DOI:
10.1039/D3AN02109B
(Paper)
Analyst, 2024,
149, 2664-2670
Liquid electron ionization-mass spectrometry as a novel strategy for integrating normal-phase liquid chromatography with low and high-resolution mass spectrometry†
Received
6th December 2023
, Accepted 10th February 2024
First published on 16th February 2024
Abstract
Normal-phase liquid chromatography (NPLC) plays a pivotal role in the rapid separation of non-polar compounds, facilitating isomer separation and finding applications in various crucial areas where aprotic solvents are necessary. Similar to reversed-phase liquid chromatography (RPLC), NPLC requires a robust and sensitive detector to unequivocally identify the analytes, such as a mass spectrometer. However, coupling NPLC with mass spectrometry (MS) poses challenges due to the incompatibility between the non-polar solvents used as the mobile phase and the primary ionization techniques employed in MS. Several analytical methods have been developed to combine NPLC with electrospray ionization (ESI), but these methods are restricted to the analysis of polar compounds. In most cases, atmospheric pressure chemical ionization (APCI) becomes necessary to expand the range of analysis applications. To overcome these limitations and fully realize the potential of NPLC-MS coupling, a technique termed liquid electron ionization-mass spectrometry (LEI-MS) can be used. LEI-MS offers a straightforward solution by enabling the effective coupling of NPLC with both low and high-resolution MS. LEI allows for the comprehensive analysis of non-polar compounds and provides a powerful tool for isomer separation and precise identification of analytes. Optimal separations, mass spectral qualities, and matches with the NIST library were obtained in both configurations, demonstrating the potential of the proposed approach.
Introduction
Liquid chromatography represents one of the most versatile analytical techniques for the separation and identification of analytes, even with vastly different chemical–physical characteristics. This versatility stems from the various combinations that can be utilized with different stationary and mobile phases. Among the different types, reversed-phase liquid chromatography (RPLC) has gained immense popularity due to its ability to address a wide range of applications. In RPLC, the stationary phase is predominantly non-polar while the mobile phase exhibits varying degrees of polarity. Despite being the earliest developed technique, normal-phase liquid chromatography (NPLC) has lost some of its analytical appeal compared to RPLC. However, NPLC, which relies on distinct retention mechanisms, can offer superior selectivity and resolution in specific applications.1 This makes it particularly advantageous when analytes are poorly separated by RPLC or when hydrophobic impurities, retained strongly in RPLC, need to be swiftly eluted to focus on the target analytes.2 In some cases, NPLC and RPLC can be combined, such as in the two-dimensional liquid chromatography approach.3,4 To achieve optimal separation of the target compounds, NPLC employs polar stationary phases such as silica-based, alumina, zirconia, or polar-embedded stationary phases. The mobile phases consist of a combination of slightly polar and non-polar solvents.5–7 Commonly used mobile phases in NPLC include hexane, heptane, ethyl acetate, and dichloromethane, which are often mixed and used in isocratic mode. This approach saves time and solvents by eliminating the need for column conditioning after consecutive analyses. Moreover, the use of non-polar solvents facilitates the solubilization of complex real samples. This enhances the loading capacity and generates low back pressure, enabling high flow rates for fast analysis time. One limitation of NPLC-based analytical methods is the inability to take full advantage of sensitive and specific detectors. UV absorbance spectrophotometric systems are commonly used in conjunction with NPLC, especially when highly non-polar solvents are employed as the mobile phase.8–11 However, these devices have limited identification capabilities and lower sensitivity compared to mass spectrometric detectors. To address these limitations, the integration of NPLC and MS (NPLC-MS) has provided high sensitivity, selectivity, and capability to ionize a diverse array of compounds.12–15 Among the various ionization techniques available, electrospray ionization (ESI) is commonly used to generate gas-phase ions from solution-phase samples. However, ESI can be challenging when low-polarity mobile phases are used, such as those typically employed in NPLC.16 As a result, atmospheric pressure chemical ionization (APCI)17–22 and atmospheric pressure photoionization (APPI)23 are preferred techniques. The use of NPLC-MS has significantly increased in various fields, including environmental, food, pharmaceutical analyses, and metabolomics. However, in terms of identification, there are currently no LC-MS spectral libraries comparable to those available for GC-MS based on electron ionization (EI). This poses limitations on qualitative analysis. This study aims to explore the possibility of coupling NPLC with EI-MS (NPLC-EI-MS). The advantages of NPLC-EI-MS can include fast separation of compounds, isomer separation, and the generation of EI spectra. Low- and high-resolution instrumentation (HRMS) were tested to assess selected ion monitoring (SIM), multiple reaction monitoring (MRM), and high-resolution full scan detection. The study utilizes an LC-MS interface called the liquid electron ionization interface (LEI), which converts the liquid phase into a gas phase for analysis in a mass spectrometer equipped with an EI ion source. The LEI interface enables targeted and untargeted identification by extending access to EI libraries. A more detailed description of the LEI concept has been discussed elsewhere.24–26 To test the proof-of-concept of NPLC-LEI-MS coupling, a selection of cannabinoids, tocopherols, phenols, and phthalates were used as model compounds due to their optimal response in NPLC and analytical relevance.27–39 Encouraging preliminary data demonstrate that NPLC-LEI-MS is a reliable alternative approach, providing high selectivity, resolution, and structural information of analytes. Preliminary experiments focused on acquiring full scan spectra and MRM transitions of the selected compounds using a triple quadrupole mass spectrometer. The compound mixtures were separated using selected ion monitoring (SIM), MRM, and HRMS modes, employing a UHPLC pump and a silica column in isocratic mode to increase sample throughput. Two different low- and high-resolution instruments were utilized to test the performance of NPLC-LEI-MS with two EI sources of different geometry. To the best of our knowledge, the experiments described in this study represent the first attempt to use non-polar LC mobile phases with EI-MS. The obtained results open new possibilities in coupling NPLC with MS and offer the potential for developing alternative methods to RPLC in the analysis of real samples, particularly in cases involving poor analyte resolution or highly hydrophobic impurities.
