
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of hydrogen-evolution reactivity in an Rh-doped SrTiO3 photocatalyst under visible-light irradiation by loading with controlled platinum nanoclusters†
Daichi
Yazaki
a,
Tokuhisa
Kawawaki
 *ab,
Tomoya
Tanaka
a,
Daisuke
Hirayama
a,
Yamato
Shingyouchi
a and
Yuichi
Negishi
*ab
*ab,
Tomoya
Tanaka
a,
Daisuke
Hirayama
a,
Yamato
Shingyouchi
a and
Yuichi
Negishi
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162–8601, Japan. E-mail: kawawaki@rs.tus.ac.jp; negishi@rs.tus.ac.jp
bResearch Institute for Science and Technology, Tokyo University of Science, 2641 2641 Yamazaki, Noda, Chiba, 278–8510, Japan
First published on 7th June 2023
Abstract
Rhodium-doped strontium titanate (SrTiO3:Rh)-based photocatalysts have long been studied because they can produce hydrogen (H2) from visible light and water in Z-scheme water-splitting systems. However, the H2-evolution reaction (HER) of SrTiO3:Rh is the rate-limiting step in such a system, so further improvement of the HER activity of SrTiO3:Rh is desired to enhance Z-scheme water splitting. In this study, we synthesize hydrophilic ∼1 nm platinum nanoclusters (Pt NCs) using a ligand-exchange method while maintaining the geometric structure of the corresponding hydrophobic ∼1 nm Pt NC, and we load the monodispersed Pt NCs onto SrTiO3:Rh. The Pt NC-loaded SrTiO3:Rh exhibits HER activity that is 30% higher than that of the Pt cocatalyst-loaded SrTiO3:Rh prepared using the conventional photodeposition method. This work also demonstrates that Z-scheme water splitting proceeds stoichiometrically using the Pt NC-loaded SrTiO3:Rh as the H2-evolution photocatalyst, bismuth vanadate as an oxygen-evolution photocatalyst, and Fe2+ as a mediator.
1. Introduction
The improvement of water-splitting photocatalysts that can produce hydrogen (H2) from water and sunlight may greatly contribute to the establishment of next-generation sustainable energy systems.1 In recent years, researchers have actively pursued the development of photocatalysts that can respond to visible light, which accounts for approximately 40% of the solar energy spectrum.2–4 In particular, rhodium-doped strontium