
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mapping of uranium particles on J-type swipes with microextraction-ICP-MS
Veronica C.
Bradley
 a,
Jacob
Burleson
a,
Hunter B.
Andrews
b,
Cyril V.
Thompson
a,
Tyler L.
Spano
c,
Daniel R.
Dunlap
a,
N. Alex
Zirakparvar
a,
Brian W.
Ticknor
a,
Cole R.
Hexel
a and
Benjamin T.
Manard
*a
a,
Jacob
Burleson
a,
Hunter B.
Andrews
b,
Cyril V.
Thompson
a,
Tyler L.
Spano
c,
Daniel R.
Dunlap
a,
N. Alex
Zirakparvar
a,
Brian W.
Ticknor
a,
Cole R.
Hexel
a and
Benjamin T.
Manard
*a
aChemical Sciences Division, Oak Ridge National Laboratory, USA. E-mail: manardbt@ornl.gov
bRadioisotope Science and Technology Division, Oak Ridge National Laboratory, USA
cNuclear Nonproliferation Division, Oak Ridge National Laboratory, USA
First published on 20th February 2024
Abstract
A microextraction liquid sampling system coupled to a quadrupole inductively coupled plasma-mass spectrometer (ICP-MS) was utilized to spatially discern uranium particles, isotopically, on a cellulose-based swipe material (i.e., J-type swipe). These types of swipes are often used by the International Atomic Energy Agency (IAEA) as part of their environmental sampling program. A grid was created such that extraction locations covered the center circle (n = 34 without overlapping). Uranium (U) particulates (<20 μm) of varying U isotopic abundance and chemical form (i.e., uranyl fluoride and uranyl nitrate hexahydrate) were mechanically placed on the swipes in random locations and detected via the microextraction-ICP-MS methodology. Heat maps were subsequently generated to show the placement of the particulate with their respective intensity and isotopic determination. This detection of the uranium particulates, via isotopic determination, agreed with reference values for these materials. Additionally, depleted (235U/238U = 0.002) uranium particulates were placed directly within a clay matrix, on the swipe surface, and subjected to analysis by microextraction-ICP-MS. The mapping of the swipe demonstrated, for the first time, the employment of the microextraction-ICP-MS method for extracting sample from a complex matrix, and correctly identifying the uranium isotopic composition. This example ultimately demonstrates the utility of the methodology for detecting particles of interest in complex matrices.
1. Introduction
Elemental and isotopic mapping of ultra-trace (sub-parts per million) analytes in solid samples is traditionally explored through laser ablation (LA) – based sampling methods. Laser ablation sampling allows for ∼μm spatial resolution and can provide elemental and isotopic determination when coupled to an inductively coupled plasma – mass spectrometer (ICP-MS). LA-ICP-MS has been applied to a wide range of fields, including biology, medicine, material science, geochemistry, environmental chemistry, and nuclear forensics.1–5 There are other forms of elemental mapping, such as scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS) or laser induced breakdown spectroscopy (LIBS) which allow for elemental composition mapping with high spatial resolution, however they have higher detection limits (on the order of ppm for LIBS, and in the wt% range for SEM-EDS).3To the authors’ knowledge, the only mapping demonstrated with ICP-MS detection has been accomplished with laser-based sampling. However, other rapid isotopic mapping techniques with low detection limits that can provide spatially resolved isotopic information with high accuracy and precision exist. Microextraction-based sampling is a recent technique for solid surface analysis that has been realized for isotopic determination.6,7 It was originally explored in the mid-2000s for liquid extraction of thin tissue samples8 and spots on thin layer chromatography (TLC) plates.9 The initial motivation for microextraction-ICP-MS was to eliminate bulk sample digestion, which can be a time consuming process, and instead directly sample the surface for the analysis of uranium,7 plutonium,10,11 and other trace elements.12 Subsequent efforts explored extraction of solid uranium particulates from the surface,13 with rapid isotopic determination.
