
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is the stability of folates in dried blood microsamples sufficient to perform home-sampling studies?†
Liesl
Heughebaert
 and
Christophe Pol
Stove
*
and
Christophe Pol
Stove
*
Laboratory of Toxicology, Department of Bioanalysis, Ghent University, Ottergemsesteenweg 460, 9000 Ghent, Belgium. E-mail: Christophe.Stove@UGent.be; Tel: +32 (0) 9 264 81 35
First published on 26th December 2023
Abstract
Dried blood microsampling is increasingly used for home-sampling and epidemiological studies because of its multiple advantages, including an often greatly improved analyte stability. However, a critical assessment of the stability under realistic conditions should always be performed as part of the validation, especially for unstable molecules like folates (vitamin B9). Here, the objective was to determine whether folate stability in dried blood microsamples is sufficient to allow the set-up of home-sampling studies for the monitoring of folate status in e.g., women of reproductive age. An extensive set of stability experiments was performed to evaluate the stability of the main folate vitamer 5-methyltetrahydrofolate (5MTHF), its oxidation product MeFOX and the minor non-methyl folate vitamers 10-formylfolic acid (10FoFA), 5,10-methenyltetrahydrofolate (5,10CH+THF) and tetrahydrofolate (THF) in dried blood microsamples using volumetric absorptive microsampling (VAMS) or regular dried blood spots (DBS). The evaluations included (EDTA-anticoagulated blood was collected from a single donor measured in four replicates per condition and time point): (i) the effect of temperature (−20 °C, 4 °C, ambient temperature and 37 °C), (ii) the effect of light (during drying and storage) and humidity, and (iii) the effect of storage under vacuum and pretreatment of the microsamples with stabilizing agents on folate stability. At −20 °C and 4 °C, all folate levels were within 85 to 115% of the baseline value up till two weeks of storage in both VAMS samples and DBS. However, at room temperature the stability of the analyzed folates was only consistently observed up till three days in VAMS samples, and for none of the folates at 37 °C. Humidity had a major impact on 5,10CH+THF stability, but this could be easily improved by using desiccant. Both vacuum treatment and pretreatment of microsamples with 0.1% DL-dithiothreitol and 5% butylated hydroxytoluene improved the stability at room temperature in VAMS samples, but these effects were limited at 37 °C and in DBS. Overall, the stability of the individual folate vitamers proved to be challenging and strongly temperature- and time-dependent. Nonetheless, if controlled transport (temperature and duration) can be assured, the set-up of home-sampling studies to evaluate the folate status using dried blood microsamples can still be beneficial.
Introduction
Since its introduction for newborn screening in the 1960s,1 microsampling is increasingly being integrated as part of patient-centric sampling procedures in several application fields e.g., therapeutic drug monitoring, toxicology and for epidemiological purposes.2–6 Typical analytes often measured for the latter are micronutrients such as vitamins.7–9 However, monitoring of vitamin status in such studies is often a complicated task: conventional blood draws are time-consuming and invasive, and medically trained personnel and hygienic measures are often lacking when performed in remote regions. Therefore, due to the minimal invasiveness, low blood volume needed and limited costs related to storage and transport, microsampling can overcome most of the issues related to conventional venipuncture.6 Two of the main techniques currently being used include the use of regular dried blood spots (DBS) on filter paper and volumetric absorptive microsampling (VAMS), which uses the Mitra® device that consists of a plastic handler and a polymeric tip, which absorbs a fixed volume of blood. Moreover, the recent shift of disease monitoring towards health monitoring in developed countries has increased the interest of performing home-sampling studies for the monitoring of vitamin status in both specific and general populations.10,11 To be able to perform such studies, an important prerequisite is to ensure stability of the analyte of interest, from sampling to analysis. Although increased stability of analytes is often observed in dried blood format as compared to liquid blood or plasma, a critical assessment of the stability under realistic conditions should always be performed as part of the validation procedure.12–14Folate or vitamin B9 is part of the group of water-soluble vitamins and plays a key-role in one-carbon metabolism, for DNA and RNA synthesis and for amino acid conversions.15,16 Therefore, folate requirements increase during periods of growth, development and reproduction, hence, the influence of folate status on neural tube defects e.g., spina bifida. Consequently, monitoring of folate status in women of reproductive age is of utmost importance.17 In this context, the set-up of home-sampling studies using microsampling could be beneficial. Apart from their important role within the human body, folates are also well-known for their instability e.g., after blood sampling, which is in essence primarily a chemical phenomenon, as elaborately discussed elsewhere.15 Also here, the use of microsampling could offer a solution.6,15,18–20 Indeed, already in the 90's relatively good stability was observed in DBS compared to liquid whole blood using a microbiological assay for total folate determination. Of note, caution should still be paid as with increasing temperatures increased degradation was observed.21,22 Later on, Zimmerman et al. reported on the results of a long-term stability study of folate in dried cord blood spots. Also here, a decrease in folate concentrations was observed with increasing temperature and higher humidity after 9 months of storage.23 More recently, Kopp and Rychlik evaluated the stability of the main folate vitamer 5-methyltetrahydrofolate (5MTHF) in DBS and VAMS samples using liquid chromatography tandem mass spectrometry (LC-MS/MS).24,25 However, only stability at −20 °C and the effect of drying on VAMS samples was included and no evaluations were done at room or higher temperatures. Thus, based on the available data, it remains unclear whether the observed stability is adequate to perform e.g., home-sampling studies. Interestingly, during pre-validation of our VAMS- and DBS-based LC-MS/MS method for folate determination in dried blood microsamples, we encountered a decreased stability during storage experiments performed in summertime (i.e., higher ‘room’ temperature as compared to wintertime). This observation, in combination with the lack of stability data on the individual folate vitamers during different storage conditions, triggered us to perform an in-depth stability study aiming at answering the following question: ‘Can folate stability in microsamples be considered sufficient to obtain reliable results when performing home-sampling studies?’.
