
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supported H8PV5Mo7O40 on activated carbon: Synthesis and Investigation of influencing factors for catalytic performance†
Anne
Wesner
a,
Max P.
Papajewski
a,
Leon
Schidowski
a,
Charlotte
Ruhmlieb
b,
Maximilian J.
Poller
 a and
Jakob
Albert
*a
a and
Jakob
Albert
*a
aInstitute of Technical and Macromolecular Chemistry, University of Hamburg, Bundesstraße 45, 20146 Hamburg, Germany
bInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: jakob.albert@uni-hamburg.de
First published on 30th July 2024
Abstract
Polyoxometalates (POMs), in particular the Keggin-type HPA-5 (H8PV5Mo7O40) are widely established as effective catalysts for acid- and redox-catalyzed reactions. Yet, they are mainly used as homogeneous catalysts, which poses challenges regarding catalyst separation. This study explores the synthesis of supported HPA-5, and its application as a heterogeneous catalyst for biomass conversion, focusing on activated carbons with diverse chemical and physical properties as support materials. Characterization of these carbons gives insights into the influence of surface area, oxygen content, and acidity on HPA adsorption and stability. Activated carbon CW20, was found to be the best support due to its high vanadium loading and effective preservation of the HPA-5 structure. It underwent various pre- and post-treatments, and the obtained supported catalysts were evaluated for their catalytic performance in converting glucose under both oxidative (OxFA process) and inert (Retro-Aldol condensation) conditions. Notably, HPA-5 supported on CW20 emerged as an exceptional catalyst for the retro-aldol condensation of glucose to lactic acid, achieving a selectivity of 15% and a conversion rate of 71%, with only minimal vanadium leaching.
Introduction
Biomass emerges as a sustainable and environmentally carbon neutral alternative to traditional fossil resources, including oil, coal, or natural gas. The development of efficient catalysts for the conversion of biomass into valuable chemicals is a pivotal step advancing towards a sustainable economy.1,2In this context, polyoxometalates (POMs) are highly promising materials. They represent a group of anionic metal–oxide clusters, exhibiting a wide array of electronic and structural characteristics. They are well established as catalysts for acid- or redox-catalyzed reactions.3–7 One of the most commonly used POM subclasses in this context are Keggin-type heteropoly acids (HPAs), which are defined by the formula [XM12O40]n− (X = P, Si, V etc. and M = Mo, W, V etc.).6,8,9 These HPAs have demonstrated a high efficiency and versatility in the conversion of lignocellulosic biomass into monosaccharides and subsequently into various valuable platform chemicals.10–14
Keggin-type Mo–V–P HPAs, especially HPA-5 (H8PV5Mo7O40), have been successfully applied as catalysts for the selective oxidation of biomass to formic acid (known as OxFA process). This process involves partial, catalytic and selective oxidation of biogenic substrates under mild conditions (90 °C, ≤ 30 bar oxygen), using O2 as an oxidant and H2O as solvent.15–17 Formic acid is a product with versatile applications, primarily used in feed industry and silage aid, but also in textile, pharmaceutical, leather, and rubber sectors. It also serves as storable and transportable secondary energy carrier, which can be decomposed either into CO and H2O, or CO2 and H2 for energy and material utilization.18 Furthermore, the same HPAs have been employed for the formation of lactic acid under mild conditions and nitrogen atmosphere, which is identified as one of NREL's top 12 value-added chemicals derived from biomass. Lactic acid is applied as a polymer precursor for polylactic acid, a biodegradable and bio-based plastic, in pharmaceutical and cosmetic applications as well as for the fermentation of food.19–21
Due to their good solubility in water, HPAs are typically employed as homogeneous catalysts. While beneficial for selectivity and conversion, this poses challenges for their separation and reuse. Consequently, several efforts have been undertaken to heterogenize HPAs. The primary strategies for heterogenizing HPAs involve complexation with inorganic or organic cations or immobilization onto supports, e.g., via covalent binding (grafting), encapsulation in porous structures, or impregnation.22–25
Activated carbon is particularly effective as a support for HPAs due to its high surface area, diverse porosity, cost-effectiveness, accessibility, recyclability, and pH stability. Consequently, this material has been frequently used for the heterogenization of HPAs due to its advantageous properties.24,26–31 H4SiW12O40, when supported on activated carbon, has demonstrated efficient hydrolysis of cellobiose and seaweed into sugars under microwave irradiation.32 Furthermore, H4SiW12O40/activated carbon, has been effectively employed for the selective hydrolysis of starch to glucose.33 In a similar vein, the H3PW12O40/activated carbon composite has been successfully used in catalyzing the hydrolysis of cellulose and hemicellulose.34,35 Nevertheless, the potential of carbon-supported HPAs in biomass conversion beyond these examples remains largely unexplored.23 Furthermore, the nature of the adsorption of HPA to activated carbon, including if it is of chemical or physical nature, still remains unclear.26,35,36
In this study, HPA-5, well-known for its bifunctionality both as acid and redox catalyst, was systematically impregnated onto a variety of activated carbons, varying in different physical (such as surface area, pore volume and morphology) and chemical characteristics (including elemental composition, surface acid sites, and functional groups). This was done to assess the impact of these properties on the suitability as a support material for HPA-5. Subsequently, the most suitable activated carbon was selected for impregnation using different synthetic methods. The catalytic activity and leaching stability of this system were then evaluated in two different multiphasic reactions, specifically the oxidation of glucose into formic acid as well as the retro-aldol condensation of glucose into lactic acid.
Experimental
Materials and catalyst preparation
Unless specified otherwise, all chemicals were obtained commercially and used as received without further purification. A range of activated carbons was purchased from Cabot, including Norit SXPlus (NSXPlus), Norit A Supra Eur (NASEur), Norit GSX (NGSX), Norit CASP F (NCASPF), and Norit Darco KBG (DKB-G), while CW20 was acquired from Silcarbon.The HPA-5 bulk catalyst was synthesized according to a previously published procedure.9,37 The successful synthesis was confirmed by IR spectroscopy and elemental analysis (see Fig. S1 and Table S1†).
Initially, for testing different activated carbons as support materials (chapter: Supporting HPA-5 on different activated carbons), wet impregnation was employed to support HPA-5 onto all activated carbons (experimental series 1, E1). For this process, 4.99 g (3.11 mmol) HPA-5 was dissolved in 250 ml of H2O adjusted to a pH range of 2–3, and 7.02 g of the selected activated carbon was suspended into the solution. The suspension was stirred at 50 °C for 3 hours at 100 rpm using a rotary evaporator. The solid was subsequently filtered off, washed with H2O until neutral pH was achieved, and then dried overnight (see Fig. 6).
Additionally, activated carbon CW20 was chosen to test various synthesis methods (experimental series) for impregnation (chapter: Supporting HPA-5 on CW20 using various synthesis methods). Different pre- and post-treatments additionally to the general wetness-impregnation procedure (see Fig. 6) were utilized. In experimental series 2 (E2), oxidative pre-treatment was performed in concentrated 65 wt% HNO3 at 90 °C for 3 hours as pre-treatment. Series 3 (E3) incorporated an additional post-treatment, heating the catalyst to 200 °C at 2 K min−1 for 5 hours. Series 4 (E4) applied the same post-treatment as E3, but without any pre-treatment of the activated carbon. Finally, series 5 (E5) used reductive pre-treatment, heating the carbon to 400 °C for 4 hours under an atmosphere of 95% N2/5% H2 (100 l h−1).
Catalyst characterization
The elemental analysis of all catalysts was conducted using Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES). Powder X-Ray Diffraction (XRD) techniques were utilized to investigate their crystalline structures. Nitrogen physisorption measurements were applied to ascertain the porosity, pore volume, and overall surface area. Catalyst morphology and metal distribution were explored through Scanning Electron Microscopy (SEM) along with Energy-Dispersive X-ray Spectroscopy (EDX). The acidity parameters of the synthesized catalysts were assessed via NH3-Temperature Programmed Desorption (NH3-TPD), Boehm titration method, and Point of Zero Charge analysis. Furthermore, Infrared (IR) and Raman spectroscopy were employed for structural characterization. Comprehensive descriptions of all characterization methods are given in the ESI.†Catalytic evaluation and determination of catalytic parameters
Catalytic experiments were carried out in a three-fold plant in stainless steel reactors. Analysis of both liquid and gaseous products were conducted using High Performance Liquid Chromatography (HPLC) and Gas Chromatography (GC), respectively. The examination of the utilized catalysts was carried out via ICP-OES and IR. Detailed descriptions of the catalytic evaluation, as well as the equations used for calculating catalytic parameters, such as conversion (X), yield (Y) and selectivity (S), are provided in the ESI.†Results and discussion
Characterization of activated carbons
Initially, all activated carbons were characterized according to their textural and chemical features. X-Ray diffraction (Fig. S2†) confirms the presence of crystalline graphitic carbon with characteristic diffraction patterns between 15–30° (002 reflection) and 40–50° (001 reflection).38,39 Elemental analysis indicates negligible impurities with minor traces of Al, Fe, and P (Table 1). The activated carbons exhibit varying carbon, hydrogen, and oxygen contents. Oxygen amount ranges from 3.49 wt% (NSXPlus) to 17.22 wt% (DKB-G), the carbon amount accordingly from 88.74 wt% (NSXPlus) to 76.80 wt% (DKB-G). Hydrogen content varies from 0.68 wt% (NSXPlus) to 3.18 wt% (NCASPF) (Table 1).| NSXPlus | NASEur | NGSX | CW20 | NCASPF | DKB-G | |
|---|---|---|---|---|---|---|
| Elemental analysis | ||||||
| Al (wt%) | 0.95 | — | 0.09 | 0.05 | — | — |
| Fe (wt%) | — | — | 0.07 | —- | — | — |
| P (wt%) | — | — | — | 1.50 | 1.25 | 1.47 |
| C (wt%) | 88.74 | 94.38 | 88.35 | 82.61 | 76.97 | 76.80 |
| H (wt%) | 0.68 | 0.46 | 1.33 | 2.21 | 3.18 | 2.89 |
| N (wt%) | — | — | — | — | — | — |
| O (wt%) | 2.47 | 3.49 | 5.18 | 8.74 | 13.93 | 17.22 |
| Textural properties | ||||||
| Specific surface area (m2 g−1) | 923 | 1771 | 736 | 1500 | 1544 | 1341 |
| Pore volumina (mL g−1) | 0.65 | 0.90 | 0.62 | 1.32 | 1.22 | 1.45 |
| ∅ pore diameter (nm) | 7.35 | 1.69 | 7.21 | 16.48 | 14.93 | 11.61 |
| Boehm titration | ||||||
| Surface acidity (mmol g−1) | 55.0 | 55.5 | 69.0 | 188.0 | 248.0 | 303.5 |
| Surface basicity (mmol g−1) | 57.0 | 82.0 | 61.5 | 45.0 | 0.0 | 0.0 |
| Carboxylic groups (mmol g−1) | 5.0 | 0.0 | 5.5 | 39.0 | 94.0 | 102.5 |
| Lactonic groups (mmol g−1) | 9.0 | 7.0 | 32.5 | 113.0 | 118.0 | 185.5 |
| Phenolic groups (mmol g−1) | 41.0 | 51.5 | 31.0 | 36.0 | 36.0 | 15.5 |
| NH 3 -TPD acidity | ||||||
| Low (150–500 °C) | 0.04 | 0.08 | 0.11 | 1.00 | 2.15 | 2.49 |
| High (500–700 °C) | 0.00 | 0.06 | 0.07 | 1.00 | 1.61 | 2.45 |
| Total | 0.01 | 0.07 | 0.08 | 1.00 | 1.79 | 2.46 |
| Point of zero charge (pH) | 8.80 | 8.65 | 7.40 | 5.74 | 2.87 | 2.62 |
Specific surface areas range from 736 m2 g−1 (NASEur) to 1544 m2 g−1 (NCASPF), with pore diameters spanning from 1.69 nm (NASEur) to 16.48 nm (CW20) (Table 1, Fig. S2 and S3†). Microscopy data (Fig. S5–10†) indicate the presence of agglomerates comprising nanoscopic substructures across all samples. Therefore, it is essential to differentiate between various surface types: firstly, the outermost surface of the sample, followed by the surface area formed by interstices between agglomerates and substructures, and lastly, nanoporous regions within the samples. These features are important to evaluate the inner surface area of the sample and are discernible in certain areas under SEM examination.
The points of zero charge range from pH 2.62 (DKB-G) to pH 8.80 (NSXPlus). Determined surface acidities range from 55 μmol g−1 for NSXPlus to 304 μmol g−1 for DKB-G, surface basicities from 0 μmol g−1 for DKB-G to 57 μmol g−1 for NSXPlus (Table 1). Notably, there is a correlation between oxygen content, point of zero charge, and surface characteristics (Fig. 2 and Fig. S11†). Activated carbon contains various surface oxide groups with different acidities such as strongly acidic carboxylic acids, acidic phenolic groups, or weakly acidic lactonic groups (Table S2†).40,41 These functional groups can also be separately determined using the Boehm method.42,43 An increase in surface acidity aligns with a higher amount of carboxylic and lactonic groups, while the quantity of phenolic groups remains relatively constant (Table 1). NH3-TPD for activated carbons with an oxygen amount under 6 wt% (NSXPlus, NASEur, NGSX) indicates negligible amounts of interactions with NH3, for activated carbons, however, with a significantly higher amount of oxygen over 6 wt% (CW20, NCASPF, DKB-G) weak and middle (150–500 °C) as well as strong (500–700 °C) interactions can be seen (Fig. 1). NH3-TPD measurements agree with the results of point of zero charge measurements and acidity determined by Boehm method (Fig. 2). Therefore, it is hypothesized that the surface acidity is directly related to the amount of oxygen groups on the surface (Fig. 2).
 | ||
| Fig. 1 NH3-TPD acidity (left) and Infrared spectra of pure activated carbons (right). | ||
 | ||
| Fig. 2 Correlation between oxygen content and acidity (top) and between Boehm and NH3 surface acidity (bottom). | ||
Raman spectroscopy analysis shows the presence of the D-band at approximately 1330 cm−1, indicative of disordered sp2-hybridized rings in carbon (Fig. S12†).44,45 The IR spectra (Fig. 1) for activated carbons with over 6 wt% oxygen content (CW20, NCASPF, DKB-G) display bands of various surface groups. For instance, bands indicating the presence of alcohol, phenol, and carboxylic groups are observed between 1000–1300 cm−1, as well as for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and surface quinones between 1500–1700 cm−1.46 The exact positions of surface groups in activated carbons are listed in Table S3.† Due to the overlap of all bands, precise assignment of surface groups is challenging. Interestingly, the IR spectra of the groups with lower oxygen content (less than 6 wt%, for NSXPlus, NASEur, NGSX), exhibit very high absorption, resulting in either no or only very weak bands.
C bonds and surface quinones between 1500–1700 cm−1.46 The exact positions of surface groups in activated carbons are listed in Table S3.† Due to the overlap of all bands, precise assignment of surface groups is challenging. Interestingly, the IR spectra of the groups with lower oxygen content (less than 6 wt%, for NSXPlus, NASEur, NGSX), exhibit very high absorption, resulting in either no or only very weak bands.
IR spectra show that the impregnation on CW20, NCASPF, and DKB-G was successful (Fig. 3). The concise IR bands of HPA-5 can be distinguished: 1049–1060 cm−1 (PO vibration), 945–962 cm−1 (MtO), 866–877 cm−1 (M–O–Mvertex), 643–767 cm−1 (M–O–Medge).9 For NSXPlus, NASEur and NGSX no reflexes could be detected, which can either be due to no HPA-5 being present on these activated carbons because of overall high absorption, or because of decomposition of the Keggin structure through the non-acidic character of these activated carbons.47 Positively charged surfaces can adsorb anionic HPA clusters due to electrostatic interactions.48 Consequently, since successful deposition of HPA-5 on these activated carbons could not be confirmed, they were excluded from further investigations.
 | ||
| Fig. 3 IR-spectra of HPA-5 impregnated carbons, red – impregnated, black – pure activated carbon. | ||
Furthermore, Raman spectroscopy and XRD were employed for analytical investigations. Raman spectra of pure HPA 5 displayed the characteristic vibrational bands indicative of the Keggin structure, as well as additional bands below 800 cm−1, which correspond to M–O–M vibrations (Fig. S13†).9 XRD analysis revealed distinct diffraction patterns at 27.6°, 27.7°, and 27.9° for the pure HPA-5 catalyst (Fig. S14†), corresponding to the Miller indices [3 1 4], [2 4 0], and [3 1 1], respectively, as determined from single-crystal X-ray diffraction data in a previous study.9 However, using Raman and XRD, no clear signals for HPA-5 were observed on the impregnated samples at all (Fig. S13 and S14†), suggesting that these methods are not suitable for this kind of investigation.
SEM reveals no significant alteration in morphology due to impregnation (Fig. S16–21†). SEM-EDX analysis and ICP-OES confirm the successful impregnation of HPA-5 onto the supports. SEM-EDX mapping shows a microscopic homogeneous distribution of Mo, V, and P (Fig. S15†). Table 2 presents the ICP-OES results, listing the content of Mo, P, and V for each activated carbon compared to the desired HPA-5 loading (right column). Additionally, the molar ratios of Mo to V for each sample have been calculated and are also displayed in the table in comparison to the desired ratio of HPA-5 (right column). The impregnation process achieves approximately 35% of the targeted HPA-5 loading on the supports (CW20 and NASEur), with the remainder not adsorbed and leached during subsequent washing. All activated carbons exhibit a molar ratio of Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V of approximately 8:4, which suggests the presence of HPA-4 (H7PV4Mo8O40) instead of HPA-5. The reason for this might be the differences in stability and charge of the HPA resulting in different interaction strengths with the carbon surface. Literature indicates that Keggin structures experience a reduction in stability as the degree of substitution increases. The preferential formation of HPA-4 over HPA-5 might be due to the enhanced dissociation of HPA-5, which can also facilitate the formation of lower substituted POMs.37
:V of approximately 8:4, which suggests the presence of HPA-4 (H7PV4Mo8O40) instead of HPA-5. The reason for this might be the differences in stability and charge of the HPA resulting in different interaction strengths with the carbon surface. Literature indicates that Keggin structures experience a reduction in stability as the degree of substitution increases. The preferential formation of HPA-4 over HPA-5 might be due to the enhanced dissociation of HPA-5, which can also facilitate the formation of lower substituted POMs.37
| Loading | NSXPlus | NASEur | NGSX | CW20 | NCASPF | DKB-G | HPA-5 (H8PV5Mo7O40) |
|---|---|---|---|---|---|---|---|
| Mo (wt%) | 5.85 | 7.45 | 4.39 | 8.06 | 7.64 | 6.7 | 17.4 |
| P (wt%) | 0.23 | 0.26 | 0.17 | 1.17 | 0.64 | 0.69 | 0.8 |
| V (wt%) | 1.74 | 2.34 | 1.22 | 2.32 | 2.03 | 1.82 | 6.6 |
| Molar ratio Mo:V |
7.7:4.3 |
7.6:4.4 |
7.9:4.1 |
7.8:4.2 |
8.0:4.0 |
7.9:4.1 |
7.0:5.0 |
The quantities of adsorbed HPA-5 vary depending on the used carbon material, with CW20 and NASEur showing the highest loadings. Interestingly, there is no overall correlation observable between the oxygen content and the loading of the catalysts (Fig. 4). Other studies have suggested that surface oxygen groups play a stabilizing role,35 however, this was not confirmed in this study, which involved a wide variety of activated carbons. Additionally, some studies indicate that the stabilization of supported HPAs may also be influenced by van der Waals forces, hydrogen bonding, or the exchange of the terminal oxygen atom (Ot) in the HPA structure.35,49 Vanadium loading on the activated carbons shows a correlation with their surface area (Fig. 4). As the surface area increases, the vanadium loading also rises from 1.22 wt% for NGSX with a surface area of 736 m2 to twice as much at 2.34 wt% for NASEur, corresponding to a surface area of 1771 m2. The increased POM loading with higher porosity was also observed in previous studies.35,50 Interestingly, we observed that CW20, compared to NASEur, even with a smaller surface area of only 1500 m2 exhibits the equal vanadium loading of 2.32 wt% V. When the vanadium content is related to the surfaces of the two activated carbons, CW20 shows a higher loading of 15.47 wt V m−2 g−1 compared to NASEur with 13.21 wt V m−2 g−1 (Fig. 5).
 | ||
| Fig. 4 Correlation between amount of oxygen and vanadium loading (top) and between surface area and vanadium loading (bottom). | ||
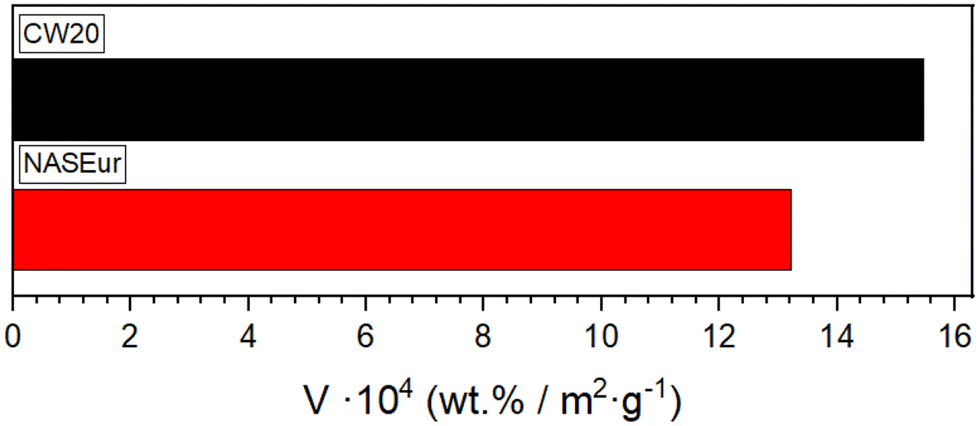 | ||
| Fig. 5 Vanadium loading of CW20 and NASEur related to surface area. | ||
As a conclusion, it can be stated that the specific surface area has a greater influence on the impregnation efficiency than the surface oxygen or the acidity of the surface. CW20 and NASEur show the highest loadings of HPA. However, the low surface acidity of NASEur could have led to the destruction of the Keggin structure of the POMs. While the individual elements Mo, V, P could be detected by elemental analysis, the Keggin structure could not be conclusively identified using spectroscopic and X-ray analytical methods for this activated carbon. Since CW20 exhibited the highest vanadium loading as well as preservation of the POM structure, this activated carbon was chosen for subsequent experiments to try out advanced synthesis techniques.
 | ||
| Fig. 6 Different experimental series with pre- and post-treatments for impregnation of activated carbon. | ||
For the experimental investigation, CW20 underwent diverse pretreatment protocols in distinct experimental series. Specifically, CW20 was applied in its untreated form, subjected to oxidative pretreatment using 65 wt% HNO3 for 3 hours (E2), and alternatively, reductively pretreated at 400 °C for 4 hours (E5) using a reductive gas mixture composed of 5% hydrogen and 95% nitrogen.
Due to the oxidative pretreatment, the surface area and pore volume decrease from 1500 m2 g−1 and 1.32 mL g−1 (CW20) to 357 m2 g−1 and 0.26 mL g−1 (CW20ox), respectively (Table 3, Fig. S22 and S23†). The oxygen content increases from 8.74 wt% to 35.88 wt%, while the carbon content correspondingly decreases from 82.61 wt% to 58.05 wt%. It is observed that the acidity of the activated carbon increases. The point of zero charge shifts from pH 5.74 to pH 1.69 and NH3-TPD reveals a more than fivefold increase of acidity compared to pure CW20. Measurements according to Boehm indicate that the density of acidic functional groups increases from 188 to 500 μmol g−1. There is an increased formation of carboxyl groups, while the relative proportion of phenolic and lactonic groups decreases. IR spectra indicate an increase in bands in the range between 1500–2000 cm−1, which can be attributed to the increase in carboxyl groups (Table 3 and Fig. 7). The SEM images (Fig. S24–26†) reveal that the pure CW20 is composed of nanosized grains and agglomerates whereas the structures of the oxidized CW20 are in the order of micrometers in size. Therefore, the overall roughness of the oxidized sample appears significantly larger. Interestingly, the images of the reduced CW20 again resemble the size distribution and roughness of the pure CW20.
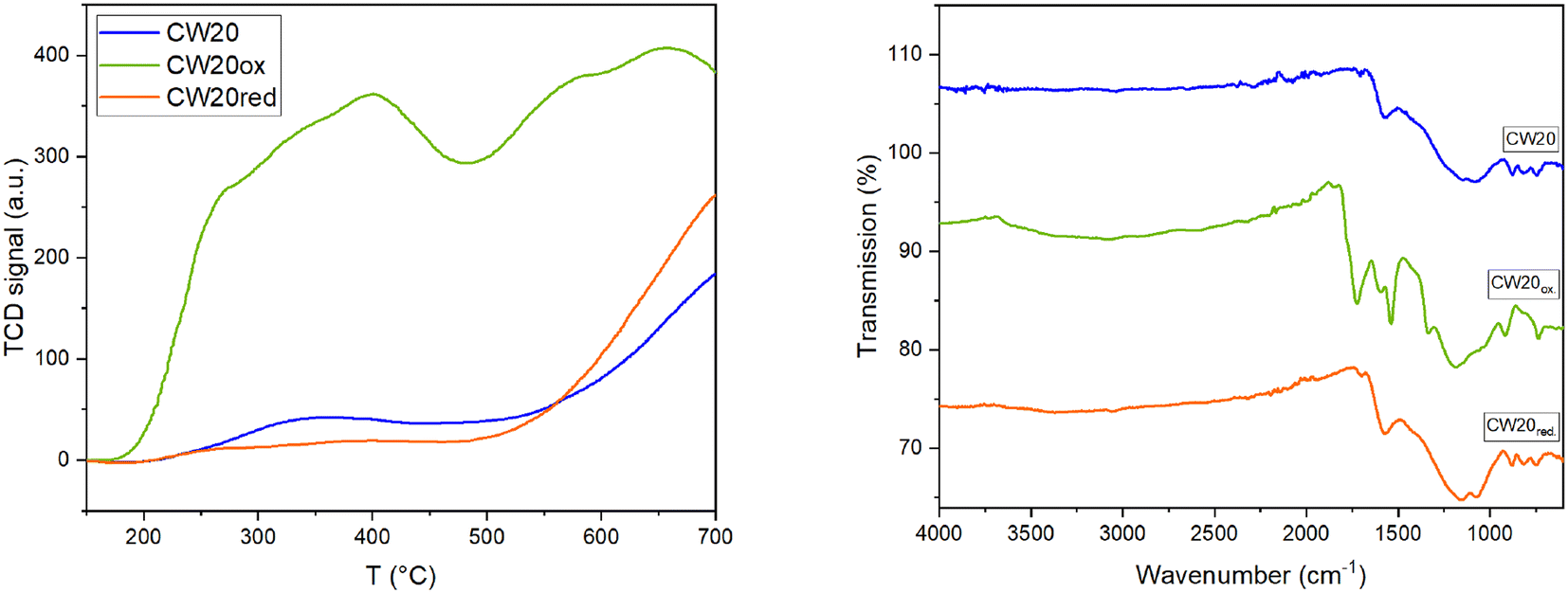 | ||
| Fig. 7 NH3-TPD acidity (left) and IR-spectra (right) of untreated CW20 and pretreated CW20ox and CW20red. | ||
| CW20 | CW20ox | CW20red | |
|---|---|---|---|
| Elemental analysis | |||
| C (wt%) | 82.61 | 58.05 | 82.58 |
| H (wt%) | 2.21 | 2.28 | 2.18 |
| N (wt%) | — | 0.3 | — |
| O (wt%) | 8.74 | 35.88 | 9.44 |
| Textural properties | |||
| Specific surface area (m2 g−1) | 1500 | 357 | 1527 |
| Pore volumina (mL g−1) | 1.32 | 0.26 | 1.38 |
| ∅ pore diameter (nm) | 16.48 | 2.61 | 20.42 |
| Boehm titration | |||
| Surface acidity (μmol g−1) | 188.0 | 500.0 | 182.5 |
| Surface basicity (μmol g−1) | 45.0 | 0.00 | 28.0 |
| Carboxylic groups (μmol g−1) | 39.0 | 473.5 | 29.5 |
| Lactonic groups (μmol g−1) | 113.0 | 58.5 | 380.5 |
| Phenolic groups (μmol g−1) | 36.0 | 0.00 | 0.00 |
| NH 3 -TPD acidity | |||
| Low (150–500 °C) | 1.00 | 9.63 | 0.47 |
| High (500–700 °C) | 1.00 | 4.03 | 1.28 |
| Total | 1.00 | 5.84 | 1.02 |
| Point of zero charge (pH) | 5.74 | 1.69 | 5.44 |
Due to the reductive pretreatment, there are neither significant changes in the textural properties, surface area nor the pore volumina remain the same. The oxygen content rises slightly to 9.44 wt%, possibly as a result of the removal of weakly bound impurities from the surface. Furthermore, the reductive pretreatment also results in the adsorption of nitrogen onto the surface of CW20red (0.3 wt%), which is present in the reductive gas, and could lead to nitrogen containing surface groups (Table 3). The acidity change is not significant within the margin of measurement error. The point of zero charge decreases from 5.74 to 5.44 and NH3-TPD shows a slightly change to higher acidity (Table 3 and Fig. 7) from 1 to 1.28. Reductive pretreatment shows no significant changes in the IR spectrum compared to CW20 (Fig. 7).
For all experimental series, SEM-EDX mapping shows a microscopic homogeneous distribution of Mo, V, and P (Fig. S30†). In the impregnation process of both untreated and pretreated activated carbon, differences are observed in the adsorbed quantity of HPA-5 (Table 4). For the untreated (E1) and reductively pretreated activated carbon (E5), vanadium amounts of 2.32 wt% (E1) and 2.54 wt% (E5) are detected, with a molar ratio of Mo:V of 7.8:4.2 (E1) and 7.4:4.6 (E5) respectively. The Keggin structure is preserved according to IR analysis (Fig. 8) for untreated (E1) as well as reductively pretreated activated carbon (E5). However, the oxidatively pretreated activated carbon (E2) results in only a 1.67 wt% V loading, with a Mo:V molar ratio of 1.5:10.5. This is attributed to the low adsorption of molybdenum, with just 0.46 wt%. The lack of successful impregnation is likely due to the significantly reduced surface area of CW20ox, which is only 357 m2 g−1 and the change of surface oxygen groups. Furthermore, IR spectra show the absence of a discernible Keggin structure.
 | ||
| Fig. 8 IR-spectra of CW20 after HPA-5 impregnation for different experimental series. | ||
| E1 | E2 | E3 | E4 | E5 | HPA-5the. | |
|---|---|---|---|---|---|---|
| Before reaction | ||||||
| Mo (wt%) | 8.06 | 0.46 | 15.88 | 12.94 | 7.61 | 17.4 |
| P (wt%) | 1.17 | 0.64 | 1.20 | 1.43 | 1.22 | 0.8 |
| V (wt%) | 2.32 | 1.67 | 7.58 | 7.03 | 2.54 | 6.6 |
| Molar ratio Mo:V |
7.8:4.2 |
1.5:10.5 |
6.3:5.7 |
6.0:6.0 |
7.4:4.6 |
7.0:5.0 |
| After formic acid formation | ||||||
| Mo (wt%) | 6.33 | <1 | 5.62 | 4.57 | 4.18 | |
| P (wt%) | 0.94 | 0.43 | 1.14 | 0.85 | 0.69 | |
| V (wt%) | 0.59 | <0.5 | 2.46 | 2.27 | 0.54 | |
| Molar ratio Mo:V |
10.1:1.9 |
— | 6.6:5.4 |
6.2:5.8 |
9.7:2.3 |
|
| Leaching of V (%) | 75 | 100 | 68 | 68 | 79 | |
| After lactic acid formation | ||||||
| Mo (wt%) | 7.07 | 0.43 | 6.41 | 5.97 | 6.77 | |
| P (wt%) | 0.86 | 0.45 | 0.76 | 0.90 | 0.21 | |
| V (wt%) | 1.92 | 1.62 | 5.98 | 5.85 | 2.00 | |
| Molar ratio Mo:V |
7.9:4.1 |
1.5:10.5 |
4.4:7.6 |
4.2:7.8 |
7.7:4.3 |
|
| Leaching of V (%) | 17 | 3 | 21 | 17 | 21 | |
The experimental series involving CW20ox was further expanded by incorporating a post-treatment step (E3), where, following the initial impregnation, the activated carbon and HPA-5 were heated to 200 °C for 3 hours before washing. The same procedure was applied to untreated CW20 for comparison (E4). The results indicate that post-treatment led to a significantly higher vanadium loading of 7.58 wt% V (E3) and 7.03 wt% V (E4), compared to standard impregnation, with a molar ratio of Mo:V of 6.3:5.7 (E3) and 6.0:6.0 (E4), respectively. This exceeded the anticipated vanadium amount of 6.6 wt%. These findings demonstrate the substantial impact of post-treatment on the impregnation efficiency. IR spectra indicate a preserved Keggin structure. The TGA analysis revealed that the majority of mass loss occurred during the heating phase up to 200 °C. Mass-spectroscopic analysis indicates that this loss predominantly corresponds to a molecular mass of 18 g mol−1 (Fig. S27–29†), suggesting it to be water loss. The enhancement in loading can be further attributed to the removal of hydroxyl groups from the activated carbon surface, facilitating the formation of ether or ester-like bonds between the surface oxygen of the activated carbon and the HPA. The release of an additional oxygen atom from water detachment not only contributes to the overall mass reduction but also potentially leads to higher POM loading and the observed elevated Mo:V ratio.
Simplified reaction pathways for the oxidative conversion and retro-aldol reaction are illustrated in Fig. 9. The conversion of glucose to formic acid and lactic acid, respectively, proceeds via dihydroxyacetone and glyceraldehyde, catalyzed by different vanadium species. Detailed descriptions of the mechanisms for both reactions are extensively documented in the literature.51–56
 | ||
| Fig. 9 Simplified reaction pathways for conversion of glucose to formic acid or lactic acid.51–56 | ||
Oxidative conversion of glucose analogous to the OxFA process
For the oxidative conversion of glucose, the supported HPA-5 catalyst was suspended in an aqueous solution of glucose, the mixture was heated to 90 °C under an atmosphere of 20 bar O2 in a steel pressure reactor for 6 hours (see ESI†), analogous to the commercial OxFA process. After the reaction, the composition of the gas phase was investigated with GC, while the liquid phase was analyzed with HPLC. The results are summarized in Fig. 10.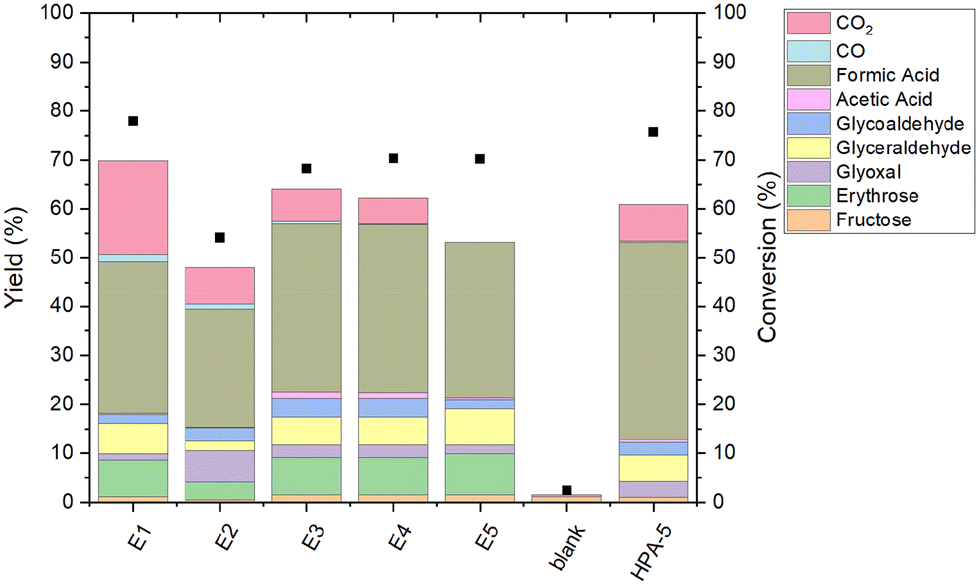 | ||
| Fig. 10 Catalytic performance of HPA-5/CW-20 for glucose oxidation to formic acid. Reaction conditions: glucose 3.603 g (20 mmol), catalyst 1.821 g (pure HPA-5: 1.14 mmol), 45 ml H2O, 90 °C, 20 bar O2, 1000 rpm, 6 h. | ||
The homogeneous HPA-5 as a benchmark shows a glucose conversion of 76% after 6 h reaction time, with a selectivity towards formic acid of 54% and a FA-yield of 41%. This is in very good alignment with previous literature, evidencing the successful execution of the experiments in this study.57
The conversion of glucose for all heterogeneous catalysts is between 68% to 78%, except for E2 with just 54%. This experiment also shows the lowest yield of formic acid with just 24% likely due to unsuccessful impregnation in this series as mentioned before (Table 4). The yield towards formic acid for the other supported catalysts is between 31% to 34% (E3), and therefore below the homogeneous HPA-5. The selectivity to formic acid for all catalysts ranges from 40% (E1) to 51% (E3). Interestingly, despite the reduced vanadium content in the supported catalysts compared to pure homogeneous HPA-5, the results demonstrate a catalytic activity comparable to that of homogeneous HPA-5.
Due to the promising results for E1, this experimental series was reproduced. Data showed sufficient reproducibility (Table S5†).
With respect to catalyst stability, leaching of vanadium exceeds 68% across all experimental series (Table 4), probably due to reaction conditions that disrupt the bond between HPA and the activated carbon support. The initial pH value above 2.5 decreases to below 1.4 by the end of the reaction (Table S4†), matching the pH of pure HPA-5 in solution, where HPA exists in a dissociated state. The Keggin structure of HPA-5 remains intact post-reaction in all catalysts, except, as expected, in E2, where it was not initially present (Fig. S36†).
In summary, the use of HPA-5 supported on CW-20 activated carbon demonstrates promising catalytic activity in the OxFA process for oxidatively converting glucose to formic acid, but is not suitable due to the high leaching of vanadium.
Inert conversion of glucose analogous to retro-aldol-condensation
For the retro-aldol condensation of glucose, the HPA-5/CW-20 catalyst was suspended in an aqueous solution of glucose, the mixture was heated to 160 °C under an atmosphere of 20 bar N2 in a steel (1.4571) pressure reactor for 1 hour (see ESI†). After reaction, the composition of the gas phase was investigated with GC, while the liquid phase was analyzed with HPLC. The results are summarized in Fig. 11. | ||
| Fig. 11 Catalytic performance of HPA-5/CW-20 for glucose conversion to lactic acid. Reaction conditions: glucose 1.032 g (20 mmol), catalyst 0.406 g (pure HPA-5: 1.14 mmol), 40 ml H2O, 160 °C, 20 bar N2, 1000 rpm, 1 h. | ||
As a benchmark, the homogeneous HPA-5 shows a glucose conversion of 71%, with a selectivity towards lactic acid of 15% and a LA-yield of 10% (Fig. 11). In literature, conversion of 28% and a yield of lactic acid of 13% was achieved under identical reaction conditions but for 3 hours reaction time and use of 1 mmol glucose and 0.1 mmol HPA-5, respectively.51
For all heterogeneous catalysts, the glucose conversion ranges between 65% to 71%, the selectivity towards lactic acid between 9% to 15%, the yield consistently exceeds 6% (Table S6†). Interestingly, the HPA-5/CW-20 catalyst synthesized without pre- and post-treatment (E1) closely matches the catalytic performance of the homogeneous HPA-5, showing a conversion of 71% and a lactic acid yield of 11%, whereby only low leaching occurs (see below). This comparison indicates that the reaction is heterogeneously catalyzed, rather than being driven solely by the leached fraction of HPA. Additionally, the lower amount of HPA on the catalyst's surface resulted in overall higher activity than the homogeneous HPA-5.
Infrared spectroscopy further verifies the preservation of the Keggin structure across all other catalysts (E3–E5). Across all experimental series, the leaching related to vanadium is below 22% (Table 4). Furthermore, in experimental series with post-treatments (3 and 4), more than half of the molybdenum is leached, indicating weaker bonding than vanadium. Compared to the oxidative conditions of the OxFA process, significantly less leaching occurs, also indicating disruptive behavior of oxidative conditions for the HPA–AC bonds (Table 4). The initial pH value of 2.9 decreased to no lower than 2.41 in all catalysts, suggesting less dissociation of the POM under these conditions than in the OxFA process with a final pH of around 1.39 (Table S4†). Collectively, both the elemental analysis and IR spectroscopy demonstrate that the supported catalysts are substantially more stable under the conditions of the retro-aldol condensation than under oxidative conditions. Interestingly, the increased temperature of 160 °C, in comparison to the OxFA process at 90 °C, does not significantly affect the stability of the catalyst compared to the pH value. The higher decrease in pH value in the OxFA process is predominantly attributable to the accumulation of acidic reaction products, notably formic and acetic acid, which contribute to a decrease in pH. Subsequently, when developing supported HPAs, it is important to consider the post-reaction pH value to ensure catalyst stability.
Conclusions
In this study, HPA-5 was successfully supported on various activated carbons using a simple wetness impregnation method. SEM analyses show a homogeneous dispersion of HPA on the various carbon supports. For activated carbons, IR spectra confirmed the preservation of the Keggin structure. Elemental analysis indicated that not HPA-5, but lower substituted POMs could be adsorbed onto activated carbon, probably due to a decrease in stability with an increasing degree of substitution by vanadium.The activated carbon CW20 was chosen for its ability to retain a high vanadium content and preserve the POM structure. It underwent various pre- and post-treatments aimed at optimizing the synthesis methodologies. Remarkably, CW20, when subjected to simple wetness impregnation without additional pre- or post-treatment, exhibited catalytic activity comparable to the homogeneous HPA-5 catalyst. This resulted in a selectivity for lactic acid of 15% and a glucose conversion of 71%.
Author contributions
Anne Wesner performed catalyst synthesis and characterization, data interpretation and conceptual planning of the experimental workflow, and wrote the manuscript draft. Leon Schidowski and Max Papajewski carried out the catalytic experiments. Maximilian J. Poller participated in planning and supervision of the experimental workflow, interpretation of data, and reviewing the manuscript. Charlotte Ruhmlieb provided valuable insights for data interpretation through scientific discussions, especially for SEM studies. Jakob Albert was responsible for acquisition of funding and resources, as well as project supervision and manuscript review.Data availability
The data that support the findings of this study are available on request from the corresponding author, JA.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Cabot for providing activated carbon samples. The central analytical services of the Chemistry Department at UHH are gratefully acknowledged for carrying out ICP-OES, SEM, Raman, and XRD measurements. Furthermore, we thank Moritz Hilgers for carrying out point of zero charge measurements during his research internship. This research was partially funded by OxFA GmbH.References
- P. Sudarsanam, R. Zhong, S. van den Bosch, S. M. Coman, V. I. Parvulescu and B. F. Sels, Functionalised heterogeneous catalysts for sustainable biomass valorisation, Chem. Soc. Rev., 2018, 47, 8349–8402 RSC.
- V. K. Vaithyanathan, B. Goyette and R. Rajagopal, A critical review of the transformation of biomass into commodity chemicals: Prominence of pretreatments, Environ. Challenges, 2023, 11, 100700 CrossRef CAS.
- J. Zhong, J. Pérez-Ramírez and N. Yan, Biomass valorisation over polyoxometalate-based catalysts, Green Chem., 2021, 23, 18–36 RSC.
- N. I. Gumerova and A. Rompel, Synthesis, structures and applications of electron-rich polyoxometalates, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- I. V. Kozhevnikov, Catalysis by Heteropoly Acids and Multicomponent Polyoxometalates in Liquid-Phase Reactions, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- T. Okuhara, N. Mizuno and M. Misono, Catalytic Chemistry of Heteropoly Compounds, in Advances in Catalysis, Elsevier, 1996, pp. 113–252 Search PubMed.
- M. T. Pope, Heteropoly and isopoly oxometalates, in Inorganic Chemistry Concepts Vol. 8, Springer, Berlin, Heidelberg, New York, Tokyo, 1983 Search PubMed.
- J.-C. Raabe, M. J. Poller, D. Voß and J. Albert, H8 PV5 Mo7 O40 - A Unique Polyoxometalate for Acid and RedOx Catalysis: Synthesis, Characterization, and Modern Applications in Green Chemical Processes, ChemSusChem, 2023, 16, e202300072 CrossRef CAS PubMed.
- Y. Ogasawara, S. Itagaki, K. Yamaguchi and N. Mizuno, Saccharification of natural lignocellulose biomass and polysaccharides by highly negatively charged heteropolyacids in concentrated aqueous solution, ChemSusChem, 2011, 4, 519–525 CrossRef CAS PubMed.
- J. Tian, J. Wang, S. Zhao, C. Jiang, X. Zhang and X. Wang, Hydrolysis of cellulose by the heteropoly acid H3PW12O40, Cellulose, 2010, 17, 587–594 CrossRef CAS.
- K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya and A. Satsuma, Effects of Brønsted and Lewis acidities on activity and selectivity of heteropolyacid-based catalysts for hydrolysis of cellobiose and cellulose, Green Chem., 2009, 11, 1627 RSC.
- L. M. Sanchez, H. J. Thomas, M. J. Climent, G. P. Romanelli and S. Iborra, Heteropolycompounds as catalysts for biomass product transformations, Catal. Rev., 2016, 58, 497–586 CrossRef CAS.
- W. Deng, Q. Zhang and Y. Wang, Polyoxometalates as efficient catalysts for transformations of cellulose into platform chemicals, Dalton Trans., 2012, 41, 9817–9831 RSC.
- J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Selective oxidation of complex, water-insoluble biomass to formic acid using additives as reaction accelerators, Energy Environ. Sci., 2012, 5(7), 7956–7962 RSC.
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Spectroscopic and electrochemical characterization of heteropoly acids for their optimized application in selective biomass oxidation to formic acid, Green Chem., 2014, 16(1), 226–237 RSC.
- J. Albert and P. Wasserscheid, Expanding the scope of biogenic substrates for the selective production of formic acid from water-insoluble and wet waste biomass, Green Chem., 2015, 17(12), 5164–5171 RSC.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, Formic Acid, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2003, pp. 1–22 Search PubMed.
- D. Voß, R. Dietrich, M. Stuckart and J. Albert, Switchable Catalytic Polyoxometalate-Based Systems for Biomass Conversion to Carboxylic Acids, ACS Omega, 2020, 5, 19082–19091 CrossRef PubMed.
- M. J. Biddy, C. Scarlata and C. Kinchin, Chemicals from Biomass: A Market Assessment of Bioproducts with Near-Term Potential, NREL/TP-5100-65509, Denver, 2016 Search PubMed.
- R. Datta and M. Henry, Lactic acid: recent advances in products, processes and technologies—a review, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- E. Rafiee and S. Eavani, Heterogenization of heteropoly compounds: a review of their structure and synthesis, RSC Adv., 2016, 6, 46433–46466 RSC.
- L. Hombach, N. Simitsis, J. T. Vossen, A. J. Vorholt and A. K. Beine, Solidified and Immobilized Heteropolyacids for the Valorization of Lignocellulose, ChemCatChem, 2022, 14, e202101838 CrossRef CAS.
- Y. Wu, X. Ye, X. Yang, X. Wang, W. Chu and Y. Hu, Heterogenization of Heteropolyacids: A General Discussion on the Preparation of Supported Acid Catalysts, Ind. Eng. Chem. Res., 1996, 35, 2546–2560 CrossRef CAS.
- Y. Zhou, G. Chen, Z. Long and J. Wang, Recent advances in polyoxometalate-based heterogeneous catalytic materials for liquid-phase organic transformations, RSC Adv., 2014, 4, 42092–42113 RSC.
- M. A. Schwegler, P. Vinke, M. van der Eijk and H. van Bekkum, Activated carbon as a support for heteropolyanion catalysts, Appl. Catal., A, 1992, 80, 41–57 CrossRef CAS.
- Y. Izumi and K. Urabe, Catalysis of heteropoly acids entrapped in activated carbon, Chem. Lett., 1981, 10, 663–666 CrossRef.
- M. N. Timofeeva, M. M. Matrosova, T. V. Reshetenko, L. B. Avdeeva, E. A. Paukshtis, A. A. Budneva, A. L. Chuvilin and V. A. Likholobov, Adsorption of H3PW12O40 by porous carbon materials, Russ. Chem. Bull., 2002, 51, 243–248 CrossRef CAS.
- Y. Izumi, Catalysis by heterogeneous supported heteropoly acid, J. Catal., 1983, 84, 402–409 CrossRef CAS.