To date, microextraction-ICP-MS studies focused on extracting actinides from 10 × 10 cm cotton swipes. These environmental sample (ES) swipes are commonly collected by the International Atomic Energy Agency (IAEA) during inspections of nuclear facilities to aid in drawing conclusions about the absence of nuclear materials and activities in a given lcoation.14,15 An example of ES could involve sampling inside a nuclear facility with woven cotton swipes or, in the case of hot cells, a small cellulose J-type swipe.16 These J-type swipes (photograph included in Fig. 1) are also used to check inspectors’ clothing or hands before entering a nuclear facility; this is a form of quality control called a pre-inspection check sample (PIC), and is used to check for potential cross-contamination on the inspectors themselves.16 The IAEA has an international Network of Analytical Laboratories (NWAL) that analyzes the actinide content/isotopics of cotton swipes through both bulk digestion and particle analysis.17 J-type swipes are traditionally analyzed via neutron activation analysis (NAA) measurements. The NAA method is fast and high throughput, but it requires a nuclear reactor with a high flux, is not particularly sensitive for low level U detection, and is not well suited for detection of some minor U isotopes (i.e.234U). Additionally, NAA does not provide information on the spatial distribution nor individual particle isotopic content for actinide-containing particles present on the swipe surface. The work presented here focuses on the development of a microextraction-ICP-MS method to map solid samples. Specifically, the method was used for analyzing (and mapping) uranium particulates, of varying isotopic and chemical composition, on J-type swipes for determination of 234U, 235U, 236U, and 238U. A rapid, robust, and sensitive technique to map the uranium content/isotopics of an entire J-type swipe sample could be useful for analysis of IAEA ES swipes, or other applications where swipe-based analysis is warranted (i.e., industrial hygiene). The work presented here offers a method for J-type swipe mapping of uranium isotopes with a microextraction-ICP-MS system, and to the author's knowledge is the first example of ICP-MS uranium mapping with a sample introduction system other than laser ablation. All prior work has focused on targeted extraction locations; this is the first in which the entire sample has been analyzed (mapped).
 | ||
| Fig. 1 Illustration of the microextraction-ICP-MS set-up for J-type swipe mapping. | ||
2. Materials and methods
2.1 Materials and reagents
Optima grade nitric acid, purchased from Fisher Scientific was used throughout, without further purification. All dilutions were made using ASTM Type 1 water (>18.0 MΩ cm) from a Thermo Scientific Barnstead GenPure xCAD Plus water purification system. Certified reference materials (CRMs) for U were obtained from the Joint Research Center of the European Commission (JRC-Geel), formerly the Institute for Reference Materials and Measurements (IRMM-2025). Solid samples of reagent-grade uranyl fluoride (UO2F2) and uranyl nitrate hexahydrate (UO2(NO3)2·6H2O; UN) were obtained from International Bioanalytics (Boca Raton, Florida, USA). J-type swipe samples were obtained from Chiyoda Technol Corporation (Yokohama, Japan). The isotopic content of the uranyl fluoride (UO2F2) and uranyl nitrate hexahydrate (UO2(NO3)2·6H2O; UN) particles were previously determined via MC-ICP-MS.13 An AxisPro Microsupport micromanipulator (Shizuaka City, Shizuaka, Japan) fitted with two 0.5 μm tungsten probes was used to place solid uranium particulates with diameters between 5 μm and 20 μm onto the surface of J-type swipes.2.2 Microextraction – ICP – MS
A commercial off-the-shelf Plate Express microextraction system (Advion, Ithaca, New York, USA) was retrofitted to include an automated XY stage that can be programmed to save sampling locations, and ultimately raster the sample under the microextraction probe. This modification has been described elsewhere.18 Previously, the microextraction system was implemented to analyze 10 × 10 cm cotton swipes; a new holder was designed and constructed at Oak Ridge National Laboratory to hold J-type swipes in place on the XY stage to enable automated sampling. The holder, shown in Fig. 1, has three components: a polyether ether ketone (PEEK) square bottom layer to hold the other components together, a Teflon sheet mid layer, which helps the microextraction probe form a seal on the sample, and a PEEK top layer with three indentations on either side to secure the wings of the J-type swipe sample. The sample holder can hold three swipes at any given time.A method using the automated microextraction stage was developed to measure the uranium content in a full J-type swipe by extracting in a grid pattern until all locations on the swipe have been sampled. The general concept of the experimental set-up can be seen in Fig. 1. The extraction locations were set so that there was no overlap from one spot to the next: this totaled 34 sampling locations when mapping the entire J-type swipe. The probe head consists of a 2 × 4 mm stainless steel oval ring with a “knife edge”. Upon lowering onto the solid surface with an applied force (300 N), a seal is formed, and the extracting solvent is delivered to the sample surface. The solvent is delivered via an isocratic pump (200 μL min−1) through a 6-port injection valve which directs the flow to the ICP-MS or the microextraction probe, depending on the positioning. When the microextraction probe is engaged on the sample surface, the solvent is delivered down onto the sample surface, and then subsequently upwards in a separate capillary, back to the 6-port valve, and to the ICP-MS nebulizer (perfluoroalkoxy, PFA, microflow, Elemental Scientific Inc, Omaha, NE) housed within a Peltier-cooled quartz cyclonic spray chamber. Here, a Thermo Scientific (Bremen, Germany) triple quadrupole ICP-MS (TQ-ICP-MS) instrument was used. The instrument was tuned daily for optimal sensitivity. In brief, the typically sensitivity of the day was determined to be ∼300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cps for a 1 ng mL−1 signal. As mentioned above, the solution was delivered at 200 μL min−1 and the respective nebulizer gas was 0.9 L min−1. Uranium isotopes were monitored (234U, 235U, 236U, and 238U) at 10 ms dwell times. Regarding measuring the matrix of the Flint Clay (NBS SRM 97b, Flint Clay), matrix isotopes (85Rb, 88Sr, 137Ba) and were also monitored at 10 ms.
