
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic behavior of amino compound oxidation on NiCo catalyst and energy conversion†
Wei
Xu
 *a,
Zhaozhao
Yan
b,
Chunhong
Liu
a,
Xu
Yang
c,
Hua
Yu
b,
Hongchao
Chang
b,
Jiarong
Zang
b,
Guangyao
Xu
b,
Linmin
Du
d and
Binbin
Yu
*b
*a,
Zhaozhao
Yan
b,
Chunhong
Liu
a,
Xu
Yang
c,
Hua
Yu
b,
Hongchao
Chang
b,
Jiarong
Zang
b,
Guangyao
Xu
b,
Linmin
Du
d and
Binbin
Yu
*b
aZhejiang Baima Lake Laboratory Co., Ltd, Hangzhou 310053, P. R. China. E-mail: 21114078@zju.edu.cn
bSchool of Pharmaceutical and Chemical Engineering, Taizhou University, Taizhou 318000, P. R. China. E-mail: yubinbin2004@126.com
cDepartment of Environmental Engineering, State Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310058, P. R. China
dTaizhou Research and Design Institute of Environmental Science, Taizhou 318000, P. R. China
First published on 18th September 2023
Abstract
Carbon-supported Ni and NiCo catalysts have been prepared for amino compound electrooxidation. Their electrocatalytic behaviors toward 8 kinds of amino acids (glycine, alanine, histidine, methionine, proline, serine, tyrosine, and cysteine) were investigated via electrochemical methods and in situ Raman spectroscopy. The Ni and NiCo catalysts demonstrated catalytic activity for amino acid electrooxidation, and the current densities differ according to the R side chain group. Compared to Ni/C, NiCo/C is superior in terms of onset potential and anodic current density. Electrochemical in situ Raman characterization indicated that NixCo1–xOOH was generated above the onset potential. The amino compounds were electrooxidized after the onset potential by the NixCo1–xOOH active species. However, the anodic current of cysteine was observed at a more negative potential than the onset potential, caused by the electrooxidation of sulfur in the R side chain. A direct alkaline fuel cell was fabricated with glycine as the fuel. The fuel cell with NiCo/C anode catalyst achieved a maximum power density of 3.0 mW cm−2 at 60 °C, 1.5 times higher than that of the fuel cell with the Ni/C anode catalyst.
Introduction
Biomass is a promising energy resource because of its abundance and carbon-neutral properties.1–4 Proteins and amino acids with the amino groups are capable of hydrogen storage, and they are abundant in biomass, accounting for about 10%.5,6 However, the utilization of biomass for biofuel production is only focused on nitrogen-free compounds. About 9.1 million tons of protein by-products were generated from corn ethanol production in 2010.5 These protein by-products are treated as biomass waste due to the lack of energy conversion strategies.7 Apart from biomass, a recent study has demonstrated that amino acids can be directly produced via the electroreduction of CO2 with NH3.8 Amino acids can be promising energy storage materials if there is an efficient way to convert amino acids to electricity.Chemical fuel cell systems have the potential feasibility to directly produce electricity from proteins and derived amino acids. There are 20 amino acids with different R side-chain groups, including glycine (aliphatic), proline (cyclic), serine (alcohol), methionine (sulfur), tyrosine and phenylalanine (aromatic), etc. If an amino acid fuel cell (AAFC) system can function successfully with all 20 amino acids as fuels, the energy utilization will be significantly simplified without the need for separation. To achieve this purpose, one of the key points is the development of anode catalysts for amino acid fuel cells.
Noble metals (e.g. Pt, Au, Ag) are commonly used as anode catalysts, however, they often suffer from poisoning due to the adsorption of poisonous products on the surfaces.9,10 Transition metal Ni-based electrocatalysts have shown the high catalytic activity of electro-oxidation reactions toward organics containing NH2 groups, such as urea, amino acids and peptides.10–13 They have wide applications in electrolysis and electrooxidation as anode catalysts. The amino acids are oxidized by Ni-based electrocatalysts according to the following reaction mechanism:10,14
| Ni(II) ⇌ Ni(III) + e− | (1) |
| Ni(III) + amino acids ⇌ Ni(II) + products | (2) |
Accordingly, the consequence of the electrooxidation of amino acids (EAA) on nickel catalyst is largely dependent on the formation of Ni(III) species. A more negative or less positive onset potential of Ni(II) to Ni(III) indicates a lower overpotential of EAA. However, research has shown that the electro-oxidation of alanine and lysine on Ni(OH)2 series catalysts occurred at potentials above ∼0.54 V vs. SHE, where the Ni(III) species NiOOH was formed.10 For histidine, the starting potential even increased to about 0.7 V vs. SHE.14 These starting potentials of EAA are in accordance with the redox potentials of Ni(III)/Ni(II) in alkaline media. Hence, it is of great significance to reduce the redox potential of Ni(III)/Ni(II) to enhance the catalytic activity for the EAA.
One strategy to improve catalyst activity is the application of element doping.15–17 Botte et al. developed nickel-cobalt bimetallic hydroxide catalysts for urea electrooxidation. The onset potential of urea oxidation decreased by 150 mV as compared to the nickel monometallic hydroxide catalyst.18 Zhang et al. prepared NiCo bimetallic electrocatalysts for the methanol oxidation reaction via a hydrogen evolution-assisted electrodeposition process.19 The results showed that the NiCo bimetallic catalysts had better electroactivity and resistance to CO poisoning because the introduction of Co into Ni increased the surface coverage of the redox species and weakened the adsorption of CO. Bimetallic Ni–Fe impurities within carbon nanotubes for arginine electrooxidation were studied by Pumera et al., demonstrating a larger oxidation current at pH 7.4.20 Cobalt catalysts were also proved to have activity toward EAA reactions.21 Reports showed that the incorporation of cobalt into nickel could result in more exposed active sites, less polarization and better electrical conductivity.18,22–24 However, nickel-cobalt bimetallic catalysts have seldom been employed for EAA reactions. Thus, it is significant to determine whether nickel-cobalt catalysts have catalytic activity for all these amino acids. Based on the above considerations, herein, we report the catalytic activity of a nickel-cobalt bimetallic catalyst toward the electrooxidation of different amino acids in alkaline solutions and test the fuel cell performance with this catalyst.