Material and methods
Standards and reagents
LC-MS grade hexane (HEX), isopropanol (IPA), ethyl acetate, ethanol (EtOH) and formic acid were purchased from VWR International, part of Avantor (Milan, Italy). Standards of (+)-α-tocopherol and (+)-δ-tocopherol (purity ≥98%) were purchased from Sigma-Aldrich (Milan, Italy), (+)-β-tocopherol (purity ≥98%) was supplied by Extrasynthese (Genay, France). Solutions of cannabinoids ((−)-delta9-trans-tetrahydrocannabinol 100 mg L−1 in methanol (MeOH); cannabidiol 1000 mg L−1 in MeOH) were provided by and manipulated at the Toxicology Laboratory facilities A.S.T. AV1, (Pesaro, Italy). Phthalates (diisodecyl phthalate; diethyl phthalate; dimethyl phthalate, purity ≥99%), and phenols (phenol; catechol; resorcinol; hydroquinone, purity ≥99%) were a gift of the Department of Biomolecular Sciences of the University of Urbino. Pure standards were stored at −20 °C.
Standard solution preparation
Stock solutions of tocopherols and phthalates were volumetrically prepared at a concentration of 1000 mg L−1 in HEX and stored in dark vials (VWR International, part of Avantor Milan, Italy) at 4 °C. Stock solutions of phenols were gravimetrically prepared at a concentration of 1000 mg L−1 in EtOH and stored in dark vials at 4 °C. Working standard solutions of tocopherols were volumetrically prepared as combined suites (100 μL of α-tocopherol at 1000 mg L−1, 100 μL of β-tocopherol at 1000 mg L−1, and 100 μL of δ-tocopherol at 1000 mg L−1) at a concentration of 333 mg L−1 each in HEX. Working standard solutions of cannabinoids were volumetrically prepared as combined suites (200 μL of THC at 100 mg L−1 and 100 μL of CBD at 1000 mg L−1) at a concentration of 67 mg L−1 for THC and 333 mg L−1 for CBD in MeOH. The cannabinoid solutions were prepared at the Toxicology Laboratory facilities A.S.T. AV1, (Pesaro, Italy). Working standard solutions of phthalates were volumetrically prepared as combined suites (100 μL of diisodecyl phthalate at 1000 mg L−1, 100 μL of diethyl phthalate at 1000 mg L−1, and 100 μL of dimethyl phthalate at 1000 mg L−1) at a concentration of 333 mg L−1 each in HEX. Working standard solutions of phenols were volumetrically prepared as combined suites (100 μL of phenol at 1000 mg L−1, 100 μL of catechol at 1000 mg L−1, 100 μL of resorcinol at 1000 mg L−1, and 100 μL of hydroquinone at 1000 mg L−1) at a concentration of 250 mg L−1 each in EtOH.
LC-LEI-QQQ-MS/MS system for low-resolution experiments
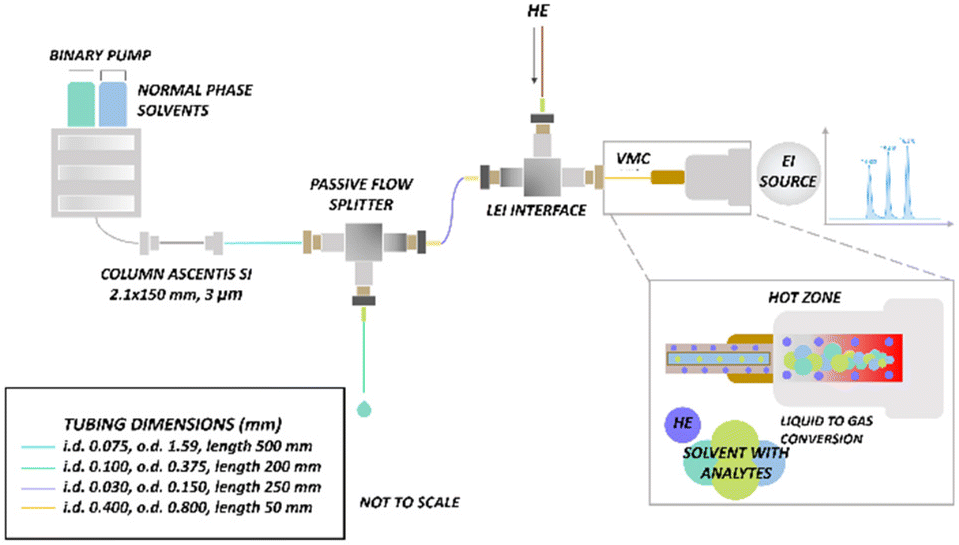
Low-resolution experiments were performed with an Agilent 1290 Infinity II UHPLC (Agilent Technologies, Santa Clara, CA, USA) with an Ascentis silica column (2.1 i.d. x 150 mm, 3 μm particle size) (Sigma-Aldrich-Supelco, Milan, Italy) coupled with an Agilent triple quadrupole (QQQ) 7010 B mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with an LEI interface. The LC pump flow rate was set at 100 μL min−1, but a post-column passive flow splitter (PFS) was used for decreasing the flow rate to 500 nL min−1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 split ratio) to accommodate LEI requirements. A detailed description of the post-column PFS is provided elsewhere.40Fig. 1 shows the schematic of the system for low- and high-resolution experiments. The injection volume was 1 μL for cannabinoids and phenols, and 4 μL for tocopherols and phthalates. The analyses were carried out in isocratic mode using different mobile phase compositions as indicated in Table 1. Agilent OpenLab CDS ChemStation software was used for UHPLC instrument control. The core of LEI is the vaporization microchannel (VMC) where the liquid phase is mixed with a helium flow, heated, and converted into the gas phase. VMC was constructed modifying the standard GC transfer line, totally lined with a 0.4 mm i.d., 0.8 mm o.d. removable fused silica capillary. The mobile phase and the analytes flow into the VMC through the inlet capillary, a narrower 150 μm o.d., 30 μm i.d. fused silica capillary that penetrates in the first portion of the VMC. A detailed description of the interface is provided elsewhere.24–26 The ion source and quadrupoles were maintained at temperatures of 280 °C and 150 °C, respectively. To ensure the rapid and efficient vaporization of the analytes without thermal degradation, different VMC temperatures were tested (250, 300, 350, and 400 °C). Specific VMC temperatures were set for the different classes of compounds to promote rapid and efficient vaporization, while avoiding thermal degradation. Data acquisitions of tocopherols, cannabinoids, and phthalates were carried out in MRM. For phenols, data acquisition was performed in SIM (Table 1). Flow injection analyses (FIA) were conducted using an external manual injector with an internal loop of 0.1 μL (Vici, Switzerland). Agilent Mass Hunter GC/MS acquisition software (Version B.07.04.2260) and Agilent Mass Hunter Qualitative Analysis software (Version B.07.00) were employed for data acquisition and processing.
