
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved pH measurement of mobile phases in reversed-phase liquid chromatography†
Agnes
Heering
 *,
Markus
Lahe
,
Martin
Vilbaste
,
Jaan
Saame
,
John Paulo
Samin
and
Ivo
Leito
*
*,
Markus
Lahe
,
Martin
Vilbaste
,
Jaan
Saame
,
John Paulo
Samin
and
Ivo
Leito
*
Institute of Chemistry, University of Tartu (UT), Ravila Street 14a, 50411 Tartu, Estonia. E-mail: agnes.heering@ut.ee; ivo.leito@ut.ee
First published on 31st January 2024
Abstract
Mobile phase pH is a critically important parameter in reversed-phase liquid chromatographic (RPLC) separations involving analytes that display acidic or basic properties in the pH range used for the mobile phase. The main problem in measuring mobile phase pH lies in the fact that RPLC mobile phases are typically aqueous–organic mixtures. In addition to experimental difficulties, the pH values refer to different aqueous–organic compositions that cannot be correctly compared. Given this situation, the unified pH (wabspH, also termed as  ) based on the absolute chemical potential of the solvated proton has been proposed as a rigorous way of characterising mobile phase acidity that is fully inter-comparable between mobile phases of any composition. Here we report the wabspH values of 78 reversed-phase liquid chromatography-mass spectrometry mobile phases that were carefully measured by potential differences in a symmetric cell with two glass electrode half-cells and almost ideal ionic liquid triethylamylammonium bis((trifluoromethyl)sulfonyl)imide [N2225][NTf2] salt bridge with multiple overlapping measurements. The system of altogether 300 ΔwabspH values was anchored to the pH value of standard pH 7.00 aqueous buffer solution. The consistency standard deviation of the whole set of measurements was 0.09 pH units. In addition to the differential potentiometric reference method, simpler measurement methods that use double junction reference or double junction combined electrodes were tested and were found suitable for routine laboratories if high accuracy is not required.
) based on the absolute chemical potential of the solvated proton has been proposed as a rigorous way of characterising mobile phase acidity that is fully inter-comparable between mobile phases of any composition. Here we report the wabspH values of 78 reversed-phase liquid chromatography-mass spectrometry mobile phases that were carefully measured by potential differences in a symmetric cell with two glass electrode half-cells and almost ideal ionic liquid triethylamylammonium bis((trifluoromethyl)sulfonyl)imide [N2225][NTf2] salt bridge with multiple overlapping measurements. The system of altogether 300 ΔwabspH values was anchored to the pH value of standard pH 7.00 aqueous buffer solution. The consistency standard deviation of the whole set of measurements was 0.09 pH units. In addition to the differential potentiometric reference method, simpler measurement methods that use double junction reference or double junction combined electrodes were tested and were found suitable for routine laboratories if high accuracy is not required.
1. Introduction
Liquid chromatography (LC), especially its reversed-phase (RP) variant, and its combination with mass spectrometry (LC-MS) are the workhorses in proteomics,1 metabolomics,2 environmental analysis3etc. Mobile phase pH is a critically important parameter in separations involving analytes that display acidic or basic properties in the pH range used for the mobile phase (meaning the majority of analytes) and is used in retention modelling.4,5 pH is also crucial for LC column stability, and manufacturers state the operational pH ranges of columns but usually refer to the aqueous pH values.6The difficulty in measuring mobile phase pH is that RPLC mobile phases are typically aqueous–organic mixtures, and mobile phase composition changes during the run if gradient elution is used (which is the case in most separations). The pH quoted for mobile phases is usually the pH of the aqueous component (denoted as wwpH)7 or the pH of the mixed mobile phase measured with pH electrode calibrated in aqueous standards (denoted as swpH).7 Neither of these is rigorous in the context of RPLC.
wwpH characterises the pH of only the aqueous component of the mobile phase. Obviously, the acidity will change upon adding the organic component. True, if the buffer system in the mobile phase and the analytes are of a similar chemical nature, then the change will at least partially be offset by the parallel changes in the pKa values of the analytes and buffer substance. swpH expresses pH in the actual mobile phase, but the way it is measured neglects the potentially quite significant liquid junction potential (LJP) difference between the aqueous calibration buffers and aqueous–organic mobile phases and therefore such a measurement may yield a value that is significantly biased from the actual pH (in terms of H+ activity) in the solution.
The solvent-specific pH scales (sspH) do not have such flaws but are rarely used due to (a) lack of calibration standards, (b) limited knowledge of the pKa values of analytes in mixed solvents and (c) (in the case of gradient elution) changing mobile phase composition during elution (meaning that in principle at any moment in the gradient run a different pH scale holds).7 pKa values of analytes in different solvents can be quite well estimated8–12 and if pH is known for the eluent composition in the beginning, in the middle and at the end of the gradient, then pH can be reasonably estimated in other parts of the gradient profile. There still remains the issue of lack of calibration standards; sspH does not solve the comparability problem, and all the pH values are still solvent-specific.
The comparability problem can be solved by the unified acidity scale (pHabs scale), which is based on solvent-independent standard state (absolute chemical potential of the solvated proton) and thus enables rigorous comparability of pHabs values between any solvent systems.13 The pHabs scale has been experimentally realised via measurement of potential difference between two glass electrodes immersed in two solutions (connected via a salt bridge) which pHabs values are compared and LC mobile phases were the first systems where it was used.14 For convenience, the pHabs scale was shifted and aligned with the aqueous pH scale to be termed wabspH (also termed as  ).14 This way of writing means that the wabspH values measured in any solvent/medium are directly comparable to the aqueous pH scale. i.e.wabspH 7.0 measured in any medium refers to the same chemical potential of the solvated proton as in aqueous solution with the conventional pH of 7.0. This also means that the acidities of LC mobile phases with various solvent compositions can be directly compared to the conventional aqueous pH scale.14
).14 This way of writing means that the wabspH values measured in any solvent/medium are directly comparable to the aqueous pH scale. i.e.wabspH 7.0 measured in any medium refers to the same chemical potential of the solvated proton as in aqueous solution with the conventional pH of 7.0. This also means that the acidities of LC mobile phases with various solvent compositions can be directly compared to the conventional aqueous pH scale.14
While rigorous in its essence, measurement of wabspH is typically carried out via differential potentiometry, which involves liquid junctions. It is, therefore, still influenced by the LJP. The first wabspH measurement method14 used a bridge electrolyte of 0.05 M Et4NClO4 solution in MeCN and a sophisticated (but still approximate) LJP estimation approach. This LJP estimation approach needed experimental data from the literature. In the (frequent) cases of non-availability of data, estimates were used instead. This led to high uncertainties of the estimated LJP values and, therefore, high uncertainties of the wabspH values.
The more advanced measurement method15 employed in this work uses a novel ionic liquid salt bridge (ILSB) with triethylamylammonium bis((trifluoromethyl)sulfonyl)imide [N2225][NTf2]. Because of the high similarity of the involved ions – both by mobilities and solvation Gibbs energies in a wide variety of solvents – this IL has been demonstrated to effectively cancel the LJP out within standard deviation of 6.3 mV, corresponding to 0.11 pH units,16–19 thus, significantly simplifying the measurement and decreasing the uncertainty of the obtained wabspH values. The ILSB approach can be used with essentially any solvent or mixture. Another advantage is that in the case of ILSB, the shape of the junction does not matter, unlike in the case of the traditional KCl salt bridge.20
Any commercially available glass electrode half-cell is suitable for differential potentiometric measurement21 and any instrument capable of measuring potentials in the millivolt range, like a pH meter, potentiostat and electrometer, is suitable if the input impedance is >1013 Ω.15
The method based on measuring potential differences between two glass electrodes might be too complex for routine laboratories. Therefore, the same approach was applied in a more conventional and simpler-to-use manner using a glass electrode half-cell with a double junction reference electrode or a double junction combined pH electrode. The outer compartment of the reference electrode was filled with the abovementioned ionic liquid. In contrast, the inner compartment was filled with the traditional 3 M or a saturated aqueous solution of KCl.
In this paper, we present wabspH values of a large number of LC mobile phases measured with improved method. Some earlier published values were remeasured to compare the previous values obtained with LJP estimation14 to the more accurate values obtained with the new method utilising the specially designed ILSB, which enables cancellation of LJP.19 In addition, the new wabspH measurement approach was applied with a double junction combined pH electrode to demonstrate the easy applicability of the method adaption by practitioners in routine laboratories.
2. Experimental
2.1. Materials and instrumentation
Different types of electrodes were used depending on the electrochemical cell. Electrodes from different manufacturers were used to test the robustness of the method. The used electrodes and instruments are given in Table 1.| Electrode type | Electrode | Diaphragm | Used in cell | Instrument | |
|---|---|---|---|---|---|
| A | Half-cell, solid metal contact | EST-0601, Izmeritelnaya Tekhnika | None | I | Metrohm 713 pH meter; Keysight B2987A |
| B | Half-cell, liquid filled | DG300-SC, Mettler-Toledo | None | II.1 | Keysight B2987A |
| C | Half-cell, liquid filled | 6.0150.100, Metrohm | None | II.1 | Keysight B2987A |
| D | Double junction combined electrode | 6.0269.100, Metrohm | Fixed ground-joint | II.2 | Elmetron CP-411 pH meter; Metrohm 744 pH meter |
| E | Double junction combined electrode | WD-35805-09, Oakton | Flushable PTFE | II.2 | Elmetron CP-411 pH meter; Metrohm 744 pH meter |
Aqueous standard buffers 4.01 ± 0.02 (Mettler Toledo), 7.00 ± 0.02 (Mettler Toledo), 9.00 ± 0.02 (Hach), 10.01 ± 0.02 (Mettler Toledo) were used for calibrating the electrodes (Table 1) and for anchoring the wabspH values. The ionic liquid [N2225][NTf2] (99%, Iolitec) and KCl (p.a., LachNer) were used as salt bridge electrolytes.
Chemicals for mobile phases: acetonitrile (CHROMASOLV, ≥99.9%, for LC-MS, Honeywell Riedel-de-Haën), methanol (CHROMASOLV, for HPLC, ≥99.9%, Honeywell Riedel-de-Haën), formic acid (puriss, p.a., ACS reagent, Reag. Ph. Eur, ≥98%, Honeywell Fluka), ammonium hydroxide solution (eluent additive for LC-MS, ≥25% in H2O, Honeywell Fluka), ammonium acetate (BioUltra, for molecular biology, ≥99.0%), 1,1,1,4,4,4-hexafluoro-2,3-bis(trifluoromethyl)butane-2,3-diol (perfluoropinacol, PP), 1,1,1,3,3,3-hexafluoro-2-methylisopropanol (HFTB), 2,2,2-trifluoroethanol (TFE) were LC-MS grade and obtained from Sigma-Aldrich, 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) was obtained from Fluka. All chemicals were used without further purification.