Experimental
Chemicals
All amino acids were L-amino acids (except for glycine) with 99% purity and were purchased from Aladdin Reagent Company (Shanghai, China). 3-Mercaptopropionic acid was also obtained from Aladdin Reagent Company with 99% purity. Other chemicals, including NiCl2·6H2O, CoCl2·6H2O, NaBH4, NaOH, C3H8O (isopropanol) and Na3C6H5O7 (trisodium citrate dehydrate) were reagent grade and were purchased from Keshi Chemical Co. Ltd (Chengdu, China). Carbon black (Cabot Vulcan XC-72R) was used as a support material.Preparation of NiCo/C catalysts
The carbon-supported nano NiCo catalyst (NiCo/C) was prepared via the NaBH4 reduction method. In a typical procedure, 120 mg of NiCl2·6H2O, 30 mg of CoCl2·6H2O and 100 mg of trisodium citrate dehydrate were added into 350 mL of deionized water to obtain the precursor solution at room temperature. The solution was pretreated by aeration with nitrogen gas for 15 minutes. After 10 min of vigorous agitation, 5 mL of fresh NaBH4 solution (4 wt%) was introduced dropwise into the mixture for the synthesis of nano NiCo particles, slowly turning the color of the solution to black. After three hours, 37 mg of carbon black was added to the bottle. After another three hours of stirring, the mixture was filtered and rinsed with de-ionized water 3–5 times. The precipitate was dried overnight in a vacuum oven at 50 °C. The carbon-supported Ni catalyst (Ni/C) and Co catalyst (Co/C) were prepared similarly, with just the absence of CoCl2·6H2O or NiCl2·6H2O in the precursor solution.Characterization
The crystal form of the prepared catalyst was detected by X-ray diffraction (XRD) using an X-ray diffractometer (XRD, D8 advanced, Bruker) with a Cu Kα source (λ = 1.54056 Å). The scanning electron microscopy (SEM) images were obtained with an S-4800 Carl Zeiss Microscope, Hitachi (cold field emission electron gun, equipped with STEM detectors). In situ Raman spectroscopy experiments were carried out with a Raman spectrometer (LabRAM HR evolution) under desired potentials controlled by the electrochemical workstation. A 50× magnification long working distance objective was used. The excitation wavelength was 532 nm from a He–Ne laser. The Raman spectrometer was calibrated using a Si wafer. A custom-made Teflon electrolytic cell was employed with a thin quartz glass plate as a cover to protect the objective. Catalyst ink as mentioned above was spread on Au foil as the working electrode. Electrochemically roughened gold foil was employed as the substrate for superior Raman scattering. The procedure to electrochemically roughen Au foil has been reported previously.25 The time of acquisition was 60 seconds, and the time of applied potential before acquisition was 100 seconds.Electrochemical measurements
The catalytic activities of the Ni/C and NiCo/C electrocatalysts for amino acid oxidation were tested by cyclic voltammetry and chronoamperometry in a conventional three-electrode H-cell using an electrochemical workstation (CHI 660D Shanghai Chenhua). Electrochemical characterization of the obtained catalysts was conducted on a glassy carbon electrode (GCE). The preparation of the working electrode was as follows. An ink mixture of 5 mg of Ni/C or NiCo/C catalyst with 0.1 ml of 5 wt% Nafion (DE520, DuPont, USA) and 0.4 ml of isopropanol was prepared by ultrasonic action for 30 min. Then, 6 μl of the ink was spread on the glassy carbon electrode (3 mm in diameter), which was polished to a mirror-like surface with 50 μm alumina powder and then washed with ethanol and deionized water before use. The GCE was dried for 20 min at 40 °C in a vacuum and the Ni or NiCo loading was about 0.4 mg cm−2. High-purity nitrogen (99.999%) was bubbled for 15 min through the solution in the cell to deoxygenize before all electrochemical measurements. A platinum foil (0.3 cm2) was used as the counter electrode. An Ag/AgCl (sat. KCl) electrode was used as the reference electrode.Fuel cell tests
Anode catalysts (Ni/C or NiCo/C) were mixed with deionized water, Nafion solution (5 wt%) and isopropanol to obtain the catalyst ink, which was coated on carbon paper using a paintbrush with metal loading of ∼10 mg cm−2. The carbon paper was dried in air for at least 24 h prior to its use as the anode. The geometric area of the carbon paper was 4.0 (2.0 × 2.0) cm−2. Cathode catalyst ink was prepared similarly using commercial Pt/C and was cast on carbon paper with a Pt loading of ∼0.8 mg cm−2 to make the cathode. An anion exchange membrane (AHA type, ASTOM, Japan) was used as a separator.Alkaline solution (1 M glycine with 1 M NaOH) was fed into the flow channel as the anode fuel at a flow rate of 2.0 mL min−1 by peristaltic pumps, and they were kept in a constant temperature water bath. At the cathode, wet oxygen (relative humidity of 100% at the operating temperature) was used as an electron acceptor. The power density was tested by a linear sweep voltammetry method at a scan rate of 1 mV s−1via an electrochemical workstation.
Results and discussion
Characterization of NiCo and Ni catalysts
Our previous work showed that Ni-based bimetallic component catalysts usually exhibit good activity in electrocatalytic oxidation. Here, we prepared a NiCo bimetal catalyst as an efficient electrocatalyst for the amino acid oxidation reaction. As shown in Fig. 1(a) and (b), the as-prepared NiCo catalyst exhibits a spherical structure with an approximate diameter of 30 nm. The SEM images in Fig. 1(c) and (d) demonstrate that the morphology of the Ni catalyst is similar to that of the NiCo catalyst. | ||
| Fig. 1 (a) and (b) SEM images of the NiCo catalyst. (c) and (d) SEM images of the Ni catalyst. | ||
XRD patterns of the Ni catalyst and the NiCo catalyst are demonstrated in Fig. 2. The red line is the XRD pattern of the Ni catalyst. The strong diffraction at 2θ = 44.5° was identified as the Ni (111) peak according to PDF#45-1027, indicating the formation of metallic nickel.26 When cobalt was added, the peak at 2θ = 44.5° became much smaller and almost disappeared from the NiCo catalyst pattern. This indicates that the incorporation of Co leads to low crystallization properties.
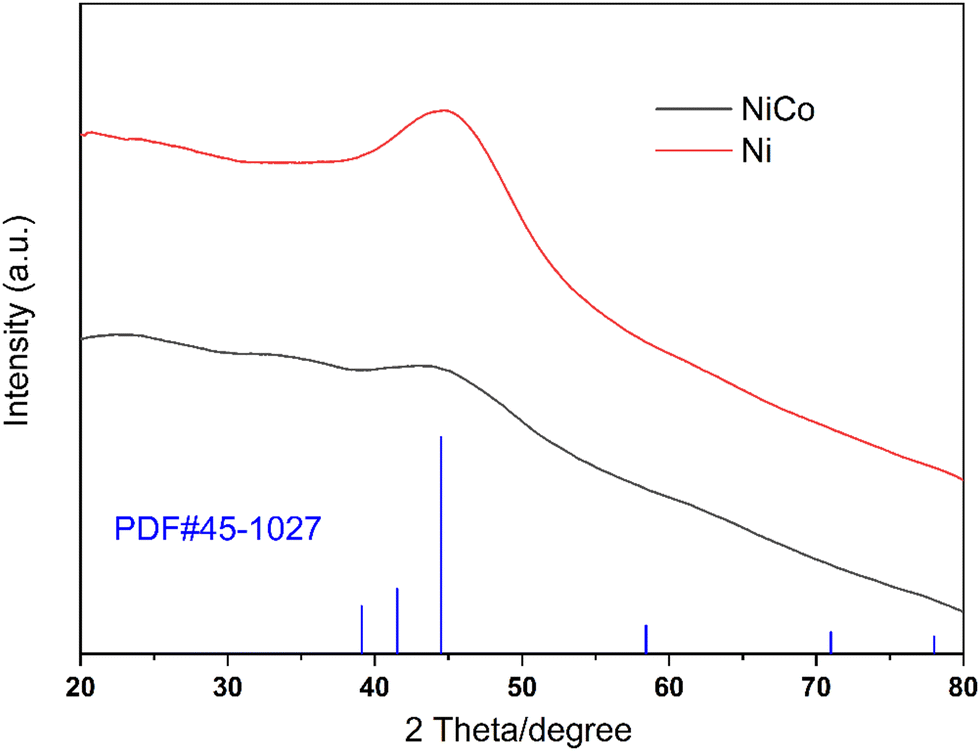 | ||
| Fig. 2 XRD patterns of the NiCo catalyst and the Ni catalyst. | ||
Electrochemical characterization
Cyclic voltammograms (CVs) of NiCo/C and Ni/C catalysts in alkaline electrolytes are recorded in Fig. 3a. The curve of the Ni/C catalyst shows an anodic peak in the positive sweep at a peak potential of about 0.42 V. In the negative sweep, a cathodic peak at a potential of about 0.22 V was observed with a similar shape. The anodic peak current density (Jpa) and cathodic peak current density (Jpc) were 26.2 and 21.3 mA cm−2, respectively. As a result, the ratio of Jpa/Jpc for Ni/C catalyst was 1.2, very close to 1. This demonstrates the reversibility of the redox peaks, which was attributed to the oxidation and reduction reactions for the Ni(III)/Ni(II) redox couple.| Ni(OH)2 + OH− ⇌ NiOOH + H2O+ e− |
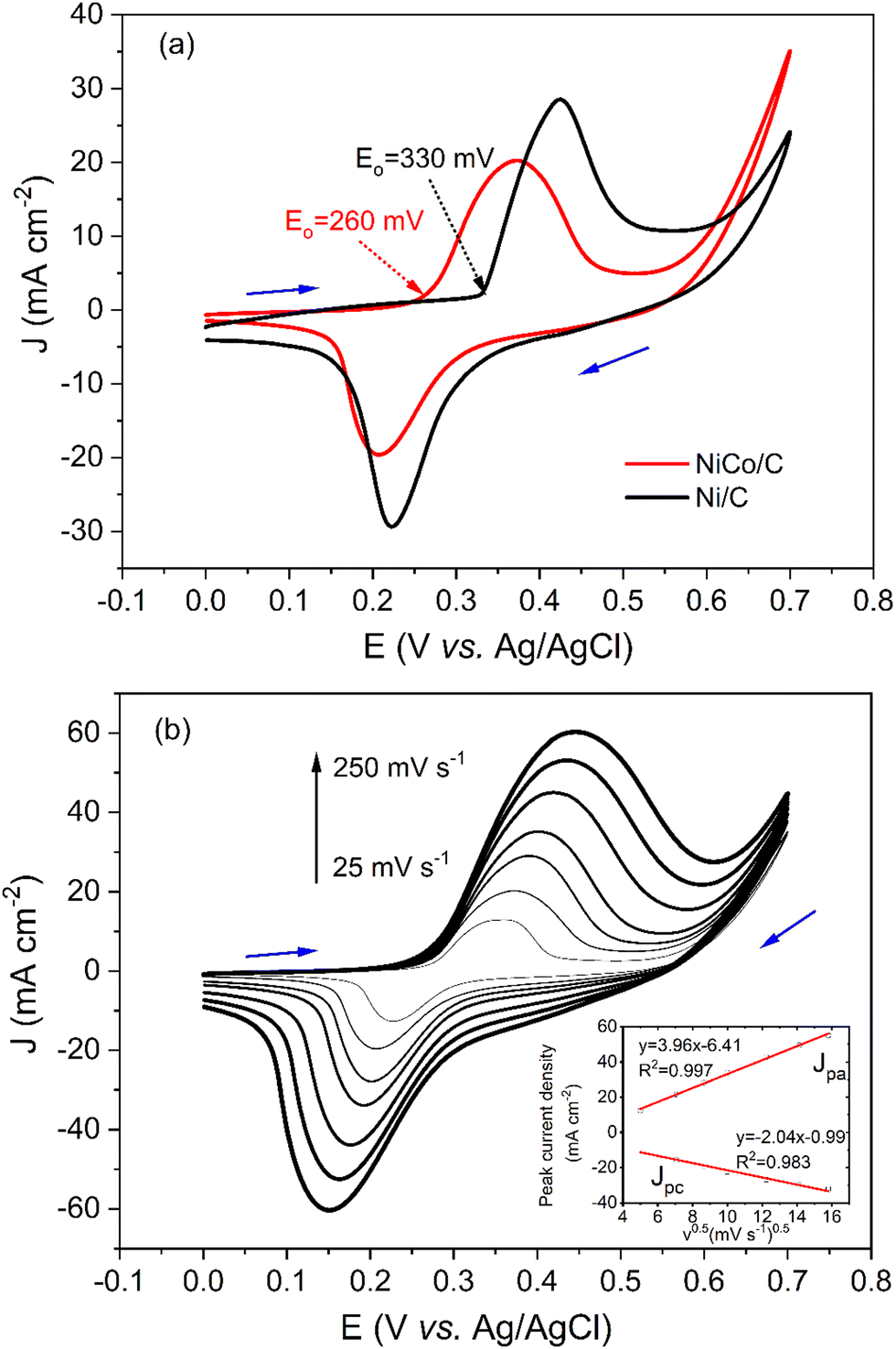 | ||
| Fig. 3 (a) CVs of glassy carbon electrode (GCE) modified with Ni/C (black) or NiCo/C (red) catalysts in 1 M NaOH electrolyte at the scan rate of 50 mV s−1. (b) CVs of GCE modified with NiCo/C in 1 M NaOH electrolyte at different scan rates. Scan rates (from inner to outer) are as follows: 25, 50, 75, 100, 150, 200, 250 mV s−1. Inset plots are peak currents on the square roots of scan rates. | ||
The curve of the NiCo/C catalyst also shows a pair of reversible redox peaks with Jpa/Jpc of 1.4. Note that the redox peaks of NiCo/C appear at more negative potentials. The anodic peak and cathodic peak of NiCo/C were achieved at potentials of about 0.37 V and 0.21 V, respectively. The onset potential of electrooxidation is defined as the potential at which 10% of the current density at the peak potential is reached.27 The onset potential of the Ni/C catalyst was about 330 mV (black line). In contrast, the onset potential of the NiCo/C catalyst was 260 mV, 70 mV more negative than the Ni/C catalyst. This is because the formation of the high-valence nickel-cobalt species becomes more facile when Co is incorporated, which is in favor of decreasing the overpotential of EAA reactions.
Fig. 3b demonstrates that the peak current densities of CV curves on NiCo/C modified GCE are proportional to the square root of sweep rates (v1/2) from 25 to 250 mV s−1. This means that the oxidation of NiCo and reduction of NiCo oxides in alkaline solutions are diffusion-controlled processes. Besides, the peak potential (Ep) is related to peak current and uncompensated resistance28 and also demonstrates larger shifts as the scan rates become higher.
To investigate the catalytic activity of the NiCo/C catalyst toward amino acids electrooxidation, electrochemical tests were performed in an alkaline solution in the presence of amino acids. When glycine was added to 1 M NaOH, a significant increase in the current density between 0.4 V to 0.6 V was observed in the CV plots of NiCo/C (Fig. S1, ESI†). Fig. 4a demonstrates the CV curves of glycine electrooxidation on NiCo/C and Ni/C catalysts. The value of Jpa/Jpc for Ni/C was about 2.2, which is larger than the Jpa/Jpc of 1.2 in the absence of glycine (result from Fig. 3a). Similarly, the Jpa/Jpc for NiCo/C in the presence and absence of glycine was 3.0 and 1.4, respectively. The change in peak current ratio indicates that glycine has gone through irreversible electrooxidation on Ni/C and NiCo/C catalysts. The NiCo/C curve exhibited an onset potential of 300 mV. This is 50 mV smaller than the onset potential of the Ni/C curve. This indicates that the NiCo/C bimetal catalyst could be transformed to the oxidized state more easily, thus leading to a smaller polarization. The result is in accordance with the CV curves recorded in the absence of glycine in Fig. 3a. The reduction of the onset potential shows that the electrooxidation of glycine can operate significantly more efficiently. Besides, when Co is incorporated into Ni, this bimetallic catalyst can achieve a larger anodic current density than monometallic Ni, indicating a superior catalytic activity in the aspect of glycine electrooxidation. We conducted the CV test of the Co/C catalyst in 1 M NaOH electrolyte in the absence and presence of 0.2 M glycine, and the results are shown in Fig. S2 (ESI†). The results indicate that the Co/C catalyst is active for the EAA. The Co/C catalyst has a lower onset potential as compared to Ni/C but the current density of Co/C is inferior to that of Ni/C. When compared with noble metal catalysts such as Pd/C and Pt/C, the NiCo/C reached an even higher current density (Fig. S3, ESI†).
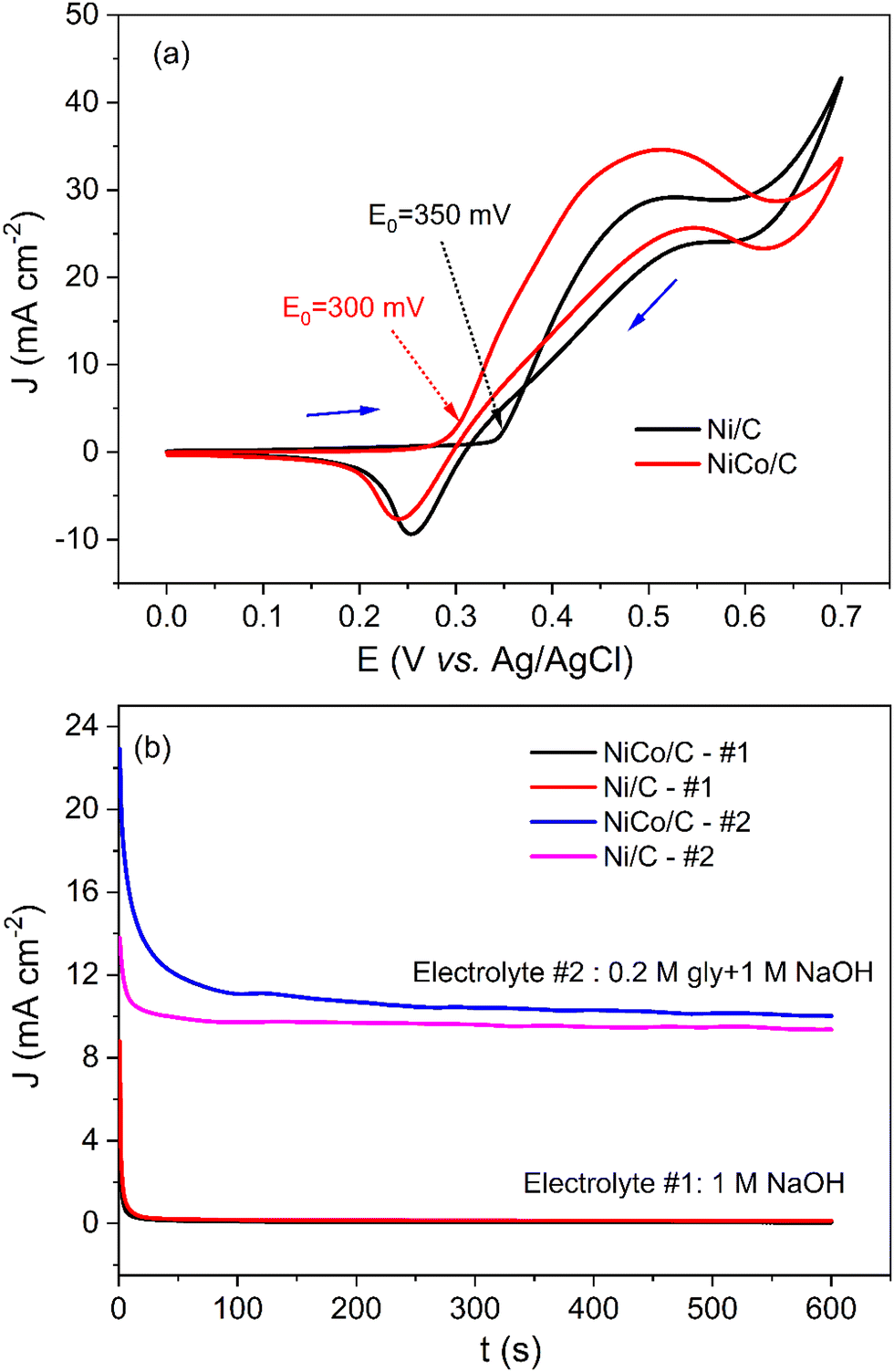 | ||
Fig. 4 (a) CVs of Ni/C (black) and NiCo/C (red) catalysts in 1 M NaOH electrolyte with 0.2 M glycine at a scan rate of 50 mV s−1. (b) Chronoamperograms of NiCo/C and Ni/C catalysts at 0.38 V vs. Ag/AgCl. Electrolyte #1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 M NaOH solution. Electrolyte #2:1 M NaOH with 0.2 M glycine. :1 M NaOH solution. Electrolyte #2:1 M NaOH with 0.2 M glycine. | ||
Fig. 4b shows that a larger current density can also be observed when using the NiCo/C catalyst in the chronoamperogram curves. The current density of glycine electrooxidation is proportional to 1 divided by the square root of time with R2 = 0.988, as shown in Fig. S4a (ESI†). Therefore, this reaction is a diffusion-controlled process according to the Cottrell equation.29 The catalytic rate constant k can be obtained from the slope of the equation J2/J1 = (πkC)1/2t1/2.11 As shown in Fig. S4b (ESI†), current density J2 was from electrolyte #2, and current density J1 was from electrolyte #1. C refers to the concentration of glycine, and t is the electrolysis time. The k for NiCo/C and Ni/C catalysts were 492.9 and 82.1 L mol−1 s−1, respectively.
Fig. 5 shows a series of Raman spectra for the NiCo catalyst in 1 M NaOH with 0.2 M glycine, acquired as a function of the applied potential. At the potentials of 0.00 V and 0.10 V, a peak appeared at 497 cm−1. The peak at 497 cm−1 is best assigned to defective or disordered Ni(OH)2,30 generated from Ni metal in the alkaline electrolyte. At 0.10 V and 0.25 V, a weak peak centered at ca. 589 cm−1 indicates the presence of Co(OH)2 from Co metal.31 The 589 cm−1 peak was found to shift negatively to 575 cm−1 when the applied potential increased to 0.30 V, indicating the transformation of Co(OH)2 to CoOOH species.32 After the applied potential reached 0.30 V (the onset potential) or higher, peaks centered at 497 cm−1 and 589 cm−1 disappeared. Two strong peaks were observed at 465 cm−1 and 545 cm−1. The occurrence of doublet peaks at 476/558 cm−1 implies that nickel and cobalt hydroxides were oxidized in situ to NixCo1–xOOH.32 The NixCo1–xOOH acts as an active intermediate for the EAA reaction. The valence change of catalysts from Raman spectra is in accordance with the onset potential of 0.30 V from the CV measurement in Fig. 4a.