:200 split ratio) to accommodate LEI requirements. A detailed description of the post-column PFS is provided elsewhere.40Fig. 1 shows the schematic of the system for low- and high-resolution experiments. The injection volume was 1 μL for cannabinoids and phenols, and 4 μL for tocopherols and phthalates. The analyses were carried out in isocratic mode using different mobile phase compositions as indicated in Table 1. Agilent OpenLab CDS ChemStation software was used for UHPLC instrument control. The core of LEI is the vaporization microchannel (VMC) where the liquid phase is mixed with a helium flow, heated, and converted into the gas phase. VMC was constructed modifying the standard GC transfer line, totally lined with a 0.4 mm i.d., 0.8 mm o.d. removable fused silica capillary. The mobile phase and the analytes flow into the VMC through the inlet capillary, a narrower 150 μm o.d., 30 μm i.d. fused silica capillary that penetrates in the first portion of the VMC. A detailed description of the interface is provided elsewhere.24–26 The ion source and quadrupoles were maintained at temperatures of 280 °C and 150 °C, respectively. To ensure the rapid and efficient vaporization of the analytes without thermal degradation, different VMC temperatures were tested (250, 300, 350, and 400 °C). Specific VMC temperatures were set for the different classes of compounds to promote rapid and efficient vaporization, while avoiding thermal degradation. Data acquisitions of tocopherols, cannabinoids, and phthalates were carried out in MRM. For phenols, data acquisition was performed in SIM (Table 1). Flow injection analyses (FIA) were conducted using an external manual injector with an internal loop of 0.1 μL (Vici, Switzerland). Agilent Mass Hunter GC/MS acquisition software (Version B.07.04.2260) and Agilent Mass Hunter Qualitative Analysis software (Version B.07.00) were employed for data acquisition and processing.
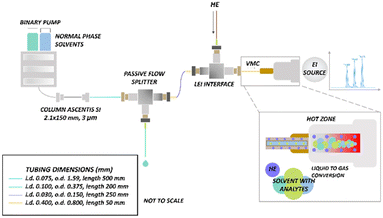 |
| | Fig. 1 Scheme of the LC-LEI-MS/MS and LC-LEI-QTOF-HRMS systems. | |
Table 1 Operational parameters for NPLC-LEI-QQQ-MS (low-resolution) separations of tocopherols, cannabinoids, phthalates, and phenols
| Compound name |
Mobile phase |
Injection volume |
Acquisition mode |
m/z MRM transitions/selected ions (collision energies) |
VMC T °C |
| α-Tocopherol |
HEX:IPA 98:2 (v:v) |
4 μL |
MRM |
430–165 (15 eV) 430–205 (5 eV) |
250 |
| β-Tocopherol |
416–151 (20 eV) 191–135 (10 eV) |
| δ-Tocopherol |
402–137 (15 eV) 402–177 (10 eV) |
| THC |
HEX:EtOH 95:5 (v:v) |
1 μL |
MRM |
314–299 (5 eV) 231–174 (20 eV) |
400 |
| CBD |
246–174 (25 eV) 231–174 (20 eV) |
| Diisodecyl phthalate |
HEX:IPA 90:10 (v:v) |
4 μL |
MRM |
149–93 (15 eV) 149–121 (10 eV) |
350 |
| Diethyl phthalate |
177–149 (5 eV) 222–149 (5 eV) |
| Dimethyl phthalate |
163–133 (5 eV) |
| Phenol |
HEX:EtOH 80:20 (v:v) |
1 μL |
SIM |
94 |
350 |
| Catechol |
110 |
| Resorcinol |
110 |
| Hydroquinone |
110 |
| Flow rate: 100 μL min−1 split to 500 nL min−1 (1:200 split ratio) |
| Column: Ascentis Si 2.1 × 150 mm, 3 μm particle size |
| Ion source temperature: 280 °C |
| Quadrupoles temperature: 150 °C |
LC-LEI-QTOF-HRMS system
An Agilent quadrupole-time of flight (QTOF) 7250 mass spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA) equipped with an LEI interface was used for the detection of the analytes in high-resolution experiments. Also in this case, the UHPLC instrument was an Agilent 1290 Infinity II (Agilent Technologies Inc., Santa Clara, CA, USA). To facilitate the comparison with the low-resolution setup, the same conditions were applied in terms of column, flow rate, injection volume, mobile phase composition, and VMC temperature. The ion source temperature was kept at 260 °C for all high-resolution experiments. Data acquisition was performed in full scan mode for each compounds’ class. The acquisition range was set from m/z 90 to m/z 500 to avoid detection of interfering ions originating from HEX (MW 86.18 g mol−1). The data extraction window was set at 25 ppm. Operational parameters used for high-resolution experiments are presented in Table 2. After each analysis, QTOF mass calibration was performed, during which no mobile phase was admitted into the ion source. Data were acquired with the Agilent Mass Hunter GC/MS acquisition software (Version 10.1.49.0) and processed with the Agilent Mass Hunter Qualitative Analysis software (Version 10.0).
Table 2 Operational parameters for NPLC-LEI-QTOF-HRMS (high-resolution) separations of tocopherols, cannabinoids, phthalates, and phenols
| Compound name |
Mobile phase |
Injection volume |
VMC T °C |
| α-Tocopherol |
HEX:IPA 98:2 (v:v) |
4 μL |
250 |
| β-Tocopherol |
| δ-Tocopherol |
| THC |
HEX:EtOH 95:5 (v:v) |
1 μL |
400 |
| CBD |
| Diisodecyl phthalate |
HEX:IPA 90:10 (v:v) |
4 μL |
350 |
| Diethyl phthalate |
| Dimethyl phthalate |
| Phenol |
HEX:EtOH 80:20 (v:v) |
1 μL |
350 |
| Catechol |
| Resorcinol |
| Hydroquinone |
| Flow rate: 100 μL min−1 split at 500 nL min−1 (1:200 split ratio) |
| Column: Ascentis Si 2.1 × 150 mm, 3 μm particle size |
| Ion source temperature: 260 °C |
| Acquisition mode: full scan from m/z 90 to m/z 500 |
Results and discussion
Low-resolution experiments
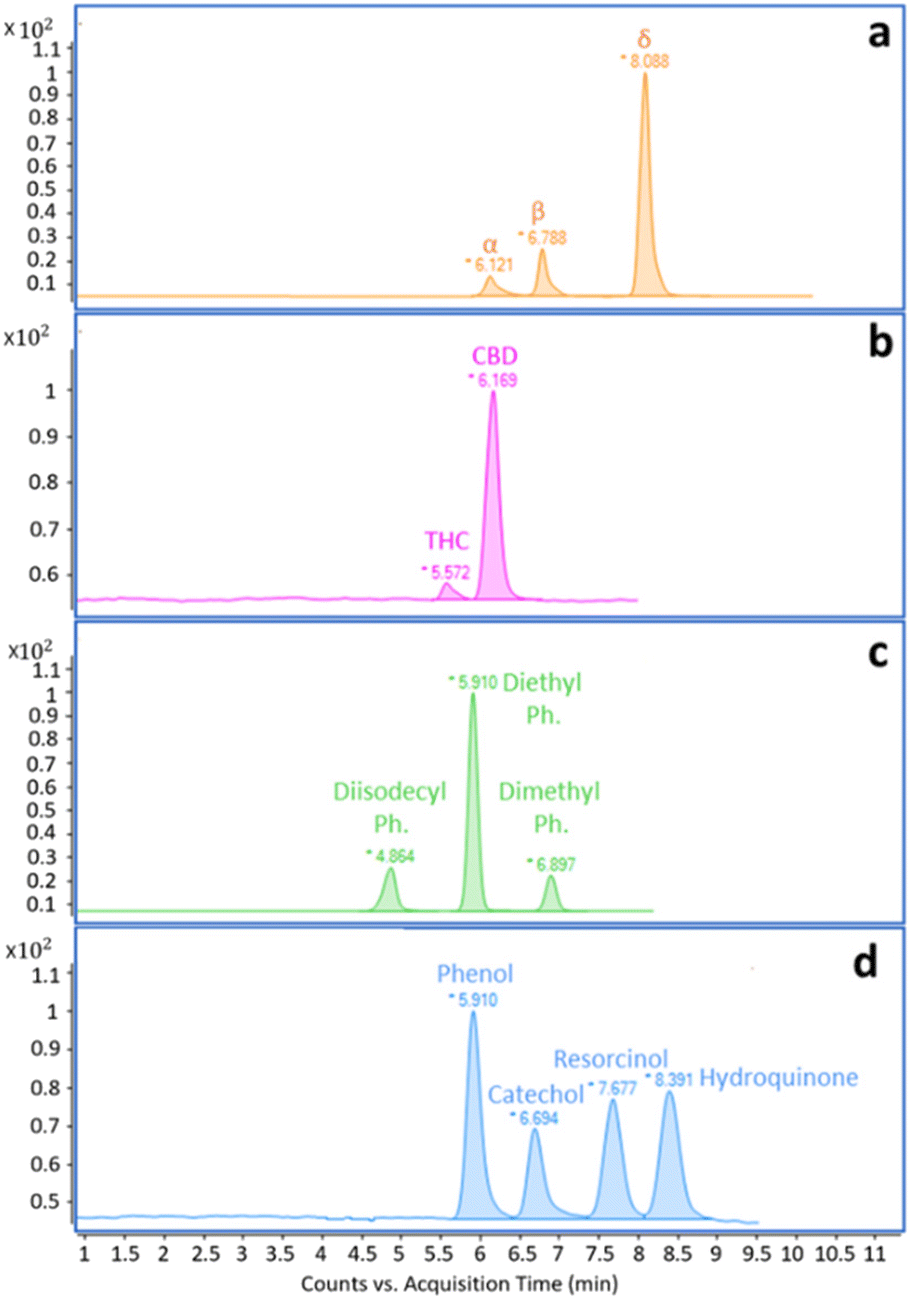
The initial experiments focused on acquiring low-resolution EI full scan mass spectra of all targeted analytes. The results were obtained using FIA. Each compound was injected and analyzed separately using the following solutions: α-, β-, and δ-tocopherol at 1000 mg L−1 in HEX; THC and CBD in MeOH at 100 mg L−1 and 1000 mg L−1, respectively; diisodecyl-, diethyl, and dimethyl phthalate at 1000 mg L−1 in HEX; phenol, catechol, resorcinol, and hydroquinone at 1000 mg L−1 in EtOH. The LC pump flow rate was set at 100 μL min−1, split at 500 nL min−1 (1:200 split ratio), using different mobile phase compositions in isocratic mode. The scan range was set from m/z 90 to m/z 500, and the VMC temperature was set to 250 °C for tocopherols, 400 °C for cannabinoids, and 350 °C for phthalates and phenols. The ion source temperature was kept at 280 °C. In Fig. 2a–c, low-resolution spectra of tocopherols were compared with the reference spectra present in the NIST library. α-tocopherol exhibited a matching factor of 740, and reverse match of 870; β-tocopherol showed a matching factor of 910, and reverse match of 950; δ-tocopherol demonstrated a matching factor of 880, and reverse match of 900. Fig. S1–S3† present the low-resolution spectra of cannabinoids, phthalates, and phenols along with NIST library spectra and matching data. An evident drawback of the use of aprotic solvents like HEX is the requirement to start the scan interval from m/z 90. This poses a challenge when dealing with compounds that exhibit intense and characteristic ions below this threshold, as it negatively impacts the automatic search in the database, resulting in slightly lower match and reverse match values. Experimental and library spectra are comparable, demonstrating that the typical NPLC mobile phases do not affect spectral quality. Based on the recorded spectra, MRM and SIM methods were developed and applied to separate the selected mixtures. Although MRM is more specific and sensitive than SIM, in the case of phenols, the response in MRM was not satisfactory due to the lack of product ions, and SIM mode was therefore chosen. The NPLC separations obtained using the conditions described in Table 1 are shown in Fig. 3a–d. All separations were performed in isocratic mode, as it often occurs in NPLC. Isocratic elution, avoiding column re-conditioning, offers a clear advantage when analyzing a large number of consecutive samples, saving both time and solvents.