000 cps for a 1 ng mL−1 signal. As mentioned above, the solution was delivered at 200 μL min−1 and the respective nebulizer gas was 0.9 L min−1. Uranium isotopes were monitored (234U, 235U, 236U, and 238U) at 10 ms dwell times. Regarding measuring the matrix of the Flint Clay (NBS SRM 97b, Flint Clay), matrix isotopes (85Rb, 88Sr, 137Ba) and were also monitored at 10 ms.
The transient data, collected via the ICP-TQ-MS, was processed through Qtegra™ software using the ICSI peak detection algorithm. In short a 10-point moving mean was utilized with 5 passes, and the subsequent peak was integrated and used for isotopic determination, as previously demonstrated.7,11,13,19 Isotopic maps of the measured swipes were produced in an in-house developed Python script which relates the probe head shape, XY coordinates of the measurement points, and measured ICP-MS signals. The program is adjustable to different scan patterns, different probe heads, and varying analytes.
2.3 Laser ablation – ICP-MS
LA-sampling was performed on an Elemental Scientific Lasers (ESL, Bozeman, MT, USA) imageGEOLIBS. This commercial system is equipped with a 193 nm excimer laser which is focused through an XYR beam aperture (providing square ablation spots) into a helium (ultra-high purity, 99.994%, Airgas, Radnor, PA, USA)-purged (1000 mL min−1) TwoVol3 ablation chamber. For the LA-ICP-MS measurements, a 20 × 20 μm aperture was employed with a laser fluence of 13.2 J cm−2 at 200 Hz and a scan speed of 500 μm s−1. Here, the He carrier gas (800 mL min−1) transported the ablated sample via Tygon tubing into a DCI2 duel concentric injector integrated within the Thermo Scientific triple quadrupole ICP-MS (TQ-ICP-MS).3. Results and discussion
3.1 Optimization of microextraction for mapping
One of the motivations of this work is the ability to quickly map the sample surface; hence effort was placed into minimizing sample transport time, dead volume, and signal transients. While the microextraction sampling does not fall within the short transient analysis time (e.g., <100 ms), such as LA-based introduction, it also does not possess long transient characteristics such as traditional ion chromatographic introduction systems (which are on the order of 1–5 min), and in fact the typical transient from microextraction introduction is ∼30 s7. The size of the inner diameter of tubing transporting the extracted analyte can affect the peak size of the analyte. Four different inner diameters (0.005, 0.01, 0.02, and 0.03 in) of polyetheretherketone (PEEK) tubing were explored to find the optimal tube size while holding other factors, such as tubing length and solvent flow rate (200 μL min−1), constant. J-type swipe samples were doped with 1 ng of IRMM 2025.20 Extractions of 30 s were performed on the doped swipe, and the extracted analyte (containing uranium) was delivered to the ICP-MS nebulizer though PEEK tubing. The microextraction system contains PEEK tubing in two places, connecting the isocratic pump to the 6-port valve and then from the 6-port valve to the ICP-MS nebulizer. For this study, the tubing from the pump to the probe head was not changed, only the second set of tubing was switched out. Fig. 2 shows the uranium extraction profiles performed in triplicate with the average transient plotted by the solid line and the deviation of the extractions as shaded area surrounding the solid lines. | ||
| Fig. 2 The 238U extraction profiles (with respective standard deviation depicted by shading) at a 200 μL min−1 flow of 5% (v/v) HNO3 using four different tubing sizes for transport. | ||
Smaller tubing (e.g., 0.005 in) had a faster elution profile, 15 s, compared with 100 s for the largest tubing (e.g., 0.03 in), indicating that band broadening is strongly related to increasing tube inner diameter. Although the two narrowest tubes (0.005 and 0.01 in.) had similar peak shapes, the 0.01 in tubing was observed to perform slightly better (regarding sealing), leading to more robust and repeatable measurements as shown in the deviation plotted in Fig. 2. For all subsequent experiments, the 0.01 in tubing, plotted in blue, was used.
