wt. In order to achieve an ultralow detection limit of just a few copies of mt target, a second amplification step is employed where POPC liposomes of 200 nm diameter are injected and captured by the immobilized mt products via the cholesterol-end. The latter causes large change in the acoustic signal leading to the detection of immobilized DNA.
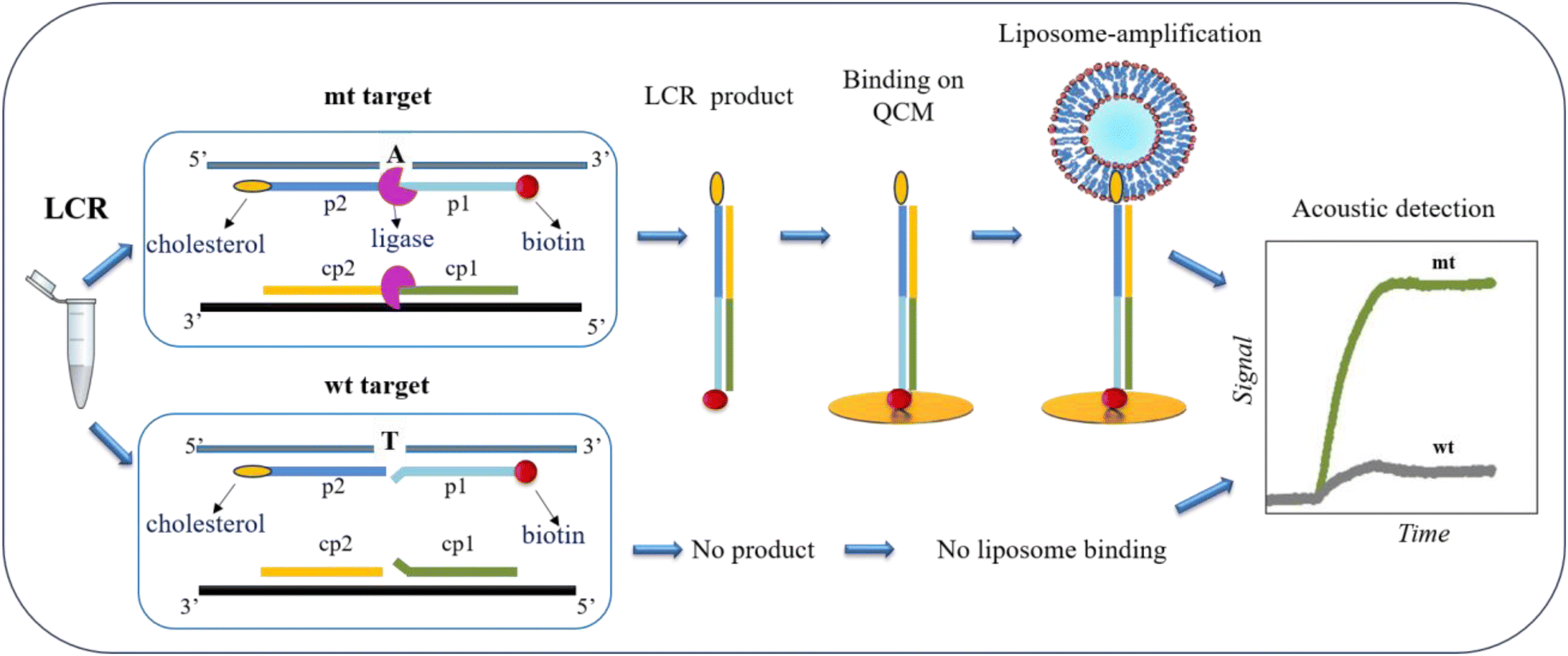 | ||
| Fig. 1 Schematic illustration of LCR coupled with acoustic analysis for the detection of the BRAF V600E point mutation. The two pairs of probes (p2/p1 and cp2/cp1) are complementary to the two mt strands and to each other, while the p2/p1 probes are labeled with a biotin and a cholesterol at each end, respectively. Due to full complementarity, only the mutant produces LCR-amplicons which, subsequently can bind to a NAv-modified (not shown) QCM Au-surface. The addition of liposomes results in cholesterol-binding and significant enhancement of the acoustic dissipation signal in the case of the mt target. The binding of the biotinylated probes (not shown here) can also take place leading to a distinctively smaller background response as shown in the acoustic real-time graph. | ||
Surface modification for capturing and detection of the LCR products
A key advancement in the successful acoustic detection of the LCR was the direct immobilization of the DNA products on a NAv-coated gold sensor obviating any purification step followed by the addition of liposomes. Overall, two protein substrates were investigated; NAv adsorbed on the gold surface and NAv bound to pre-adsorbed b-BSA (Fig. S1A and B†). Frequency and dissipation changes as well as the acoustic ratio measured during the adsorption of NAv on gold or binding to the b-BSA are presented in Fig. 2A–C. The lower ΔF and ΔD signals obtained from the NAv binding on the b-BSA surface (215 ± 22 Hz, 1.80 ± 0.21 × 10−6) are characterized by a slightly better reproducibility compared to NAv directly on gold (−424 ± 50 Hz, 2.88 ± 0.62 × 10−6). Overall, the b-BSA/NAv substrate showed more consistent results compared to just NAv on gold. This is likely because NAv molecules bind specifically to the b-BSA. This specific binding reduces the aggregation of NAv molecules on the surface. As for the acoustic ratio, NAv on b-BSA showed a slightly higher value (0.008 ± 0.001) compared to NAv on gold (0.006 ± 0.001). This increase might be due to NAv being more suspended when attached to b-BSA. Moreover, both surfaces were tested for their specificity during liposome injection as well as for their stability upon the addition of a crude LCR mix. For the liposome-based detection of DNA, we used a synthetic ssDNA of 50 nt length modified by a biotin at the 5′ end and cholesterol at the 3′ end. Note that, the addition of the DNA on the NAv-coated sensor resulted in no dissipation or frequency changes, since the DNA concentration was below the threshold for acoustic detection which for the acoustic sensor used here is 25 nM of dsDNA (data not shown). Fig. 2D shows the changes in dissipation during liposome binding as a function of the DNA concentration for the two NAv surfaces. According to this figure, both the overall response and detection limit (50 pM) on the b-BSA/NAv were higher than for the NAv surface (Fig. 2D and S2A†). The difference in the dissipation response was attributed possibly to the presence of the biotin linker in the BSA which may lead to the formation of more suspended and, thus, less sterically hindered NAv molecules above the BSA. Regarding the frequency response, this was proven to be less sensitive than the dissipation (Fig. S2B†). Finally, the surfaces were tested with the addition of a no template control (NTC) LCR mixture. As shown in Fig. 2E & F, the b-BSA/NAv substrate was significantly more stable and reproducible. The addition of the reaction on the NAv-coated surface resulted in partial removal of the NAv and a higher non-specific interaction of the liposomes with the surface (Fig. 2E).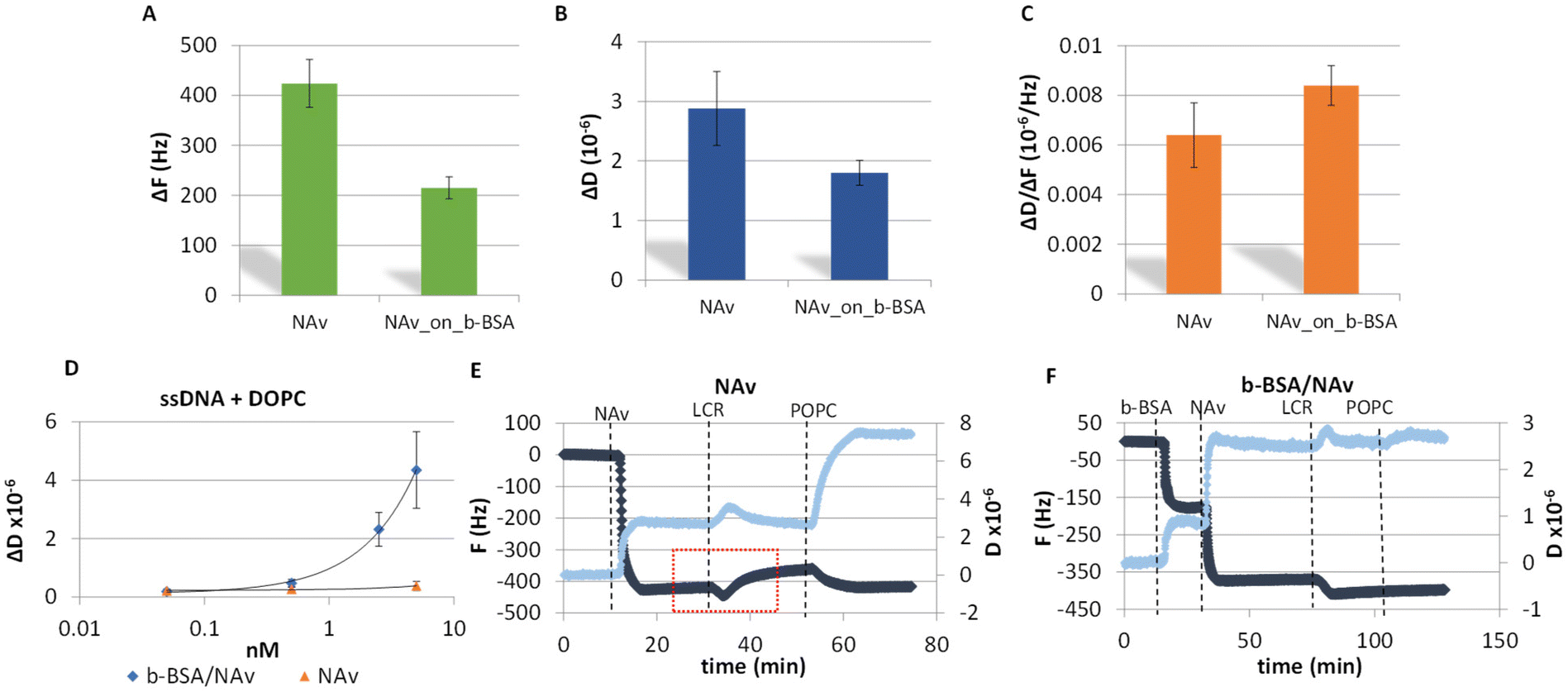 | ||