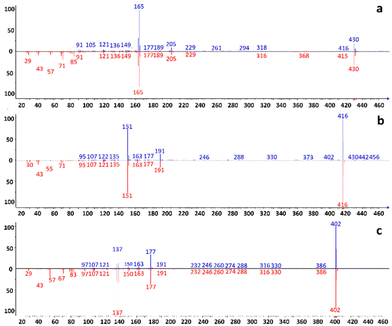 |
| | Fig. 2 (a–c). Low-resolution experiments. Comparison between the recorded and the NIST library reference spectra of (a) α-tocopherol, (b) β-tocopherol, and (c) δ-tocopherol. Top: experimental spectrum; Bottom: NIST reference spectrum. | |
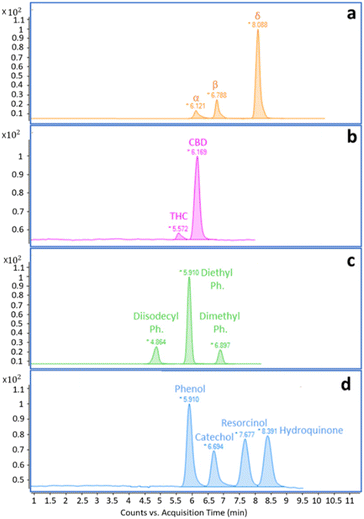 |
| | Fig. 3 (a–d). NPLC MRM and SIM reconstructed ion chromatograms of the selected model compounds: (a) tocopherols; (b) cannabinoids; (c) phthalates; (d) phenols. Analytical conditions are reported in Table 1. | |
High-resolution experiments
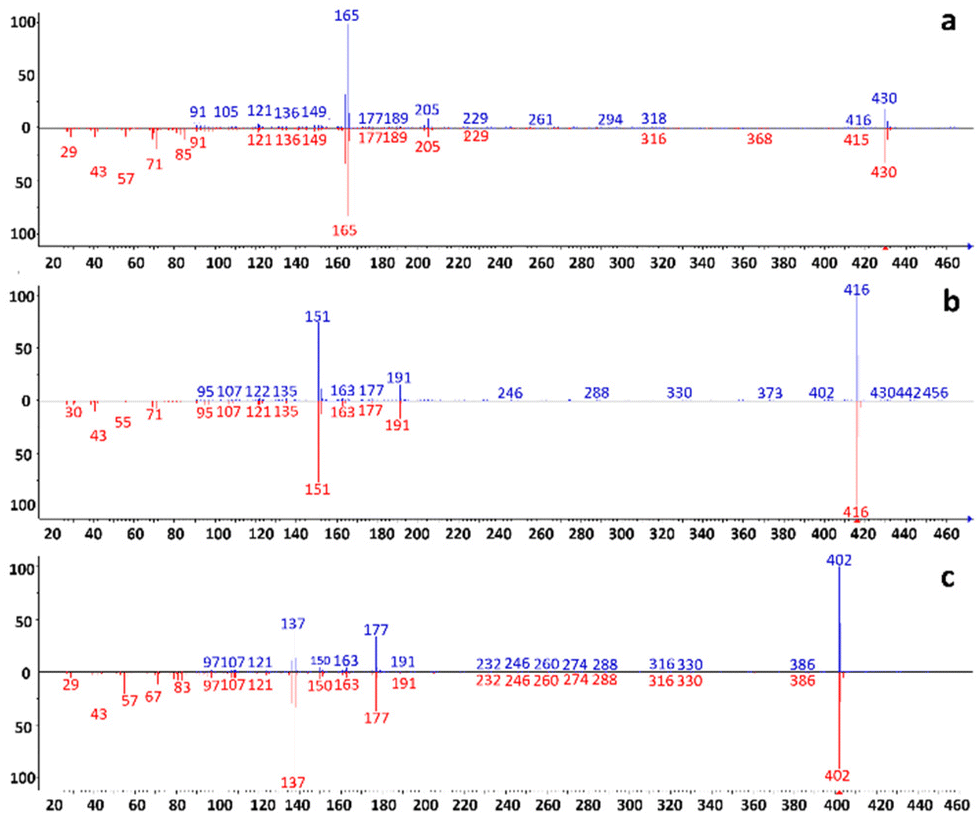
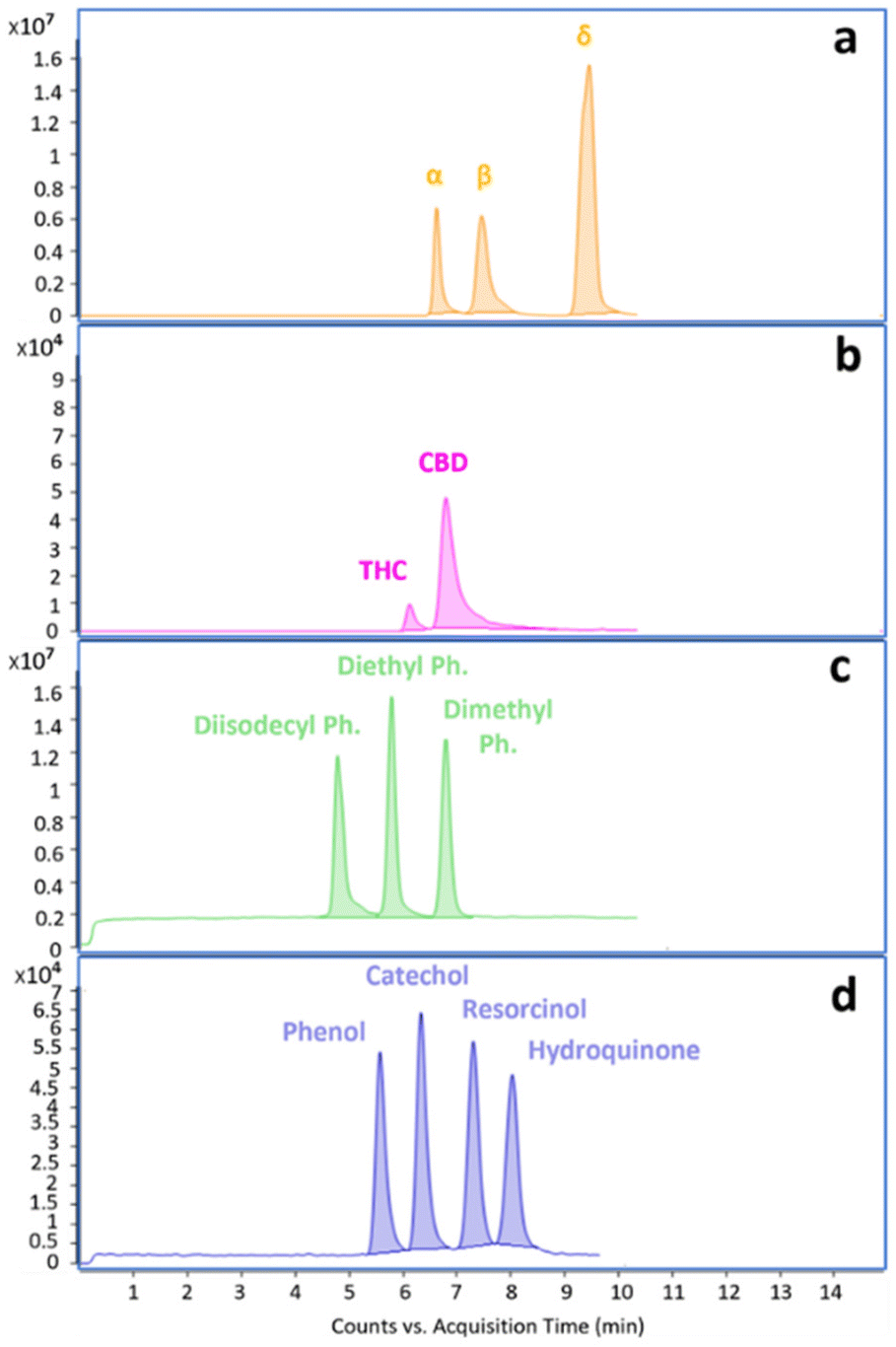
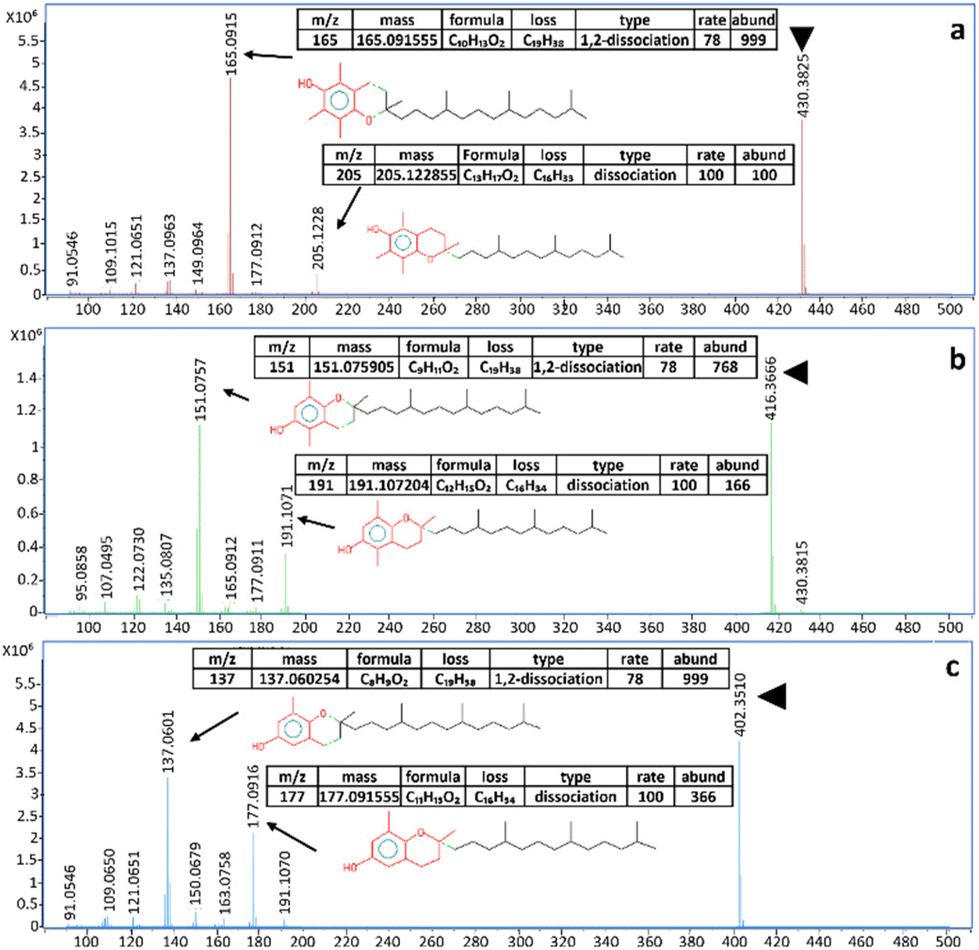
The NPLC-LEI system was installed on a QTOF-HRMS to replicate the sets of experiments on a high-resolution instrument with a different ion source geometry, ensuring results repeatability for superior performance. The separations of the same model compounds were carried out without modifying the previously optimized chromatographic and mass spectrometric parameters, but the acquisitions were performed in full scan mode. This was possible because the QTOF-HRMS offers greater sensitivity in this mode, eliminating the need for FIA to acquire mass spectra. The separations are reported in Fig. 4a–d. The use of a source with a different geometry, especially with a different volume, could impede the evacuation of the solvent vapors used in NPLC, leading to chemical ionization phenomena. However, this phenomenon, which has been previously studied and solved with the typical RPLC solvents, was not observed in either the QQQ or QTOF-HRMS source, as evidenced by the perfectly comparable mass spectra. The recorded high-resolution mass spectra and main fragments assignment of tocopherols are presented in Fig. 5a–c. High-resolution mass spectra of the other model compounds can be found in Fig. S4–S6.† The software supplied with the QTOF-HRMS automatically identifies the exact masses of the main fragments by considering the possible cleavages of the molecular bonds, facilitating unambiguous molecule identification. As described in the introduction, NPLC can also serve as a rapid elution method for highly hydrophobic and highly retained interferences in RPLC, effectively separating them from the analytes of interest. Fig. 6a and b shows the chromatograms obtained from the analysis of a non-disclosable commercial mixture where secondary components required characterization. In RPLC, these components elute after the main compound, with a retention time of approximately 50 min, making their identification a time-consuming and challenging process. However, in NPLC, the same separation demonstrates a completely different trend, as the secondary components swiftly exit the chromatographic column in less than 6 min and before the elution of the main compound. The intensity of the two peaks appears different, but their areas are comparable.
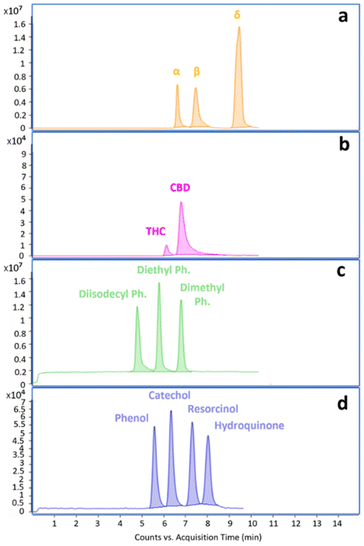 |
| | Fig. 4 (a–d). NPLC full scan chromatograms of the selected model compounds: (a) tocopherols; (b) cannabinoids; (c) phthalates; (d) phenols. Analytical conditions are reported in Table 2. | |
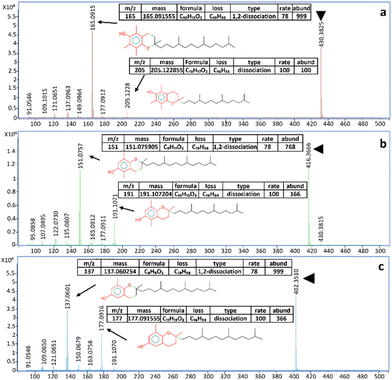 |
| | Fig. 5 (a–c). Recorded EI high-resolution full scan spectra of (a) α-tocopherol, (b) β-tocopherol, and (c) δ-tocopherol and assignment of the main fragments according to the MS interpreter utility developed by NIST. Molecular ions are indicated by the green triangle. | |
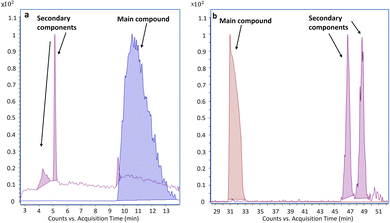 |
| | Fig. 6 (a and b). Extracted ion chromatograms of secondary components and main compound present in a commercial mixture obtained using the following separation conditions: (a) NPLC. A: HEX, B: ethyl acetate; gradient: 1–40% B in 25 min, 40–90% in 1 min, 90% held for 10 min; column: Ascentis Si 2.1 × 150 mm, 3 μm particle size; flow rate: 100 μL min−1; (b) RPLC. A: water + 0.3% formic acid; B: ACN + 0.3% formic acid; gradient: 0–100% B in 30 min, 100% B held for 20 min; column: Waters Acquity UHPLC C18 1.7 μm; flow rate: 100 μL min−1. | |
Conclusions
The results of this preliminary study demonstrate, thanks of the LEI interface, the feasibility and advantage of using MS with NPLC. By combining the unique separation capabilities of NPLC with a high-performance detector like MS with an EI source, both sensitivity and specificity are enhanced compared to traditional spectrophotometric detectors. These characteristics show the potential for future applications in targeted and untargeted analysis of complex matrices, especially with a high-resolution analyzer. NPLC-LEI-MS has proven to be a promising technique that offers an alternative approach to RPLC-MS based on soft ionization techniques. The application of NPLC-LEI-MS to the analysis of selected model compounds and a real sample has shown that the mass spectrometric results remain unaltered, as evidenced by the comparison of the experimental EI spectra with those recorded in the NIST library. The presented separations also indicate that SIM or MRM analyses can be performed without particular precautions. The next phase of the research will focus on developing methods for analyzing a variety of samples, taking advantage of the potential of NPLC-LEI-MS.
Author contributions
N. M.: Investigation, formal analysis. G. G.: Investigation. A. A.: Validation, methodology, project administration. G. F.: Methodology, writing-original draft, writing—review & editing, project administration. M. A: Drug of abuse—sample preparation. C. R.: Drug of abuse—sample preparation. P. P.: Methodology, Writing—review & editing, project administration. A. C.: Supervision, writing—review & editing, funding acquisition, project administration, conceptualization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Agilent Technologies for providing the analytical instrumentation.
References
- A. W. Salotto, E. L. Weiser, K. P. Caffey, R. L. Carty, S. C. Racine and L. R. Snyder, J. Chromatogr., 1990, 498, 55–65 CrossRef CAS.
-
L. R. Snyder, J. J. Kirkland and J. W. Dolan, Introduction to modern liquid chromatography, Wiley, Hoboken, 3rd edn, 2010 Search PubMed.
- P. Dugo, O. Favoino, R. Luppino, G. Dugo and L. Mondello, Anal. Chem., 2004, 76(9), 2525–2530 CrossRef CAS PubMed.
- K. Arena, E. Trovato, F. Cacciola, L. Spagnuolo, E. Pannucci, P. Guarnaccia, L. Santi, P. Dugo, L. Mondello and L. Dugo, Molecules, 2022, 27(5), 1727 CrossRef CAS PubMed.
-
L. R. Snyder, High-performance liquid chromatography - Advances and perspectives, ed. C. S. Horvath, Academic Press, New York, 1980, p. 207 Search PubMed.
-
W. T. Cooper, Encyclopedia of Analytical Chemistry: Application, Theory, and Instrumentation, ed. R. A. Meyers and J. G. Dorsey, Wiley Online Library, 2006 Search PubMed.
-
A. A. Younes, C. Galea, D. Mangelings and Y. Vander Heyden, Analytical Separation Science, ed. J. L. Anderson, A. Berthod, V. Pino and A. M. Stalcup, Wiley-VCH, Weinheim, 2015, pp. 227–244 Search PubMed.
- L. Jiahao, X. Xiyue, M. Ming, C. Bo, C. Yingzhuang and J. S. Oliver, J. Chromatogr. A, 2023, 1691, 463821 CrossRef PubMed.
- M. Zhengyue, S. Panpan, L. Delu, N. Yangyang, L. Yanli, G. Xinyuan, W. Benben, B. Ligai and Q. Xiaoqiang, Chirality, 2023, 35(9), 636–644 CrossRef PubMed.
- Z. You-Ping, L. Kuan, X. Ling-Xiao, W. Bang-Jin, X. Sheng-Ming, Z. Jun-Hui and Y. Li-Ming, J. Chromatogr. A, 2022, 1683, 463551 CrossRef PubMed.
- A. A. Boyd-Boland and J. B. Pawliszyn, Anal. Chem., 1996, 68(9), 1521–1529 CrossRef CAS PubMed.