3.2 One-dimensional line scan optimization
Individual particles of UO2F2 and UN were placed along a horizontal line in the center of the swipe for a total of 2–3 particles per swipe. Extractions were performed along this line (five extractions per swipe, plus a blank extraction on each wing), in triplicate. Fig. 3 shows each of the line scans conducted, where the oval shapes (2 × 4 mm) denote the extraction locations. The respective isotopic results are presented in Table 1. Fig. 3a–c contain only UO2F2 in varying locations within the line scan, but with varying degrees of separation. Two particles of different isotopic composition were placed onto the J-type swipe with ∼8 mm separation. The first is depleted in the 235U isotope, followed by a particle of natural isotopic abundance. The analysis of this line scan is presented in Fig. 3d. Clearly the microextraction-ICP-MS mapping method was able to discern these two particles. In Fig. 3e, the spot in location 3 contains a particle of the depleted UN, location 4 contains one particle each of UO2F2 (natural isotopic) and UN (depleted), and location 5 contains a particle of UO2F2. The primary motivation of this experiment was not only to demonstrate the applicability to discern different isotopic systems (natural and depleted), but also to illustrate the situation in which two particles, of different isotopic composition, would exist in the extraction location. The two single particle (UO2F2) extractions were accurately determined such that the average (n = 18 particles) 235U/238U and 234U/238U had a % relative difference (RD), seen in eqn (1), of 0.84% and 0.52%, respectively. Fig. 3 shows that microextraction can detect individual particles and determine their isotope ratio with high accuracy. The depleted uranium and natural uranium samples are clearly differentiable, and when multiple particles with differing isotope ratios are present in one spot, this result is reflected in the measured isotope ratio. | (1) |
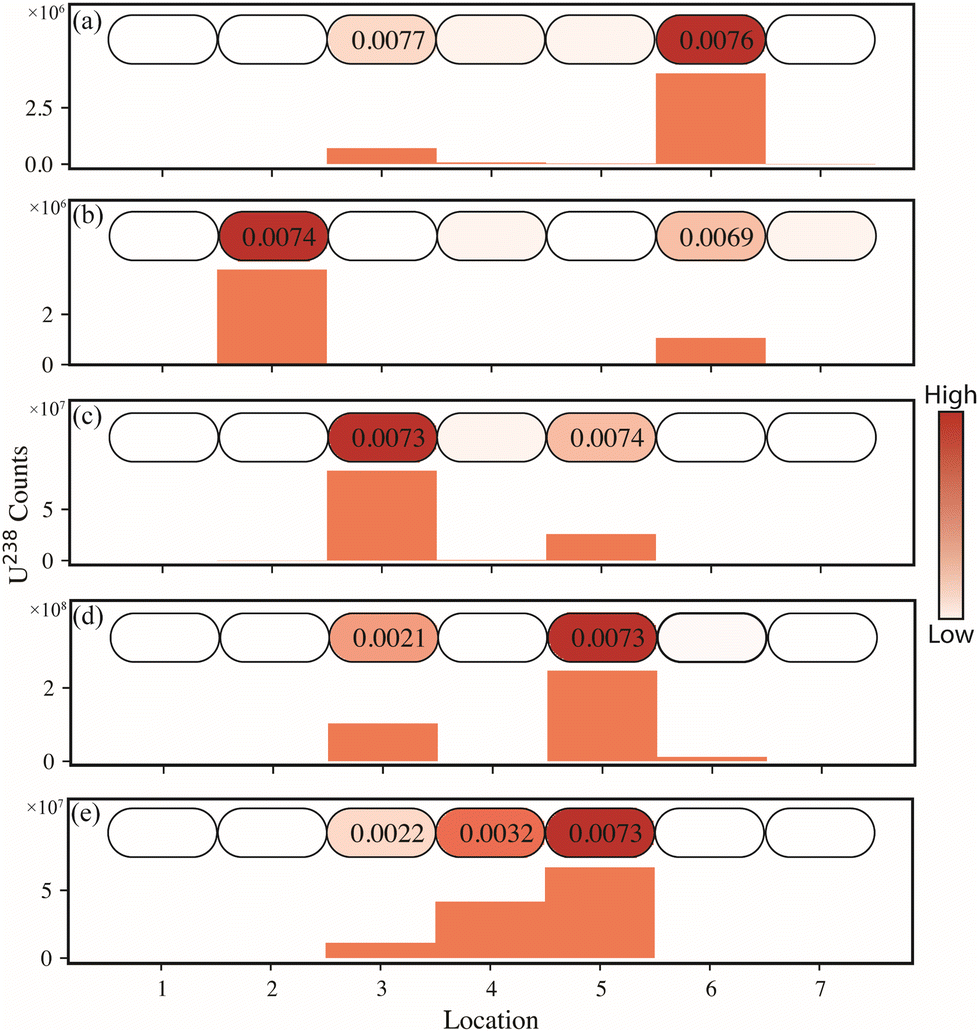 | ||
| Fig. 3 J-Type swipe line scans for 5 different particle placements (a–e). Each oval corresponds to an extraction location. Below each line map, the measured uranium signal is shown as a bar graph. The 235U/238U isotope ratios are shown in the ovals where signal was observed. | ||
| Isotope ratio | UO2F2_ _ _UO2F2 | %RD | UO2F2_ _ _UO2F2 | %RD | UO2F2 _ UO2F2 | %RD | UN_UO2F2 | %RD |
|---|---|---|---|---|---|---|---|---|
| Note: (A) denotes the first particle measured in each line scan. (B) denotes the second particle measured in each line scan. Underscore denotes number of blank locations between particle A and particle B | ||||||||
| 234U/238U (A) | 0.000053 (10) | −4.0 | 0.0000503 (34) | −8.5 | 0.000050 (22) | −9.2 | 0.0000088 (15) | −12 |
| 235U/238U (A) | 0.00728 (29) | 2.4 | 0.00746 (54) | 4.9 | 0.00726 (27) | 2.1 | 0.00219 (91) | −1.7 |
| 236U/238U (A) | <LOD | N/A | <LOD | N/A | <LOD | N/A | 0.0000273 (16) | −23 |
| 234U/238U (B) | 0.000060 (18) | 8.6 | 0.0000594 (80) | 8.0 | 0.000060 (17) | 8.5 | 0.0000589 (76) | 7.1 |
| 235U/238U (B) | 0.00715 (42) | 0.61 | 0.00734 (44) | 3.3 | 0.00735 (22) | 3.4 | 0.00729 (11) | −0.58 |
| 236U/238U (B) | <LOD | N/A | <LOD | N/A | <LOD | N/A | <LOD | N/A |