| Fig. 2 (A) Comparison of ΔF average values of NAv when added directly on gold or a b-BSA pre-coated QCM surface. (B) Same as A but for ΔD. (C) Same as A but for the ΔD/ΔF. (D) Dissipation changes observed upon addition of 200 nm DOPC liposomes on various concentrations of a 50 nt ssDNA pre-attached on NAv and b-BSA/NAv coated surfaces. (E) Investigation of the non-specific binding of the liposomes on the sensor surface. Addition of the LCR reaction on a NAv-modified substrate results in some protein being washed away (red box) leading to the subsequent non-specific binding of liposomes. (F) Same as (E), but for b-BSA/NAv. The b-BSA/NAv substrate remains stable after LCR addition, exhibiting only minor non-specific binding. In both figures, the light and dark blue curves represent changes in dissipation and frequency, respectively. | ||
Sensitivity and specificity of the 99-cycles LCR combined with acoustic detection
As described in section A and shown in Fig. 1, for the acoustic detection we need to use probes modified with biotin and cholesterol for the binding and detection of the LCR products on the acoustic sensor. However, to optimize the assay firstly we tested the sensitivity of the 99 cycles LCR using various amounts of BRAF dsDNA as a template and unmodified oligonucleotides. Using gel electrophoresis, a detection limit of 1.67 × 105 copies of mt DNA was clearly observed, while the same amount of wt copies and NTC controls produced no detectable products (Fig. S3†). Similarly, we investigated the sensitivity and specificity of the 99 cycles LCR of various amounts of mt dsDNA i.e., from 1.00 × 105 to 1.67 × 107 mt molecules and of 1.67 × 106 wt copies as a control and used the acoustic sensor for detection. In this case, probes modified with biotin (p1) and cholesterol (p2) were used for the binding of the LCR products on the b-BSA/NAv-coated resonator and of liposomes on the immobilized DNAs respectively. Firstly, we investigated whether the ΔF and ΔD or the resulted ΔD/ΔF caused by the direct immobilization of the 100 bp LCR product could differentiate between the mt and wt reactions. Results showed no discrimination between the mt and wt LCR reactions as well as from the NTC (Fig. S4†). The recorded changes were related to the non-specific adsorption of the LCR cocktail components (i.e., BSA, ligase, biotinylated probe and its complementary strand, etc.) instead of the biotinylated LCR amplicons. To achieve specific acoustic detection of the amplicons, it was necessary to add 200 nm liposomes which caused significantly higher changes in the acoustic signal in the case of the mt target (Fig. 3A). Fig. 3B, presents the measured ΔD values for a range of wt concentration. Based on this figure, the LOD was estimated at 1.00 × 105 mt copies which resulted in ΔD = 1.24 ± 0.36 × 10−6 compared to the control of 1.67 × 106 wt copies (ΔD = 0.5 ± 0.3 × 10−6). Regarding the other two acoustic responses, i.e., the ΔF and the acoustic ratio, the first although gave the same pattern as the ΔD, was found to be less sensitive with a LOD of 1.67 × 105 copies (Fig. S5†). Finally, the acoustic ratios gave similar values for all the mt and the mt–wt samples (Fig. S5†) as expected since the acoustic ratio indicates the geometry of the bound entity rather than the amount bound to the surface.32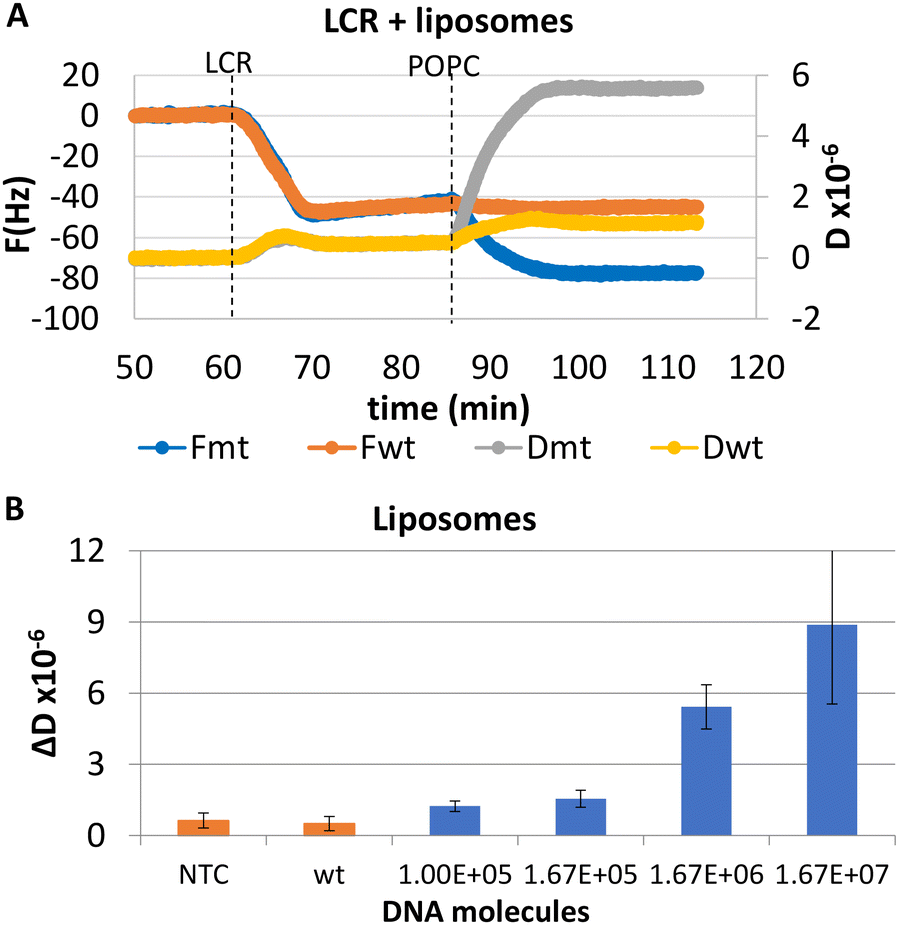 | ||