The aqueous phase compositions are given in Table 2. The used ammonia concentration is not common in liquid chromatography mobile phases but was used to compare results obtained with the previously used method.14
| Aqueous phase composition | Short name | wwpH |
|---|---|---|
| 0.1 vol% HCOOH | None | 2.68 |
| 1 mM NH3 | None | 9.75 |
| 5 mM CH3COONH4 titrated with HCOOH or NH4OH | Acetate | 5.00; 6.00; 7.00; 9.50 |
| 0.1 vol% HCOOH titrated with NH4OH | Formate | 4.00; 4.50; 5.00; 5.50 |
| 5 mM 1,1,1,3,3,3-hexafluoro-2-methyl-2-propanol titrated with NH4OH | HFTB | 5.00; 6.00; 7.00; 9.50 |
| 50 mM Na2HPO4 + 50 mM KH2PO4 | Phosphate | 7.50 |
| 5 mM perfluoropinacol titrated with NH4OH | PP | 5.00; 6.00; 7.00 |
| 5 mM 2,2,2-trifluoroethanol titrated with NH4OH | TFE | 8.50; 9.00; 10.00 |
| 5 mM 1,1,1,3,3,3-hexafluoro-2-propanol titrated with NH4OH | HFIP | 8.50; 9.00; 10.00 |
wwpH values were measured with Oakton WD-35801-09 connected with Evikon E6115 pH meter or Elmetron EPP-1 connected with Elmetron CP-411 pH meter. The combined pH electrode was calibrated daily with standard buffers with pH values of 7.00 and 4.01 or 10.01, depending on the pH values of the prepared aqueous solutions.
2.2. Methods
wabspH measurements were performed with two methods. One was the potential measurement between two glass electrodes (GE) (cell I). This is the so-called “reference method” for wabspH. The other method, aiming to be the method of choice for routine use, measures the potential between a glass and a double-junction reference electrode (cell II) or a double junction combined pH electrode. This method is termed the “routine method”. The above-mentioned cells are as follows:| GE 2|Solution 2|[N2225]+[NTf2]−|Solution 1|GE 1 | (I) |
| Ag|AgCl|sat. KCl(aq)|[N2225]+[NTf2]−|Solution|GE | (II) |
The reference method with the cell I used a Metrohm 713 pH meter or a Keysight B2987A electrometer to measure the potential difference between two glass electrodes immersed in the two solutions that are compared, separated by a salt bridge containing the ionic liquid [N2225]+[NTf2]− (see Fig. S2 and S3 in ESI†).
A Pt wire was used as an auxiliary electrode for measurements with Metrohm 713 pH meter.14 Metal solid-contact glass electrodes (electrode A, Table 1) were used. Radiometer K401 saturated calomel reference electrode was used to calibrate the metal solid-contact glass electrodes. Measurements were thermostated at 25.0 °C (±1.0 °C) using a thermostat MLW U2C and later Julabo Corio CD-200F. These measurements were carried out in a special water-jacketed glass cell15 from Gebr. Rettberg (Fig. S1†), which was placed in a Faraday cage (VistaShield, Gamry).
Measurements with the cell I give “relative” wabspH values (ΔwabspH values). When overlapping measurements are in excess, these values can be combined into a “ladder”. A least-squares minimization technique15 assigns “absolute” wabspH values to mobile phases. One of the solutions with a known pH value is used as the “anchor” solution. In this case, it is the standard aqueous buffer solution with a pH value of 7.00. MS Excel Solver is used to carry out the least-squares minimization.
Although every effort was made to choose GE 1 and GE 2 with as close a slope and intercept as possible, there are still some minor differences between them. The most straightforward approach to minimize the effect of these differences is taking an average of two measurements where the electrode positions have been switched between the solutions.15 Although simple, this approach is labour-intensive.
Therefore, this was not done with all measurements but with a set of 31 mobile phase pairs measured with both polarities. Minimizations were done with potentials from measurements of Solution 1–Solution 2, Solution 2–Solution 1 and the average potential of both polarities. The root mean square difference between the three obtained sets of values was 0.04. Also, averaging did not give better consistency than using individual differences. Thus, making the main bulk of measurements with only one polarity of the electrodes was considered sufficient.
The aim was to measure a large wabspH data pool to validate the method and define quality requirements for the measured data. Data points were collected at 3 s (Metrohm) or 10 s (Keysight) intervals. Initially, the data for one measurement series was collected for 30 minutes and later for 60 minutes. Most of the data was collected for 60 min. One measurement was 10 h to test the stability. In all cases, only the points from a continuous 15 min time interval giving the most stable signals were used for averaging to get the ΔpHabs values displayed on the arrows in Fig. S7.† The quality criteria for stable reading were set retrospectively to retain only reliable measurements. The criteria are given in section 3.1.
The routine method (Fig. S4†) uses cell II to measure the potential difference between a glass and a double junction reference electrode. These measurements were made at room temperature, around 21 °C in our case, and a magnetic stirrer was used for mixing.
The method using cell II can be operated with any mainstream pH meter. This method was realised (a) separately with a glass half-cell pH electrode (electrodes B and C, Table 1) together with a double junction Metrohm Ag/AgCl reference electrode 6.0729.100 (DJ ref) designated as cell II.1 (see Fig. S4A†), and (b) as a combined electrode with double junction pH electrodes D and E (Table 1), designated as cell II.2 (see Fig. S4B†). Electrodes B and C were used because glass electrodes with metal contact (electrode A) are challenging to obtain.
Measurements with cell II were intended to be similar to routine measurements. Measurements with cell II.1 were carried out with Keysight B2987A because it enables the connection of a separate glass and a reference electrode. This method is new and there is no information about the temporal nature of the data. Points were collected for 600 s with 5 s intervals to ensure that the plateau had been reached. Points from 285 s to 315 s were averaged where the stable reading had been reached. Glass electrode half-cell measurements against a reference electrode reach a plateau faster than measurements against another glass electrode half-cell. Most measurements with cell II.2 were carried out during 30 s to 120 s with an Elmetron CP-411 pH meter to mimic routine pH measurements and were single values. CP-411 resolution is lower. Some of the measurements with cell II.2 were done with Metrohm 744 pH meter to investigate the behaviour of combined electrodes.
The inner solution of the double junction reference electrode was saturated aqueous KCl solution, and the outer filling was [N2225][NTf2]. Combined electrodes were filled with 3 M aqueous KCl and [N2225][NTf2]. In the case of an ionic liquid salt bridge, the LJP does not depend on the shape of the junction.20 Cell II.1 has been previously tested with TRIS-buffered artificial seawater22 and water–ethanol mixtures.23,24 This is the first successful use of cell II.2. Two-point calibration25 using standard aqueous buffers was used to measure wabspH values.
3. Results and discussion
3.1. Overview of the results
Overall, the wabspH values of 78 mobile phases were measured with cell I (Table 3), connected by ΔpHabs measurements conducted over two years. Based on the typical measurement results, the maximum standard deviation of the used data points of 1 mV and maximum drift of 4 mV h−1 were set as quality limits. Measurement series that failed to meet these criteria were omitted from data analysis. Eventually, 300 measurement series out of the overall 427 were used. 90% of measurements done with MeOH mixtures passed the quality control, but only 60% of MeCN-based mixtures passed. This is due to specific experimental problems related to mobile phases with 70% to 80% acetonitrile by volume. Fig. S7 in ESI† presents the full set of measurement results.| Mobile phasea | wabspHb | Earlier value |
|---|---|---|
a Composition given as organic phase/aqueous phase. The pH values indicated in compositions refer to the aqueous phase, before mixing.
b This work and the most accurate value.
c ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ref. 14.
d Ref. 26.
e Ref. 27.
f Ref. 28. Ref. 14.
d Ref. 26.
e Ref. 27.
f Ref. 28.
|
||
| MeCN/1 mM NH3 80/20 | 10.81 | 10.47c |
| MeOH/pH 10 (TFE) 25/75 | 10.49 | 9.73d |
| MeOH/pH 10 (HFIP) 25/75 | 10.29 | 9.87d |
| MeCN/1 mM NH3 50/50 | 10.21 | 8.70c |
| MeOH/pH 9.5 (HFTB) 25/75 | 10.14 | |
| Aqueous buffer pH 10.01 | 10.05 | |
| MeOH/1 mM NH3 80/20 | 9.95 | 8.89c |
| MeOH/pH 9.5 (acetate) 25/75 | 9.93 | |
| MeOH/pH 9.5 (PP) 25/75 | 9.93 | |
| MeOH/1 mM NH3 20/80 | 9.92 | |
| MeOH/1 mM NH3 50/50 | 9.90 | 10.07c |
| MeCN/1 mM NH3 20/80 | 9.85 | |
| MeOH/pH 9 (TFE) 25/75 | 9.61 | 7.38d |
| MeOH/pH 9 (HFIP) 25/75 | 9.54 | 9.18d |
| 9.62e | ||
| MeOH/pH 9 (HFTB) 25/75 | 9.39 | 9.20d |
| 9.40e | ||
| MeOH/pH 8.5 (HFIP) 25/75 | 9.30 | 8.83d |
| 9.36e | ||
| MeOH/pH 8.5 (TFE) 25/75 | 9.23 | 7.23d |
| MeCN/pH 5 (acetate) 80/20 | 9.14 | 8.99c |
| MeOH/pH 8.5 (HFTB) 25/75 | 8.98 | 8.85d |
| 9.03e | ||
| MeOH/pH 7.5 (phosphate) 50/50 | 8.98 | |
| Aqueous buffer pH 9.00 | 8.96 | |
| MeOH/pH 9 (acetate) 25/75 | 8.96 | 8.70d |
| 9.00e | ||
| MeOH/pH 9 (PP) 25/75 | 8.91 | 8.68e |
| 8.87e | ||
| MeCN/pH 5.5 (formate) 80/20 | 8.63 | 8.59f |
| MeOH/pH 7 (HFTB) 80/20 | 8.52 | |
| MeCN/pH 5 (formate) 80/20 | 8.50 | 8.43f |
| MeOH/pH 8.5 (acetate) 25/75 | 8.49 | 8.17d |
| 8.48e | ||
| MeOH/pH 8.5 (PP) 25/75 | 8.40 | 8.20d |
| 8.47e | ||
| MeOH + 5 mM HFTB/pH 7 (HFTB) 80/20 | 8.37 | |
| MeCN/pH 4.5 (formate) 80/20 | 8.23 | 8.10f |
| MeOH/pH 5 (acetate) 90/10 | 8.11 | |
| MeOH/pH 7.5 (phosphate) 20/80 | 7.96 | |
| MeOH + 5 mM HFTB/pH 7 (HFTB) 40/60 | 7.86 | |
| MeCN/pH 4 (formate) 80/20 | 7.84 | 7.64f |
| MeOH/pH 7 (HFTB) 40/60 | 7.83 | |
| MeOH/pH 7 (HFTB) 25/75 | 7.79 | |
| MeOH/pH 5 (acetate) 80/20 | 7.70 | |
| MeOH/pH 7 (PP) 25/75 | 7.65 | |
| MeOH/pH 5.5 (formate) 80/20 | 7.57 | |
| MeOH/pH 7 (HFTB) 5/95 | 7.48 | |
| MeOH + 5 mM HFTB/pH 7 (HFTB) 5/95 | 7.46 | |
| MeOH/pH 7 (acetate) 25/75 | 7.34 | |
| MeOH/pH 6 (HFTB) 25/75 | 7.33 | |
| MeOH/pH 5 (formate) 80/20 | 7.22 | |
| MeCN/pH 5 (acetate) 50/50 | 7.19 | 7.50c |
| MeCN/pH 5.5 (formate) 50/50 | 7.09 | 7.49f |
| MeOH/pH 6 (PP) 25/75 | 7.02 | |
| Aqueous buffer pH 7.00 | 7.00 | |
| MeOH/pH 6 (acetate) 25/75 | 6.82 | |
| MeOH/pH 5 (HFTB) 25/75 | 6.82 | |
| MeCN/pH 5.5 (formate) 40/60 | 6.78 | 7.11f |
| MeCN/pH 4 (formate) 75/25 | 6.69 | 7.32f |
| MeCN/pH 5 (formate) 50/50 | 6.59 | 7.05f |
| MeOH/pH 5.5 (formate) 50/50 | 6.56 | |
| MeCN/pH 4 (formate) 70/30 | 6.53 | 7.03f |
| MeOH/pH 5 (acetate) 50/50 | 6.50 | 6.47c |
| MeOH/pH 5 (PP) 25/75 | 6.23 | |
| MeCN/pH 5 (formate) 40/60 | 6.19 | 6.62f |
| MeOH/pH 5 (formate) 50/50 | 6.08 | |