Table 1 lists the average isotope ratio and uncertainty that were determined from three line scan replicates from separately prepared swipes. The uranium samples used in this study were not isotopic standard reference materials (SRMs), so there is no certified isotope ratio; however, the material was dissolved and analyzed by thermal ionization mass spectrometry (TIMS) (described in detail elsewhere21), which determined the UN material isotope ratios to be 0.00001009 (26) for 234U/238U, 0.0022277 (12) for 235U/238U, and 0.0000353 (11) for 236U/238U. The UO2F2 isotope ratios were 0.00005501 (71) for 234U/238U and 0.0072480 (12) for 235U/238U. The 236U in the UO2F2 material was below the TIMS detection limit. These isotope ratios were used as reference values to investigate the accuracy of the isotope ratios determined via the microextraction method. Measured and reference values were compared based on the % RD between the two. The relative differences from the reference ratio for all measured 235U/238U ratios were less than 5%. The relative differences for the minor isotope ratios were primarily less than 9%.
3.3 Mapping of full J-type swipe
Swipes were imaged using 34 extractions across the surface. Briefly, the swipes are mapped with a sequence which involves performing an extraction on the Teflon surface (to washout out the lines between samples), followed by a J-type swipe blank extraction (to establish a method blank), and finally the location on the swipe surface. This procedure is repeated 34× and requires ∼45 min to map the swipe. For this study, the focus was to demonstrate the capability of this method for rapid and accurate single particle analysis on swipe materials. In this preliminary investigation, ∼74% of the swipe is analyzed. Future work, including newly designed probe heads, will focus on increasing the mapped area of the swipe. If this methodology was to be employed as a screening-type method, one could imagine single replicates being subjected to this method, or the top half of the swipe being rapidly characterized. This would allow for guidance for sample down selecting (out of 100s) and swipes of interest could still be subjected to a more traditional analysis regime, including bulk ashing, digestion, separation, etc.22 Regarding data processing, in each instance the blank extraction is subtracted from the sample, similarly to blank subtraction with traditional solution-based analysis. Fig. 4a shows a representative photograph of a J-type sample (post microextraction mapping). | ||
| Fig. 4 Image of a J-type swipe (a) and data acquired microextraction-ICP-MS mapping of J-type swipes loaded with 5 (b) and 10 (c) uranium particulates. The color inside each oval shows the intensity of the U signal, and the measured isotope ratios shown in each circle (for U determination above LOQ). | ||
Two swipes were doped with varying quantities of uranium particles and mapped via the microextraction-ICP-MS method. A 2-D representation of the 238U signal at each extraction point in the swipe is presented in Fig. 4b and c, and the 235U/238U isotope ratio measured when the signal was above the detection limit is presented within the respective location. The first swipe had 5 particulates placed on it (Fig. 4b), four of which were UO2F2 (natural isotopic abundance), and one particulate of the UN (depleted isotopic abundance) material. The second swipe was a heavier load with 10 particulates (Fig. 4c). Fig. 4 demonstrates that the microextraction-ICP-MS method can directly analyze a swipe and accurately differentiate between natural and depleted uranium in the 2-dimensional space. In fact, Fig. 4c, row 5, has an instance where a depleted and natural particle were in adjacent extraction locations (<8 mm apart). The average 235U/238U isotope ratios, for UN and UO2F2 particles detected in Fig. 4b and c (n = 15), was 0.00292 (21) and 0.00746 (11), respectively. The 234U/238U for the UN and UO2F2 was 0.0000214 (81) and 0.000051 (17), respectively. It should be noted that no mass bias correction was employed for the ICP-MS analysis. In the future, a mass bias measurement completed at the start of each analysis could help correct bias in the isotope ratio measurements.