Methods
Chemicals and materials
All internal standards (IS) were obtained from Merck & Cie (Schaffhausen, Switzerland) and included (6S)-13C5-5MTHF calcium salt, (6S)-13C5-tetrahydrofolate (THF), (6R)-13C5-5,10-methenylTHF (5,10CH+THF) hydrochloric acid, (6S)-13C5-MeFOX and (6S)-13C5-5-formylTHF (5FoTHF) calcium salt. Sodium phosphate, ascorbic acid (AA), DL-dithiothreitol (DTT) and butylated hydroxytoluene (BHT) were purchased from Merck Life Science (Overijse, Belgium). Ultrapure water was generated using a Synergy® UV water purification system from Merck Life Science. LC-MS grade acetonitrile (ACN) was purchased from Biosolve (Valkenswaard, the Netherlands). LC-MS grade formic acid (FA) (98–100%) was obtained from Chem-lab (Zedelgem, Belgium). Male rat serum was acquired from Envigo RMS bv (Horst, the Netherlands). Ten microliter VAMS devices (brand name Mitra®) were purchased from Neoteryx/Trajan (Torrance, CA, USA). Whatman 903 filter paper was obtained from GE Healthcare (Dassel, Germany). Amicon ultra 0.5 centrifugal filters (3 kDa) and desiccant packages (Minipax® absorbent packets, 5 g) were obtained from Merck Life Science. For vacuum treatment, a Lava V.100® vacuum sealer (Landig + Lava, Bad Saulgau, Germany) and aluminium-coated vacuum bags (Marmelot, Dalfsen, the Netherlands) were used.Sample collection and preparation
To assess selectivity and matrix effects (ME), EDTA-anticoagulated blood was collected from six healthy volunteers (approved by the Ethics Committee of Ghent University Hospital – EC-BC 07324). All experiments were performed in accordance with the Guidelines for Good Clinical Practices (ICH/GCP) and in accordance with the Declaration of Helsinki. Informed consent was obtained from all human subjects included in the study. For the evaluation of the below-specified storage conditions, distinct EDTA-anticoagulated blood was used, collected from another male healthy volunteer. For the latter, the approximate endogenous folate levels were 450 nM for the major folate vitamer 5MTHF and 65 nM for its oxidation product MeFOX. For the minor non-methyl folate vitamers, levels were approximately 10 nM for 10FoFA and 30 nM for 5,10CH+THF and THF. Of note, although initially aimed for, we were unable to monitor 5FoTHF, since the low endogenous levels precluded monitoring of any stability-related decreases. The collected anticoagulated blood was stored at 4 °C prior to preparing dried blood microsamples within 24 h after blood collection. Based on the previously determined stability of folates in whole blood15 and the fact that the focus of the manuscript is on the comparative evaluation of the stability of the different folates in dried blood microsamples, with dried blood microsamples freshly prepared from liquid blood being considered the ‘baseline’ condition, we considered the possible impact of the time frame between blood collection and dried blood microsample generation (and, hence, the possible impact of differences in oxidative and enzymatic stressors between fresh and 1-day old blood) negligible. VAMS samples were prepared by dipping the tip in whole blood at an angle of 45° for 6 s, as recommended by the manufacturer. After complete filling of the tip, sampling was finished after waiting 3 additional seconds. DBS were generated by pipetting 30 μL of blood onto Whatman 903 filter paper. Once prepared, both sample types were dried for 2.5 h at ambient temperature (monitored at 21.0 ± 1.2 °C, further referred to as room temperature (RT)) before being stored at the respective test conditions. Unless otherwise specified, samples (n = 4 per condition, per time point and per sample type) were put in aluminum-wrapped zip-locked bags containing one package (5 g) of desiccant.Selectivity and matrix effects
To ensure that the stability results were not confounded by methodological issues, selectivity and ME were assessed. As folates are endogenously present, blank matrix is not available. Therefore, selectivity was first assessed by comparing the ion ratios of standards dissolved in neat solvent vs. those of folates present in extracts prepared from dried blood microsamples. Here, ‘ion ratio’ is defined as the ratio of the qualifier ion (i.e., the lower abundant daughter ion) and the quantifier ion (i.e., the higher abundant daughter ion) (Table S-1†). This was done for freshly prepared dried blood microsamples (i.e., whole blood VAMS samples or DBS dried for 2.5 h before storage at −80 °C) from 6 different donors (Hct range 40.0 to 44.5 L L−1) that had been spiked post-extraction at low concentrations, namely concentrations close to 75–100% of the endogenous level (Table S-2†) (n = 6). Depending on the ion ratio of each of the monitored folate vitamers and based on the identification criteria of the WADA Technical Document – TD2021IDCR, absence of interference was accepted when the mean ion ratio of the native samples spiked at low concentrations was within ±20% (relative; applicable for 5MTHF, 5,10CH+THF and THF, with ion ratios of 0.37, 0.39 and 0.31, respectively), ±0.05 (absolute; applicable for MeFOX, with an ion ratio of 0.20) or ±0.10 (absolute; applicable for 10FoFA, with an ion ratio of 0.52) of the ratio of the neat standard solutions.26,27 Second, since a high variation in internal standard signals was observed depending on the storage condition and time, ME were assessed after drying (‘fresh’ samples, t0) and after 1 week of storage at 37 °C (‘aged’ samples) using the same whole blood samples as those that were used for the evaluation of selectivity, with inclusion of one additional low (19.4 L L−1) and high Hct (61.6 L L−1) sample (n = 8). The evaluation was based on the method suggested by Matuszewski et al. and as previously described by Verstraete et al. for endogenous analytes.26,28 First, both fresh and aged blank (unspiked, containing endogenous folate, n = 8 × 3) VAMS and DBS samples were extracted using the extraction protocol explained below (but without addition of the IS). Thereafter, to allow correction for endogenous folate levels, each of three replicate extracts was treated differently. To one extract only IS was spiked (B0), while the other two extracts were spiked with a mixture of analyte (at either a low or high concentration, (B)) and IS (Table S-2†). Finally, the extraction solvent (i.e., neat solvent) was spiked with the same low and high concentrations (A), as well as with the IS. The absolute ME was calculated by first correcting the peak area of (B) for the endogenous peak area (B0). Thereafter, the corrected area [(B) − (B0)] was divided by the area of (A). The relative ME was calculated by the following formula: {[(B) − (B0)]/IS}/(A/IS).26 For the relative ME, the deviation should be within ±15% and also the coefficient of variation (CV, %) should not exceed 15%.12,13 To rule out any between-batch effects, all samples were processed within the same analytical batch.Storage and treatment conditions
To reflect real-life use, the timeframe of the stability evaluations was limited to two weeks, as this should allow sufficient time between self-sampling at home by the participant and sending the collected samples via regular mail to an analysing laboratory. Even longer timeframes were considered irrelevant. In addition, because of practical feasibility and the high number of conditions, time points and replicates per condition and time point (n = 4), the stability evaluation was restricted to blood from a single donor (no biological replication), as stability ex vivo can in essence be considered a chemical phenomenon.15 A schematic overview of the study design is shown in Fig. S-1–4.†Short-term stability
For the evaluation of short-term storage without any pretreatment, VAMS samples and DBS were stored at −20 °C, 4 °C, RT (as specified above) and 37 °C for 1, 2 and 3 days and 1 and 2 weeks, after which they were stored at −80 °C until analysis. Since similar trends were observed in both VAMS samples and DBS after this first experiment (see Results section short-term stability), further focus lay on stability in VAMS samples only, except for the evaluation of the effect of several stabilizing agents, which was evaluated in both types of microsamples. In addition, as no stability issues were observed at 4 °C or lower, further experiments were limited to storage of samples at RT and at 37 °C i.e., temperatures at which stability proved to be challenging in previous experiments.Effect of light (during drying and storage)
Next, the effect of light after drying for 2.5 h or storing freshly prepared VAMS samples in unprotected conditions (i.e., on a bench in a regular laboratory environment away from direct sunlight) was evaluated. For the latter, VAMS samples were stored in transparent zip-locked bags with desiccant for 1, 2 and 3 days and 1 and 2 weeks. VAMS samples stored at RT and protected from light were used as a reference.Effect of humidity
The effect of humidity was assessed by storing VAMS samples at 28 °C and 80% relative humidity (RH) for 1, 2 and 3 days and 1 week, either with or without desiccant. Again here, VAMS samples stored at RT were used as a reference.Effect of vacuum treatment
In a first attempt to improve the stability, VAMS samples were stored in aluminum vacuum-treated bags for 1, 2 and 3 days and 1 and 2 weeks. The VAMS samples stored at either RT or at 37 °C without treatment were used as a reference.Effect of pretreatment with stabilizing agents
Secondly, the VAMS tips and the Whatman 903 filter paper were pretreated with three different stabilizing agents (and three increasing concentrations) and two mixtures: (i) 0.5%, 1% and 2% AA, (ii) 0.1%, 0.5% and 1% DTT, (iii) 1%, 5% and 10% BHT, (iv) 1% AA combined with 0.5% DTT (cf. composition of the extraction solvent) and (v) 1% AA and 0.5% DTT combined with 5% BHT. The collection devices were pretreated by pipetting 30 μL of reagent onto DBS filter paper or by immersing the VAMS tips in the respective stabilizing agent solution until complete filling of the tip. After 24 h of drying, dried blood microsamples were prepared as described above and dried again for 2.5 h. Also here, both VAMS samples and DBS were stored at RT and at 37 °C for 1, 2 and 3 days and 1 week, and the samples stored at either RT or at 37 °C without treatment were used as a reference.Data analysis
To be able to comparatively evaluate the different conditions and to rule out any within-batch variability, each analyte-IS ratio was compared to the analyte-IS ratio of the baseline value (i.e., the samples stored at −80 °C immediately after drying, t0), analysed within the same analytical run. Since – as mentioned earlier – it was practically impossible to analyse all samples within a single analytical batch, between-batch variability was controlled by including replicates of the same untreated conditions (samples stored at either RT and/or at 37 °C at the different time points) in each of the experiments, as schematically shown in Fig. S-1–4.†Samples were considered stable if the mean analyte![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IS ratio (further on referred to as “recovery” – but to be distinguished from extraction recovery) relative to t0 was within 85% to 115% (lower and upper limit of acceptance (LLOA and ULOA), respectively). Important to note, we historically determined linearity of the analyte:IS response for all the monitored folates within a 5 to 1000 nM range. As in the present study, the maximum endogenous approximate folate concentration was below 500 nM, we can confidently state that a 15% decrease in analyte:IS response corresponds to a similar decrease in concentration.
:IS ratio (further on referred to as “recovery” – but to be distinguished from extraction recovery) relative to t0 was within 85% to 115% (lower and upper limit of acceptance (LLOA and ULOA), respectively). Important to note, we historically determined linearity of the analyte:IS response for all the monitored folates within a 5 to 1000 nM range. As in the present study, the maximum endogenous approximate folate concentration was below 500 nM, we can confidently state that a 15% decrease in analyte:IS response corresponds to a similar decrease in concentration.
Sample extraction
10 μL VAMS tips or 6 mm DBS punches were first transferred to 2 mL amber Eppendorf tubes (VWR, Leuven, Belgium). Before the start of each experiment, extraction solvent (50 mM sodium phosphate buffer, pH 7.4, containing 1% AA and 0.5% DTT and containing the IS at a concentration of 74 nM for 13C5-5MTHF, 28 nM for 13C5-5,10CH + THF and 13C5-THF and 11 nM for 13C5-MeFOX and 13C5-5FoTHF (stock solutions of each IS at approximately 200 μM were stored at −80 °C)) was freshly prepared and 165 μL was added after which the samples were extracted for 90 min at 25 °C and 1200 rpm. To deconjugate the folate polyglutamates present in the red blood cell fraction of the blood, the extract was incubated with 35 μL charcoal-stripped rat serum (prepared as described by Kiekens et al.29) for 1 h at 37 °C. Following deconjugation, 160 μL of extract was transferred to a centrifugal filter and centrifuged twice for 15 min at 14000 g and 4 °C. Finally, 70 μL eluate was transferred to an amber vial containing a glass insert before injection onto the LC-MS/MS system. All manipulations were performed under subdued light.
LC-MS/MS analysis
Analysis was performed using a Waters Acquity UPLC® coupled to a XEVO TQ-S mass spectrometer (Waters, Milford, MA, USA) controlled by the Waters MassLynx software. A Waters Acquity UPLC® HSS T3 column (1.8 μm, 2.1 × 150 mm) equipped with an HSS T3 VanGuard precolumn (1.8 μm, 2.1 × 50 mm) was used for chromatographic separation of the different folate vitamers: 5MTHF, its oxidation product MeFOX (a pyrazino-s-triazine derivative of 4′-hydroxy-5MTHF), 10-formylfolic acid (10FoFA), 5,10CH+THF and THF. Baseline separation of all analytes was achieved (Fig. S-5†). The column oven was set at 60 °C while the autosampler temperature was held at 8 °C. Using water and ACN, both containing 0.1% FA, as mobile phase A and B, respectively, the gradient was as follows: 0.00 min (100% A and 0% B, curve initial), 0.30 min (100% A and 0% B, curve 1), 1.60 min (90% A and 10% B, curve 6), 2.25 min (80% A and 20% B, curve 6), 2.50 min (5% A and 95% B, curve 6), 3.00 (5% A and 95% B, curve 6), 3.01 (100% A and 0% B, curve 6), resulting in a total run time of 4 min with a constant flow of 0.60 mL min−1. The mass spectrometer was operated in positive electrospray ionization mode and multiple reaction monitoring by monitoring two characteristic precursor-to-product ion transitions per folate vitamer and per corresponding IS. Details about the transitions as well as other compound specific parameters are listed in Table S-1.† Instrument-specific parameters were: capillary voltage of 0.4 kV, desolvation temperature was 600 °C at a gas flow of 1000 L h−1 (nitrogen) and cone gas flow of 150 L h−1 (nitrogen). Argon was used as collision gas at a flow rate of 0.12 mL min−1.Results
Selectivity and matrix effects
The observed ion ratios in neat solvent and matrix (VAMS samples and DBS) are displayed in Table S-3.† Absence of interference could easily be accepted as the ion ratios in matrix fell within ±8%, ±3% and ±10% (which is well within the relative acceptance criterion of 20% mentioned earlier) and within ±0.02 and ±0.05 (acceptance criteria of ±0.05 and ±0.10, respectively) of the ion ratios obtained in neat solvent for 5MTHF, 5,10CH+THF, THF, MeFOX and 10FoFA, respectively. In addition, the CV on the observed ion ratios was ≤16% in neat solvent, ≤11% in VAMS samples and ≤14% in DBS. Although a clear difference was observed in absolute ME in fresh and aged samples for some of the folates, all IS-corrected ME were within 86% to 111%, with CVs ≤11%, thereby meeting the pre-set acceptance limits (Table S-4†).12,13Short-term stability
Fig. 1 displays the short-term stability results for the different folate vitamers obtained using VAMS samples (panels A, C, E, G and I) and DBS (panels B, D, F, H and J). When storing VAMS samples at −20 °C or 4 °C all signals lay within ±15% of those obtained for t0, indicating that no relevant degradation was observed for any of the folate vitamers studied after two weeks of storage. In contrast, at RT, consistent stability (i.e., compared across the different experiments, cf. Fig. S-1–4†) could only be observed up till three days in VAMS samples. For the 1-week time point, the ±15% criterion was sometimes but not always met, with THF being the folate most sensitive to instability. When increasing the temperature to 37 °C, none of the folates showed consistent stability, readily failing the acceptance criterion after one day of storage when using VAMS samples (cf. variation in instability observed in Fig. 1vs.Table 1, untreated condition). Moreover, when comparing these results to those obtained for DBS, similar instability could be observed, except for 10FoFA, which showed stability in DBS up till 1 to 2 weeks of storage at 37 °C, in contrast to its variable stability observed in VAMS samples (Fig. 1vs.Table 1). | ||
| Fig. 1 Evaluation of the stability of 5MTHF (blue, panels A and B), MeFOX (green, panels C and D), 10FoFA (purple, panels E and F), 5,10CH+THF (red, panels G and H) and THF (yellow, panels I and J) at −20 °C, 4 °C, RT and 37 °C after drying (baseline value, t0), 1, 2 and 3 days and 1 and 2 weeks of storage (n = 4 per condition, mean ± CV). Results obtained using VAMS samples (panels A, C, E, G and I) and DBS (panels B, D, F, H and J) are illustrated. Dotted black lines represent the lower and upper limit of acceptance of 85% and 115%, respectively. | ||

|

|
Effect of light (during drying and storage)
Typically, all manipulations with respect to sample processing for folate determinations are performed under subdued light. Indeed, in a controlled laboratory environment, freshly prepared microsamples can be dried under subdued light or even in a dark room. However, when considering the application of microsampling for the determination of a person's folate status in a home-sampling setting, drying will typically be performed in uncontrolled conditions (i.e., in the presence of artificial light or sunlight). Therefore, we examined whether drying or storing VAMS samples in the presence of light would affect the folate levels (Fig. 2). This revealed that, when VAMS samples were dried unprotected, caution should be paid for 5MTHF and 10FoFA, as only a mean recovery of 81 ± 1% and 77 ± 2% compared to t0 (i.e., analyte:IS ratio obtained for samples dried in the dark) was found. For the other folate vitamers, all analyte:IS ratios were within ±15% of the baseline value. When storing VAMS samples for two weeks at unprotected conditions after drying in the dark, the mean recoveries relative to t0 were 81 ± 2%, 88 ± 3%, 65 ± 3%, 69 ± 2% and 64 ± 7% for 5MTHF, MeFOX, 10FoFA, 5,10CH+THF and THF, respectively.
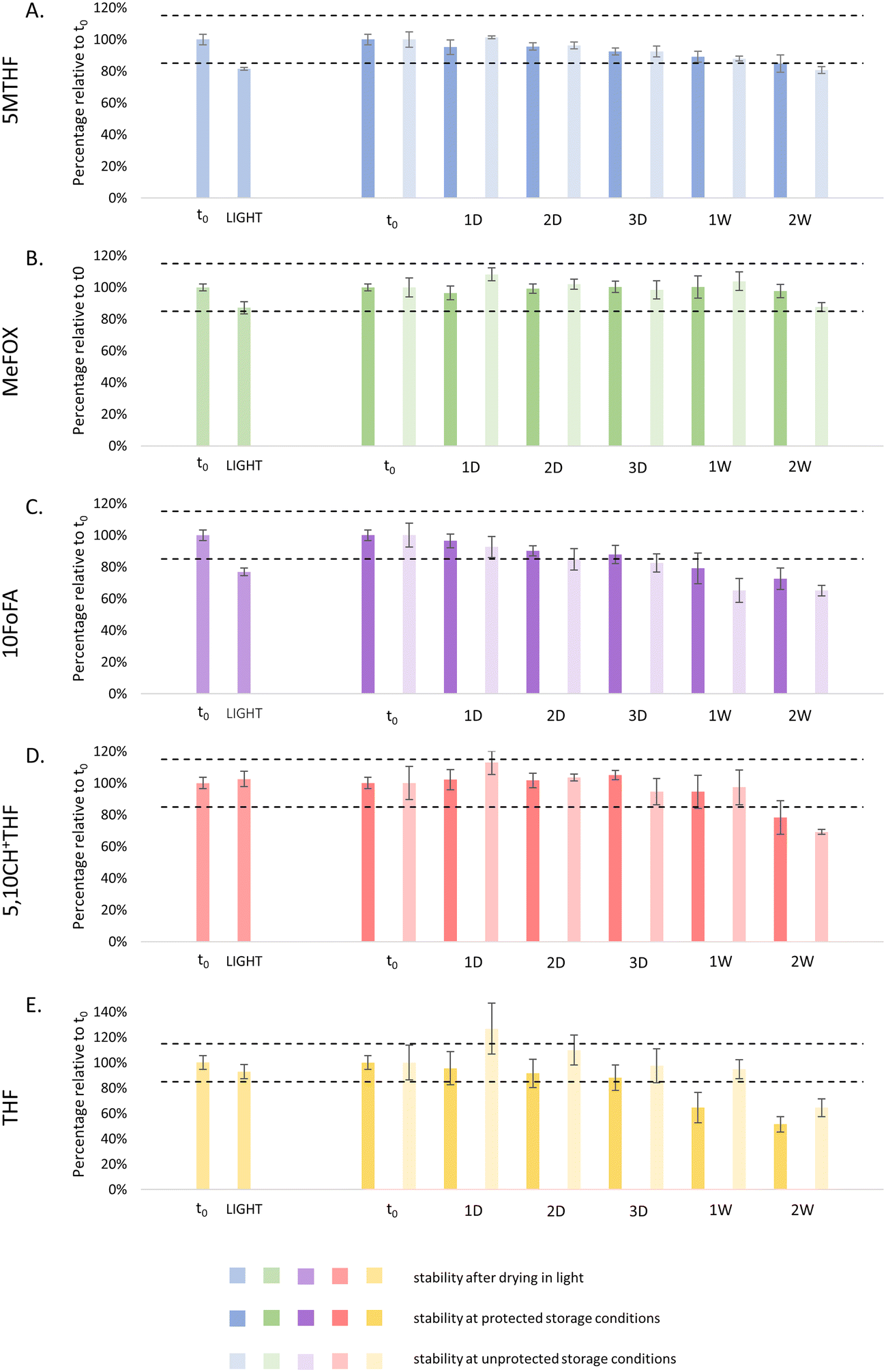 | ||
| Fig. 2 Evaluation of the effect of light during drying (left part of the graphs) and storage (right part of the graphs) for 5MTHF (blue, panel A), MeFOX (green, panel B), 10FoFA (purple, panel C), 5,10CH+THF (red, panel D) and THF (yellow, panel E) in VAMS samples. Samples were dried for 2.5 h or stored at RT for 1, 2 and 3 days and 1 and 2 weeks either protected or unprotected (i.e., in the presence of light) (n = 4 per condition, mean ± CV). Note the difference in scale on the y-axis for THF in panel E. Dotted black lines represent the lower and upper limit of acceptance of 85% and 115%, respectively. | ||
Two-week storage at light-protected conditions yielded values of respectively 85 ± 5%, 98 ± 4%, 73 ± 7%, 78 ± 11% and 51 ± 6% of t0. Overall, this suggests a slight negative impact of storage under light conditions (except for THF), although the differences remained limited, certainly when also taking into consideration the CVs.
Effect of humidity
The effect of humidity was assessed by storing VAMS samples both desiccated (as the reference condition) and non-desiccated at 28 °C and 80% RH (Fig. 3, panels A, C, E, G and I). This revealed that particularly 5,10CH+THF was sensitive to a negative impact of humidity, with already after one day of storage at 28 °C and 80% RH a drop to 60 ± 6% of t0, compared to 115 ± 7% and 112 ± 9% for VAMS samples stored desiccated at RT and high humidity, respectively (Fig. 3, panel G). For the other folates no relevant trends could be discerned with respect to the effects of high humidity. Another interesting observation here is related to the impact of a slightly higher temperature (28 °C vs. RT). More specifically, when looking at the one-week time point, the stability of samples stored either desiccated or non-desiccated at high humidity was similar, and consistently lower compared to the reference condition (RT, desiccated) for all folates except 10FoFA. This illustrates again that temperature (here only a limited increase of approximately 6 °C compared to the reference condition) is a very important parameter influencing folate stability.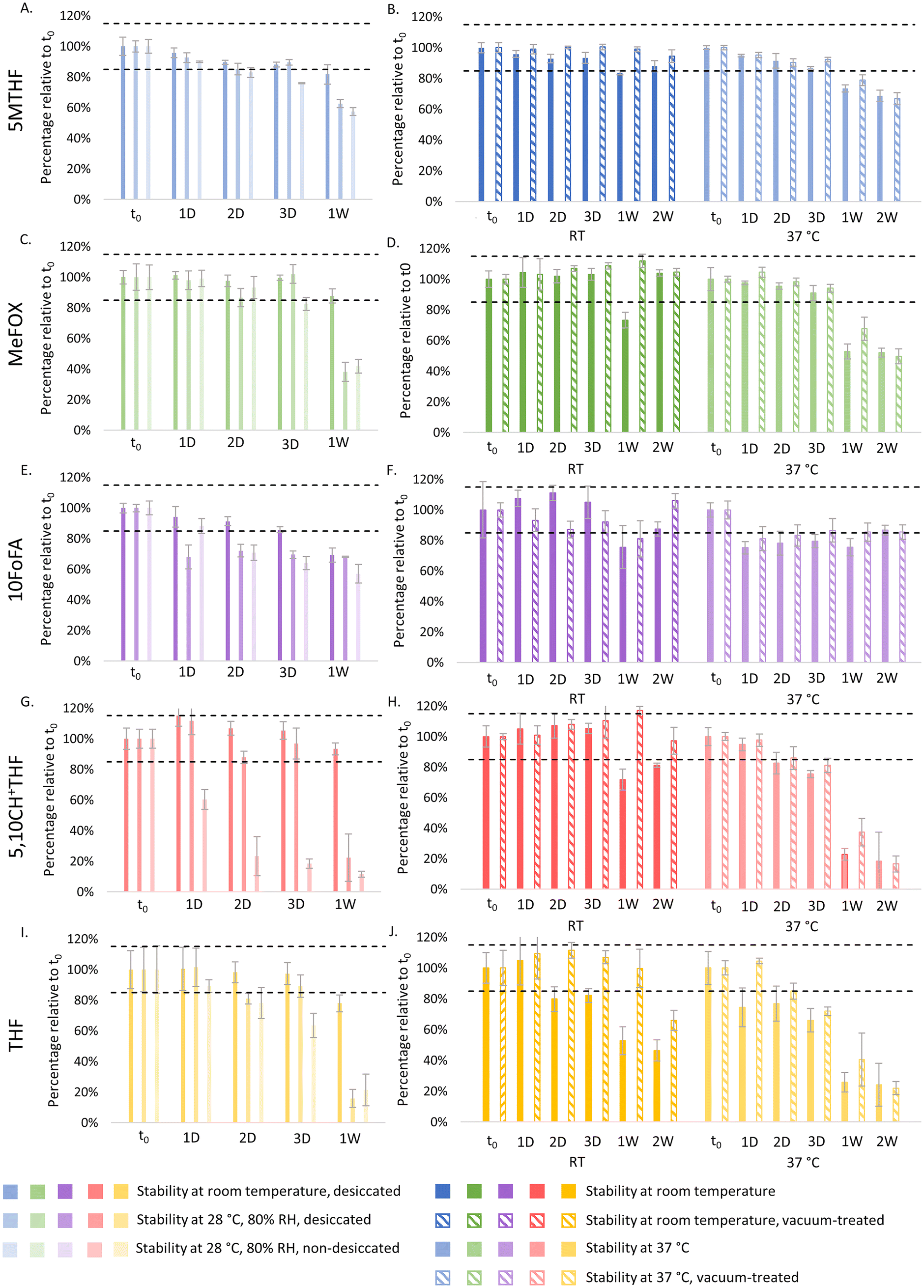 | ||
| Fig. 3 Evaluation of the effect of humidity (panels A, C, E, G and I) and vacuum treatment (panels B, D, F, H and J) for 5MTHF (blue, panels A and B), MeFOX (green, panels C and D), 10FoFA (purple, panels E and F), 5,10CH+THF (red, panels G and H) and THF (yellow, panels I and J) in VAMS samples (n = 4 per condition, mean ± CV). Dotted black lines represent the lower and upper limit of acceptance of 85% and 115%, respectively. | ||
Effect of vacuum treatment
In a first attempt to improve stability, we tried to reduce the impact oxygen may have by storing VAMS samples in vacuum-treated bags immediately after drying at RT and at 37 °C (Fig. 3, panels B, D, F, H and J). After one week of storage at RT, a recovery (relative analyte:IS ratio compared to t0) of 99 ± 2%, 81 ± 12%, 117 ± 3% and 100 ± 12% was found for 5MTHF, 10FoFA, 5,10CH+THF and THF, respectively. Comparing this to the stability data at the one-week time point without treatment (84 ± 1%, 76 ± 14%, 72 ± 7% and 53 ± 9% for 5MTHF, 10FoFA, 5,10CH+THF and THF, respectively), overall, a relevant stabilizing effect could be observed for the major and all minor folates, except 10FoFA (Fig. 3, panels B, F, H and J, left part of the graphs). However, when prolonging storage to two weeks, the stabilizing effect was minor for the major folate vitamer 5MTHF (94 ± 4% vs. 88 ± 4% for vacuum- and non-treated storage, respectively) and insufficient for the minor folate THF, as the mean recovery relative to t0 dropped below the LLOA of 85% (66 ± 6% vs. 46 ± 7% for vacuum- and non-treated storage, respectively). With respect to the degradation product MeFOX (Fig. 3, panel D, left part of the graph), an apparent inconsistency in mean recovery was observed between the one- and two-week time point of the untreated condition at RT (73 ± 5% vs. 104 ± 2%, respectively), implying that caution should be paid when interpreting these data. When repeating this experiment at 37 °C, the elevation in storage temperature compromised the mild stabilizing effect of vacuum treatment, with after 1 week of storage only the minor folate 10FoFA deviating less than 15% from t0 (though borderline, at 85 ± 6%). For the other two minor folate vitamers 5,10CH+THF and THF, three days of storage at 37 °C was sufficient to fail the acceptance criterion, despite vacuum treatment (Fig. 3, panels B, D, F, H and J, right part of the graphs).