| MeCN/pH 4.5 (formate) 50/50 | 6.06 | 6.56f |
| MeOH/pH 5 (acetate) 25/75 | 6.05 | |
| MeCN/pH 4 (formate) 60/40 | 5.98 | |
| MeCN/pH 4.5 (formate) 40/60 | 5.76 | 6.14f |
| MeOH/pH 5.5 (formate) 20/80 | 5.75 | |
| MeOH/pH 5 (acetate) 20/80 | 5.50 | |
| MeCN/0.1% HCOOH 80/20 | 5.48 | 5.37c |
| MeOH/pH 5 (acetate) 10/90 | 5.36 | |
| MeOH/pH 5 (formate) 20/80 | 5.29 | |
| MeCN/pH 4 (formate) 40/60 | 5.25 | |
| MeOH/0.1% HCOOH 90/10 | 4.95 | |
| MeCN/pH 4 (formate) 25/75 | 4.73 | |
| MeOH/0.1% HCOOH 80/20 | 4.69 | 4.79c |
| 0.1% HCOOH MeOH/0.1% HCOOH 90/10 | 4.60 | |
| 0.1% HCOOH MeOH/0.1% HCOOH 80/20 | 4.36 | |
| MeCN/0.1% HCOOH 50/50 | 3.97 | 4.39c |
| Aqueous buffer pH 4.01 | 3.96 | |
| MeOH/0.1% HCOOH 50/50 | 3.68 | 3.89c |
| 0.1% HCOOH MeOH/0.1% HCOOH 50/50 | 3.60 | |
| MeOH/0.1% HCOOH 20/80 | 2.95 | |
| 0.1% HCOOH MeOH/0.1% HCOOH 20/80 | 2.88 | |
| MeOH/0.1% HCOOH 10/90 | 2.74 | |
| 0.1% HCOOH MeOH/0.1% HCOOH 10/90 | 2.68 | |
Mobile phases with acetonitrile (MeCN) are more complicated to measure than the ones with methanol (MeOH) because the ionic liquid (IL) solubility is better in MeCN. The most difficult-to-measure mobile phases were those with 70% to 80% acetonitrile by volume. With mobile phase composition in this region, droplets were formed on the IL-solution interface (Fig. S5†), and their movement disturbed the signal stability. This results in the corresponding mobile phase wabspH measurements more often failing the quality check and being left out from least squares minimization (Fig. S6†).
Assigning absolute wabspH values to individual solutions using the obtained ΔpHabs values via least-squares minimization is the same as in literature15,29 except for the modified data analysis.30
The complete set of ΔpHabs measurement results are shown in Fig. S7.† As described above, the wabspH values were anchored to an aqueous standard buffer with conventional pH (and thus also wabspH) equal to 7.00. The wabspH values of the other three aqueous standard buffers were allowed to change during the least squares minimization. The good agreement between their reference values and the values assigned as a result of minimization (3.96 vs. 4.01; 8.96 vs. 9.00 and 10.05 vs. 10.00) gives additional evidence that the measurement method works well. The consistency standard deviation15 of the whole set of measurements was 0.09 pH units. The uncertainties are in the range of 0.12 to 0.13, the largest contribution coming from LJP cancellation.29
When the same solution is on both sides of cell I, which means no pH difference between the solutions, the measured pH difference can be up to 0.07 instead of zero for all tested mobile phases.
3.2. Relations between wabspH values and other ways of expressing pH of mobile phases
There is a weak correlation between wwpH values and wabspH values when different buffers and organic solvent compositions are involved (Fig. 1A)—the changes in the pKa values of the buffer components with adding the organic solvent cause this. The pKa changes more in the case of neutral acids compared to cationic acids. However, there is a reasonable correlation between wwpH values and wabspH values within a fixed organic solvent percentage (Fig. 1B) because the change in pKa is constant for the given solvent composition. | ||
| Fig. 1 (A) Relation between wwpH values and wabspH values (cell I) in all measured mobile phases. (B) Relation between wwpH values and wabspH values (cell I) in mobile phases with 25% MeOH and varying buffer component. PP is perfluoropinacol, HFTB is 1,1,1,3,3,3-hexafluoro-2-methyl-2-propanol. | ||
There is a correlation between swpH values and wabspH values. Still, they do not differ simply by a constant value as was previously assumed26 (Fig. 2A). Some of the swpH measurements were repeated, and the values differed up to 0.3 pH units. Solubility issues might have caused this difference. KCl solubility in methanol31 and acetonitrile31 and their aqueous mixtures32 is substantially lower than in water, and KCl might partially precipitate in the diaphragm of the combined electrode during measurements. These findings suggest that the δ parameter33–35 proposed for such pH conversions is not a constant value, and the difference seems more significant for MeCN-based mobile phases.
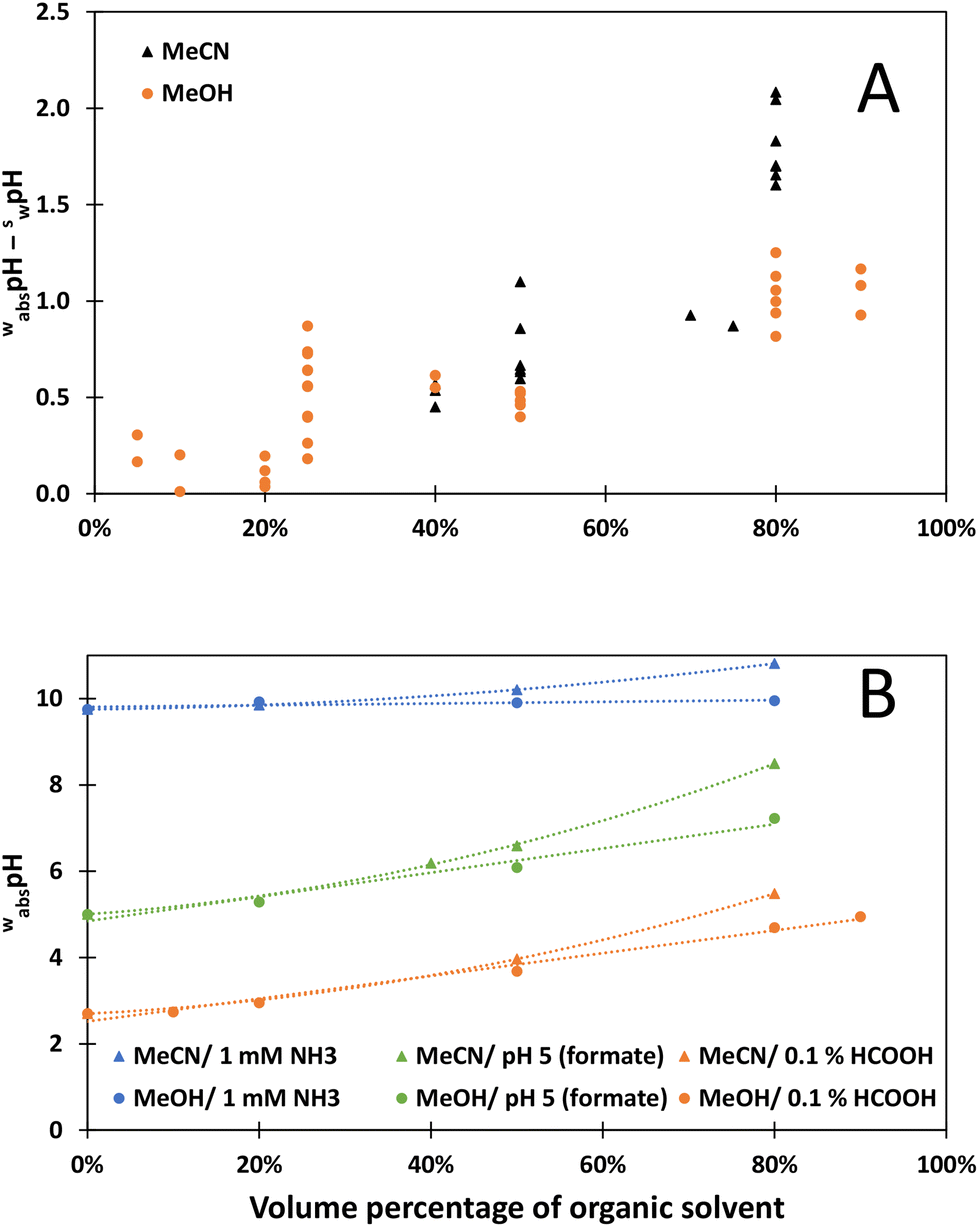 | ||
| Fig. 2 (A) Relation between swpH and wabspH values (cell I). (B) Dependence of the wabspH value on organic component volume percentage. | ||
The dependence of mobile phase acidity on the organic component volume percentage (Fig. 2B) is the same as previously reported.14,36,37 The change in pH is similar for both MeCN and MeOH-based mobile phases up to 50 volume per cent of the organic component. At higher percentages than 50% by volume of organic component, the acidity changes linearly upon adding MeOH, but not when adding MeCN. Interestingly, the ammonia-based mobile phases have rather similar wabspH. This might be because changes in the pKa compensate for the changes in solvent acidity.
Changes in pKa are very similar for carboxylic acids. Therefore, the pH changes of acetate and formate buffers should be very similar when adding the same amount of the same organic phase. The difference between acetate and formate buffer pH 5 is due to the concentrations of the used acetate (5 mM) and formate buffers (26.5 mM).
According to the wabspH values, all aqueous–organic mobile phases are more basic than the respective aqueous phases. The most extreme example is acetate buffer with wwpH 5, which after adding 80% MeCN in volume, becomes a basic solution with wabspH 9.14. The reason for this is a significant increase in pKa values of carboxylic acids when MeCN is added to water. Both the aqueous pH and the volume per cent of the aqueous phase play a role in mobile phase acidity. Different mobile phase compositions can achieve similar wabspH value. For example, the pH of MeCN/pH 5 (formate) 50/50, MeCN/pH 4 (formate) 70/30, MeOH/pH 5.5 (formate) 50/50 and MeOH/pH 5 (acetate) 50/50 are all in the range of 6.50 to 6.59.
The possibility of measuring wabspH values of mobile phases with ordinary pH measurement equipment was investigated. Although the reference method cell I provides good accuracy, it is complicated to set up (it needs a specialised glass cell) and operate. Moreover, to obtain the wabspH of a solution, it is necessary to measure it against at least two reference solutions, increasing the workload. Therefore, it is not expected that routine laboratories will widely adopt it.
Measurements with cell II were done to find an alternative for the reference method (cell I) suitable for routine laboratories. Cell II uses a glass electrode half-cell and a double junction reference electrode or a double junction combined pH electrode. Measurements are done the conventional way – the potential of the glass electrode against the reference electrode is measured. The cell was calibrated using the standard aqueous buffers. 45 mobile phases out of 78 were measured with cell II.1 (glass and a reference electrode), and 23 with cell II.2 (combined electrode). Fluoroalcohol-based mobile phases were left out. The differences between the results obtained with the two cells are presented in Fig. 3.
 | ||
| Fig. 3 Difference between results obtained with cell I and II. (A) MeOH-based mobile phases. (B) MeCN-based mobile phases. Glass electrodes are given in Table 1. DJ ref is a double junction reference electrode. | ||
The results show that the suitability of cell II depends on electrodes and the measured system. Cell II.1 worked somewhat better but occasionally displayed discrepancies from the cell I results, amounting to around 0.5 pH units and, in a few cases, even more. Cell II.2 performance differs strongly: electrode D is unsuitable for these measurements, while electrode E behaves the same as cell II.1.
The reasons for the discrepancies might be the different types of the junction, possibly insufficient electrical contact, the temperature difference (cell I was thermostated at 25 °C; cell II was at room temperature, not thermostated), stirring was used with cell II but not with the cell I. We have not studied the double junction electrodes enough to confidently state that double junction electrodes are, as a rule, suitable for this work. However, the present results give an indication that these electrodes may work. The reason for discrepancies between cells needs further investigation.
Measurements with a separate glass electrode half-cell and a double junction reference electrode (B or C vs. DJ ref, cell II.1) agree satisfactorily with the results of cell I, and there is no difference between glass electrodes. In the case of the double junction combined pH electrode (D or E, cell II.2), the agreement depends on the used electrode. Results with electrode E agree with the results of cell I and with measurements made with separate electrodes (cell II.1).
Electrode D gave unacceptable results. Some measurements were repeated with a second pH meter. There was no difference in results with electrode E between the two pH meters, but with electrode D the results differed by up to 0.2 pH units. This indicates that the diaphragm type of the reference electrode plays an important role.
Electrode E has a flushable PTFE junction, and the DJ ref has a moveable ground-joint diaphragm. Hence the wetting of the diaphragm is ensured. Electrode D has a fixed ground-joint, and the IL's leakage is insufficient for good contact between IL and the test solution.
These results show that wabspH can be routinely measured with a double junction combined pH electrode if high accuracy is not required. Still, care must be taken when choosing the junction of the reference electrode. The larger the leakage rate of the diaphragm, the better the connection between the solutions and the more accurate the results.
4. Conclusions
Overall 427 relative measurements were made to assign unified acidity (wabspH) values to 78 mobile phases. Based on the predefined quality limits (standard deviation not more than 1 mV and drift not more than 4 mV h−1), 300 relative measurements out of 427 were used in the data assignment of pH values. A novel method based on a double junction reference electrode filled with ionic liquid was tested to be adequate for routine measurements. This concept was tested as a double junction combined electrode for the first time. Results show that the design of the junction is an important factor in deciding if the electrode can be used for unified acidity measurements. The effect of electrode design on the quality of results needs further investigation.Author contributions
Agnes Heering: conceptualization, methodology, writing – original draft preparation, investigation, formal analysis, writing – review & editing; Markus Lahe: methodology, investigation; Martin Vilbaste: investigation, formal analysis, writing – review & editing; Jaan Saame: investigation; John Paulo Samin: investigation, formal analysis; Ivo Leito: conceptualization, resources, formal analysis, writing – review & editing.Data availability
The data sets of this work are available at https://doi.org/10.5281/zenodo.7913050.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded from the EMPIR programme (project 17FUN09 “UnipHied”) co-financed by the Participating States and from the European Union's Horizon 2020 research and innovation programme; by the EU through the European Regional Development Fund under project TK141 “Advanced materials and high-technology devices for energy recuperation systems” (2014-2020.4.01.15-0011) and by the Estonian Research Council grant (PRG690). This work was carried out using the instrumentation at the Estonian Center of Analytical Chemistry (TT4, https://www.akki.ee).References
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- H. G. Gika, G. A. Theodoridis, R. S. Plumb and I. D. Wilson, J. Pharm. Biomed. Anal., 2014, 87, 12–25 CrossRef CAS PubMed.
- M. Petrovic, M. Farré, M. L. de Alda, S. Perez, C. Postigo, M. Köck, J. Radjenovic, M. Gros and D. Barcelo, J. Chromatogr. A, 2010, 1217, 4004–4017 CrossRef CAS PubMed.
- C. Zisi, A. Pappa-Louisi and P. Nikitas, J. Chromatogr. A, 2020, 1617, 460823 CrossRef CAS PubMed.
- M. J. den Uijl, P. J. Schoenmakers, B. W. J. Pirok and M. R. van Bommel, J. Sep. Sci., 2021, 44, 88–114 CrossRef CAS PubMed.
- T. Alvarez-Segura, X. Subirats and M. Rosés, Anal. Chim. Acta, 2019, 1050, 176–184 CrossRef CAS PubMed.
- M. Rosés, J. Chromatogr. A, 2004, 1037, 283–298 CrossRef PubMed.
- A. Kütt, S. Selberg, I. Kaljurand, S. Tshepelevitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef.
- X. Subirats, M. Rosés and E. Bosch, Sep. Purif. Rev., 2007, 36, 231–255 CrossRef CAS.
- F. Rived, I. Canals, E. Bosch and M. Rosés, Anal. Chim. Acta, 2001, 439, 315–333 CrossRef CAS.
- J. L. Beltrán, N. Sanli, G. Fonrodona, D. Barrón, G. Özkan and J. Barbosa, Anal. Chim. Acta, 2003, 484, 253–264 CrossRef.
- F. Z. Erdemgil, S. Şanli, N. Şanli, G. Özkan, J. Barbosa, J. Guiteras and J. L. Beltrán, Talanta, 2007, 72, 489–496 CrossRef CAS PubMed.
- D. Himmel, S. K. Goll, I. Leito and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 6885–6888 CrossRef CAS PubMed.
- A. Suu, L. Jalukse, J. Liigand, A. Kruve, D. Himmel, I. Krossing, M. Rosés and I. Leito, Anal. Chem., 2015, 87, 2623–2630 CrossRef CAS PubMed.
- A. Heering, D. Stoica, F. Camões, B. Anes, D. Nagy, Z. Nagyné Szilágyi, R. Quendera, L. Ribeiro, F. Bastkowski, R. Born, J. Nerut, J. Saame, S. Lainela, L. Liv, E. Uysal, M. Roziková, M. Vičarová and I. Leito, Symmetry, 2020, 12, 1150 CrossRef CAS.
- V. Radtke, A. Ermantraut, D. Himmel, T. Koslowski and I. Leito, Angew. Chem., Int. Ed., 2018, 57, 2344–2347 CrossRef CAS PubMed.
- A. Ermantraut, V. Radtke, N. Gebel, D. Himmel, T. Koslowski and I. Leito, Angew. Chem., Int. Ed., 2018, 57, 2348–2352 CrossRef CAS PubMed.
- V. Radtke, N. Gebel, D. Priester, A. Ermantraut, M. Bäuerle, D. Himmel, R. Stroh, T. Koslowski, I. Leito and I. Krossing, Chem. – Eur. J., 2022, e202200509 CrossRef CAS PubMed.
- V. Radtke, D. Priester, A. Heering, C. Müller, T. Koslowski, I. Leito and I. Krossing, Chem. – Eur. J., 2023, e202300609 CrossRef CAS PubMed.
- Handbook of Reference Electrodes, ed. G. Inzelt, A. Lewenstam and F. Scholz, Springer-Verlag, Berlin, Heidelberg, 2013 Search PubMed.
- A. Heering, F. Bastkowski and S. Seitz, J. Sens. Sens. Syst., 2020, 9, 383–389 CrossRef.
- S. Lainela, I. Leito, A. Heering, G. Capitaine, B. Anes, F. Camões and D. Stoica, Water, 2021, 13, 2522 CrossRef CAS.
- L. Deleebeeck, A. Snedden and D. Stoica, Anal. Chim. Acta: X, 2022, 10, 100085 CAS.
- F. Bastkowski, A. Heering, E. Uysal, L. Liv, I. Leito, R. Quendera, L. Ribeiro, L. Lisa Deleebeeck, A. Snedden, D. Nagy, Z. N. Szilágyi, F. Camões, B. Anes, M. Roziková and D. Stoica, Anal. Bioanal. Chem., 2024, 416, 461–465 CrossRef CAS PubMed.
- F. G. K. Baucke, Anal. Bioanal. Chem., 2002, 374, 772–777 CrossRef CAS PubMed.
- R. Veigure, K. Lossmann, M. Hecht, E. Parman, R. Born, I. Leito, K. Herodes and K. Kipper, J. Chromatogr. A, 2020, 1613, 460667 CrossRef CAS PubMed.
- K. Lossmann, R. Hecht, J. Saame, A. Heering, I. Leito and K. Kipper, J. Chromatogr. A, 2022, 1666, 462850 CrossRef CAS PubMed.
- P. Liigand, A. Heering, K. Kaupmees, I. Leito, M. Girod, R. Antoine and A. Kruve, J. Am. Soc. Mass Spectrom., 2017, 28, 2124–2131 CrossRef CAS PubMed.
- R. J. N. Bettencourt da Silva, J. Saame, B. Anes, A. Heering, I. Leito, T. Naykki, D. Stoica, L. Deleebeeck, F. Bastkowski, A. Snedden and M. F. Camões, Anal. Chim. Acta, 2021, 1182, 338923 CrossRef PubMed.
- A. Heering, I. Leito, M. Lahe, M. Vilbaste and J. P. Samin, protocols.io, 2023, DOI:10.17504/protocol.
- Y. Marcus, Solubility and solvation in mixed solvent systems, 1990, vol. 62 Search PubMed.
- M. Li, D. Constantinescu, L. Wang, A. Mohs and J. Gmehling, Ind. Eng. Chem. Res., 2010, 49, 4981–4988 CrossRef CAS.
- R. G. Bates, Determination of pH; Theory and Practice, John Wiley & Sons, New York, London, Sydney, Toronto, 2nd edn, 1973 Search PubMed.
- C. B. Castells, C. Ràfols, M. Rosés and E. Bosch, J. Chromatogr. A, 2003, 1002, 41–53 CrossRef CAS PubMed.
- L. G. Gagliardi, C. B. Castells, C. Ràfols, M. Rosés and E. Bosch, Anal. Chem., 2007, 79, 3180–3187 CrossRef CAS PubMed.
- D. v. McCalley, J. Chromatogr. A, 2017, 1483, 71–79 CrossRef CAS PubMed.
- X. Subirats, E. Bosch and M. Rosés, J. Chromatogr. A, 2007, 1138, 203–215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison with previous values, photographs of the experimental setup and the full ladder scheme with all differential pH measurements. See DOI: https://doi.org/10.1039/d3an02029k |
| This journal is © The Royal Society of Chemistry 2024 |