Generally, swipe samples are not in pristine condition when they are sent for analysis. The swipes often contain dirt and other contamination that must be considered when testing the robustness of an analysis method. To ensure that environmental contamination would not interfere with the mapping of uranium particles on a J-type swipe, a small amount of a matrix (NBS SRM 97b, Flint Clay) containing various trace elements which could be found in real-world samples (e.g., aluminum, barium, cerium, rubidium, strontium, and thorium) was smeared across the lower half of a J-type swipe. Fig. 5 shows the swipe with this matrix placed on it.
 | ||
| Fig. 5 J-type swipe with images from a uranium particulate in a pristine location (A) and embedded within the matrix (B). | ||
Particles containing depleted UN were placed at various locations on the swipe, both outside of the main matrix area (n = 2), and among the SRM 97b (n = 2) matrix, totaling 4 UN particles. Fig. 5a shows a UN particle (∼6 μm diameter) placed on a pristine location as well as a UN particle (∼15 μm) placed among the much larger matrix particles (Fig. 5b). Particles of this size (i.e., matrix) have never been previously extracted with the microextraction-based introduction system. In particular, these types of geological materials likely contain minerals that are more difficult to dissolve than the uranium compounds used as target analytes so far in this study.
Fig. 6 shows the map of the signal intensity of uranium (Fig. 6a) and three trace impurities (Fig. 6b–d, rubidium, strontium, and barium, respectively) for the swipe containing UN particles plus NBS SRM 97A. Fig. 6a shows that uranium was detected within 4 extraction locations, with elevated uranium signal in the expected locations and depleted in 235U. If the uranium signal were coming from the matrix, then the 235U content should be natural abundance. The average 235U/238U ratio from the 4 locations was determined to be 0.00215 (6), which has a 3.5% RD from the expected value. The average 234U/238U and 236U/238U for those particles was 0.0000097 (37) and 0.0000248 (43), respectively. Additionally, there was no statistical difference in determining the isotopic abundance from uranium particles which were in pristine conditions in comparison to the ones embedded within the matrix. Fig. 6b–d show maps of the rubidium, strontium, and barium signals, respectively. These isotope intensities align well with the location of the NBS SRM 97b shown in Fig. 6. Each extraction was completed in less than a minute, and a full swipe can be imaged in 45 min.
 | ||
| Fig. 6 Microextraction-ICP-MS map of J-type sample with results reported for uranium (a), rubidium (b), strontium (c), and barium (d). The 235U/238U isotope ratios are shown in each circle (for U determination above LOQ). | ||
3.4 Investigation of method blanks
A low level of uranium is ubiquitous in the environment, complicating measurements of very small quantities of target uranium particles. Therefore, it is vital to have a strong understanding of the background levels of uranium and the method limits of detection (LODs). J-type swipes are manufactured from cellulose; samples appear to contain trace levels of uranium. To determine the background uranium signal, many extractions were performed on various locations on a J-type swipe and sample holder. Table 2 shows the average uranium signal intensity for blank extractions of Teflon, which is the material that the samples are placed on during the extractions, the cellulose-based swipe itself, and finally the portion of the swipe where a number is printed in black ink. As the table shows, the uranium signal in the instrument blank (an extraction on Teflon) is drastically different (19×) from the method blank (an extraction onto the surface of the cellulose swipe). Furthermore, in the areas where an identifying number is printed (to differentiate individual swipes in each lot), the uranium signal is even higher, suggesting elevated uranium concentrations in the ink.| Material | Sample size | Average 238U integrated counts | Standard deviation | RSD (%) | LOD (pg) |
|---|---|---|---|---|---|
| Note: RSD is the relative standard deviation | |||||
| Teflon | 19 | 2400 | 800 | 34 | 0.71 |
| Swipe | 102 | 46000 |
29000 |
62 | 19 |
| Swipe (ink area) | 12 | 145000 |
19000 |
13 | 30 |
To confirm these findings, a J-type was subjected to analysis by LA-ICP-MS to monitor the 238U distribution. In fact, it is clear that the ink on the J-type swipe does indeed contain higher concentration of U than the J-swipe itself, as shown in Fig. 7. It should be noted that the analysis time for the LA-ICP-MS measurement was 90 min. Historical ORNL data for bulk analysis of J-type swipe samples suggests that the uranium content in a J-type swipe is approximately 80–100 pg, although to our knowledge no previous testing has been done to differentiate uranium coming from the ink as opposed to the cellulose component of the swipe. The microextraction-ICP-MS LOD for a given location (Teflon, swipe, ink) were determined by dividing the average of the integrated signal (+3σ) by the relative response for a known extraction of uranium (1 ng). The LOD changes depending on the sampling location because the background uranium counts are different in different locations. In most sampling locations on the swipe surface, the LOD is 19 pg, whereas over the inked labels, the LOD increases to 30 pg. These LODs are both lower than the LOD determined for cotton swipes (∼50 pg),10,18 which have a higher uranium content than the J-type swipes.