Effect of pretreatment with stabilizing agents
An alternative strategy that was explored to improve folate stability in microsamples was the pretreatment of the VAMS tips with different types of stabilizing agents. Table 1 displays the results, with a color-code indicating whether folate recoveries were within ±15% of t0 (green), lay between 75%–85% or 115%–125% of t0 (orange), or deviated more than 25% from t0 (red). Already after one day of storage of VAMS samples at RT, a remarkable negative impact on stability was apparent for all folates in the samples collected with AA-pretreated VAMS tips, independent of the concentration used. One exception to this trend was the minor folate vitamer 10FoFA, which seemed to be less affected, with even increased levels when using 0.5% and 1% of AA as a pretreatment. This overall negative impact was also observed with both the evaluated mixtures, which also contain AA at a concentration of 1%. Surprisingly, at elevated temperature, the use of 2% AA-pretreated VAMS tips showed an apparent (partial) stabilizing effect for the major folate vitamer 5MTHF only. Pretreatment of the VAMS tips with 0.1% DTT had a relevant stabilizing effect, as all folate mean recoveries remained above 91% of t0 after one week of storage at RT. Also when using 5% BHT-pretreated VAMS tips to collect the samples, a stabilizing effect was seen at RT, although some deviating results were obtained for 5,10CH+THF (folate levels above the ULOA of 115%). Upon increasing the storage temperature to 37 °C, the stabilizing effect of 0.1% DTT decreased to two days of storage, except for the minor folate vitamer 10FoFA, which again remained stable. At this elevated temperature, 5% BHT appeared to be the better choice, as this specific pretreatment resulted in acceptable stability data for all folates after three days of storage. Also, pretreatment of the VAMS tips with 1% or 10% BHT resulted in an increased stability compared to no pretreatment, although not to the same extent as pretreatment with 5% BHT. Because of these promising results when using VAMS devices, the experiment was repeated in DBS. However, at both RT and 37 °C, none of the tested conditions could sufficiently improve the stability, with readily after two days storage at RT or after one day at 37 °C all folates, except for 10FoFA, failing the acceptance criterion. Of note, similar to the observation in VAMS samples, also in DBS, the use of 2% AA-pretreated DBS filter paper showed an apparent stabilizing effect for the major folate vitamer 5MTHF at elevated temperature.Discussion
The findings reported here expand and confirm earlier observations by O'Broin et al.21,22 and Zimmerman et al.23 for total folate in DBS, revealing that the stability of the major folate vitamer 5MTHF, its oxidation product MeFOX, and the minor non-methyl folate vitamers 10FoFA, 5,10CH+THF and THF in dried blood microsamples is temperature- and time-dependent (Fig. 1). At frozen and refrigerated conditions, acceptable stability was shown for all folates measured up till two weeks using either VAMS samples or DBS. At RT (which was monitored at 21.6 ± 1.2 °C), consistent stability could only be ensured up till three days in VAMS samples and for two days in DBS (Fig. 1, Table 1). Importantly, by increasing the temperature to 37 °C, this decreasing trend was, as expected, more pronounced (except for the minor folate vitamer 10FoFA which was stable in DBS but showed variable stability in untreated samples stored at 37 °C in VAMS samples across the different experiments) (Fig. 1 panel F, Table 1). This study, which focused on the measurement of multiple individual folates rather than on total folate levels, not only confirms the findings of older studies, but also provides insight into what happens with the individual folate vitamers. In addition, this study also allowed to compare the stability of folates when applying two commonly used microsampling techniques, being VAMS and regular DBS sampling, demonstrating stability issues in both. This is relevant, as differences in stability, possibly related to the type of microsample used, have already been reported e.g., for miltefosine.30 Overall, for the individual folate vitamers, similar instability can be observed in VAMS samples and DBS.An important point of attention when collecting samples in a home-sampling setting is the fact that samples will be collected, and dried, in the presence of light. This is especially relevant for folates, as they are well-known for their light sensitivity.15,31 However, our analyses revealed that drying and/or storage in the presence of light did not relevantly affect the individual folate levels when using VAMS samples. Only for the major folate vitamer 5MTHF and the minor folate vitamer 10FoFA a decrease was observed compared to drying in protected conditions, however the mean recovery relative to t0 was still above 75% (Fig. 2, panels A and C, left part of the graphs). Although not previously tested in DBS, these results are in line with a previous report by O'Broin et al., who focused on total folate in dried serum spots.32 Moreover, also the folate levels obtained after unprotected storage did not relevantly differ from those obtained after protected storage, indicating that the presence of light does not contribute to additional stability issues after drying (Fig. 2, right part of the graphs).
Although the effects of higher humidity are less of a concern when performing a home-sampling study in countries like Belgium, where this study was conducted, there is still a variation to be expected according to the season and the country wherein the study is set up.33 However, surprisingly, the effects of higher humidity remained limited in VAMS samples, as only for one minor folate vitamer i.e., 5,10CH+THF, an immediate effect was seen, which was easily controlled using desiccant. A more important observation – which is in line with the ‘summer effect’ observed during our pre-validation experiments – is the decrease in stability observed for all folate vitamers, except 10FoFA, after one week of storage at 28 °C compared to RT (which was only about 6 degrees lower, Fig. 3, panels A, C, E, G and I). In this study, RT was closely monitored, however if samples would be collected at participants’ homes, the definition of RT will no longer be a fixed value but rather a range of temperatures, which, as shown within this stability study, could have a major impact on the resulting folate levels. Especially when dried blood microsamples would be exposed to elevated temperatures, this may be detrimental for the reliability of the results – and hence, the conclusions drawn about someone's ‘folate status’.
To overcome the above-mentioned challenges related to stability, two strategies were evaluated, both aiming at reducing the effects of oxygen, in addition to the use of desiccant packages and aluminum foil, which readily eliminated possible effects of moisture and light, respectively. Because of its stabilizing effect for vitamin B1, B2 and B6 in DBS at RT, a first strategy included storage under vacuum.34 Unfortunately, in this prior study no direct comparison with untreated samples was performed, hence, the net effect of vacuum treatment remained unknown. Here, a direct comparison was performed in VAMS samples at both RT and at 37 °C, and a stabilizing effect was observed, although solely relevant at RT (Fig. 3, panels B, D, F, H and J). The limited effects at 37 °C aside, the question arises whether this approach is feasible in a home-sampling setting. Although pocket-sized vacuumizers do exist (mainly used in the field of food preservation), making such devices available to all prospective users would increase the costs related to conducting field studies involving home-sampling. A more straightforward and less costly alternative to vacuum treatment could be the use of oxygen scavengers, although this would need to be thoroughly evaluated as well, especially since a negative impact on the stability of a wide range of metabolites has been previously reported.35
A second strategy that was evaluated in both VAMS samples and DBS focused on the elimination of the effects of oxygen using several stabilizing agents. Based on literature, three types of stabilizing agents were chosen: (i) AA, (ii) DTT and (iii) BHT.21,22,32,36 First, vitamin C or AA is well-known for its antioxidant properties and has already been successfully used to stabilize folates in dried plasma spots.32 Surprisingly, when using ascorbate-treated filter paper for DBS, folate stability was greatly diminished. O'Broin and Gunter hypothesized that this was caused by AA contributing to red blood cell lysis at the time of spotting and by enhancing deconjugation of red blood cell polyglutamates, resulting in an increased level of (less stable) folate monoglutamates.15,32 This hypothesis implies that when red blood cells dry naturally on filter paper a greater stability would be achieved compared to using AA-treated paper. This is in contrast to serum, where folates are already present as monoglutamates. Since the use of AA as a stabilizer was not part of the evaluation by Kopp and Rychlik in DBS nor in VAMS for 5MTHF,24,25 we decided to still include this antioxidant to evaluate whether the previously reported results could be reproduced and, more importantly, whether the observed effect would be similar for all folate vitamers monitored in this study. Indeed, an overall deteriorating effect could be seen at all concentrations tested, which was most pronounced for the oxidation product MeFOX and the minor non-methyl folate vitamers 5,10CH+THF and THF in both VAMS samples and DBS (Table 1). Overall, we reproduced the negative impact of AA on folate stability in both VAMS samples and DBS, collecting detailed information on the effect on multiple individual folate vitamers. Secondly, because the extraction solvent also contained DTT to stabilize folates by capturing the formaldehyde formed by the ascorbate anions,37 the stabilizing properties of DTT on VAMS samples and plain filter paper (i.e., DBS) was tested as well. The lowest concentration of DTT (0.1%) showed very promising stabilizing effects at RT in VAMS samples, which, however, could not be reproduced in DBS and at higher temperatures. Based on the promising results reported by Zhang et al. for retinol, a third and last stabilizing agent that was evaluated was BHT.36 Similar as for 0.1% DTT, a stabilizing effect could be seen at RT using a 5% concentration of BHT for all folates, except 5,10CH+THF which had slightly increased values (above 115% but below 125%). Using the same concentration of 5% BHT, folate stability in VAMS samples at 37 °C could still be achieved up till three days. Unfortunately, also here, these results could not be reproduced in DBS. Finally, to test possible synergistic effects when combining multiple agents, two mixtures were evaluated, including (i) a combination of AA and DTT and (ii) a combination of all three. Nonetheless, the stability observed using these mixtures was clearly determined by the effects of AA and could not be improved by the addition of another stabilizing agent.