- S. Lababidi, S. K. Panda, J. T. Andersson and W. Schrader, Anal. Chem., 2013, 85(20), 9478–9485 CrossRef CAS PubMed.
- S. Lababidi and W. Schrader, Rapid Commun. Mass Spectrom., 2014, 28(12), 1345–1352 CrossRef CAS PubMed.
- E. V. S. Maciel, A. L. de Toffoli, E. Sobieski, C. E. D. Nazario and F. M. Lanças, Anal. Chim. Acta, 2020, 1103, 11–31 CrossRef PubMed.
- K. Mejía-Carmona, J. S. da Silva Burato, J. V. Basolli Borsatto, A. L. de Toffoli and F. M. Lanças, TrAC, Trends Anal. Chem., 2020, 122, 115735 CrossRef.
- P. Kalo, A. Kemppinen and V. Ollilainen, Lipids, 2009, 44(2), 169–195 CrossRef CAS PubMed.
- N. Strobel, S. Buddhadasa, P. Adorno, K. Stockham and H. Greenfield, Food Chem., 2013, 138(2–3), 1042–1047 CrossRef CAS PubMed.
- M. Mejean, A. Brunelle and D. Touboul, Anal. Bioanal. Chem., 2015, 407, 5133–5142 CrossRef CAS PubMed.
- T. Saldanha, A. C. Sawaya, M. N. Eberlin and N. Bragagnolo, J. Agric. Food Chem., 2006, 54(12), 4107–4113 CrossRef CAS PubMed.
- R. K. Khajuria, V. Bhardwaj, R. K. Gupta, P. Sharma, P. Somal, P. Mehta and G. N. Qazi, J. Chromatogr. Sci., 2007, 45(8), 519–523 CAS.
- S. Kemmo, V. Ollilainen, A. M. Lampi and V. Piironen, Food Chem., 2007, 101(4), 1438–1445 CrossRef CAS.
- R. Schex, V. M. Lieb, V. M. Jiménez, P. Esquivel, R. M. Schweiggert, R. Carle and C. B. Steingass, Food Res. Int., 2018, 105, 645–653 CrossRef CAS PubMed.
- C. H. Grün and S. Besseau, J. Chromatogr. A, 2016, 1439, 74–81 CrossRef PubMed.
- V. Termopoli, G. Famiglini, P. Palma, M. Piergiovanni and A. Cappiello, Anal. Chem., 2017, 89(3), 2049–2056 CrossRef CAS PubMed.
- V. Termopoli, G. Famiglini, P. Palma, M. Piergiovanni, P. Rocio-Bautista, M. F. Ottaviani, A. Cappiello, M. Saeed and S. Perry, J. Chromatogr. A, 2019, 1591, 120–130 CrossRef CAS PubMed.
-
A. Cappiello, G. Famiglini, M. Piergiovanni and V. Termopoli, Machine for Chemical Analysis Comprising the Combination of Electron Ionization Mass Spectrometry with Liquid Chromatography, International Application Patent No. PCT/IB2018/057970, US Application Patent No16/755, 2020, 332 Search PubMed.
- G. Stefkov, I. Cvetkovikj Karanfilova, V. Stoilkovska Gjorgievska, A. Trajkovska, N. Geskovski, M. Karapandzova and S. Kulevanova, Molecules, 2022, 27(3), 975 CrossRef CAS PubMed.
- C. De Luca, A. Buratti, Y. Krauke, S. Stephan, K. Monks, V. Brighenti, F. Pellati, A. Cavazzini, M. Catani and S. Felletti, Anal. Bioanal. Chem., 2022, 414, 5385–5395 CrossRef CAS PubMed.
- R. F. X. Klein, Microgram J., 2015, 12, 1–4 CAS.
- T. Onishi and W. J. Umstead, Pharmaceuticals, 2021, 14(12), 1250 CrossRef CAS PubMed.
- L. Huang, J. Li, Y. Bi, Y. Xu, Y. Wang, J. Wang and D. Peng, J. Food Compos. Anal., 2021, 104, 104132 CrossRef CAS.
- L. Barros, D. M. Correia, I. C. F. R. Ferreira, P. Baptista and C. Santos-Buelga, Food Chem., 2008, 110(4), 1046–1050 CrossRef CAS PubMed.
- A. Bouymajane, Y. O. El Majdoub, F. Cacciola, M. Russo, F. Salafia, A. Trozzi, F. R. Filali, P. Dugo and L. Mondello, Molecules, 2020, 25(22), 5428 CrossRef CAS PubMed.
- P. D. Boes, S. R. Elleman and N. D. Danielson, Separations, 2023, 10(2), 70 CrossRef CAS.
- M. Zhang, A. K. Mallik, M. Takafuji, H. Ihara and H. Qiu, Anal. Chim. Acta, 2015, 887, 1–16 CrossRef CAS PubMed.
- E. Guerrero-Hurtado, A. Gutiérrez-Docio, R. Fiedorowicz, E. Mollá, G. Reglero and M. Prodanov, J. Chromatogr. A, 2023, 1696, 463957 CrossRef CAS PubMed.
- W. Cuijie, Z. Ling, L. Xinglin, Y. Ajuan and Z. Shusheng, Talanta, 2020, 218, 121155 CrossRef PubMed.
- C. L. Flakelar, P. D. Prenzler, D. J. Luckett, J. A. Howitt and G. Doran, Food Chem., 2017, 214, 147–155 CrossRef CAS PubMed.
- T. Lechhab, F. Salmoun, W. Lechhab, Y. O. El Majdoub, M. Russo, M. R. Testa Camillo, E. Trovato, P. Dugo, L. Mondello and F. Cacciola, Eur. Food Res. Technol., 2021, 247(12), 2993–3012 CrossRef CAS.
- N. Marittimo, G. Famiglini, P. Palma, A. Arigò and A. Cappiello, J. Chromatogr. A, 2022, 1681, 463479 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Genny
Grasselli
a,
Adriana
Arigò
a,
Giorgio
Famiglini
a,
Marco
Agostini
b,
Caterina
Renzoni
b,
Pierangela
Palma
ac and
Achille
Cappiello
a,
Genny
Grasselli
a,
Adriana
Arigò
a,
Giorgio
Famiglini
a,
Marco
Agostini
b,
Caterina
Renzoni
b,
Pierangela
Palma
ac and
Achille
Cappiello