 | ||
| Fig. 7 Stitched photograph of a J-type swipe (a) and subsequent 238U distribution from the J-type swipe from analysis by laser ablation – inductively coupled plasma – mass spectrometry (b). | ||
4. Conclusions
A method was developed for microextraction-ICP-MS that enables rapid, spatially resolved interrogation of ES samples for particles of interest. Specifically, this method was able to successfully map J-type swipes loaded with μm scale uranium particles of varying chemical form and U isotopic composition. Uranium particles were detected on the swipe surface, and isotope ratios were determined with less than 5% RD from reference values for the major isotope ratios, and <9% RD for minor isotope ratios. In fact, presented here, the microextraction-ICP-MS method could readily distinguish two neighboring (∼4 mm apart) particles had different characteristic isotope abundances and chemical forms. The methods presented here are robust and were successfully employed for analysis of U particles in a heavy matrix on a J-type swipes. In fact, these particles were accurately analyzed (235U/238U) within 3.5% RD from the expected value. A custom program was developed to visualize these mapped swipes in terms of both analyte signal and isotopic ratios. A full swipe can be imaged in less than 45 min, showing that this method is very rapid, especially compared with bulk digestion, which can take several weeks and loses isotopic information from individual particles. The microextraction method shows significant promise as a high-throughput, low-cost screening method for both elemental and isotopic content on a variety of sample types. The cellulose-based sample matrix studied here is currently in use by IAEA for PIC samples, but this method could be expanded to directly sample surfaces of other types of environmental samples, including filters and/or electrostatic plates from active air samplers. The ability to automate the sampling of a certain region of the material surface ensures that the untouched portion of the sample is still available for submission to traditional high-sensitivity, high precision, but slower and lower throughput methods. Future studies will aim to extend this method to new sample types, as well as more fully demonstrate its utility as a reliable screening method for routine, operational sample streams.Notes
This manuscript has been authored in part by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Oak Ridge National Laboratory, managed by UT-Battelle for the Department of Energy under contract DE-AC05-000R22725. This work was funded by the United States National Nuclear Security Administration's Office of Defense Nuclear Nonproliferation Research & Development. The authors would like to acknowledge Jacquelyn DeMink (ORNL) for assistance with graphics. The authors would also like to acknowledge the Department of Defense (DoD) SkillBridge Program.References
- A. Limbeck, P. Galler, M. Bonta, G. Bauer, W. Nischkauer and F. Vanhaecke, Recent advances in quantitative LA-ICP-MS analysis: challenges and solutions in the life sciences and environmental chemistry, Anal. Bioanal. Chem., 2015, 407(22), 6593–6617 CrossRef CAS PubMed.
- A. Sussulini, J. S. Becker and J. S. Becker, Laser ablation ICP-MS: Application in biomedical research, Mass Spectrom. Rev., 2017, 36(1), 47–57 CrossRef CAS PubMed.
- D. Chew, K. Drost, J. H. Marsh and J. A. Petrus, LA-ICP-MS, imaging in the geosciences and its applications to geochronology, Chem. Geol., 2021, 559, 119917 CrossRef CAS.
- B. T. Manard, C. D. Quarles Jr., E. M. Wylie and N. Xu, Laser ablation – inductively couple plasma – mass spectrometry/laser induced break down spectroscopy: a tandem technique for uranium particle characterization, J. Anal. At. Spectrom., 2017, 32(9), 1680–1687 RSC.
- J. S. Becker, Applications of inductively coupled plasma mass spectrometry and laser ablation inductively coupled plasma mass spectrometry in materials science, Spectrochim. Acta, Part B, 2002, 57, 1805–1820 CrossRef.
- H. W. Paing, B. T. Manard, B. W. Ticknor, J. R. Bills, K. A. Hall, D. A. Bostick, P. Cable-Dunlap and R. K. Marcus, Rapid Determination of Uranium Isotopic Abundance from Cotton Swipes: Direct Extraction via a Planar Surface Reader and Coupling to a Microplasma Ionization Source, Anal. Chem., 2020, 92(12), 8591–8598 CrossRef CAS PubMed.