Although part of the tested agents could improve folate stability in VAMS samples, it should be noted that besides providing evidence that sufficient stability can be achieved with a certain type of stabilizing agent, a thorough validation of the pretreatment conditions is required, especially when aiming at performing home-sampling studies. Hence, as a next step, studies would be needed to evaluate the performance of the stabilizing agent under realistic conditions, including for example the stability of the agent on the microsampling device following storage of the pretreated device at different temperatures and different times. Of note, after pretreatment of the VAMS tips with BHT, differences in blood absorption rate as compared to untreated tips were observed (i.e., the higher the BHT concentration, the lower the blood absorption rate). However, the latter did not negatively impact the precision as the difference in mean CVs (i.e., calculated from all the obtained CVs for all time points and folates, Table 1) was not statistically significant (mean CV of 5% vs. 6% for the untreated and the 5% BHT pre-treated VAMS tips at RT, respectively, p = 0.37940). In addition to the impact on precision, it should also be evaluated whether the pretreatment could impact the accuracy (e.g., due to a difference in absorbed blood volume caused by the pretreatment). This is important, as it should be known whether for preparing the calibrators and quality controls also pretreated VAMS tips should be used. Overall, based on our data we conclude that whereas none of the stabilizing agents could relevantly improve folate stability in DBS, the use of VAMS tips for sample collection could be considered, provided these are pretreated with 0.1% DTT or 5% BHT and that the sample transport time is kept relatively short (i.e., <3 days).
Importantly, the stability study conducted here was based on EDTA-anticoagulated venous blood collected from a single blood donor. Although we have no scientific basis to believe that the folate stability in dried non-anticoagulated samples (as directly collected via finger prick) would be different from that in dried EDTA-anticoagulated samples, or that stability ex vivo would differ across multiple donors, future studies wishing to explore folate determination in capillary dried blood microsamples should ideally take these aspects into account.
Furthermore, the evaluation of folate ‘recovery’ following storage of dried blood microsamples, as performed here (via analyte:IS ratios), should be done with caution, as this implies that the detector response should be linear (a requirement which we historically demonstrated – cf. Methods section). Second, small variations in the amount of IS added during extraction, and hence, small variations in analyte:IS ratios cannot be ruled out. On the one hand, this implies the requirement to take along appropriate controls in every batch of samples that is analysed in a stability study, on the other hand, this inherently results in some additional variation and sometimes apparently conflicting results – especially when stability is ‘borderline OK’. Third, the use of an arbitrary (in)stability cut-off inherently implies that the variability associated with any analysis (which is the result of a multitude of (pre-)analytical factors, including stability, extractability etc.) is in essence not fully considered (e.g., in stability experiments 86% is arbitrarily classified as ‘stable’, whereas 84% is classified as ‘unstable’). Therefore, based on these limitations and the fact that some experimental variation was observed for the untreated conditions across the different experiments, statements were mainly limited to the relative stability within a given experiment.
Conclusions
This study is the first to provide an extensive insight into the stability of five folate vitamers, representing the major (namely 5MTHF and its oxidation product MeFOX) and the minor folate fraction (namely non-methyl folate vitamers 10FoFA, 5,10CH+THF and THF) in dried blood microsamples (both DBS and VAMS samples). Moreover, this is the first study evaluating the effect of multiple stabilizing strategies on folate stability in different dried blood microsample types.Although 5MTHF is the main folate vitamer, which correlates best to the overall folate status in the human body, the monitoring of multiple folate vitamers is relevant to allow to distinguish between low 5MTHF levels related to folate deficiency and low 5MTHF levels related to the presence of a genetic polymorphism.15 In addition, the side-by-side comparison of two of the main microsampling techniques currently being used, VAMS and DBS sampling, provided relevant information on folate stability related to the type of microsample. On the one hand, when using untreated VAMS tips and plain filter paper, the stability of the examined folates was similar. On the other hand, when using different stabilizing agents as a pretreatment, stability of all folates but one (i.e., 10FoFA) was superior in VAMS samples compared to DBS. Based on these data, we hypothesize that there may be a combined effect of the pretreatment and the interaction of the analyte with the polymeric tip of the VAMS device on folate stability, an effect which is not present when using filter paper. This underscores the importance of including, as part of the method validation, both the evaluation of stability at the expected storage and transport conditions and the evaluation of the exact type of microsample(s) that will be used during a study.
Both vacuum treatment (tested in VAMS samples only) and pretreatment with stabilizing agents (tested in both VAMS samples and DBS) were found to result in an improved folate stability. However, the stabilizing effects were compromised by storing samples at elevated temperatures. Moreover, a negative impact on folate stability in VAMS samples was seen when the temperature increased with only 6 degrees. As higher temperatures can be expected when using regular postal services for sample transport (e.g., the inside of a postal box can easily reach high temperatures in summertime), it should be concluded that in the absence of temperature control measures performing home-sampling studies by sending samples via regular mail to the laboratory is not feasible for the follow-up of someone's folate status, unless close to next-day delivery of the samples can be guaranteed. The latter is typically not the case with regular postal services – at least, not in Belgium. E.g., a delivery time between 2–5 days was observed by Verstraete et al.38 Indeed, an inherent variable in home-sampling studies is the temperature, and this is a variable which cannot be fully controlled. As a consequence, one risks that studies to determine the folate status in a home-sampling context may not provide accurate information on the folate status within a certain population. More particularly, falsely high percentages of folate deficiency may be scored, owing to the stability issues reported here.
Theoretically, the monitoring of degradation products, formed stoichiometrically from the individual folates, could allow ‘correction’ for the folate losses during storage. However, this requires that these degradation products should remain stable themselves. Focusing on MeFOX, a well-known oxidation product stemming from 5-MTHF, this proved not to be the case. Moreover, quantitative monitoring of other degradation products is hampered by the absence of stably labelled internal standards.
Taking all the above into account, the question ‘Can folate stability in microsamples be considered sufficient to obtain reliable results when performing home-sampling studies?’ should be answered with caution. Although the use of microsamples can improve folate stability to a limited extent, at least partially controlled transport conditions (in terms of temperature and duration) should be guaranteed for successful application of microsampling for the determination of a person's folate status in a home-sampling setting. Finally, the additional efforts needed to ensure folate stability should be weighed against the numerous advantages associated with microsampling, including its patient centricity and convenience.
Author contributions
LH designed and conducted the research, carried out data analysis, drafted the initial manuscript, reviewed and revised the manuscript. CS designed the research, and critically reviewed the manuscript for important intellectual content. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank all volunteers who participated in the study. LH would like to thank the Special Research Fund (BOF) from Ghent University for granting her a PhD fellowship (01D05220).References
- R. Guthrie and A. Susi, Pediatrics, 1963, 32, 338–343 CrossRef CAS PubMed.
- H. Y. Tey and H. H. See, J. Chromatogr. A, 2021, 1635, 461731 CrossRef CAS PubMed.
- N. Sadones, S. Capiau, P. M. M. De Kesel, W. E. Lambert and C. P. Stove, Bioanalysis, 2014, 6, 2211–2227 CrossRef CAS.
- M. P. Wong, M. A. Meas, C. Adams, S. Hernandez, V. Green, M. Montoya, B. M. Hirsch, M. Horton, H. L. Quach, D. L. Quach, X. R. Shao, I. Fedrigo, A. Zermeno, J. Huffaker, R. Montes, A. Madden, S. Cyrus, D. McDowell, P. Williamson, P. Contestable, M. Stone, J. Coloma, M. P. Busch, L. F. Barcellos and E. Harris, Microbiol. Spectrum, 2022, 10, e0247121 CrossRef PubMed.
- M. N. Handel, P. Frederiksen, A. Cohen, C. Cooper, B. L. Heitmann and B. Abrahamsen, Am. J. Clin. Nutr., 2017, 106, 155–161 CrossRef PubMed.
- J. Verstraete, L. Boffel and C. Stove, TrAC, Trends Anal. Chem., 2020, 132, 116057 CrossRef CAS.
- N. Densupsoontorn, C. Srisawat, K. Chotipanang, S. Junnu, S. Kunnangja, R. Wongarn, W. Sriboonnark, H. Tirapongporn and P. Phuangphan, Asia Pac. J. Clin. Nutr., 2019, 28, 116–121 CAS.
- S. Mohammadi, Z. Hajhashemy and P. Saneei, Crit. Rev. Food Sci. Nutr., 2022, 62, 8178–8198 CrossRef CAS PubMed.
- C. M. Pfeiffer, M. R. Sternberg, Z. Fazili, D. A. Lacher, M. Zhang, C. L. Johnson, H. C. Hamner, R. L. Bailey, J. I. Rader, S. Yamini, R. J. Berry and E. A. Yetley, Br. J. Nutr., 2015, 113, 1965–1977 CrossRef CAS PubMed.
- H. Veenhof, J. F. M. van Boven, A. van der Voort, S. P. Berger, S. J. L. Bakker and D. J. Touw, Br. J. Clin. Pharmacol., 2020, 86, 1357–1366 CrossRef CAS PubMed.
- U. Hoeller, M. Baur, F. F. Roos, L. Brennan, H. Daniel, R. Fallaize, H. Forster, E. R. Gibney, M. Gibney, M. Godlewska, K. Hartwig, S. Kolossa, C. P. Lambrinou, K. M. Livingstone, J. A. Lovegrove, A. L. Macready, Y. Manios, C. F. M. Marsaux, J. A. Martinez, C. Celis-Morales, G. Moschonis, S. Navas-Carretero, C. B. O'Donovan, R. San-Cristobal, W. H. M. Saris, A. Surwillo, I. Traczyk, L. Tsirigoti, M. C. Walsh, C. Woolhead, J. C. Mathers, P. Weber and F. M. Project, Br. J. Nutr., 2016, 115, 202–211 CrossRef CAS PubMed.
- European Medicines Agency, Guideline on bioanalytical method validation https://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed 28/12/2021).
- U.S. Department of Health and, Human Services Food and Drug Administration, 2018.
- S. Capiau, H. Veenhof, R. A. Koster, Y. Bergqvist, M. Boettcher, O. Halmingh, B. G. Keevil, B. C. P. Koch, R. Linden, C. Pistos, L. M. Stolk, D. J. Touw, C. P. Stove and J. W. C. Alffenaar, Ther. Drug Monit., 2019, 41, 409–430 CrossRef CAS PubMed.
- J. Verstraete, F. Kiekens, S. Strobbe, H. De Steur, X. Gellynck, D. Van Der Straeten and C. P. Stove, Anal. Bioanal. Chem., 2019, 411, 4383–4399 CrossRef CAS PubMed.
- R. Green, Am. J. Clin. Nutr., 2011, 94, 666s–672s CrossRef PubMed.
- S. C. Tinker, H. C. Hamner, Y. P. Qi and K. S. Crider, Birth Defects Res., Part A, 2015, 103, 517–526 CrossRef CAS PubMed.
- A. Faller, B. Richter, M. Kluge, P. Koenig, H. K. Seitz and G. Skopp, Int. J. Leg. Med., 2013, 127, 603–610 CrossRef PubMed.
- B. W. Adam, E. M. Hall, M. Sternberg, T. H. Lim, S. R. Flores, S. O'Brien, D. Simms, L. X. Li, V. R. De Jesus and W. H. Hannon, Clin. Biochem., 2011, 44, 1445–1450 CrossRef CAS PubMed.
- J. Bjorkesten, S. Enroth, Q. J. Shen, L. Wik, D. M. Hougaard, A. S. Cohen, L. Sorensen, V. Giedraitis, M. Ingelsson, A. Larsson, M. Kamali-Moghaddam and U. Landegren, Mol. Cell. Proteomics, 2017, 16, 1286–1296 CrossRef PubMed.
- S. D. O'Broin, B. P. Kelleher, A. Davoren and E. W. Gunter, Am. J. Clin. Nutr., 1997, 66, 1398–1405 CrossRef PubMed.
- S. D. O'Broin and E. W. Gunter, Am. J. Clin. Nutr., 1999, 70, 359–367 CrossRef.
- R. K. Zimmerman, M. E. Slater, E. K. Langer, J. A. Ross and L. G. Spector, J. Pediatr., 2013, 163, 596–597E1 CrossRef CAS PubMed.
- M. Kopp and M. Rychlik, PLoS One, 2015, 10, e0143639 CrossRef PubMed.
- M. Kopp and M. Rychlik, Front. Nutr., 2017, 4, 9 CrossRef PubMed.
- J. Verstraete and C. Stove, Anal. Chem., 2021, 93, 2660–2668 CrossRef CAS PubMed.
- World Anti-Doping Agency, Identification criteria for qualititative assays – technical document TD2021IDCR, https://www.wada-ama.org/sites/default/files/resources/files/td2021idcr_final_eng_0.pdf.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS PubMed.
- F. Kiekens, J. Van Daele, D. Blancquaert, D. Van Der Straeten, W. E. Lambert and C. P. Stove, J. Chromatogr. A, 2015, 1398, 20–28 CrossRef CAS PubMed.
- A. E. Kip, K. C. Kiers, H. Rosing, J. H. M. Schellens, J. H. Beijnen and T. P. C. Dorlo, J. Pharm. Biomed. Anal., 2017, 135, 160–166 CrossRef CAS PubMed.
- V. De Brouwer, G. F. Zhang, S. Storozhenko, D. Van Der Straeten and W. E. Lambert, Phytochem. Anal., 2007, 18, 496–508 CrossRef CAS.
- S. O'Broin and E. Gunter, Clin. Chem., 2002, 48, 1128–1130 CrossRef.
- A. Wypych, B. Bochenek and M. Rozycki, Atmosphere, 2018, 9, 18 CrossRef.
- M. Zhang, H. Liu, X. Huang, L. Shao, X. Xie, F. Wang, J. Yang, P. Pei, Z. Zhang, Y. Zhai, Q. Wang, T. Zhang, J. Huang and X. Cui, J. Chromatogr. B: Biomed. Sci. Appl., 2019, 1112, 33–40 CrossRef CAS PubMed.
- K. Li, J. C. Naviaux, J. M. Monk, L. Wang and R. K. Naviaux, Metabolites, 2020, 10, 82 CrossRef CAS PubMed.
- M. Zhang, Y. Wu, F. Wang, H. Liu, S. Zhang, Z. Zhang, L. Shao, J. Yang, X. Cui, Y. Zhang, T. Zhang, J. Huo and J. Wu, Anal. Bioanal. Chem., 2019, 411, 8073–8080 CrossRef CAS PubMed.
- V. De Brouwer, S. Storozhenko, J. C. Van de Steene, S. M. R. Wille, C. P. Stove, D. Van Der Straeten and W. E. Lambert, J. Chromatogr. A, 2008, 1215, 125–132 CrossRef CAS PubMed.
- J. Verstraete and C. Stove, Am. J. Clin. Nutr., 2021, 114, 1200–1207 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional tables and figures. See DOI: https://doi.org/10.1039/d3an01004j |
| This journal is © The Royal Society of Chemistry 2024 |