| Fig. 3 Detection of BRAF V600E point mutation by a 99-cycles exponential LCR. (A) Real time acoustic detection of the 1.67 × 106 mt copies together with a wt control sample (1.67 × 107 DNAs). Only the sample containing the mt gave a signal change upon addition of liposomes. (B) Acoustic detection of LCR of various amounts of mt DNA (1.00 × 105–1.67 × 107) through 200 nm POPC liposomes. Orange columns represent the wt (1.67 × 106) and NTC reactions. | ||
However, the overall efficiency of the assay remained low compared to the number of the DNA molecules that can be presented in real DNA samples provided for SNV detection. For this reason, we tried to further decrease the LOD of the LCR by increasing the cycles to 149 for the detection of 3.3 × 104 mt, 3.3 × 104 wt and NTC samples. Following gel electrophoresis, a background signal of the same molecular weight and intensity as of the expected LCR product was observed not only in the mt but also in the wt and NTC samples. This background signal probably was caused by target-independent blunt-end ligation of the p1/cp1 and p2/cp2 duplexes rather than contamination. Fewer number of cycles, i.e., 125 were also tested and compared to the 99 cycles; background signal was again obtained but only when 125 cycles were performed (Fig. S6†). We conclude that this non-specific signal becomes significant when LCR exceeds the 99 cycles. Our findings confirm previous studies evaluating LCR and the AmpLigase used here, where non-specific ligation became predominant when LCRs of more than a number of cycles were run.11,39,40
Optimized 30-cycles LCR combined with acoustic detection for enhanced sensitivity and specificity
To eliminate blunt-end ligation and increase the efficiency of the overall assay, we redesigned both the LCR and the acoustic method. Regarding LCR, new probes were employed in the reaction where the mismatch discrimination nucleotide of the cp1–cp2 pair of probes was transferred from the 5′ end of cp1 to the 3′ end of cp2 probe; this was based on manufacturer instructions where ligation selectivity increases when the mismatch discrimination nucleotide is at the 3′ end. Moreover, assuming that the cholesterol in the p2-probe may affect the efficiency of the LCR reaction since cholesterol tends to aggregate, we removed the cholesterol from the corresponding probe and instead a 18 nt non-complementary to the target or the probes DNA tail was added at the 3′ end of the p2 probe. Cholesterol is added during the acoustic assay following LCR's injection through a 20 nt cholesterol-modified DNA complementary to the 18nt tail (Fig. 4A).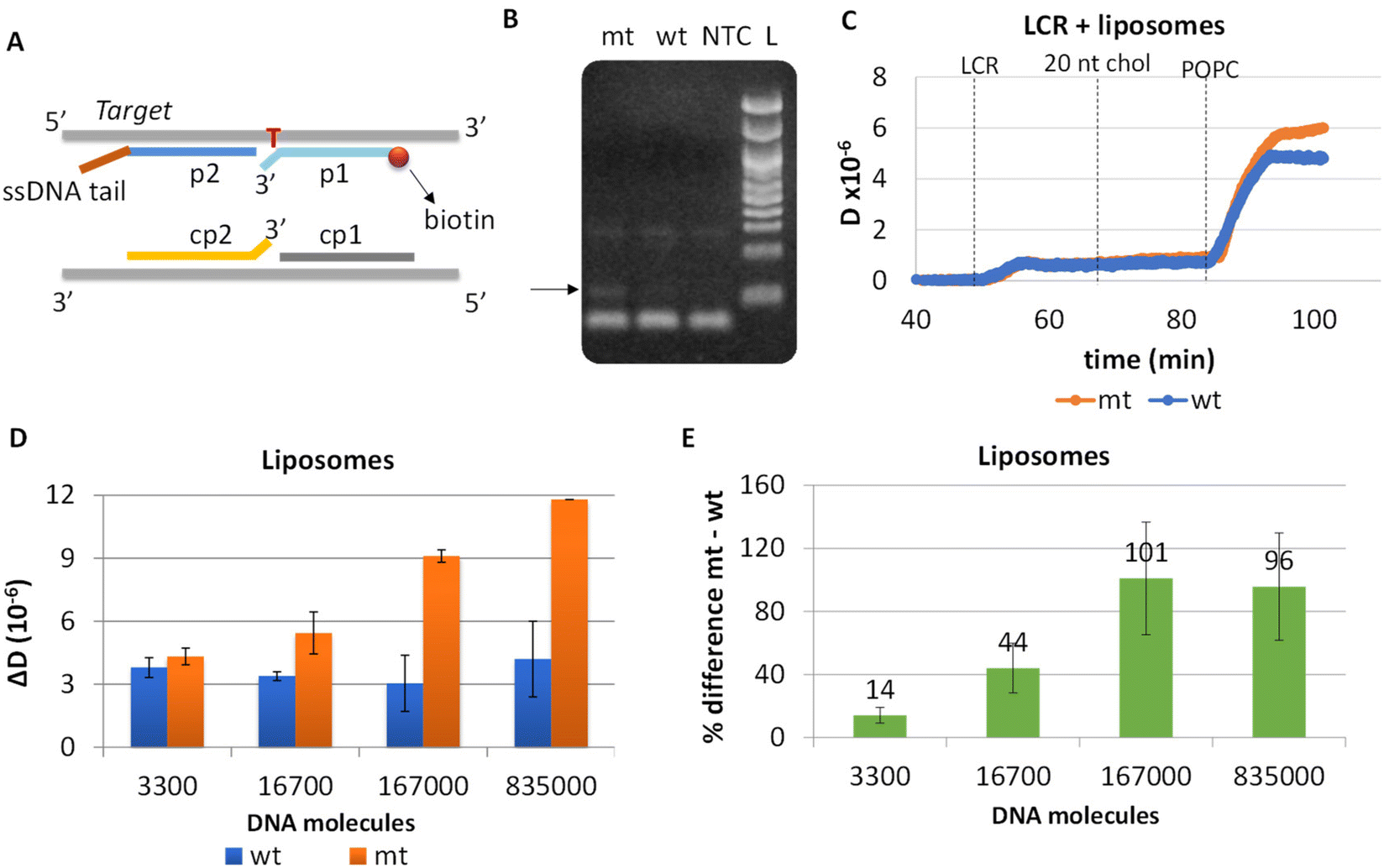 | ||
| Fig. 4 (A) Illustration of the new probe design. In both pairs of probes (p1–p2 & cp1–cp2), the mismatch discrimination nucleotide is located at the 3′ end. In addition, p2 probe was modified by a ssDNA tail complementary to the 20 nt chol DNA and non-complementary to the target. (B) LCR's gel electrophoresis of 1.67 × 105 BRAF V600E (mt) copies, 3.34 × 105 wt and no template control (NTC). The black arrow shows the ∼100 bp LCR product. A 100 bp DNA ladder (L) was used. (C) Real time acoustic detection of an LCR of 3300 mt copies together with a wt control (Cwt = 2 × Cmt). (D) Comparison of ΔD changes observed between the mt/wt reactions following the capturing of POPC liposomes on various LCR reactions and their corresponding wt controls. (E) % difference between mt and wt reactions of the same LCR preparation and acoustic experiment. | ||
Apart from the probes, LCR's conditions (ligase and probes concentration, ligation temperature and time and no of cycles) were optimized and the products were analyzed by gel electrophoresis. Finally, we ended-up with a 30-repeats protocol of denaturation at 92 °C for 5 s and 5 s ligation at 65 °C. By this protocol, 1.67 × 105 mt molecules were successfully detected when compared with 3.34 × 105 wt copies; visualized products were observed exclusively in the case of mt target, indicating the high specificity of the assay (Fig. 4B). Interestingly, despite the improvements in the ligation protocol, template-independent ligation again appeared when >30 cycles were performed (Fig. S7†). It is worth mentioning, that as the number of cycles increases up to 30, LCR by-products are also observed at higher molecular weights (∼300 bp).
LCR was then combined with the improved acoustic method. As described above, for the capturing of the liposomes a 20 nt cholesterol-modified probe (20 nt chol-probe) was firstly added in high concentration (500 nM). To verify the specificity of the new detection approach, we flowed the 20 nt chol-probe through the b-BSA/NAv coated surface followed by the addition of liposomes. This step caused no significant ΔD or ΔF changes (0.25 × 10−6 and 2.3 Hz, respectively), confirming the specificity of the acoustic approach (Fig. S8†). LCRs were performed for several concentrations of mt template, i.e., 3.30 × 103, 1.67 × 104, 1.67 × 105 & 8.35 × 105 number of mt molecules (Cmt). As a control, samples containing only the wt target in a concentration of Cwt = 2 × Cmt were used. Acoustic analysis of the LCR products, without the addition of liposomes, resulted in no considerable changes in the generated acoustic values (ΔD, ΔF and ΔD/ΔF) between the mt samples and their corresponding wt controls (Fig. S9†). However, upon addition of the cholesterol probe and the liposomes, a significant ΔD and ΔF change were measured. Note that, although the wt control gave a background signal, for the same LCR preparation and the same acoustic experiment the method's detection limit was 3.30 × 103 copies of mutant target (N = 6) since the mt could always be detected in the real time graph (Fig. 4C). This low detection capability is not clearly reflected when the average ΔD response is plotted (Fig. 4D). In addition, as expected for a higher amount of starting mt template a greater ΔD signal was measured. Regarding frequency response, the ΔF was shown to be less sensitive and with large error bars (Fig. S10A†). For better representation of the above results, the % difference 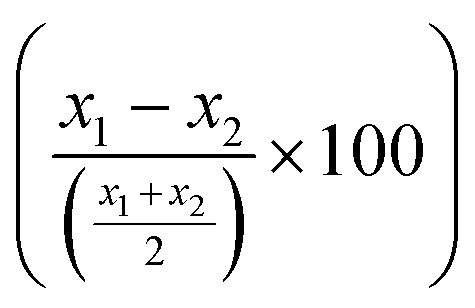 of the ΔD (Fig. 4E) and ΔF (Fig. S10B†) between the same set of mt/wt samples was calculated and plotted. Overall, the new protocol showed a significant improvement in the detection efficiency achieving a detection limit that is two orders of magnitude lower after only 30 cycles of LCR and acoustic detection through liposomes. It's important to note that this enhanced detection capability is not due to the changes made to the LCR protocol itself; when comparing the sensitivity of the new LCR protocol to the old one using gel electrophoresis, both protocols showed equal sensitivity, detecting as low as 1.67 × 105 copies of the BRAF V600E mt gene. Instead, the lower detection limit stems from advancements in the acoustic detection technique, specifically the introduction of the cholesterol probe during the acoustic detection to bind the highly dissipative liposomes. It is worth mentioning that the background signal produced from the wt samples remained constant regardless of the initial amount of the wt DNA target used (Fig. 4D). The constant background suggested that a significant amount of the LCR products was produced through non-specific and template-independent ligation; acoustic analysis of NTC LCRs created a ΔD of 3.05 ± 0.77 (N = 4). Specifically, the average acoustic signal of all the wt reactions of 6.6 × 103–1.67 × 106 copies prior to LCR was calculated at ΔD = 3.57 ± 0.51 × 10−6, quite similar with those derived from negative non-template control LCRs (NTC) which was ΔD = 3.05 ± 0.77 × 10−6. The above result demonstrated that the LCR by-products are mainly formed through target-independent ligation, which is one of the method's shortcomings.
of the ΔD (Fig. 4E) and ΔF (Fig. S10B†) between the same set of mt/wt samples was calculated and plotted. Overall, the new protocol showed a significant improvement in the detection efficiency achieving a detection limit that is two orders of magnitude lower after only 30 cycles of LCR and acoustic detection through liposomes. It's important to note that this enhanced detection capability is not due to the changes made to the LCR protocol itself; when comparing the sensitivity of the new LCR protocol to the old one using gel electrophoresis, both protocols showed equal sensitivity, detecting as low as 1.67 × 105 copies of the BRAF V600E mt gene. Instead, the lower detection limit stems from advancements in the acoustic detection technique, specifically the introduction of the cholesterol probe during the acoustic detection to bind the highly dissipative liposomes. It is worth mentioning that the background signal produced from the wt samples remained constant regardless of the initial amount of the wt DNA target used (Fig. 4D). The constant background suggested that a significant amount of the LCR products was produced through non-specific and template-independent ligation; acoustic analysis of NTC LCRs created a ΔD of 3.05 ± 0.77 (N = 4). Specifically, the average acoustic signal of all the wt reactions of 6.6 × 103–1.67 × 106 copies prior to LCR was calculated at ΔD = 3.57 ± 0.51 × 10−6, quite similar with those derived from negative non-template control LCRs (NTC) which was ΔD = 3.05 ± 0.77 × 10−6. The above result demonstrated that the LCR by-products are mainly formed through target-independent ligation, which is one of the method's shortcomings.
LCR combined with a microfluidic fluidized bed DNA extraction technology
The final goal of this work was to assess the capability of the method to detect DNA carrying single nucleotide variants in patients’ crude samples. For this reason, the performance of our assay was tested in plasma samples. Moreover, in order to automate this part of the procedure the method was combined with a new DNA extraction technology based on the use of a microfluidic fluidized bed (Fig. 5A).41 Briefly, this method could be especially advantageous for the capturing of DNA since it is characterized by high surface to volume ratio and constant mixing potentially enhancing capture efficiency.42 Most importantly, it is possible the extraction of only the DNA targets of interest with fewer steps and limited manual handling and sample transfer compared to the commercial extraction kits, thus decreasing both the overall process complexity and the risk of cross contamination.43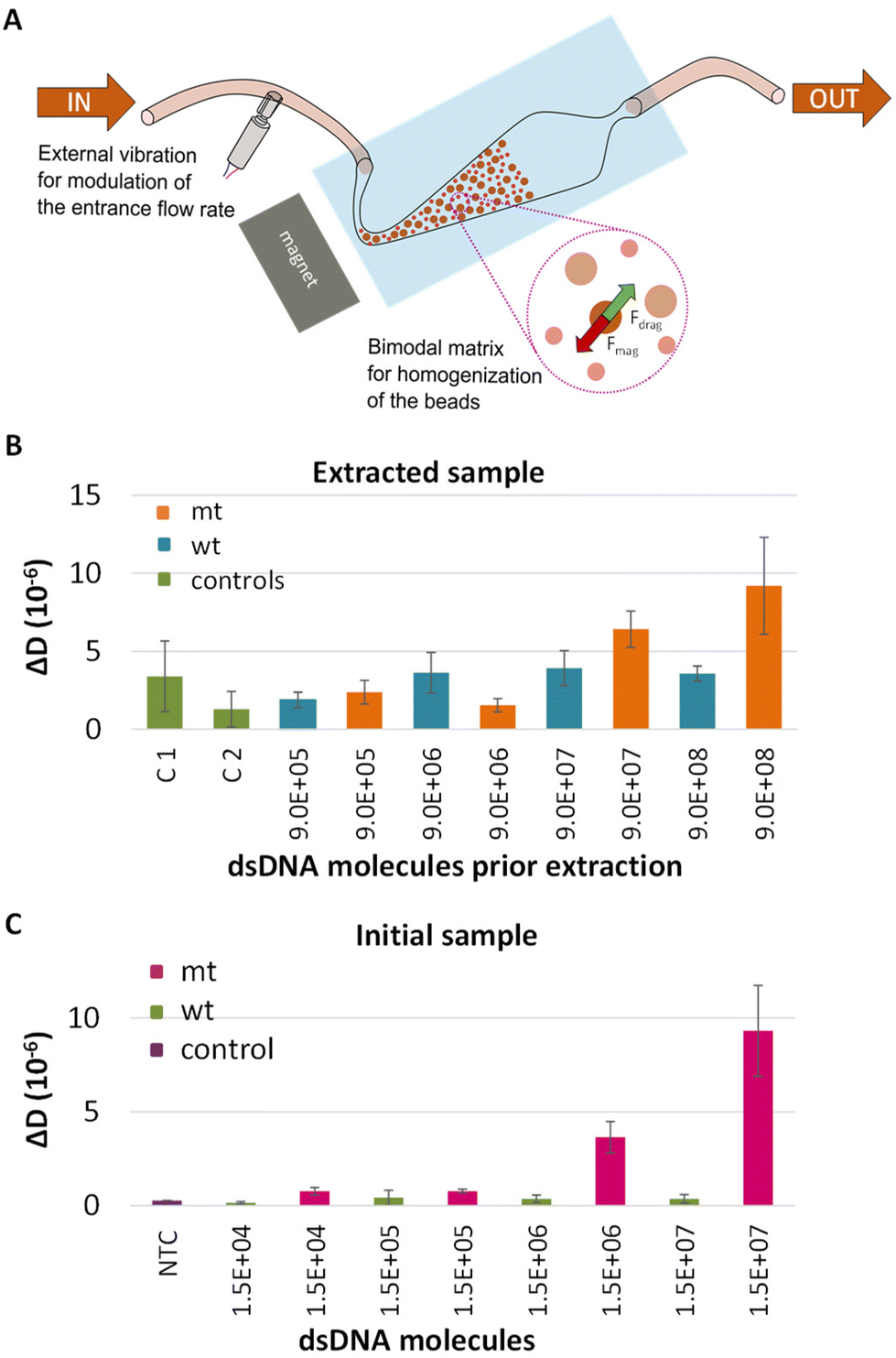 | ||
| Fig. 5 (A) Schematic representation of the fluidized bed with a close-up view of the bimodal matrix of beads and the equilibrium between the drag forces (Fdrag) and the magnetic forces (Fmag) applied on the beads. (B) Comparison of ΔD changes recorded at the POPC liposome step during the acoustic analysis of LCR derived from dsDNA BRAF mt/wt targets spiked in human plasma samples following capturing on FB and (C) Same as (B) but for LCR derived from 2.5 μL human plasma samples spiked in with dsDNA 277 bp BRAF mt/wt targets at various concentrations ranging from 10 fM–10 pM (otherwise 1.5 × 104–1.5 × 107 copies). C1 and C2 correspond to negative control plasma samples with and without capture probes, while NTC to a LCR with plasma sample without DNA. | ||
The characteristics of the FB and the experimental process are described in detail in Alexandre L. et al., 2023.41 Here, plasma was spiked in with 10 fM (9 × 105 copies)–10 pM (9 × 108 copies) synthetic mt or wt BRAF dsDNA and processed with the fluidized bed. Following the capturing on magnetic beads, the beads were collected, heated to release the target and the sample was analyzed by LCR and acoustic detection. In all cases the capture efficiency was expected to be more than 70% (Fig. S11†). To verify the specificity of the technique, except from the wt control, negative controls, including plasma samples without any DNA template, passed from the FB. ΔD and ΔF values were measured at the liposome step and presented in Fig. 5B and Fig. S12,† respectively. According to the plot, a detection limit of 1 pM (9 × 107 copies) mt target was recorded when compared to the corresponding wt control. As a control, we also analyzed LCRs derived from 2.5 μL initial sample i.e., plasma spiked in with mt or wt dsDNA 277 bp at a concentration of 10 fM–10 pM. While LCR's detection limit was the 3.3 × 103 copies when target was spiked in buffer, the overall efficiency of the assay was negatively affected by the sample source, since only the 1.5 × 106–1.5 × 107 (otherwise 2.5 μL × 1 pM–2.5 μL × 10 pM) were detected (Fig. 5C). Apart from the sample source, the high detection limit of the assay could be attributed to various parameters such as the capture and the release efficiency of the target. While, more research is required for the application of the overall method, i.e., FB combined with LCR to blood samples for ctDNA extraction, the proposed technique has potential for the extraction and detection of other kinds of targets like miRNAs presented in plasma or serum, DNA from pathogens or human genomic DNA for the determination of single nucleotide polymorphisms linked with genetic diseases. Regarding FB, notable advancements of this technology are the ability to extract in a fast and simple manner only the DNA targets of interest from a heterogonous DNA population; work with crude samples like serum, plasma etc.; and enrich and concentrate specific areas of the DNA target. All the above could significantly favor the downstream analysis.
Comparison of the combined LCR/acoustic method to current state-of-the-art LCRs and ligased-based biosensors
Although LCR is often beneficial for allelic discrimination, its inherent limitations, such as the target-independent ligation, reduce its effectiveness in accurately detecting SNVs or cancerous point mutations, particularly when these are present in extremely low quantities. In a large variety of publications, to achieve a better detection limit, LCR is either combined with other amplification techniques like typical PCR,2 qPCR4 and Rolling Cycle Amplification (RCA)7,11 or is converted to ligase-based detection techniques consisted of a preamplification PCR step followed by Ligase Detection Reaction (LDR),6,44,45 oligonucleotide ligation assay (OLA)10 or gap-LCR,5 thus increasing the overall complexity of the assay. To our knowledge, the best-reported detection limit of ligase chain reaction is the 120 copies, where 160 LCR cycles are combined with FRET real-time detection.46 Note that, in this approach non-specific signal is also detected in the wt and blank controls, however, in lower values compared to the mt target. Here we report a detection limit of 3300 copies or 220 aM or 5.5 zmol using only 30 cycles of LCR and liposome amplification and in a total duration of ∼2.7 hours. Our method carries a better detection efficiency among improved variations of LCR, such as LCR coupled with RCA & fluorescence detection (220 aM vs. 1 fM),11 Gap-LCR coupled with quantum dots detection (5.5 zmol vs. 170 zmol),47 PCR followed by Ligase Detection Reaction (LDR) and Capillary gel electrophoresis (CGE) – Laser induced fluorescence (LIF) detection (220 aM vs. 0.1 nM)6 and PCR followed by Oligonucleotide Ligation Assay (OLA) and electrochemilluminescence (ECL) (5.5 zmol vs. 10 fmol).48 Finally, LCR and acoustic detection had a significantly improved detection limit (220 aM) compared to surface ligation biosensing approaches combined with an enhancing step like gold nanoparticles,21 silver deposition20 or enzymatic amplification (horse peroxidase)22 (0.9 pM, 80 fM and 1 pM, respectively). Note that single nucleotide mutations of less than 100 copies are detected only through LCR coupled with a 2nd amplification molecular reaction like Hyperbranch-RCA or qPCR, which is one of the method's limitations. Last but not least, compared to other DNA array or biosensing platforms usually employed in DNA detection, the proposed acoustic technology allows the processing of non-purified samples and avoids the issues arising during the detection of dsDNA targets, i.e., the need for sample heating and the prevention of DNA strands reassociation.Conclusions
In this work, we report the combination of the LCR with acoustic detection for the analysis of SNVs. We applied the method for the detection of the V600E point mutation in the BRAF gene reaching a LOD of 3300 copies or 220 aM or 5.5 zmol. We tried to overcome the general low efficiency of the LCR by developing a well optimized detection surface to capture the DNA amplicons as well as employing a 2nd amplification step during the acoustic analysis based on liposomes. By this addition, the detection limit was improved by two orders of magnitude compared to traditional gel electrophoresis analysis. Furthermore, we combined LCR and acoustic detection with a cutting-edge DNA extraction technique based on the microfluidic fluidized bed, allowing us to detect the BRAF target in real plasma samples. Overall, we faced limitations due to target-independent ligation and overall LCR's low efficiency, consistent with previous reports in the bibliography.2 While the application of LCR in certain domains, such as the highly demanding field of liquid biopsy, may pose challenges, it demonstrates excellent potential for analyzing other point mutations prevalent in genetic diseases and pathogens providing high level specificity and reliable results in the fields of precision medicine and diagnostics.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the European Union's Horizon H2020-FETOPEN-1-2016-2017 under grant agreement no. 737212 (CATCH-U-DNA).References
- Y. Li, X. Wang, M. Wang, M. Liu, H. Wang and W. Xia, et al. Advances in ligase-based nucleic acid amplification technology for detecting gene mutations: a review, in Molecular and Cellular Biochemistry, Springer, 2023. Vol. 478, pp. 1621–1631 Search PubMed.
- A. A. Gibriel and O. Adel, Advances in ligase chain reaction and ligation-based amplifications for genotyping assays: Detection and applications, in Mutation Research - Reviews in Mutation Research, Elsevier B.V., 2017, Vol. 773, pp. 66–90. DOI:10.1016/j.mrrev.2017.05.001.
- Z. J. Liu, L. Y. Yang, Q. X. Wei, C. L. Ye, X. W. Xu and G. X. Zhong, et al., A novel ligase chain reaction-based electrochemical biosensing strategy for highly sensitive point mutation detection from human whole blood, Talanta, 2020, 216, 120966 CrossRef CAS PubMed.
- P. Yi, H. Jiang, L. Li, F. Dai, Y. Zheng and J. Han, et al. A New Genotyping Method for Detecting Low Abundance Single Nucleotide Mutations Based on Gap Ligase Chain Reaction and Quantitative PCR Assay, Cell Biochem. Biophys., 2012, 62(1), 161–167 CrossRef CAS PubMed.
- S. Jenner and D. Techel, Development of a gLCR-based KRAS mutation detection approach and its comparison with other screening methods, Tumor Biol., 2015, 36(8), 6361–6368 CrossRef CAS PubMed.
- M. Hamada, K. Shimase, K. Tsukagoshi and M. Hashimoto, Discriminative detection of low-abundance point mutations using a PCR/ligase detection reaction/capillary gel electrophoresis method and fluorescence dual-channel monitoring, Electrophoresis, 2014, 35(8), 1204–1210 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, L. Huang, Y. Zhang, Y. Li and S. Ding, et al., Ultrasensitive biosensing of low abundance BRAF V600E mutation in real samples by coupling dual padlock-gap-ligase chain reaction with hyperbranched rolling circle amplification, Sens. Actuators, B, 2019, 287, 111–117 CrossRef CAS.
- H. Wang, H. Wang, Y. Sun, X. Liu, Y. Liu and C. Wang, et al., A general strategy for highly sensitive analysis of genetic biomarkers at single-base resolution with ligase-based isothermally exponential amplification, Talanta, 2020, 212, 120754 CrossRef CAS PubMed.
- Y. Sun, B. Han and F. Sun, Ultra-specific genotyping of single nucleotide variants by ligase-based loop-mediated isothermal amplification coupled with a modified ligation probe, RSC Adv., 2021, 11(28), 17058–17063 RSC.
- M. H. Bui, G. G. Stone, A. M. Nilius, L. Almer and R. K. Flamm, PCR-oligonucleotide ligation assay for detection of point mutations associated with quinolone resistance in Streptococcus pneumoniae, Antimicrob. Agents Chemother., 2003, 47(4), 1456–1459 CrossRef CAS PubMed.
- Y. Cheng, J. Zhao, H. Jia, Z. Yuan and Z. Li, Ligase chain reaction coupled with rolling circle amplification for high sensitivity detection of single nucleotide polymorphisms, Analyst, 2013, 138(10), 2958–2963 RSC.
- J. D. Davis, P. K. Riley, C. W. Peters and K. H. Rand, A comparison of ligase chain reaction to polymerase chain reaction in the detection of Chlamydia trachomatis endocervical infections, Infect. Dis. Obstet. Gynecol., 1998, 6(2), 57–60 CAS.
- T. Kanagawa, Bias and artifacts in multitemplate polymerase chain reactions (PCR), J. Biosci. Bioeng., 2003, 96(4), 317–323 CrossRef CAS PubMed.
- M. F. Polz and C. M. Cavanaugh, Bias in template-to-product ratios in multitemplate PCR, Appl. Environ. Microbiol., 1998, 64(10), 3724–3730 CrossRef CAS PubMed.
- S. G. Acinas, R. Sarma-Rupavtarm, V. Klepac-Ceraj and M. F. Polz, PCR-induced sequence artifacts and bias: Insights from comparison of two 16s rRNA clone libraries constructed from the same sample, Appl. Environ. Microbiol., 2005, 71(12), 8966–8969 CrossRef CAS PubMed.
- A. Psifidi, C. Dovas and G. Banos, Novel quantitative real-time LCR for the sensitive detection of SNP frequencies in pooled DNA: Method development, evaluation and application, PLoS One, 2011, 6(1), 1–11 CrossRef PubMed.
- Z. Yuan, Y. Zhou, S. Gao, Y. Cheng and Z. Li, Homogeneous and sensitive detection of microRNA with ligase chain reaction and lambda exonuclease-Assisted cationic conjugated polymer biosensing, ACS Appl. Mater. Interfaces, 2014, 6(9), 6181–6185 CrossRef CAS PubMed.
- K. Feng, J. Li, J. H. Jiang, G. L. Shen and R. Q. Yu, QCM detection of DNA targets with single-base mutation based on DNA ligase reaction and biocatalyzed deposition amplification, Biosens. Bioelectron., 2007, 22(8), 1651–1657 CrossRef CAS PubMed.
- Z. S. Wu, J. H. Jiang, G. L. Shen and R. Q. Yu, Highly Sensitive DNA Detection and Point Mutation Identification : An Electrochemical Approach Based on the Combined Use of Ligase and Reverse Molecular Beacon, Hum. Mutat., 2006, 28(6), 630–637 CrossRef PubMed.
- P. Zhang, X. Chu, X. Xu, G. Shen and R. Yu, Electrochemical detection of point mutation based on surface ligation reaction and biometallization, Biosens. Bioelectron., 2008, 23(10), 1435–1441 CrossRef CAS PubMed.
- Q. Wang, L. Yang, X. Yang, K. Wang, L. He and J. Zhu, Electrochemical biosensors for detection of point mutation based on surface ligation reaction and oligonucleotides modified gold nanoparticles, Anal. Chim. Acta, 2011, 688(2), 163–167, DOI:10.1016/j.aca.2011.01.004.
- Q. Xu, K. Chang, W. Lu, W. Chen, Y. Ding and S. Jia, et al., Detection of single-nucleotide polymorphisms with novel leaky surface acoustic wave biosensors, DNA ligation and enzymatic signal amplification, Biosens. Bioelectron., 2012, 33, 274–278 CrossRef CAS PubMed.
- F. Hu, W. Zhang, W. Meng, Y. Ma, X. Zhang and Y. Xu, et al., Ferrocene-labeled and purification-free electrochemical biosensor based on ligase chain reaction for ultrasensitive single nucleotide polymorphism detection, Anal. Chim. Acta, 2020, 1109, 9–18 CrossRef CAS PubMed.
- W. Zhang, F. Hu, X. Zhang, W. Meng, Y. Zhang and Y. Song, et al., Ligase chain reaction-based electrochemical biosensor for the ultrasensitive and specific detection of single nucleotide polymorphisms, New J. Chem., 2019, 43(36), 14327–14335 RSC.