- B. T. Manard, K. T. Rogers, B. W. Ticknor, S. C. Metzger, N. A. Zirakparvar, B. D. Roach, D. A. Bostick and C. R. Hexel, Direct Uranium Isotopic Analysis of Swipe Surfaces by Microextraction-ICP-MS, Anal. Chem., 2021, 11133–11139 CrossRef CAS PubMed.
- G. J. Van Berkel and V. Kertesz, Application of a liquid extraction based sealling surface sampling probe for mass spectrometric analysis of dried blood spots and mouse whole-body thin tissue sections, Anal. Chem., 2009, 81, 9146–9152 CrossRef CAS PubMed.
- H. Luftmann, A simple device for the extraction of TLC spots: direct coupling with an electrospray mass spectrometer, Anal. Bioanal. Chem., 2004, 378(4), 964–968 CrossRef CAS PubMed.
- B. T. Manard, S. C. Metzger, K. T. Rogers, B. W. Ticknor, N. A. Zirakparvar, B. D. Roach, D. A. Bostick and C. R. Hexel, Direct analysis of cotton swipes for plutonium isotope determination by microextraction-ICP-MS, J. Anal. At. Spectrom., 2021, 36(10), 2202–2209 RSC.
- V. C. Bradley, B. W. Ticknor, D. R. Dunlap, N. A. Zirakparvar, S. C. Metzger, C. R. Hexel and B. T. Manard, Microextraction–TQ–ICP–MS for the Direct Analysis of U and Pu from Cotton Swipes, Anal. Chem., 2023, 15867–15874 CrossRef CAS PubMed.
- C. J. Stouffer and R. K. Marcus, Direct, multielement determinations from cotton swipes via plate express microextraction coupled to an inductively coupled plasma mass spectrometer (μEx-ICP-MS), J. Anal. At. Spectrom., 2023, 1943–1951 RSC.
- V. C. Bradley, T. L. Spano, S. C. Metzger, B. W. Ticknor, D. R. Dunlap, N. A. Zirakparvar, B. D. Roach, C. R. Hexel and B. T. Manard, Direct isotopic analysis of solid uranium particulates on cotton swipes by microextraction-ICP-MS, Anal. Chim. Acta, 2022, 1209, 339836 CrossRef CAS PubMed.
- D. L. Donahue, Strengthened nuclear safeguards, Anal. Chem., 2002, 74, 28A–35A CrossRef.
- D. L. Donahue, Strengthening IAEA safeguards through environmental sampling and analysis, J. Alloys Compd., 1998, 271–273, 11–18 CrossRef.
- IAEA Safeguards Glossary: 2022 edition. International Atomic Energy Agency, Vienna, Austria, 2022 Search PubMed.
- S. Boulyga, S. Konegger-Kappel, S. Richter and L. Sangely, Mass spectrometric analysis for nuclear safeguards, J. Anal. At. Spectrom., 2015, 30(7), 1469–1489 RSC.
- V. C. Bradley, T. L. Spano, C. V. Thompson, B. W. Ticknor, D. R. Dunlap, S. C. Metzger, C. R. Hexel and B. T. Manard, Analysis of solid uranium particulates on cotton swipes with an automated microextraction-ICP-MS system, Anal. Methods, 2022, 14(44), 4466–4473 RSC.
- V. C. Bradley, T. L. Spano, C. V. Thompson, B. W. Ticknor, D. R. Dunlap, S. C. Metzger, C. R. Hexel and B. T. Manard, Analysis of solid uranium particulates on cotton swipes with an automated microextraction-ICP-MS system, Anal. Methods, 2022, 14(44), 4466–4473 RSC.
- S. Richter, C. Venchiarutti, C. Hennessy, U. Jacobsson, R. Bujak, J. Truyens and Y. Aregbe, Preparation and certification of the uranium nitrate solution reference materials series IRMM-2019 to IRMM-2029 for the isotopic composition, J. Radioanal. Nucl. Chem., 2018, 318(2), 1359–1368 CrossRef CAS.
- V. C. Bradley, T. L. Spano, S. C. Metzger, B. W. Ticknor, D. R. Dunlap, N. A. Zirakparvar, B. D. Roach, C. R. Hexel and B. T. Manard, Direct isotopic analysis of solid uranium particulates on cotton swipes by microextraction-ICP-MS, Anal. Chim. Acta, 2022, 1209, 339836 CrossRef CAS PubMed.
- S. C. Metzger, K. T. Rogers, D. A. Bostick, E. H. McBay, B. W. Ticknor, B. T. Manard and C. R. Hexel, Optimization of uranium and plutonium separations using TEVA and UTEVA cartridges for MC-ICP-MS analysis of environmental swipe samples, Talanta, 2019, 198, 257–262 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |