
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExternal standard calibration method for high-repetition-rate shock tube kinetic studies with synchrotron-based time-of-flight mass spectrometry†
Fabian E.
Cano Ardila
a,
Sharath
Nagaraju
a,
Robert S.
Tranter
 b,
Gustavo A.
Garcia
c,
Anthony
Desclaux
a,
Anthony
Roque Ccacya
a,
Nabiha
Chaumeix
a and
Andrea
Comandini
*a
b,
Gustavo A.
Garcia
c,
Anthony
Desclaux
a,
Anthony
Roque Ccacya
a,
Nabiha
Chaumeix
a and
Andrea
Comandini
*a
aCNRS-INSIS, I.C.A.R.E., 1C Avenue de la recherche scientifique, 45071 Orléans cedex 2, France. E-mail: andrea.comandini@cnrs-orleans.fr
bChemical Sciences and Engineering Department, Argonne National Laboratory, 9700 S. Cass Avenue, Lemont, Illinois 60439, USA
cSynchrotron SOLEIL, L'Orme des Merisiers, St. Aubin BP 48, 91192 Gif sur Yvette, France
First published on 30th January 2024
Abstract
The signal levels observed from mass spectrometers coupled by molecular beam sampling to shock tubes are impacted by dynamic pressures in the spectrometer due to rapid pressure changes in the shock tube. Accounting for the impact of the pressure changes is essential if absolute concentrations of species are to be measured. Obtaining such a correction for spectrometers operated with vacuum ultra violet photoionization has been challenging. We present here a new external calibration method which uses VUV-photoionization of CO2 to develop time-dependent corrections to species concentration/time profiles from which kinetic data can be extracted. The experiments were performed with the ICARE–HRRST (high repetition rate shock tube) at the DESIRS beamline of synchrotron SOLEIL. The calibration experiments were performed at temperatures and pressures behind reflected shock waves of 1376 ± 12 K and 6.6 ± 0.1 bar, respectively. Pyrolytic experiments with two aromatic species, toluene (T5 = 1362 ± 22 K, P5 = 6.6 ± 0.2 bar) and ethylbenzene (T5 = 1327 ± 18 K, P5 = 6.7 ± 0.2 bar), are analyzed to test the method. Time dependent concentrations for molecular and radical species were corrected with the new method. The resulting signals were compared with chemical kinetic simulations using a recent mechanism for pyrolytic formation of polycyclic aromatic hydrocarbons. Excellent agreement was obtained between the experimental data and simulations, without adjustment of the model, demonstrating the validity of the external calibration method.
Introduction
Recent advances in chemical kinetic modeling of reactive systems have led to the development of comprehensive and detailed tools capable of accurately simulating the pyrolysis and oxidation of complex fuels starting from their components and simple fuel mixtures. The predictive capabilities of the models have also significantly improved; it is not uncommon for a simulation with a state-of-the-art model that was developed and validated at one set of conditions to accurately simulate new experimental measurements at different conditions without adjusting the model. For testing and developing models it is essential to have accurate measurements of species concentrations as part of the target set. Time-dependent measurements of stable and reactive molecules are particularly valuable. For high-temperature processes (e.g., T > 1000 K) shock tubes are often the preferred reactors due to the well-defined reaction conditions, the breadth of conditions that can be created and the near instantaneous heating of the reagent mixture.1–3Shock tubes (ST) coupled by differential pumped molecular beam sampling (MBS) to time-of flight mass spectrometers (TOF-MS) have been used to study complex reaction systems and simultaneously measure stable and radical species concentrations with high time resolution.4–6 Generally, in ST/TOF-MS cations are created by electron impact ionization (EI). While EI is efficient, typical energies are quite high, and most species fragment following ionization. Fragmentation can severely complicate analysis of the mass spectra particularly, for complex mixtures such as those in this study. In principle, fragmentation can be minimized by reducing the ionization energy (IE). However, this also severely reduces the sensitivity of the experiment and with most ST/TOF-MS apparatuses signal averaging methods cannot be used to compensate for the reduced sensitivity. The standard ST/TOF-MS method has been successfully applied to quite complex systems, but the lack of ability to discriminate between isomers and fragmentation limit the applicability to larger molecular weight components such as polycyclic aromatic hydrocarbons (PAH).
Many of the challenges posed by EI can be addressed with vacuum ultraviolet (VUV) photoionization (PI) which largely reduces fragmentation compared to EI. Furthermore, with highly tunable synchrotron sourced VUV, isomers and species of the same mass but different chemical composition can be distinguished.7–10 However, signal levels from PI-TOF-MS tend to be low and signal averaging is essential to obtain sufficient signal/noise (S/N). To take advantage of synchrotron-based VUV-PI methods miniature high repetition rate shock tubes (HRRST) were developed.11 Two of these are routinely used at synchrotron facilities. The original one developed at Argonne National Laboratory (ANL) is primarily used at the Advanced Light Source (ALS) with TOF-MS (ANL–HRRST/TOF-MS).12–15 A later HRRST was built at the Institut de Combustion Aérothermique Réactivité Environnement (ICARE)16 and is primarily used with double imaging photolelectron/photoion spectroscopy (i2PEPICO) at the DESIRS beamline of synchrotron SOLEIL.17–20 The ANL–HRRST and ICARE–HRRST are coupled by MBS systems to the charged particle analyzers. To a first order the apparatuses are identical and differ in the information content of the datasets produced. While both apparatuses have produced considerable insight into complex reaction mechanisms it has so far not been possible to extract kinetic data from them due to challenges with obtaining absolute concentrations of species. The main difficulty is a consequence of the fast changes in pressure that occur within a shock tube experiment that induce a slower increase in pressure in the ionization region of the spectrometer. This rise in pressure results in more ions being generated, even for an inert species, and the observed signal correspondingly increases. Thus, the signal for a reagent or product is a combination of the mole fraction of the species and the local pressure in the ionization region. These effects have to be decoupled to extract meaningful rate coefficients. The effect is well-understood4,5 and in EI systems is dealt with by adding a small amount of an inert gas (usually Ar, Kr or Xe) to the reagent mixture. The inert gas acts as an internal standard and allows the pressure effects to be accounted for.4,5 Implementing an internal calibration method for a shock tube experiment with PI sources is difficult, primarily due to the lack of inert species that have ionization energies in the range of most organic molecules. In the remainder of this paper, we present an alternative method for calibrating the pressure response which we refer to as an external calibration. Conceptually the method is similar to the external chemical thermometry method often employed with single pulse shock tubes.21,22 Following a discussion of the method, it is applied to two recent studies on toluene pyrolysis and ethylbenzene pyrolysis with the ICARE–HRRST/i2PEPICO experiment. The method is demonstrated for photoionization, but it should be equally applicable to other methods such as low energy electron impact and chemical ionization.
Experimental methods
ICARE–HRRST/i2PEPICO
The ICARE–HRRST and its coupling with the DELICIOUS III double imaging (i2PEPICO) spectrometer18,19 of the DESIRS beamline17 at the SOLEIL synchrotron facility has been previously described in detail.16 A brief description is provided here of the salient features. The driver section consists of a high-pressure reservoir and a fast acting, high-pressure, solenoid actuated valve.23 The driven section protrudes into the driver section and in this part the bore of the driven section is 12 mm. The driven section extends from the driver section giving the shock tube an overall length of ∼1.4 m, a schematic is provided in the ESI.† Downstream of the driver section the driven bore section is reduced to 8 mm. The driven section is terminated with a custom-made nozzle (0.4 mm orifice) which forms the entrance to the MBS interface. Shock velocities are obtained by the time taken for the incident shock wave to travel between pressure transducers (CHIMIE METAL A25L05B, 2 mm diameter) spaced 60.0 mm centre-to-centre. The final pressure transducer is 20.0 mm from the nozzle orifice and the velocity of the incident shock wave at the nozzle is obtained by extrapolation. The temperatures and pressures immediately behind the reflected shock wave, T5 and P5 respectively, are obtained from the incident shock velocity, the driven section loading pressure (P1) and the driven section temperature (T1) via the normal shock wave equations.1 The calculated T5 and P5 define the initial reaction conditions. However, wave dynamics in the HRRST can cause variations in P5 with respect to time. The P5 history was obtained in separate experiments performed in the laboratory at ICARE where the nozzle was replaced by a piezoelectric pressure transducer (PCB 113B24) covered by a thin layer of RTV paste. Space constraints prevented the nozzle and pressure transducer being installed simultaneously. However, the high reproducibility of the HRRST allowed nearly identical experiments to be performed at SOLEIL and ICARE.The driven section of the ICARE–HRRST is largely encased in the SAPHIRS chamber (primary vacuum chamber) at the DESIRS beamline.16 Gases continuously elute through the endwall nozzle into SAPHIRS forming a supersonic jet that quenches reaction. A portion from the jet centre passes through a skimmer (1 mm orifice) creating a molecular beam in an intermediate chamber. A portion from the core of the molecular beam passes through a second skimmer (2 mm orifice) into the ion source of the DELICIOUS III i2PEPICO spectrometer.19 The combination of nozzle and skimmers defines two stages of differential pumping between shock tube and ion source. The transient pressure changes in the spectrometer are around one order of magnitude (from ∼10−7 to ∼10−6 mbar) in each experimental cycle. The base pressure in the spectrometer was ∼5 × 10−8 mbar with the shock tube under vacuum.
The i2PEPICO method yields a multi-dimensional dataset from which mass spectra and the photoelectron spectrum associated with each peak in the mass spectra are extracted. Additionally, time-dependent data are obtained by providing a trigger signal that synchronizes firing of the HRRST and data acquisition with DELICIOUS III. For every experiment data were acquired from DELICIOUS III for 7.8 ms and the dataset comprised both pre-shock (non-reactive) and post-shock data. For every experimental cycle, i.e., each shock, sufficient data are recorded to allow post-processing of the dataset on a shock-by-shock basis. For example, different binning strategies can be tested, or shocks excluded to reduce standard deviations in reaction conditions. The experiments presented in this work were obtained at a repetition rate of the HRRST of 1 Hz. The photon energy was fixed at 10.0 eV for toluene (IE 8.82 eV)24 and ethylbenzene (IE 8.77 eV),24 and 14.5 eV for carbon dioxide (IE 13.78 eV).25 An argon gas filter removed high harmonic photons prior to the monochromator which was fitted with a 200 gr mm−1 low dispersion grating and delivered photon resolutions of around 25 meV at 10 eV. Details of DESIRS and DELICIOUS III in ref. 17–20.
Reagent mixtures
The reagent mixtures were prepared on-the-fly as needed for the experiments. The mixture preparation set-up comprised a bubbler for entraining vapor from liquid fuels into the argon carrier gas, a mixing station with three MKS mass flow controllers (MFC, model 1179A) and a six-liter tank to store the mixture. The flow controllers were activated by opening or closing air-actuated ball valves located at the exit of the MFCs. One of the MFCs controls the flow through the bubbler, the second the argon flow to bring the mixture to the desired dilution level. The operation of the mixing apparatus was fully automated. Similar techniques have been shown to provide excellent agreement between expected and actual fuel concentrations for single fuel mixtures.26 In the present experiments, all mixtures were composed of 0.1% fuel in argon. Toluene and ethylbenzene were purchased from Sigma Aldrich (>99.8% pure) while carbon dioxide and argon (>99.9999% pure) were supplied by Air Liquide.External calibration method
The method is conceptually similar to the external chemical thermometry method often employed with single-pulse shock tubes.21 In the external calibration method, a mixture is prepared consisting of a molecule that is inert under the target reaction conditions (calibration species) dilute in the same bath gas used for the regular experiments e.g., pyrolysis of toluene. The concentrations of the calibration species and reagent are similar. The calibration mixture is introduced into the driven section of the shock tube and shock heated to similar T and P as the reagent mixture experiences. Both the calibration and reagent mixtures are dilute. Consequently, the shock wave properties are mainly determined by the bath gas, argon, with small contributions from the calibrant and reagent. The temperature (T5) and pressure (P5) immediately behind the reflected shock wave are calculated using the ideal shock wave equations assuming frozen conditions.1 Over the range of conditions in the current work the difference in T5 for shocks in pure argon and shocks in 0.1% toluene/argon mixtures is approximately 1%. The maximum variation in the temperature due to the thermal decomposition of the fuel compared to the experiments with pure argon is obtained from chemical kinetic simulations and is also about 1%. Finally, the impact of the mixture composition on the expansion of gas through the nozzle has to be considered. The primary factor here is the specific heat ratio. This differs by about 0.25% between pure argon and the mixture and is too small to produce a noticeable difference in the supersonic jet properties. Consequently, by using dilute mixtures in the same bath gas similar T5 and P5 are obtained and similar flows through the nozzle are obtained in the calibration and reactive experiments. This results in essentially the same pressure rise in the ion source of the spectrometer in the calibration and reactive experiments. Due to the low signal intensities characteristic of VUV–PI, signals from many experiments are averaged to obtain sufficient signal/noise. An additional benefit of averaging is that statistical fluctuations in signal levels that are evident in single shot experiments are largely removed.5 The number of experiments that need to be averaged are largely dependent on the apparatus and based on experience ranges from as few as 200 for the ANL–HRRST/VUV–TOF-MS to several thousand for the ICARE–HRRST/VUV–i2PEPICO.In general, the mass spectrometer signal for species x, Sx, can be written as follows:
| Sx = σx·[x]·nexp·Dx·ηset·f(P,t) | (1) |
| SCO2 = σCO2·[CO2]·nCO2\_set·DCO2·ηCO2\_set·f(P,t) | (2) |
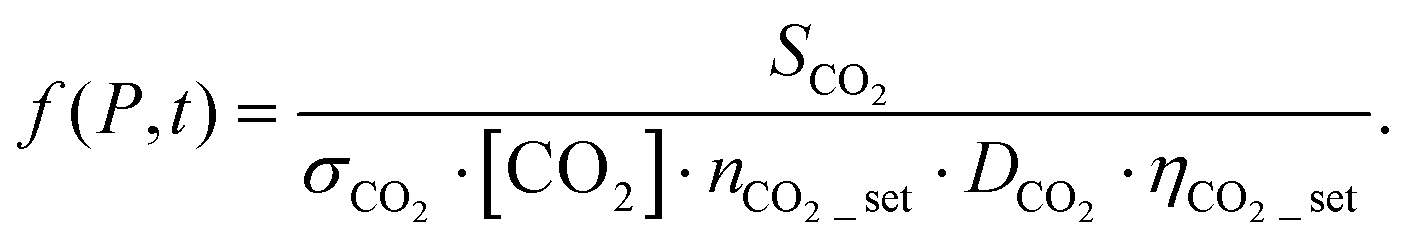 | (3) |
Substituting eqn (3) into eqn (1), the concentration of any species x can be derived from
 | (4) |
The concentration of the external standard and the number of experiments are known, the ratio between signals can be obtained from the experimental measurements, while the mass discrimination factors can be estimated based on previous investigations.27 Thus, if the photoionization cross sections are measured or calculated, the only unknown in the equation is the ratio between the transmission factors. This ratio is the same for all the species measured in a specific dataset, including the fuel molecule. Thus, its value can be derived from the following equation
 | (5) |
Experimental results and discussion
A typical mass spectrum obtained from i2PEPICO by averaging over 107![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 experiments with 0.1% toluene in argon at T5 and P5 equal to 1362 ± 22 K and 6.6 ± 0.2 bar, respectively, is shown in Fig. 1. The uncertainties represent one standard deviation from the average for all the experiments. The results include contributions of molecules coming from the pre-shock region, reflected conditions, and after the arrival of the rarefaction waves, during 7.8 ms period. In Fig. 1 several peaks between m/z 78 (benzene) and m/z 106 (ethylbenzene) correspond to single ring aromatics, heavier species correspond to multi-branched alkylated aromatics and/or PAHs, and several peaks, particularly at m/z < 92 represent fragments from dissociation of the fuel (toluene/ethylbenzene). The maximum m/z detectable is around 426. The identification and analysis of these compounds is the focus of ongoing research and is outside the scope of this paper which focuses on the development and application of the external calibration method. For peaks with single/noise ratio of at least 3:1 reliable concentration/time profiles are obtained (e.g., m/z < 350 in Fig. 1), although adequate profiles can be extracted for S/N of 2, see the ESI† for some examples.
000 experiments with 0.1% toluene in argon at T5 and P5 equal to 1362 ± 22 K and 6.6 ± 0.2 bar, respectively, is shown in Fig. 1. The uncertainties represent one standard deviation from the average for all the experiments. The results include contributions of molecules coming from the pre-shock region, reflected conditions, and after the arrival of the rarefaction waves, during 7.8 ms period. In Fig. 1 several peaks between m/z 78 (benzene) and m/z 106 (ethylbenzene) correspond to single ring aromatics, heavier species correspond to multi-branched alkylated aromatics and/or PAHs, and several peaks, particularly at m/z < 92 represent fragments from dissociation of the fuel (toluene/ethylbenzene). The maximum m/z detectable is around 426. The identification and analysis of these compounds is the focus of ongoing research and is outside the scope of this paper which focuses on the development and application of the external calibration method. For peaks with single/noise ratio of at least 3:1 reliable concentration/time profiles are obtained (e.g., m/z < 350 in Fig. 1), although adequate profiles can be extracted for S/N of 2, see the ESI† for some examples.
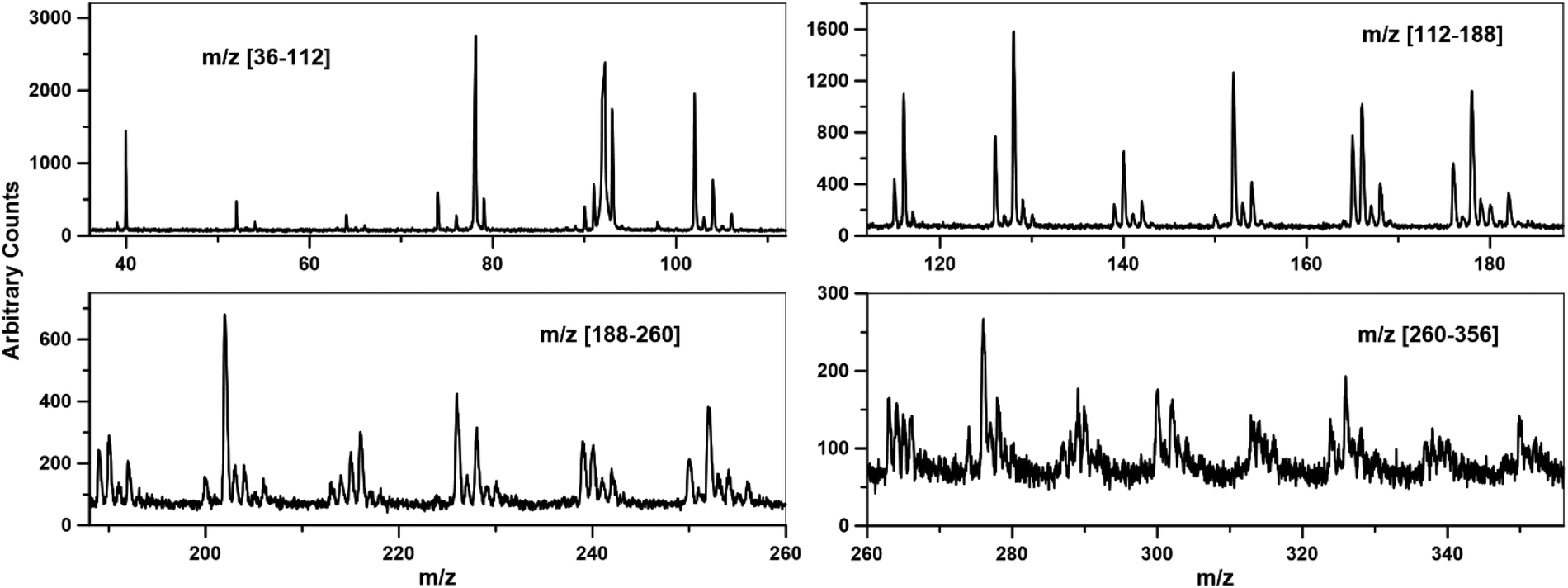 | ||
| Fig. 1 Toluene pyrolysis, 0.1% in argon at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy 10.0 eV. Average mass spectrum from 107000 experiments. | ||
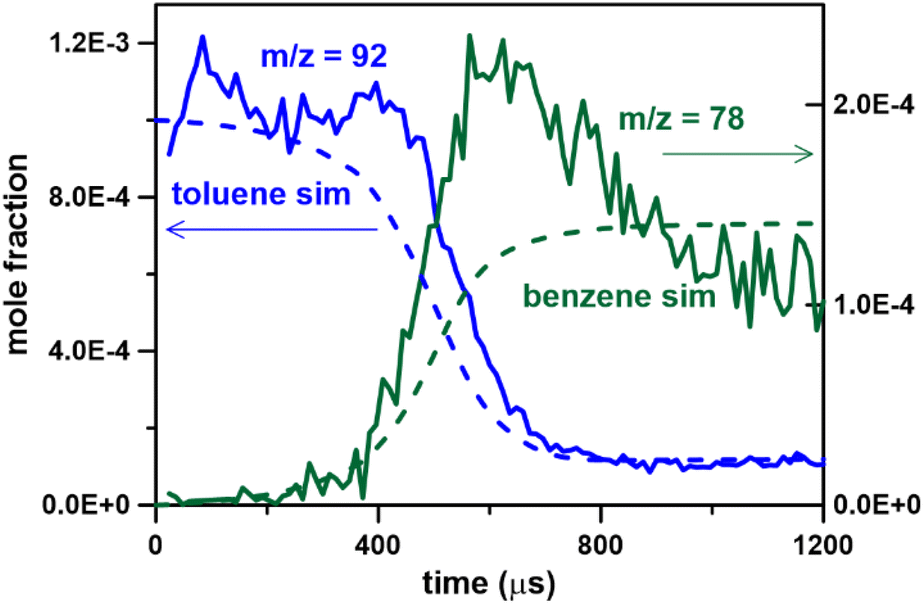
As discussed above, the mass spectrometer signal is proportional to the gas density (or pressure) inside the mass spectrometer chamber, which varies during the miniature shock tube cycle due to the large rapid changes in thermodynamic conditions in the HRRST due to the primary shock wave. However, there is also a slower and smaller pressure variation in the post-shock conditions due to wave dynamics in the HRRST. This slow variation also has to be accounted for particularly in chemical kinetic simulations of species concentrations. An example pressure trace averaged over 1000 experiments is shown in Fig. 2. In this case, the single pressure profiles were shifted in the x-direction to align the arrival times at the sensor. The pressure profile in Fig. 2 represents the thermodynamic conditions inside the shock tube, to be used in kinetic simulations. The stochastic distribution of the incident shock wave arrival times at the endwall is within ±50 μs around the average. These times are estimated with respect to the initial signals which trigger the shock tube cycle starting with the opening of the high-pressure valve. The average pressure profile over 1000 experiments from the raw data without shifting is presented in Fig. S1.† The averaging process also affects the shape of the experimental curves. Fig. 2 also contains signal intensity/time (kinetic traces) plots for m/z = 92 (fuel molecule) and m/z = 78 (one of the main peak products, mainly benzene) from the dataset presented in Fig. 1 (0.1% toluene/Ar). The kinetic data shown in Fig. 2 have not been corrected for pressure changes in the spectrometer, and the fuel profile has been translated so that the time corresponding to around 50% of its maximum value matches the zero of the pressure profile. Following the initial rise in the m/z 92 signal it stays nearly constant for around 400 μs, before dropping almost to its baseline within 200 μs. For the example shown, the rise time for the fuel signal to go from the pre-shock value to its maximum is about 150 μs. This rise time is related to the time taken for the pressure increase in the spectrometer to reach a maximum and is similar to that seen in single shot ST/MS experiments.5 The width of initial rise in the averaged pressure profile from the raw data (Fig. S1†) is around 80 μs, the pressure build-up in the mass spectrometer chamber contributes for the remaining effect. The region of stable m/z 92 signal indicates that dissociation of toluene is slow in these experiments. As the fuel signal falls, first primary and then secondary products start to appear, for example m/z = 78 in Fig. 2. After the initial sharp rise at time = 0 the pressure profile rises slowly for 600 μs before decreasing. This behavior is due to wave dynamics within the HRRST and has to be accounted for when simulating the kinetic traces. Similar behavior is seen in more conventional shock tubes and the impact of reaction temperatures has been previously analyzed.28,29 In particular, the decrease in m/z 78 (benzene) cannot be due to sudden consumption after 600 μs. Rather the observed signal is a convolution of the actual concentration and variations in signal due to non-idealities in the HRRST and pressure build up in the spectrometer. As will be seen in the following sections the inert calibration species signal exhibits similar behavior. Thus, the external calibration provides not only a correction for the build-up of the pressure inside the mass spectrometer chamber but also for the non-ideal behaviors inside the HRRST, allowing extraction of kinetic information in units of mole fractions. For this to be accurate, it is important that the conditions for the external calibration experiments and the actual ones to be corrected are similar, not only in terms of T5 and P5, but also in terms of pressure time history.
 | ||
| Fig. 2 Toluene pyrolysis, 0.1% in argon. Pressure profile from 1000 experiments; temporal species profiles from 107000 experiments at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy 10.0 eV. | ||
A CO2 mole fraction of 0.1% in argon, at an average temperature of 1376 ± 12 K and pressure of 6.6 ± 0.1 bar, and photon energy equal to 14.5 eV was used for the external calibration experiments. A total of 6042 experiments were averaged to derive the CO2 time profile, presented in Fig. 3 together with the m/z = 92 from Fig. 2. Carbon dioxide does not react at the thermodynamic conditions of the experiments, thus its mole fraction behind the reflected shock wave is constant. On the other hand, its signal varies due to the pressure variations inside the shock tube and the shape is similar to the pressure profile in Fig. 2. The initial rise in the CO2 profile is slightly faster than the case of toluene. This is probably an artifact of the large difference in the number of experiments (∼6000 vs. 107000) and the signal levels. Note that the CO2 values have been multiplied by a factor of 3 in Fig. 3.
 | ||
| Fig. 3 Temporal species profiles, m/z = 92 from toluene pyrolysis (0.1% in argon), averaged over 107000 experiments at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy = 10.0 eV; m/z = 44 from carbon dioxide pyrolysis (0.1% in argon), averaged over 6000 experiments at T = 1376 ± 12 K and P = 6.6 ± 0.1 bar, photon energy = 14.5 eV. | ||
In the following sections, the results of applying the external standard calibration technique to toluene pyrolysis and ethylbenzene pyrolysis will be presented. One of the uncertainties in the development of the external calibration technique is related to the alignment between the fuel and the CO2 profiles, e.g., Fig. 3. In Fig. 4 three methods of correcting for the small temporal misalignment between the CO2 and toluene (m/z = 92) signals are compared. The m/z = 92 signal is anchored at t = 0 as in Fig. 2 and the CO2 signal is shifted on the X-axis as follows: (i) the times of the 50% signal rises are matched (Fig. 4 red dashed lines), (ii) the times of the two main peaks at the end of the sharp increase in signal are matched (Fig. 4 solid lines, CO2 profile shifted by +36 μs), (iii) the times at which the two profiles start rising are matched (Fig. 4 light-blue dashed lines, CO2 profile shifted by −24 μs). These times refer to the profiles in Fig. 3. During the first few tens of microseconds both the CO2 and m/z = 92 signals are small and what look like minor fluctuations can introduce quite large perturbations in the corrected signal. Overall, fewer fluctuations are seen with method ii although the three methods produce essentially the same shapes in the profiles indicating that the choice of alignment method is not significant. The corrected m/z = 78 product signal is little affected by the choice of correction method. This is primarily due to the CO2 signal being relatively large and stable when the m/z = 78 signal starts to rise. For all three methods, the noise in the treated signal before time zero, pre-shock, is quite large, thus the derived information is not useful. Our experience has shown that large fluctuations are also observed in the pre-shock signal when an internal calibrant is used with electron ionization.
 | ||
| Fig. 4 Temporal species profiles from toluene pyrolysis (0.1% in argon), averaged over 107000 experiments at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy = 10.0 eV. Different matching between fuel and CO2 profiles: solid lines at the peak; red dashed lines at start of the rise; light-blue dotted lines at 50% rise. | ||
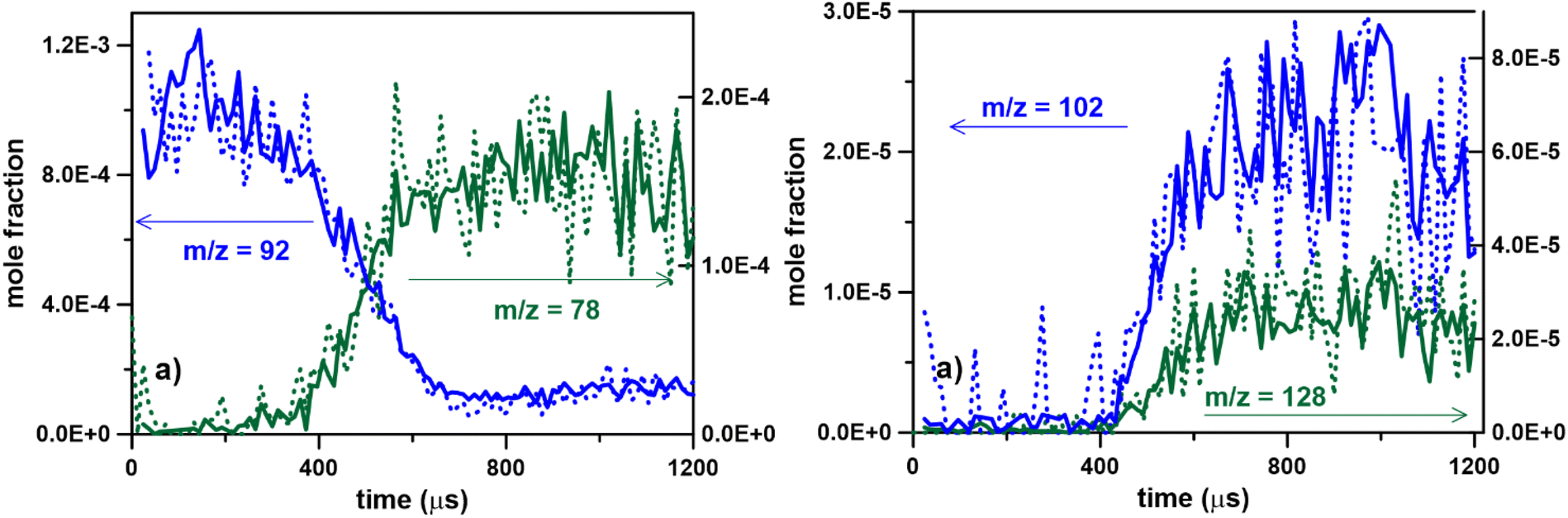
Fig. 5a–c present the temporal species profiles for the fuel and some of the main products after correction. The species shown were chosen because they could be unambiguously identified by comparing the experimental photoelectron spectra with literature ones (excluding minor contributions from isomers) and the photoionization cross sections have been measured or estimated. The mass spectral peaks at m/z 78, 92, 102 and 104 are almost entirely due to benzene, toluene, phenylacetylene and styrene, respectively, for m/z 116 indene accounts for 90% of the signal, while at m/z 128 naphthalene is about 70% of the peak. The error associated due to the difference in photoionization cross sections for the remaining isomers not considered in the current analysis is not expected to be significant. The absolute photoionization cross sections at 10.0 eV for toluene, phenylacetylene, styrene, indene, and ethylbenzene were taken from Zhou et al.,30 benzene from Rennie et al.,31 while for naphthalene the estimated values from ref. 32 were used. An uncertainty of 20% in the photoionization cross sections of the single-ring species is estimated, which increases to 30% for indene and 50% for naphthalene. These uncertainties were used to calculate the error bars for the mole fractions, Fig. 5. The time uncertainties are represented by error bars based on the maximum shift in the fuel profile from Fig. 4. The photoionization cross section for carbon dioxide at 14.5 eV was obtained from Hitchcock et al.33 by interpolation of the values at 14.0 eV and at 15.0 eV. In Fig. 5a–c, the dashed lines represent simulations obtained with the ICARE chemical kinetic mechanism35 for PAH formation and growth. This mechanism was previously validated against species profiles vs. temperature conditions from single-pulse shock tube experiments for pyrolysis of many single fuels at a nominal pressure of 20 bar. Some of the datasets used to develop the model were toluene34,35 (100–200 ppm, T5 1050–1700 K), and ethylbenzene36 (100 ppm, T5 950–1700 K) and their mixtures with small aliphatics (including reactions toluene + C2Hx37 and toluene + C3Hx35 at similar conditions). The simulations were performed with ANSYS CHEMKIN-Pro 2021 software using the batch reactor model with variable pressure. For these simulations the pressure profile in Fig. 2 was used. The error in the simulation results was obtained by considering the minimum and maximum temperatures in the T5 distribution (1362 ± 22 K). Overall there is excellent agreement between experiments and simulations for the decomposition of the fuel and the formation of the main single-ring aromatic products, including benzene (m/z = 78), styrene (m/z = 104), and phenylacetylene (m/z = 102), especially considering that the mechanism was not modified to match the experimental data. A peak at m/z = 91, corresponding to the benzyl radical, was also detected. The photoionization cross section was estimated by Li as 24.03 Mb at 9.98 eV.32 The shape of the m/z = 91 profile is correctly captured by the model. Species profiles for larger PAH products including indene (m/z = 116) and naphthalene (m/z = 128) are shown in Fig. 5c. Although the rise in the simulation profiles in Fig. 5c are delayed compared to the experiments and the absolute concentrations are not perfectly reproduced (especially in relative terms), the results are quite satisfactory considering the complexity of the chemistry involved in the PAH formation and the experimental errors related mainly to the availability and accuracy of ionization cross-sections. In particular, the simulations for the maximum naphthalene concentrations are within the experimental errors. Nevertheless, the data provide targets for future model development. The m/z 116 and m/z 128 profile would also need to be reduced by around 10% and 30% for comparison with indene and naphthalene, respectively, as additional isomers are not considered here. Thus, especially for naphthalene, the experimental profile would get closer to the simulation and substantially overpredict indene formation.
 | ||
| Fig. 5 Temporal species profiles from toluene pyrolysis (0.1% in argon), averaged over 107000 experiments at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy = 10.0 eV. Dashed lines in (a)–(c) represent the results of simulations with the ICARE PAH chemistry model. | ||
Fig. 5d contains mole fraction profiles of various main products (m/z = 78, 91, 102, and 202). In the analysis, the photoionization cross section of pyrene, estimated by Li,32 was used for m/z 202. From prior single-pulse shock tube measurements34 pyrene is one of the main isomers at m/z 202 and the small differences in photoionization cross sections of the different isomers will introduce little error. For clarity simulation results are not plotted. However, the figure shows that the corrected profiles have the shapes and relative heights that would be expected for species that are formed sequentially.
As briefly mentioned in the previous paragraphs, the external calibration method provides corrections not only for the initial pressure rise in the mass spectrometer, but also for the non-ideal behavior inside the shock tube at later times. To demonstrate the capability to compensate for long time effects, calculations were made using a constant value for the correction term, i.e. f(P,t) = f(t = 0) rather than the time dependent expression obtained from the CO2 profiles. The results are presented in Fig. 6 together with the kinetic simulations from Fig. 5 for toluene and benzene. Clearly, setting f(P,t) = f(t = 0) has a significant impact on the experimental profiles demonstrating the need to incorporate the full f(P,t) model. The use of the profiles in Fig. 6 for model validation would lead to incorrect mechanisms and kinetics.
 | ||
| Fig. 6 Temporal species profiles from toluene pyrolysis (0.1% in argon), averaged over 107000 at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy = 10.0 eV (solid lines), with f(P,t) = f(t = 0). Dashed lines the results of simulations with the ICARE PAH chemistry model. | ||
The experimental results presented in Fig. 4 and 5 were obtained by averaging around 107000 signals from experiments at a 1 Hz repetition rate which took at least 36 hours to perform (∼30 hours of experiments and time to change gas bottles, refill bubblers with reagents etc.). This large number of experiments was performed to enhance the S/N of high mass (m/z > 326) and low concentration products. Ideally, the smallest number of experiments necessary would be performed to obtain S/N of the desired level. The number of experiments needed is largely dependent on the concentration and photoionization cross section of a target species. Thus, the number of experiments needed will vary greatly depending on the overall goal of a study. To test the effects of reducing the number of runs on the signal quality, a sub-set of 27000 experiments was selected and used to derive temporal species profiles with the same data treatment procedure presented above. The number of experiments selected is the same as obtained in a subsequent study on ethylbenzene, discussed below. In this analysis, the first 27000 experiments were kept. Tests were also performed randomly selecting different batches of 27000 experiments and similar results were obtained. Selected results are presented in Fig. 7 for various m/z (dashed lines) together with the results from averaging 107000 runs (solid lines). The overall profiles are very similar in the two cases, in terms of shape and mole fractions, although the noise is considerably reduced when all the runs are considered in the analysis. This is expected as the signal to noise ratio is proportional to the square root of the number of experiments. However, apart from the increase in noise the information gained from the full dataset, Fig. 5, and the reduced set, Fig. 7, are very similar. This suggests that, depending on the overall goals, a small experimental set can produce reliable results, albeit with more noise, and allow for efficient use of limited beamtime. For example, in Fig. S2b and e,†m/z 276 and 326 are reported from the average of 27000 experiments, compared to the average from 107000 experiments in Fig. S2a and d of the ESI.† The profile for m/z 276 is still well defined, despite the fact the related S/N is now 2 (compared to 4 in Fig. S2a†). The two profiles obtained with 107000 and 27000 experiments are compared in Fig. S2c.† The initial rise in the profiles as well as the concentrations at later times (around 1–1.2 ms) are not affected by the number of experiments averaged. There is a slight discrepancy around 800 μs which is mainly due to the noise levels. On the other hand, the S/N ratio is too low for m/z 326, and no kinetic profiles could be determined with reduced number of experiments. In order to further confirm that the S/N ratio of around 2 is sufficient to obtain the kinetic profiles, the number of experiments in Fig. 7 was further reduced to 7 000. The resulting signal has a S/N ∼2 and it still reproduces the signal from averaging 107000 shocks, see Fig. 8.
 | ||
| Fig. 7 Temporal species profiles from toluene pyrolysis (0.1% in argon), averaged over 107000 (solid lines) and 27000 (dashed lines) experiments at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy = 10.0 eV. | ||
 | ||
| Fig. 8 Raw signals for m/z 102, toluene pyrolysis, at T = 1362 ± 22 K and P = 6.6 ± 0.2 bar, photon energy 10.0 eV. 107000 experiments (green); 7000 experiments (orange). | ||
The decomposition of toluene is relatively slow at 1360 K and 6 bar whereas ethylbenzene is far more reactive. The initial fuel concentration of ethylbenzene was the same as toluene (0.1% in argon) and the average temperature and pressure of the experiments was 1327 ± 18 K and 6.7 ± 0.2 bar, respectively. The small difference in temperature compared to the calibration experiments will have little impact on the mass flow through the nozzle. Consequently, the same CO2 calibration experiments were used with ethylbenzene. Approximately, 27000 experiments on ethylbenzene pyrolysis with an ionization photon energy of 10.0 eV were averaged. The corrected concentration/time profiles of ethylbenzene and toluene are compared in Fig. 9a. In the case of ethylbenzene, the profile starts decreasing right after the arrival of the shock wave at the end-wall. In addition, nearly all the ethylbenzene is consumed after 500 μs, while around 12% of toluene remains unreacted. The kinetic simulations with the ICARE pyrolysis model accurately capture the ethylbenzene consumption (Fig. 9b). Selected aromatic intermediates including benzene (m/z = 78), toluene (m/z = 92), styrene (m/z = 104), phenylacetylene (m/z = 102) and the benzyl radical (m/z = 91) are also reported in Fig. 9b and c. Overall, the experimental data are quite well reproduced by the model both in their shape and absolute values, although a small shift in the peak time is observed for toluene. Similar agreement is also observed for larger multi-ring products, such as naphthalene (m/z = 128) and indene (m/z = 116) as in Fig. 9d, where once again the relative concentrations are not perfectly simulated as for the toluene case, but the overall profiles are captured reasonably well.
 | ||
| Fig. 9 Temporal species profiles from ethylbenzene pyrolysis (0.1% in argon), average over 27000 experiments at T = 1327 ± 18 K and P = 6.7 ± 0.2 bar, photon energy = 10.0 eV; (a) includes the m/z = 92 from toluene experiments as in Fig. 4. Dashed lines in (b)–(d) the results of simulations with the ICARE PAH chemistry model. | ||
Conclusions
A new method for correction of time-history concentration profiles from miniature high-repetition-rate shock tubes coupled to synchrotron-based mass spectrometry diagnostics was developed based on the use of carbon dioxide as an inert external standard. The method corrects for both the effects of pressure build-up inside the mass spectrometer after passage of the reflected shock wave at the endwall and pressure variations inside the shock tube due to non-ideal behavior and rarefaction waves. Two test cases were considered, involving the thermal decomposition of toluene and ethylbenzene. The average T5 for ethylbenzene were 30 K lower than those of the toluene experiments. This small difference in temperature will have negligible impact on the pressure rise in the ion source of the spectrometer, although future studies will examine the range of applicability of calibration experiments obtained at T and P different to the reactive experiments. In particular, based on the difference in the pressure profiles obtained at ∼1300 K and ∼1500 K presented in ref. 16, the calibration curves are expected to be valid within a range of around ±50 K without major variation in the experimental corrected profiles. The corrected results were compared to kinetic modeling simulations performed with the ICARE PAH chemistry model. Excellent agreement between experiments and simulations was found for the fuel profiles and the formation of the main aromatic products, proving the reliability of the procedure. Different analyses were implemented to test the effects of the shift in the CO2 time profile to match different features of the toluene profile and the variation in the quality of the results with the number of experiments. The external standard method has been proven a flexible tool for extracting kinetic information from HRRST/TOF-MS experiments at synchrotrons.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 756785). R. S. T. acknowledges support from the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through Argonne National Laboratory and the Argonne/Sandia Consortium on High Pressure Combustion Chemistry. Argonne is a U.S. Department of Energy laboratory managed by UChicago Argonne, LLC, under contract DE-AC02-06CH11357. The authors would like to thank Dr L. Nahon and J. F. Gil for the support during the experiments at SOLEIL and fruitful discussions.References
- A. G. Gaydon and I. R. Hurle, The Shock Tube in High-Temperature Chemical Physics, Chapman & Hall, London, 1963 Search PubMed.
- Handbook of Shock Waves, ed. G. Ben-Dor, O. Igra and A. Lifshitz, Academic Press, New York, 2001 Search PubMed.
- Shock Waves in Chemistry, ed. A. Lifshitz, Dekker, New York, 1981 Search PubMed.
- R. D. Kern, H. J. Singh and Q. Zhang, in Mass Spectrometric Methods for Chemical Kinetics in Shock Tubes, ed. G. Ben-Dor, O. Igra, T. Elperin and A. Lifshitz, Academic Press, San Diego, CA, 2001, pp. 1–27 Search PubMed.
- R. S. Tranter, B. R. Giri and J. H. Kiefer, Shock Tube/Time-Of-Flight Mass Spectrometer for High Temperature Kinetic Studies, Rev. Sci. Instrum., 2007, 78, 034101 CrossRef PubMed.
- S. H. Dürrstein, M. Aghsaee, L. Jerig, M. Fikri and C. Schulz, A shock tube with a high-repetition-rate time-of-flight mass spectrometer for investigations of complex reaction systems, Rev. Sci. Instrum., 2011, 82, 084103 CrossRef.
- D. L. Osborn, P. Zou, H. Johnsen, C. C. Hayden, C. A. Taatjes, V. D. Knyazev, S. W. North, D. S. Peterka, M. Ahmed and S. R. Leone, The multiplexed chemical kinetic photoionization mass spectrometer: A new approach to isomer-resolved chemical kinetics, Rev. Sci. Instrum., 2008, 79, 104103 CrossRef.
- Y. Li and F. Qi, Recent applications of synchrotron VUV photoionization mass spectrometry: insight into combustion chemistry, Acc. Chem. Res., 2010, 43, 68–78 CrossRef CAS PubMed.
- F. Qi, Combustion chemistry probed by synchrotron VUV photoionization mass spectrometry, Proc. Combust. Inst., 2013, 34, 33–63 CrossRef CAS.
- N. Hansen, B. Yang and T. Kasper, Chapter 2: Synchrotron-Based VUV Photoionization Mass Spectrometry in Combustion Chemistry Research, in Synchrotron Radiation Applications, ed. X. Zhang, World Scientific, 2018, pp. 37–65 Search PubMed.
- R. S. Tranter and P. T. Lynch, A miniature high repetition rate shock tube, Rev. Sci. Instrum., 2013, 84, 094102 CrossRef CAS PubMed.
- P. T. Lynch, T. P. Troy, M. Ahmed and R. S. Tranter, Probing Combustion Chemistry in a Miniature Shock Tube with Synchrotron VUV Photo Ionization Mass Spectrometry, Anal. Chem., 2015, 87, 2345–2352 CrossRef CAS PubMed.
- C. Banyon, T. Sikes and R. S. Tranter, Reactions of propyl radicals: A shock tube–VUV photoionization mass spectrometry study, Combust. Flame, 2021, 224, 14–23 CrossRef CAS.