titanate5 (SrTiO3:Rh; i.e., STO:Rh) is known as a visible-light-driven photocatalyst that exhibits remarkable activity in the H2-evolution reaction (HER). The visible-light absorption of STO:Rh is attributed to a transition from the electron donor level formed by Rh3+ to the conduction band consisting of Ti 3d orbitals. This allows the STO:Rh to absorb visible light up to a wavelength of ∼520 nm, enabling H2 production by water splitting using not only ultra-violet light but also visible light.5–10 For example, lanthanum (La) and Rh co-doped SrTiO3 (STO:La/Rh7,8,11–14), which prevents Rh4+ substitution at Ti4+ sites, has achieved a quantum yield of 33% at visible light region and a solar-to-H2 efficiency of > 1% with the use of photocatalytic sheets.15Notably, STO:Rh-based H2-evolution photocatalysts (HEPs) can achieve water splitting under visible light when combined with oxygen (O2)-evolution photocatalysts (OEPs) to form a Z-scheme system (Fig. 1A).16–18 For the construction of Z-scheme systems, STO:Rh-based HEP has been combined with OEPs such as TiO2-rutile,10 BiVO4,9,10,13,15,19,20 SrTiO3:Rh/Sb,21 AgNbO3,10 Bi2MoO6,10 TiO2:Cr/Sb,10 TiO2:Rh/Sb,10 WO3,10,19 Ta3N5,22 Bi4NbO8Cl,23,24 La0.5Sr0.5Ta0.5Ti0.5O2N,25etc. However, in numerous reports of Z-scheme water splitting using STO:Rh-based HEP, the HER is the rate-limiting step that suppresses the water-splitting activity. Thus, further improvement of the HER activities of STO:Rh-based HEP is desired to enhance the activity of various Z-scheme water-splittings.9,11
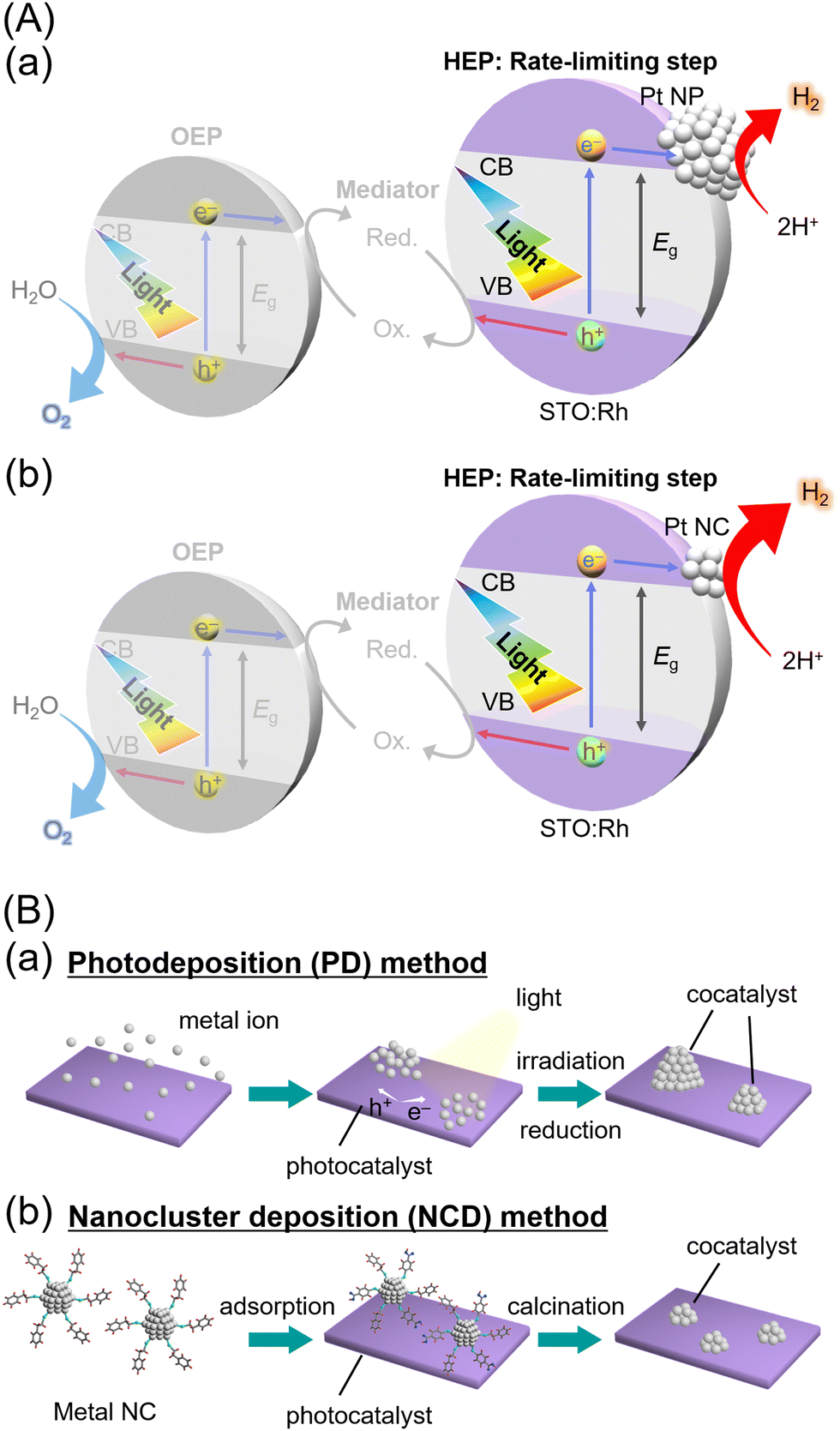 | ||
| Fig. 1 Schematic illustrations of (A) Z-scheme photocatalytic water splitting and (B) the preparation procedure of cocatalysts on the photocatalysts using the (a) conventional PD method and (b) NCD method. In (A), CB, conduction band; VB, valence band; Eg, band gap; Red, reductants; and Ox, oxidants. STO:Rh is one of the most advanced HEP. The improvement in water-splitting activity can be expected by enhancing the reaction in HEPs, which is the rate-limiting step. | ||