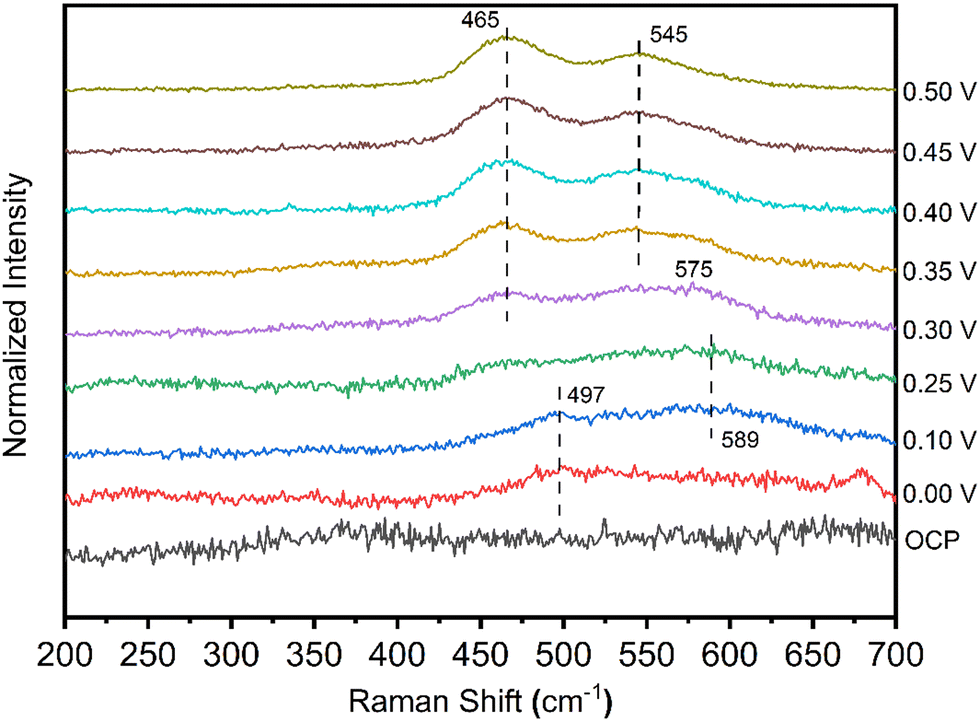 | ||
| Fig. 5 In situ Raman spectra were collected for NiCo deposited on a roughened Au substrate at the potential range from open-circuit-potential to 0.5V vs. Ag/AgCl in 1 M NaOH with 0.2 M glycine. | ||
Electrochemical impedance spectroscopy (EIS) characterization conducted was with frequencies ranging from 0.1 Hz to 1 MHz in 1 M NaOH solution with 0.2 M glycine. The Nyquist plots from the two systems using Ni and NiCo catalysts on glassy carbon electrodes were obtained, and the results are shown in Fig. S5 (ESI†). The Nyquist plots of each system displayed a semicircle in the high-frequency region, which could be related to the charge-transfer dominant process. Distinctly, the semicircle of the NiCo catalyst is smaller as compared to that of the Ni catalyst, indicating a reduced charge-transfer resistance. An equivalent circuit model was established to extract the quantitative impedance values, where Rs, CPE, Rct and Zw represent solution resistance, a constant phase element corresponding to the double-layer capacitance, the charge transfer resistance and Warburg impedance, respectively. The Rct of the Ni catalyst and NiCo catalyst were 145.5 Ohm and 90.5 Ohm, respectively. This result is consistent with the higher glycine-electrooxidation current densities using the NiCo catalyst.
Apart from glycine, the electrocatalytic behaviors of alanine, histidine, methionine, proline, serine, tyrosine, and cysteine were investigated with a similar method, and the results are shown in Fig. 6 and Table 1. The molecular structures of these amino acids are shown in Scheme S6 (ESI†). Among these amino acids, histidine and tyrosine showed relatively low electrooxidation currents. This is possibly due to the covalent ring structure of the R side chain groups. The catalytic rate constant k in Table 1 indicates that the NiCo/C bimetallic catalyst has superior catalytic activity toward the EAA reaction. Besides, the onset potentials of Ni/C for these amino acids in Fig. 6 are around 350 mV. The overpotential of EAA reactions can be reduced when using the NiCo/C bimetallic catalyst with onset potentials between 290 mV and 300 mV.
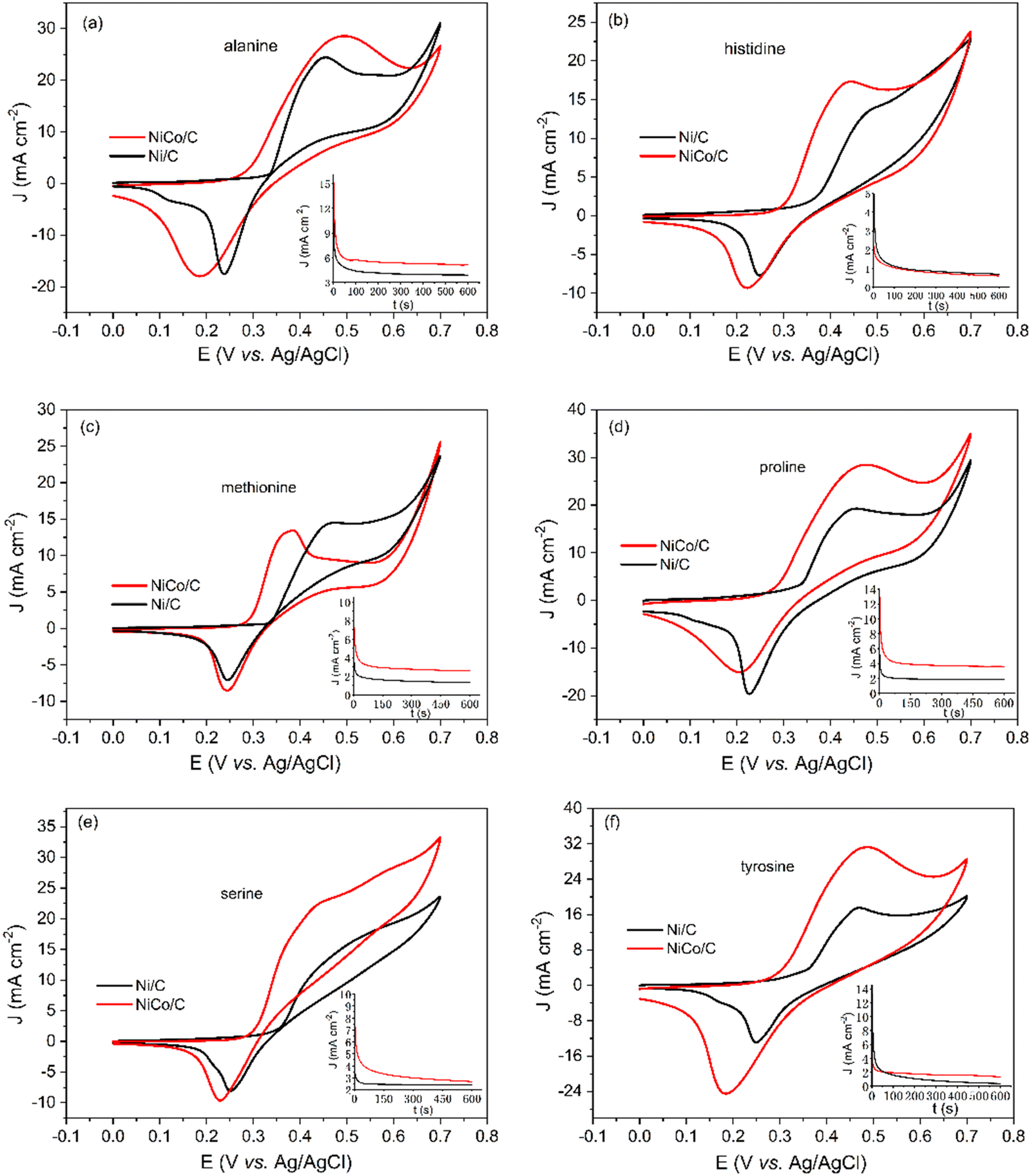 | ||
| Fig. 6 CVs of Ni/C (black) or NiCo/C (red) catalysts in 1 M NaOH electrolyte in the presence of 0.2 M (a) alanine, (b) histidine, (c) methionine, (d) proline, (e) serine, and (f) tyrosine at the scan rate of 50 mV s−1. Inset plots show the chronoamperograms of NiCo/C and Ni/C catalysts at 0.38 V vs. Ag/AgCl. | ||
| Ni/C | NiCo/C | |||
|---|---|---|---|---|
| Onset potential (mV) | Catalytic rate constant k (L mol−1 s−1) | Onset potential (mV) | Catalytic rate constant k (L mol−1 s−1) | |
| Glycine | 350 | 82.1 | 300 | 492.9 |
| Alanine | 350 | 24.6 | 290 | 136.0 |
| Histidine | 350 | 1.9 | 290 | 9.1 |
| Methionine | 350 | 3.9 | 290 | 47.6 |
| Proline | 350 | 18.4 | 290 | 85.8 |
| Serine | 350 | 6.2 | 300 | 56.5 |
| Tyrosine | 350 | 4.5 | 300 | 34.8 |
| Cysteine | — | 50.2 | — | 168.2 |
Similarly, NiCo/C demonstrated a better catalytic activity for cysteine, as shown in Table 1. The CVs for cysteine are illustrated in Fig. 7a. At a positive scan from 0.4 V to 0.6 V, NiCo/C displayed a higher current density as compared to Ni/C, indicating improved activity toward cysteine electrooxidation. However, It was observed that the electrochemical behavior of cysteine in CVs was quite different from other amino acids. An increase in the anodic current at potentials from 0 V to 0.3 V was observed with both NiCo/C and Ni/C for