- J. B. Randazzo, R. Sivaramakrishnan, A. W. Jasper, T. Sikes, P. T. Lynch and R. S. Tranter, An experimental and theoretical study of the high temperature reactions of the four butyl radical isomers, Phys. Chem. Chem. Phys., 2020, 22, 18304–18319 RSC.
- T. Sikes, C. Banyon, R. A. Schwind, P. T. Lynch, A. Comandini, R. Sivaramakrishnan and R. S. Tranter, Initiation reactions in the high temperature decomposition of styrene, Phys. Chem. Chem. Phys., 2021, 23, 18432–18448 RSC.
- S. Nagaraju, R. S. Tranter, F. E. Cano Ardila, S. Abid, P. T. Lynch, G. A. Garcia, J. F. Gil, L. Nahon, N. Chaumeix and A. Comandini, Pyrolysis of ethanol studied in a new high-repetition-rate shock tube coupled to synchrotron-based double imaging photoelectron/photoion coincidence spectroscopy, Combust. Flame, 2021, 226, 53–68 CrossRef CAS.
- L. Nahon, N. de Oliveira, G. A. Garcia, J. F. Gil, D. Joyeux, B. Lagarde and F. Polack, DESIRS: a state-of-the-art VUV beamline featuring high resolution and variable polarization for spectroscopy and dichroism at SOLEIL, J. Phys.: Conf. Ser., 2012, 425, 122004 CrossRef.
- X. Tang, G. A. Garcia, J. F. Gil and L. Nahon, Vacuum upgrade and enhanced performances of the double imaging electron/ion coincidence end-station at the vacuum ultraviolet beamline DESIRS, Rev. Sci. Instrum., 2015, 86, 123108 CrossRef PubMed.
- G. A. Garcia, B. K. Cunha de Miranda, M. Tia, S. Daly and L. Nahon, DELICIOUS III: a multipurpose double imaging particle coincidence spectrometer for gas phase vacuum ultraviolet photodynamics studies, Rev. Sci. Instrum., 2013, 84, 053112 CrossRef CAS PubMed.
- G. A. Garcia, L. Nahon and I. Powis, Two-dimensional charged particle image inversion using a polar basis function expansion, Rev. Sci. Instrum., 2004, 75, 4989–4996 CrossRef CAS.
- R. S. Tranter, R. Sivaramakrishnan, N. Srinivasan and K. Brezinsky, Calibration of reaction temperatures in a very high pressure shock tube using chemical thermometers, Int. J. Chem. Kinet., 2001, 33, 722–731 CrossRef CAS.
- Y. Han, J. M. Mehta and K. Brezinsky, Temperature approximations in chemical kinetics studies using single pulse shock tubes, Combust. Flame, 2019, 209, 1–12 CrossRef.
- R. S. Tranter and T. Sikes, Solenoid actuated driver valve for high repetition rate shock tubes, Rev. Sci. Instrum., 2020, 91, 056101 CrossRef CAS PubMed.
- J. O. Howell, J. M. Goncalves, C. Amatore, L. Klasinc, R. M. Wightman and J. K. Kochi, Electron transfer from aromatic hydrocarbons and their π-complexes with metals. Comparison of the standard oxidation potentials and vertical ionization potentials, J. Am. Chem. Soc., 1984, 106, 3968–3976 CrossRef CAS.
- F. M. Benoit and A. G. Harrison, Predictive value of proton affinity. Ionization energy correlations involving oxygenated molecules, J. Am. Chem. Soc., 1977, 99, 3980–3984 CrossRef CAS.
- A. Dalmiya, J. M. Mehta, R. S. Tranter and P. T. Lynch, High pressure, high flow rate batch mixing apparatus for high throughput experiments, Rev. Sci. Instrum., 2021, 92, 114104 CrossRef CAS PubMed.
- B. Gans, L. A. Vieira Mendes, S. Boyé-Péronne, S. Douin, G. Garcia, H. Soldi-Lose, B. K. Cuhna de Miranda, C. Alcaraz, N. Carrasco, P. Pernot and D. Gauyacq, Determination of the Absolute Photoionization Cross Sections of CH3 and I Produced from a Pyrolysis Source, by Combined Synchrotron and Vacuum Ultraviolet Laser Studies, J. Phys. Chem. A, 2010, 114, 3237–3246 CrossRef CAS PubMed.
- R. S. Tranter, K. Brezinsky and D. Fulle, Design of a high-pressure single pulse shock tube for chemical kinetic investigations, Rev. Sci. Instrum., 2001, 72, 3046–3054 CrossRef CAS.
- W. Tang and K. Brezinsky, Chemical kinetic simulations behind reflected shock waves, Int. J. Chem. Kinet., 2006, 38(2), 75–97 CrossRef CAS.
- Z. Zhou, M. Xie, Z. Wang and F. Qi, Determination of absolute photoionization cross-sections of aromatics and aromatic derivatives, Rapid Commun. Mass Spectrom., 2009, 23, 3994–4002 CrossRef CAS PubMed.
- E. E. Rennie, C. A. F. Johnson, J. E. Parker, D. M. P. Holland, D. A. Shaw and M. A. Hayes, A photoabsorption, photodissociation and photoelectron spectroscopy study of C6H6 and C6D6, Chem. Phys., 1998, 229, 107–123 CrossRef CAS.
- Edited by JiuZhong Yang and Combustion Team. Photonionization Cross Section Database (Version 2.0). https://flame.nsrl.ustc.edu.cn/database/. National Synchrotron Radiation Laboratory, Hefei, China. (2017). Estimated data provided by YuYang Li, JiuZhong Yang and ZhanJun Cheng.
- A. P. Hitchcock, C. E. Brion and M. J. Van der Wiel, Absolute Oscillator Strengths for Valence-Shell Ionic Photofragmentation of N2O and CO2 (8–75 eV), Chem. Phys., 1980, 45, 461–478 CrossRef CAS.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, Probing PAH formation chemical kinetics from benzene and toluene pyrolysis in a single-pulse shock tube, Proc. Combust. Inst., 2021, 38, 891–900 CrossRef CAS.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, Influences of propylene/propyne addition on toluene pyrolysis in a single-pulse shock tube, Combust. Flame, 2022, 236, 111799 CrossRef CAS.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, A comparative kinetic study of C8–C10 linear alkylbenzenes pyrolysis in a single-pulse shock tube, Combust. Flame, 2020, 221, 136–149 CrossRef CAS.
- W. Sun, A. Hamadi, S. Abid, N. Chaumeix and A. Comandini, Detailed experimental and kinetic modeling study of toluene/C2 pyrolysis in a single-pulse shock tube, Combust. Flame, 2021, 226, 129–142 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an00819c |
| This journal is © The Royal Society of Chemistry 2024 |