- Y. Yang, K. Chiang and N. Burke, Porous carbon-supported catalysts for energy and environmental applications: A short review, Catal. Today, 2011, 178, 197–205 CrossRef CAS.
- M. Almohalla, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Comparative study of three heteropolyacids supported on carbon materials as catalysts for ethylene production from bioethanol, Catal. Sci. Technol., 2017, 7, 1892–1901 RSC.
- S. Tsubaki, K. Oono, A. Onda, T. Ueda, T. Mitani and M. Hiraoka, Microwave-assisted hydrolysis of biomass over activated carbon supported polyoxometalates, RSC Adv., 2017, 7, 12346–12350 RSC.
- V. B. Kumar, I. N. Pulidindi and A. Gedanken, Selective conversion of starch to glucose using carbon based solid acid catalyst, Renewable Energy, 2015, 78, 141–145 CrossRef CAS.
- S. d. A. Carminati, A. C. C. Arantes, A. C. S. d. Oliveira and M. L. Bianchi, Enhancing the sugars production yieldby supporting H3PW12O40 heteropoly acid on activated carbon for use as catalyst in hydrolysis of cellulose, Matéria, 2018, 23, e12260 Search PubMed.
- L. Hombach, N. Hausen, A. G. Manjón, C. Scheu, H. Kraffczyk, M. Rose, J. Albert and A. K. Beine, Carbon supported polyoxometalates as recyclable solid acid catalysts in aqueous reactions, Appl. Catal., A, 2023, 666, 119392 CrossRef CAS.
- R. Ghubayra, R. Yahya, E. F. Kozhevnikova and I. V. Kozhevnikov, Oxidative desulfurization of model diesel fuel catalyzed by carbon-supported heteropoly acids: Effect of carbon support, Fuel, 2021, 301, 121083 CrossRef CAS.
- J.-C. Raabe, J. Aceituno Cruz, J. Albert and M. J. Poller, Comparative Spectroscopic and Electrochemical Study of V(V)-Substituted Keggin-Type Phosphomolybdates and -Tungstates, Inorganics, 2023, 11, 138 CrossRef CAS.
- X.-Y. Liu, M. Huang, H.-L. Ma, Z.-Q. Zhang, J.-M. Gao, Y.-L. Zhu, X.-J. Han and X.-Y. Guo, Preparation of a carbon-based solid acid catalyst by sulfonating activated carbon in a chemical reduction process, Molecules, 2010, 15, 7188–7196 CrossRef CAS PubMed.
- B. S. Girgis, Y. M. Temerk, M. M. Gadelrab and I. D. Abdullah, X-ray Diffraction Patterns of Activated Carbons Prepared under Various Conditions, Carbon Lett., 2007, 8, 95–100 CrossRef.
- M. S. Shafeeyan, W. M. A. W. Daud, A. Houshmand and A. Shamiri, A review on surface modification of activated carbon for carbon dioxide adsorption, J. Anal. Appl. Pyrolysis, 2010, 89, 143–151 CrossRef CAS.
- U. Zielke, K. J. Hüttinger and W. P. Hoffman, Surface-oxidized carbon fibers: I. Surface structure and chemistry, Carbon, 1996, 34, 983–998 CrossRef CAS.
- H.-P. Boehm, Functional Groups on the Surfaces of Solids, Angew. Chem., Int. Ed. Engl., 1966, 5, 533–544 CrossRef CAS.
- H. Boehm, Surface oxides on carbon and their analysis: a critical assessment, Carbon, 2002, 40, 145–149 CrossRef CAS.
- P. Kurzweil and O. K. Dietlmeier, Elektrochemische Speicher: Superkondensatoren, Batterien, Elektrolyse-Wasserstoff, Rechtliche Grundlagen, 1. Aufl. 2015, Springer Vieweg, Wiesbaden, 2015 Search PubMed.
- N. Shimodaira and A. Masui, Raman spectroscopic investigations of activated carbon materials, J. Appl. Phys., 2002, 92, 902–909 CrossRef CAS.
- P. E. Fanning and M. Vannice, A DRIFTS study of the formation of surface groups on carbon by oxidation, Carbon, 1993, 31, 721–730 CrossRef CAS.
- G. Marcì, E. García-López, M. Bellardita, F. Parisi, C. Colbeau-Justin, S. Sorgues, L. F. Liotta and L. Palmisano, Keggin heteropolyacid H3PW12O40 supported on different oxides for catalytic and catalytic photo-assisted propene hydration, Phys. Chem. Chem. Phys., 2013, 15, 13329–13342 RSC.
- W. Qi, W. Liu, S. Liu, B. Zhang, X. Gu, X. Guo and D. Su, Heteropoly Acid/Carbon Nanotube Hybrid Materials as Efficient Solid–Acid Catalysts, ChemCatChem, 2014, 6, 2613–2620 CrossRef CAS.
- J. Breibeck, N. I. Gumerova and A. Rompel, Oxo-Replaced Polyoxometalates: There Is More than Oxygen, ACS Org. Inorg. Au, 2022, 2, 477–495 CrossRef CAS PubMed.
- J. Alcañiz-Monge, G. Trautwein, S. Parres-Esclapez and J. A. Maciá-Agulló, Influence of microporosity of activated carbons as a support of polyoxometalates, Microporous Mesoporous Mater., 2008, 115, 440–446 CrossRef.
- J. Albert, M. Mendt, M. Mozer and D. Voß, Explaining the role of vanadium in homogeneous glucose transformation reactions using NMR and EPR spectroscopy, Appl. Catal., A, 2019, 570, 262–270 CrossRef CAS.
- T. Lu, M. Niu, Y. Hou, W. Wu, S. Ren and F. Yang, Catalytic oxidation of cellulose to formic acid in H 5 PV 2 Mo 10 O 40 + H 2 SO 4 aqueous solution with molecular oxygen, Green Chem., 2016, 18, 4725–4732 RSC.
- Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang and Y. Wang, Transformation of cellulose and its derived carbohydrates into formic and lactic acids catalyzed by vanadyl cations, ChemSusChem, 2014, 7, 1557–1567 CrossRef CAS PubMed.
- H. Wang, M. Wang, J. Shang, Y. Ren, B. Yue and H. He, H3PMo12O40 Immobilized on Amine Functionalized SBA-15 as a Catalyst for Aldose Epimerization, Materials, 2020, 13, 507 CrossRef CAS PubMed.
- Z. Li, P. Wu, J. Pang, X. Li, S. Zhai and M. Zheng, Catalytic Conversion of Sugars into Lactic Acid via a RuOx/MoS2 Catalyst, Catalysts, 2023, 13, 545 CrossRef CAS.
- M. Xia, W. Dong, Z. Shen, S. Xiao, W. Chen, M. Gu and Y. Zhang, Efficient production of lactic acid from biomass-derived carbohydrates under synergistic effects of indium and tin in In–Sn-Beta zeolites, Sustainable Energy Fuels, 2020, 4, 5327–5338 RSC.
- J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Biomass oxidation to formic acid in aqueous media using polyoxometalate catalysts – boosting FA selectivity by in situ extraction, Energy Environ. Sci., 2015, 8, 2985–2990 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01336k |
| This journal is © The Royal Society of Chemistry 2024 |