The water-splitting photocatalyst is generally composed of a photocatalyst support and metal/metal oxide nanoparticles (NPs) that function as cocatalysts. In the development of highly active photocatalysts, not only the improvement of the photocatalyst support but also the improvement of the cocatalyst, which works as the actual reaction site, is necessary. The cocatalysts are mainly loaded using the photodeposition26 (PD, Fig. 1Ba) and impregnation methods.27 Although these methods are simple, it is difficult to control the particle size and electronic state of the cocatalyst particles. Therefore, the improvement of cocatalyst-loading methods28–32 has also been actively studied to enhance water-splitting activity. In particular, the method33–36 of loading metal NPs with controlled size and chemical composition, which are prepared by liquid-phase synthesis, onto a photocatalyst is effective in controlling the particle size and electronic state of the cocatalyst.37–41 Furthermore, metal nanoclusters (NCs) with diameters of ∼1 nm have unique properties compared with those of metal NPs (diameters > ∼2 nm) and bulk metals. The NC deposition (NCD, Fig. 1Bb) method, which exploits the unique characteristics of metal NCs, can create a cocatalyst surface that is more favorable for the adsorption of the substrate and the desorption of the products, resulting in enhanced activity in the water-splitting reaction. The use of atomically precise gold (Au) NCs,38,42,43 ∼1 nm Au–platinum (Pt) alloy NCs,44 rhodium–chromium mixed oxide (Rh2–xCrxOy) NCs,45 Pt∼51 NCs,46 ∼1.4 nm Pt–ruthenium (PtRu),47 and Au NCs48 as cocatalysts has led to improved water-splitting and HER activity.
In this study, we attempted to further improve the HER activity of STO:Rh by employing fine Pt NCs as a cocatalyst. To this end, we established a method to load Pt NCs on STO:Rh, which has a hydrophilic surface, with a high adsorption yield by applying an appropriate hydrophilic treatment to the Pt NCs. As a result, the H2-evolution efficiency of STO:Rh was increased by 30% compared with that of the Pt-cocatalyst-loaded STO:Rh prepared using the conventional PD method. Additionally, stoichiometric overall water splitting was successfully achieved using Pt NC-loaded STO:Rh as the HEP in Z-scheme water splitting.
2. Results and discussion
2.1. Preparation of Pt NC-loaded SrTiO3:Rh
The STO:Rh was prepared by solid-state reaction with 1 mol% of Rh doping, according to a previous report.5 The Brunauer–Emmett–Teller surface area of STO:Rh (Fig. S1, ESI†) was estimated to be 2.5 m2 g−1 (Table S1, ESI†) and powder X-ray diffraction (PXRD) indicated relatively high crystallinity of the product (Fig. S2, ESI†). The strong visible-light absorption was confirmed by ultraviolet-visible (UV-Vis) absorption spectroscopy. These results confirmed that the desired STO:Rh was obtained (Fig. S2, ESI†).The Pt∼51 NC protected by 2-phenylethanethiolate (PET) and carbon monoxide (CO) (Pt∼51(PET)n(CO)m) was synthesized by combining polyol reduction with ligand exchange, similar to our previous report.49 Unfortunately, the obtained Pt∼51(PET)n(CO)m cannot efficiently adsorb on the hydrophilic surface of STO:Rh because the PET ligands are hydrophobic. Therefore, parts of the ligands on the Pt NCs were replaced by p-mercaptobenzoic acid (p-MBA), which has a hydrophilic carboxylic acid group but a similar framework structure to PET.44 Fourier transform infrared (FT-IR) spectra were obtained before and after ligand exchange (Table S2, ESI†), as shown in Fig. S3a (ESI†). The spectra of these modified NCs after ligand exchange showed peaks attributed to the C–OH stretching and O–H⋯O bending vibrations in p-MBA (1427.1, 1326.8, and 927.6 cm−1),50 which confirmed that ligand exchange proceeded. No significant changes occurred in the matrix-assisted laser desorption ionization mass (MALDI-MS) spectra (Fig. S3b, ESI†), UV-Vis absorption spectra (Fig. S3c, ESI†), Pt L3-edge X-ray absorption near edge structure (XANES) spectra (Fig. S5a, ESI†), or Fourier transform-extended X-ray absorption fine structure (FT-EXAFS) spectra (Fig. S5b, ESI†) before and after ligand exchange. These results suggest that many ligands were replaced from PET to p-MBA while maintaining the structure of the Pt NCs. The Pt NCs obtained by ligand exchange (Pt∼51(PET)n(CO)m(p-MBA)l) showed high solubility in polar solvents such as methanol and acetone (Fig. S4 and Table S3, ESI†). However, they were not soluble in water, which is a highly polar solvent, indicating that not all PET ligands were replaced by p-MBA, and some PET ligands remained on the surfaces of the Pt NCs.
The obtained Pt∼51(PET)n(CO)m(p-MBA)l was dispersed in tetrahydrofuran and stirred with STO:Rh for 3 h to adsorb on the STO:Rh surface. A 96% adsorption yield was achieved, which is higher than that of Pt NCs without ligand exchange (Pt∼51(PET)n(CO)m, 73%), due to the formation of hydrogen bonds between the carboxylic acid group (–COOH) of the p-MBA ligands on Pt∼51(PET)n(CO)m(p-MBA)l and the hydroxyl group (–OH) on STO:Rh.44,48 However, the presence of ligands suppresses the accessibility of a substance and the efficiency of electron transfer from the photocatalysts to the NCs, which leads to a decrease in catalytic activity. Therefore, the obtained Pt∼51(PET)n(CO)m(p-MBA)l-loaded STO:Rh (Pt∼51(PET)n(CO)m(p-MBA)l/STO:Rh) was calcined at 300 °C under reduced pressure to remove most of the ligands from the Pt NC surface (PtNC/STO:Rh). The calcination temperature (300 °C) was determined based on the desorption temperature of the ligands in the thermogravimetric analysis of Pt NCs.49
Fig. 2 shows the Pt L3-edge FT-EXAFS spectra of Pt NC and Pt NC-loaded STO:Rh in each preparation process (Fig. S6, ESI†). The peak located at approximately 2.0 Å, which is attributed to the Pt–S bond, was observed in both Pt∼51(PET)n(CO)m(p-MBA)l and Pt∼51(PET)n(CO)m(p-MBA)l/STO:Rh. In the spectrum of PtNC/STO:Rh, the Pt–S bond was not observed, and instead, a peak (∼1.8 Å) attributed to the Pt–O bond was observed (Fig. 2). This suggests that most of the S species in the ligands were desorbed from Pt NC by the calcination process through Pt–S dissociation. The S 2p3/2 X-ray photoelectron spectroscopy (XPS) spectrum of PtNC/STO:Rh (Fig. S7, ESI†) shows a peak attributed to a S species in a highly oxidized state. These results indicate that most of the S remains on STO:Rh after desorption from Pt NC surfaces, mainly as SO42− (Fig. S7, ESI†).48 Transmission electron microscopy (TEM) images (Fig. 3a) indicated that the average particle size of the Pt NC cocatalyst (PtNC) in PtNC/STO:Rh was 1.5 ± 0.3 nm. This indicates that there was effectively no significant aggregation of Pt∼51 during calcination and it was presumed that 92.8% of Pt∼51 on PtNC/STO:Rh has a three-dimensional structure after the calcination (Fig. S8 and S9, ESI†). Therefore, fine and monodispersed Pt NCs were loaded on STO:Rh (Fig. 3a).
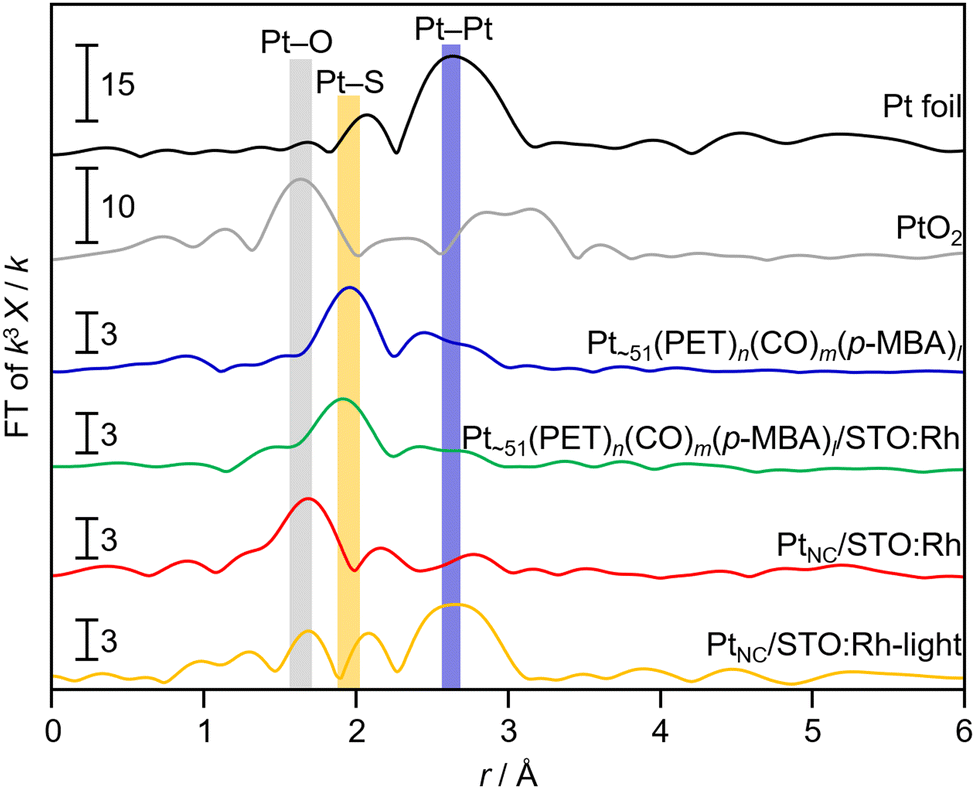 | ||
| Fig. 2 Pt L3-edge FT-EXAFS spectra of Pt∼51(PET)n(CO)m(p-MBA)l, before (Pt∼51(PET)n(CO)m(p-MBA)l/STO:Rh) and after (PtNC/STO:Rh) calcination of Pt NC-loaded STO:Rh, and after light irradiation of PtNC/STO:Rh (PtNC/STO:Rh-light). Pt foil and PtO2 powder are also shown for comparison. | ||
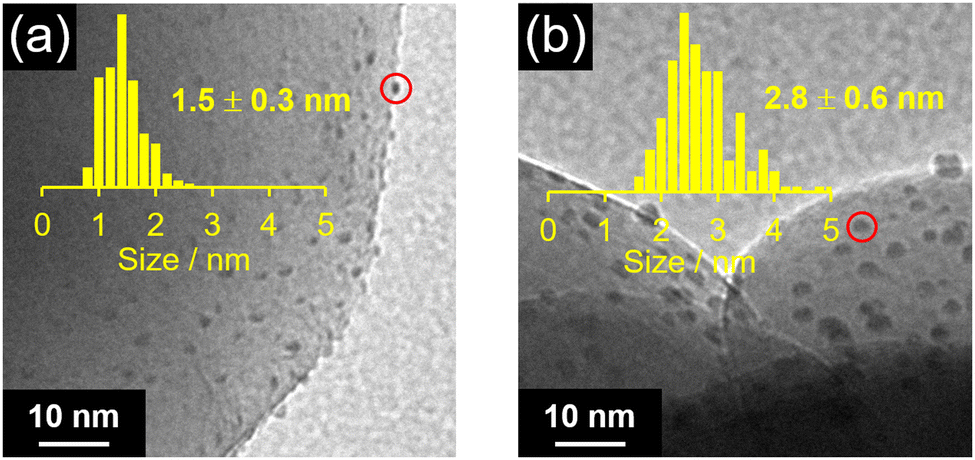 | ||
| Fig. 3 TEM images and resulting histograms (inset) for the particle-size distribution of Pt cocatalysts in (a) PtNC/STO:Rh and (b) PtPD/STO:Rh. | ||
We also used the PD method to load a Pt cocatalyst (PtPD) on STO:Rh (PtPD/STO:Rh). As a result, PtPD with an average particle size of 2.8 ± 0.6 nm was loaded on STO:Rh (Fig. 3b). This indicates that Pt cocatalysts with high monodispersity but about half the particle size can be obtained using our method (PtNC/STO:Rh) compared with the PD method (PtPD/STO:Rh). Assuming that the loaded PtNC and PtPD have a face-centered cubic structure, PtNC was estimated to have 1.58 times more active sites (i.e., Pt atoms on the surfaces of Pt cocatalysts) than PtPD (Table S4, ESI†).