cysteine. According to the results from Fig. 4a and Fig. 6, no anodic current was observed in this potential range (before the onset potential). The difference in the electrochemical behaviors between cysteine and other amino acids is probably due to the reaction mechanisms. Most amino acids follow the EC′ mechanism, proceeding as reactions (1) and (2).10 In this situation, no electrooxidation reaction happens till the transition of the Ni(III)/Ni(II) redox couple. Therefore, there is no anodic current before the formation of the intermediate catalyst. Unlike other amino acids, the CVs of cysteine indicated that the cysteine electrooxidation will not conform to the EC′ mechanism in potential regions before 0.3 V. This phenomenon is probably caused by the electrooxidation of the mercapto group.33 To confirm the electrooxidation reaction at low potential, we also recorded the CVs of 3-mercaptopropionic acid (3-MA) on NiCo/C and Ni/C catalysts in Fig. 7b and c. Here, 3-MA was chosen due to a mercapto group with a similar chemical structure to cysteine. As shown in Fig. 7d, the results revealed that 3-MA had similar electrochemical behaviors to cysteine in the potential region from −0.2 V to the onset potential of 0.30 V (Fig. 7b) or 0.35 V (Fig. 7c). This further supports that the reaction of –SH group led to different behaviors in the electrooxidation of cysteine. Finally, after the onset potential, the electrooxidation of cysteine proceeded according to the EC′ mechanism again. Thus, a sharp increase in current density was observed after the onset potential due to the electrocatalytic oxidation of cysteine on the Ni(III)/Ni(II) redox couple. The “amino compounds oxidation” refers to extracting electrons from the amino group, carbon chain and side chain groups (such as the mercapto group) for energy conversion. Fig. 6 and 7 demonstrate that there was no selectivity for amino compound oxidation on the NiCo/C catalyst since NiCo/C is active for different amino compounds with different side chain groups.
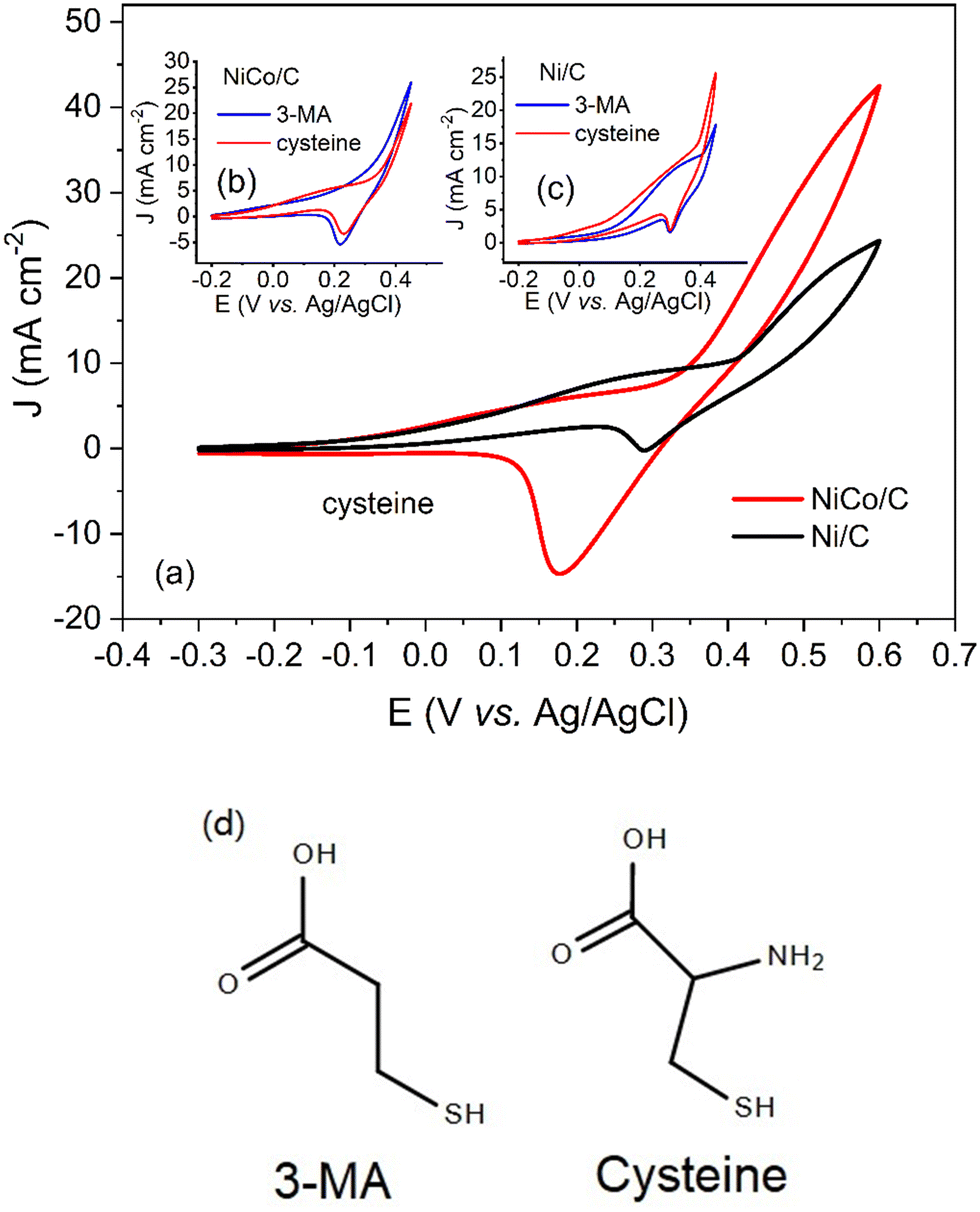 | ||
| Fig. 7 (a) CVs of the glassy carbon electrode (GCE) modified with Ni/C (black) or NiCo/C (red) catalysts in 1 M NaOH electrolyte with 0.2 M cysteine at the scan rate of 50 mV s−1. (b) CVs of NiCo/C in 1 M NaOH electrolyte with 0.2 M cysteine or 3-mercaptopropionic acid. (c) CVs of Ni/C in 1 M NaOH electrolyte with 0.2 M cysteine or 3-mercaptopropionic acid. (d) Structures of 3-Mercaptopropionic acid (3-MA) and cysteine. | ||
Fuel cell performance
The Ni/C and NiCo/C catalysts were further investigated in fuel cells. Fig. 8a demonstrates that the power density of amino acid fuel cells (AAFC) increased as the operation temperature became higher. Maximum power densities of 1.29, 1.57 and 1.9 mW cm−2 were achieved at 20 °C, 40 °C and 60 °C, with open-circuit voltages of 0.83, 0.87 and 0.88 V, respectively. In comparison, much higher power density can be obtained when NiCo/C is used, as shown in Fig. 8b. The maximum power densities at 20 °C and 40 °C are similar, reaching 1.99 and 2.18 mW cm−2, respectively. If the temperature further increases to 60 °C, the maximum power density is 3.00 mW cm−2, which is 1.5 times the values from fuel cells with Ni/C catalyst. This shows that NiCo/C has better performance than Ni/C functioning as an anode catalyst for AAFC. According to the electrochemical characterization of Ni/C and NiCo/C catalysts, NiCo/C has better electrochemical activity toward the electrooxidation of amino acids and a lower overpotential, thus it can lead to the promotion of cell electricity output.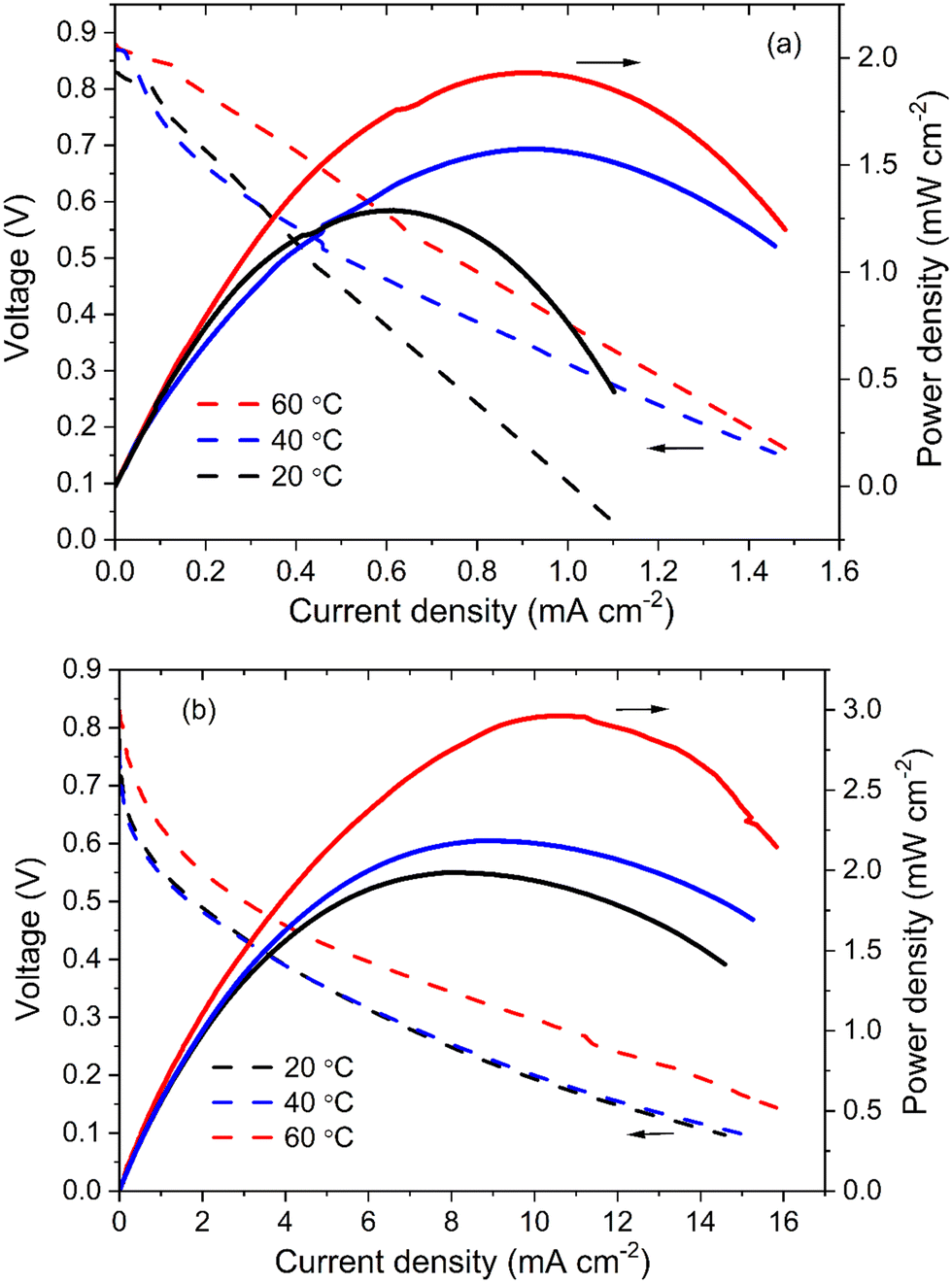 | ||
| Fig. 8 Polarization and power density curves of the amino acid fuel cells using (a) Ni/C and (b) NiCo/C as anode catalysts with 1 M glycine + 1 M NaOH as fuels at 20 °C, 40 °C and 60 °C, respectively. | ||
The AAFC performance is largely influenced by the alkalinity of the anolyte. Operating the AAFC with the anolyte pH well above neutral can greatly improve the power output due to the accelerated reaction rate of amino acid electrooxidation. Another important aspect related to AAFC performance is the concentration of amino acids in the fuels. According to the Nernst equation, a higher amino acid concentration is beneficial for achieving a more negative anode potential and higher OCV. It may also help to improve the power density due to the facilitated mass transfer. Moreover, the energy density of AAFC will be greatly increased with the increase in the amino acid percentage in the fuel. In such cases, more electricity (or electrons) will be released from the fuels under the same volume or weight conditions. However, the solubilities of 20 amino acids are quite different because of the different hydrophobicities of their side-chain groups. The soluble amino acids are Ala, Arg, Gly, Lys, Pro, and Ser; the relatively insoluble amino acids are Asn, Asp, Cys, Gln, Glu, His, Ile, Leu, Met, Phe, Thr, Trp, Tyr, and Val. Most amino acids are relatively insoluble in neutral water, leading to a limitation of protein application in AAFC as the energy density is proportional to the fuel's amino acid content. One strategy is to use alkaline AAFC because the solubility of amino acids will be greatly increased in the alkaline aqueous solution. To investigate the effects of amino acid concentration, the AAFC was operated with high and low amino acid concentrations, respectively, and the results are shown in Fig. S7 (ESI†). Met and Tyr both presented small solubilities at 25 °C of 56.2 g L−1 and 0.4 g L−1, respectively. When their concentrations were low (50 mM Met and 2.5 mM Tyr), the OCVs of AAFC were 0.67 V for Met and 0.55 V for Tyr. The maximum power densities were only 0.14 mW cm−2 at the current density of 0.47 mA cm−2 for Met and 0.053 mW cm−2 at the current density of 0.23 mA cm−2 for Tyr. Fixing the Met and Tyr contents and just increasing the pH by adding sodium hydroxide could improve the power density to 0.41 mW cm−2 for Met and 0.87 mW cm−2 for Tyr, respectively; these correspond to the results above. As power improves, the limitation of low-content fuel would be more serious at high current densities, which requires much faster mass transfer to obtain sufficient fuel. In the alkaline solution, we would be able to use Met-rich (1 mol L−1) fuel due to the improved solubility, and the results showed that the OCV further increased to 0.75 V with power density up to 0.71 mW cm−2, almost twice the value of 0.05 M Met. Similarly, higher OCV and power density of 0.83 V and 1.1 mW cm−2 were also achieved when using Tyr-rich (0.25 mol L−1) fuel. The red and blue polarization plots in Fig. S7 (ESI†) refer to the amino acids with high concentration and low concentration, respectively. This demonstrated that the two plots were close at an area of low current density (electrochemical polarization as the dominant loss), and the main difference between the two plots was observed at an area of high current density (concentration polarization as the dominant loss). These results showed that the concentration polarization could be reduced by using high-concentration fuel.