- K. Mitsakakis, A. Tsortos and E. Gizeli, Quantitative determination of protein molecular weight with an acoustic sensor; significance of specific versus non-specific binding, Analyst, 2014, 139(16), 3918–3925 RSC.
- I. Reviakine, D. Johannsmann and R. P. Richter, Hearing What You Cannot See and Visualizing What You Hear, 2011 Search PubMed.
- V. Raptis, A. Tsortos and E. Gizeli, Theoretical Aspects of a Discrete-Binding Approach in Quartz-Crystal Microbalance Acoustic Biosensing, Phys. Rev. Appl., 2019, 11(3), 1 Search PubMed.
- A. Vázquez-quesada, M. M. Schofield, A. Tsortos, P. Mateos-gil, D. Milioni and E. Gizeli, et al., Hydrodynamics of Quartz-Crystal-Microbalance DNA Sensors Based on Liposome Amplifiers, Phys. Rev. Appl., 2020, 10(1), 1 Search PubMed.
- A. Tsortos, G. Papadakis, K. Mitsakakis, K. A. Melzak and E. Gizeli, Quantitative determination of size and shape of surface-bound DNA using an acoustic wave sensor, Biophys. J., 2008, 94(7), 2706–2715 CrossRef CAS PubMed.
- G. Papadakis, A. Tsortos and E. Gizeli, Acoustic characterization of nanoswitch structures: Application to the DNA holliday junction, Nano Lett., 2010, 10(12), 5093–5097 CrossRef CAS PubMed.
- A. Tsortos, G. Papadakis and E. Gizeli, On the Hydrodynamic Nature of DNA Acoustic Sensing, Anal. Chem., 2016, 88(12), 6472–6478 CrossRef CAS PubMed.
- A. Tsortos, A. Grammoustianou, R. Lymbouridou, G. Papadakis and E. Gizeli, Detection of multiple DNA targets with a single probe using a conformation-sensitive acoustic sensor, Chem. Commun., 2015, 57, 11504–11507 RSC.
- N. Naoumi, K. Michaelidou, G. Papadakis, A. E. Simaiaki, R. Fernández and M. Calero, et al., Acoustic Array Biochip Combined with Allele-Specific PCR for Multiple Cancer Mutation Analysis in Tissue and Liquid Biopsy, ACS Sens., 2022, 7(2), 495–503 CrossRef CAS PubMed.
- D. Milioni, P. Mateos-Gil, G. Papadakis, A. Tsortos, O. Sarlidou and E. Gizeli, Acoustic Methodology for Selecting Highly Dissipative Probes for Ultrasensitive DNA Detection, Anal. Chem., 2020, 92(12), 8186–8193 CrossRef CAS PubMed.
- E. Reimhult, F. Höök and B. Kasemo, Intact vesicle adsorption and supported biomembrane formation from vesicles in solution: Influence of surface chemistry, vesicle size, temperature, and osmotic pressure, Langmuir, 2003, 19(5), 1681–1691 CrossRef CAS.
- K. A. Melzak, F. Bender, A. Tsortos and E. Gizeli, Probing mechanical properties of liposomes using acoustic sensors, Langmuir, 2008, 24(16), 9172–9180 CrossRef CAS PubMed.
- B. J. Frisken, C. Asman and P. J. Patty, Studies of vesicle extrusion, Langmuir, 2000, 16(3), 928–933 CrossRef CAS.
- J. J. J. Gillissen, J. A. Jackman, S. R. Tabaei and N. J. Cho, A Numerical Study on the Effect of Particle Surface Coverage on the Quartz Crystal Microbalance Response, Anal. Chem., 2018, 90(3), 2238–2245 CrossRef CAS PubMed.
- K. Abravaya, J. J. Carrino, S. Muldoon and H. H. Lee, Detection of point mutations with a modified ligase chain reaction (gap-LCR), Nucleic Acids Res., 1995, 23(4), 675–682 CrossRef CAS PubMed.
- I. Kälin, S. Shephard and U. Candrian, Evaluation of the ligase chain reaction (LCR) for the detection of points mutation, Mutat. Res. Lett., 1992, 283(2), 119–123 CrossRef PubMed.
- L. Alexandre, M. Araya-Farias, M. L. Nguyen, N. Naoumi, G. Gropplero and E. Gizeli, et al., High-throughput extraction on a dynamic solid phase for low-abundance biomarker isolation from biological samples, Microsyst. Nanoeng., 2023, 9(1), 109 CrossRef CAS PubMed.
- I. Pereiro, S. Tabnaoui, M. Fermigier, O. Du Roure, S. Descroix and J. L. Viovy, et al., Magnetic fluidized bed for solid phase extraction in microfluidic systems, Lab Chip, 2017, 17(9), 1603–1615 RSC.
- H. Lee, C. Park, W. Na, K. H. Park and S. Shin, Precision cell-free DNA extraction for liquid biopsy by integrated microfluidics, npj Precis. Oncol., 2020, 4(1), 3 CrossRef CAS PubMed.
- P. Yi, W. Lu, J. Guo, Q. Liu, Z. Chen and J. Han, et al., Development of a PCR/Ligase Detection Reaction/Nanogold-Based Universal Array Approach for the Detection of Low-Abundant DNA Point Mutations, Cell Biochem. Biophys., 2011, 61(3), 629–636 CrossRef CAS PubMed.
- W. Choi, G. W. Shin, H. S. Hwang, S. P. Pack, G. Y. Jung and G. Y. Jung, A multiplex single nucleotide polymorphism genotyping method using ligase-based mismatch discrimination and CE-SSCP, Electrophoresis, 2014, 35(8), 1196–1203 CrossRef CAS PubMed.
- Y. Sun, X. Lu, F. Su, L. Wang, C. Liu and X. Duan, et al., Real-time fluorescence ligase chain reaction for sensitive detection of single nucleotide polymorphism based on fluorescence resonance energy transfer, Biosens. Bioelectron., 2015, 74, 705–710 CrossRef CAS PubMed.
- Y. Song, Y. Zhang and T. H. Wang, Single Quantum Dot Analysis Enables Multiplexed Point Mutation Detection by Gap Ligase Chain Reaction, Small, 2013, 9(7), 1096–1105 CrossRef CAS PubMed.
- H. Zhou, D. Xing, D. Zhu and X. Zhou, Rapid and sensitive detection of point mutation by DNA ligase-based electrochemiluminescence assay, Talanta, 2009, 78(4–5), 1253–1258 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further technical and analytical details on devices and assays, respectively; real-time acoustic graphs; frequency and dissipation data during direct detection of target-DNA (no liposomes); gel images of LCR products; and the frequency responses obtained during the liposome step. See DOI: https://doi.org/10.1039/d3an02142d |
| This journal is © The Royal Society of Chemistry 2024 |