The obtained PtNC in PtNC/STO:Rh was more oxidized than that in Pt∼51(PET)n(CO)m(p-MBA)l and Pt∼51(PET)n(CO)m(p-MBA)l/STO:Rh (Fig. 2 and 4). This means that oxidation of the Pt NCs occurred during calcination. Therefore, we attempted to reduce PtNC with photoexcited electrons produced by irradiating the PtNC/STO:Rh dispersed in water (PtNC/STO:Rh-light). As a result, PtNC was significantly reduced and a strong Pt–Pt bond was observed (Fig. 2 and 4). Accordingly, the peaks attributed to the S species were significantly decreased in the S 2p3/2 XPS spectrum of PtNC/STO:Rh-light compared with PtNC/STO:Rh before light irradiation (Fig. S7, ESI†). This means that SO42− and other residues on the STO:Rh surface were removed by washing or oxidative desorption after photoirradiation in water. The PXRD patterns, Rh K-edge XANES spectra, FT-IR spectra, and scanning electron microscopy images of the samples (Fig. S1, S2, and S10, ESI†) confirmed that no change occurred in the STO:Rh during the series of operations required for this Pt NC-loading even except for only a charge state of Rh. The slightly oxidized electronic state of Rh by the calcination process was reduced by light irradiation (Fig. S11, ESI†). From these results, we concluded that Pt NCs with metallic electronic states were successfully loaded on STO:Rh with relatively fine and monodispersed particle sizes.