Conclusions
A direct route from amino acids to electricity has been demonstrated. The NiCo/C bimetallic catalyst was fabricated by a chemical reduction method. An electrochemical evaluation revealed that this is a promising catalyst with a lower overpotential and higher catalytic activity toward the electrooxidation of amino acids as compared to the Ni/C catalyst. The amino acids were directly oxidized by the NixCo1–xOOH active species, which was formed above the onset potential according to electrochemical Raman measurements. Sulfur in the R side chain of cysteine can be electrooxidized before the onset potential. Amino acid fuel cells with the NiCo/C anode catalyst achieved a maximum power density of 2.0 mW cm−2 at 20 °C and 3.0 mW cm−2 at 60 °C. Amino acids, with their high solubility in alkaline solution, are promising candidates for hydrogen storage in fuel cells, with large energy density and low toxicity. These advantages make AAFC commercially promising for use in portable battery replacements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by fund of National Natural Science Foundation of China (Grant No. 22202146, 22202145, 22202147), Zhejiang Provincial Natural Science Foundation of China (Grant No. LGG21B070001), Key Research and Development Plan of Zhejiang Province (Grant No. 2021C03022) and National Innovation Training program for college students (Grant No. 202210350038). The authors also thank Yan Liu in School of Earth Sciences, Zhejiang University, for assistance with Raman measurements.References
- K. Srirangan, L. Akawi, M. M. Young and C. P. Chou, Appl. Energy, 2012, 100, 172–186 CrossRef.
- S. Kagale, C. Koh, J. Nixon, V. Bollina, W. E. Clarke, R. Tuteja, C. Spillane, S. J. Robinson, M. G. Links, C. Clarke, E. E. Higgins, T. Huebert, A. G. Sharpe and I. A. P. Parkin, Nat. Commun., 2014, 5, 3706 CrossRef CAS PubMed.
- R. Slade, A. Bauen and R. Gross, Nat. Clim. Change, 2014, 4, 99–105 CrossRef.
- H. Haberl, T. Beringer, S. C. Bhattacharya, K. H. Erb and M. Hoogwijk, Curr. Opin. Environ. Sustain., 2010, 2, 394–403 CrossRef PubMed.
- Y. X. Huo, D. G. Wernick and J. C. Liao, Curr. Opin. Biotechnol., 2012, 23, 406–413 CrossRef CAS PubMed.
- B. E. Dale, M. S. Allen, M. Laser and L. R. Lynd, Biofuels, Bioprod. Biorefin., 2009, 3, 219–230 CrossRef CAS.
- P. J. Crutzen, A. R. Mosier, K. A. Smith and W. Winiwarter, Atmos. Chem. Phys., 2008, 8, 389–395 CrossRef CAS.
- Y. Fang, X. Liu, Z. Liu, L. Han, J. Ai, G. Zhao, O. Terasaki, C. Cui, J. Yang, C. Liu, Z. Zhou, L. Chen and S. Che, Chemistry, 2023, 9, 460–471 CrossRef CAS.
- Z. X. Chen, F. Jing, M. H. Luo, X. H. Wu, H. C. Fu, S. W. Xiao, B. B. Yu, D. Chen, X. Q. Xiong and Y. X. Jin, Carbon Energy, 2023, e443 Search PubMed.
- W. Huang, J. Zheng and Z. Li, J. Phys. Chem. C, 2007, 111, 16902–16908 CrossRef CAS.
- S. Majdi, A. Jabbari, H. Heli and A. A. M. Movahedi, Electrochim. Acta, 2007, 52, 4622–4629 CrossRef CAS.
- B. K. Boggs, R. L. King and G. G. Botte, Chem. Commun., 2009, 4859–4861 RSC.
- M. Vidotti, S. I. Córdoba de Torresi and L. T. Kubota, Sens. Actuators, B, 2008, 135, 245–249 CrossRef CAS.
- J. Nai, Z. Chen, H. Li, F. Li, Y. Bai, L. Li and L. Guo, Chem. – Eur. J., 2013, 19, 501–508 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- U. B. Demirci, J. Power Sources, 2007, 173, 11–18 CrossRef CAS.
- B. K. Boggs and G. G. Botte, Electrochim. Acta, 2010, 55, 5287–5293 CrossRef CAS.
- W. Yan, D. Wang and G. G. Botte, Electrochim. Acta, 2012, 61, 25–30 CrossRef CAS.
- X. Cui, W. Guo, M. Zhou, Y. Yang, Y. Li, P. Xiao, Y. Zhang and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 493–503 CrossRef CAS.
- M. Pumera, H. Iwai and Y. Miyahara, Chem. Phys. Chem., 2009, 10, 1770–1773 CrossRef CAS.
- M. Hasanzadeh, G. K. Nezhad, N. Shadjou, M. Hajjizadeh, B. Khalilzadeh, L. Saghatforoush, M. H. Abnosi, A. Babaei and S. Ershad, Anal. Biochem., 2009, 389, 130–137 CrossRef CAS.
- M. Vidotti, M. R. Silva, R. P. Salvador, S. I. Córdoba de Torresi and L. H. Dall’Antonia, Electrochim. Acta, 2008, 53, 4030–4034 CrossRef CAS.
- J. W. Kim and S. M. Park, J. Electrochem. Soc., 2003, 150, E560–E566 CrossRef CAS.
- S. I. Córdoba de Torresi, K. Provazi1, M. Malta and R. M. Torresi, J. Electrochem. Soc., 2001, 148, A1179–A1184 CrossRef.
- O. Diaz-Morales, D. Ferrus-Suspedraa and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- L. Deng, Y. Tan, C. Chen, F. Li and X. Zhang, New J. Chem., 2023, 47, 8032–8041 RSC.
- F. Maillard, A. Bonnefont, M. Chatenet, L. Guétaz, B. D. Cottignies, H. Roussel and U. Stimming, Electrochim. Acta, 2007, 53, 811–822 CrossRef CAS.
- S. W. Feldberg, J. Electroanal. Chem., 2008, 624, 45–51 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods Fundamentals and Applications, John Wiley & Sons, Inc, New York, 2nd edn, 2003 Search PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- J. A. Koza, C. M. Hull, Y. Liu and J. A. Switzer, Chem. Mater., 2013, 25, 1922–1926 CrossRef CAS.
- X. Yang, H. Zhang, B. Yu, Y. Liu, W. Xu and Z. Wu, Energy Technol., 2022, 10, 2101010 CrossRef CAS.
- E. A. Tumanova, A. Yu Safronov and A. V. Kapustin, Russ. J. Electrochem., 2001, 37, 972–975 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00368j |
| This journal is © The Royal Society of Chemistry 2023 |