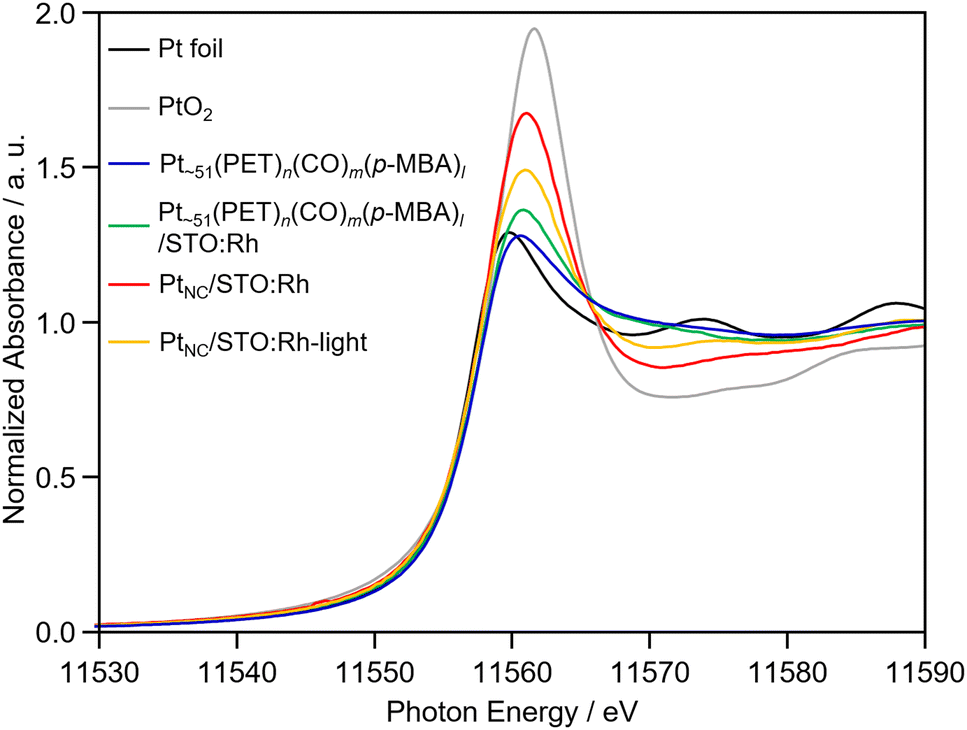 | ||
| Fig. 4 Pt L3-edge XANES spectra of Pt∼51(PET)n(CO)m(p-MBA)l, before (Pt∼51(PET)n(CO)m(p-MBA)l/STO:Rh) and after (PtNC/STO:Rh) calcination of Pt NC-loaded STO:Rh, and after light irradiation onto PtNC/STO:Rh (PtNC/STO:Rh-light). Pt foil (Pt0) and PtO2 powder (Pt4+) were also shown for comparison. A stronger and weaker absorbance indicates a more oxidized and reduced electronic state of Pt at the whiteline, respectively. | ||
2.2. H2-evolution activity of Pt NC-loaded SrTiO3:Rh
Next, we evaluated the photocatalytic activity of PtNC/STO:Rh. To estimate the function of PtNC as an HER cocatalyst more accurately, H2-evolution activity was evaluated in a half-reaction with methanol as a hole sacrificial agent (Fig. S12, ESI†). For comparison, the same measurements were performed for PtPD/STO:Rh.First, we investigated the effect of ligand removal by calcination on H2-evolution activity (Fig. 5a). From the comparison with PtNC/STO:Rh before and after calcination, we confirmed that the H2-evolution activity increased by ∼80 times after the calcination treatment. The removal of ligands from the PtNC surface contributes to this improvement by making it easier for protons to approach the PtNC cocatalyst and enhancing electron transfer from STO:Rh to the PtNC cocatalyst. For comparison, H2-evolution activity was also measured for PtPD/STO:Rh, in which the Pt cocatalyst was loaded on STO:Rh pre-calcined at 300 °C using the PD method. As a result, almost no change in the activity of PtPD/STO:Rh was observed (Fig. S13, ESI†). This indicates that the calcination treatment did not affect the activity of the photocatalyst support (i.e., STO:Rh), and the increase in activity is mainly attributed to the removal of ligands from the surface of the PtNC cocatalyst.
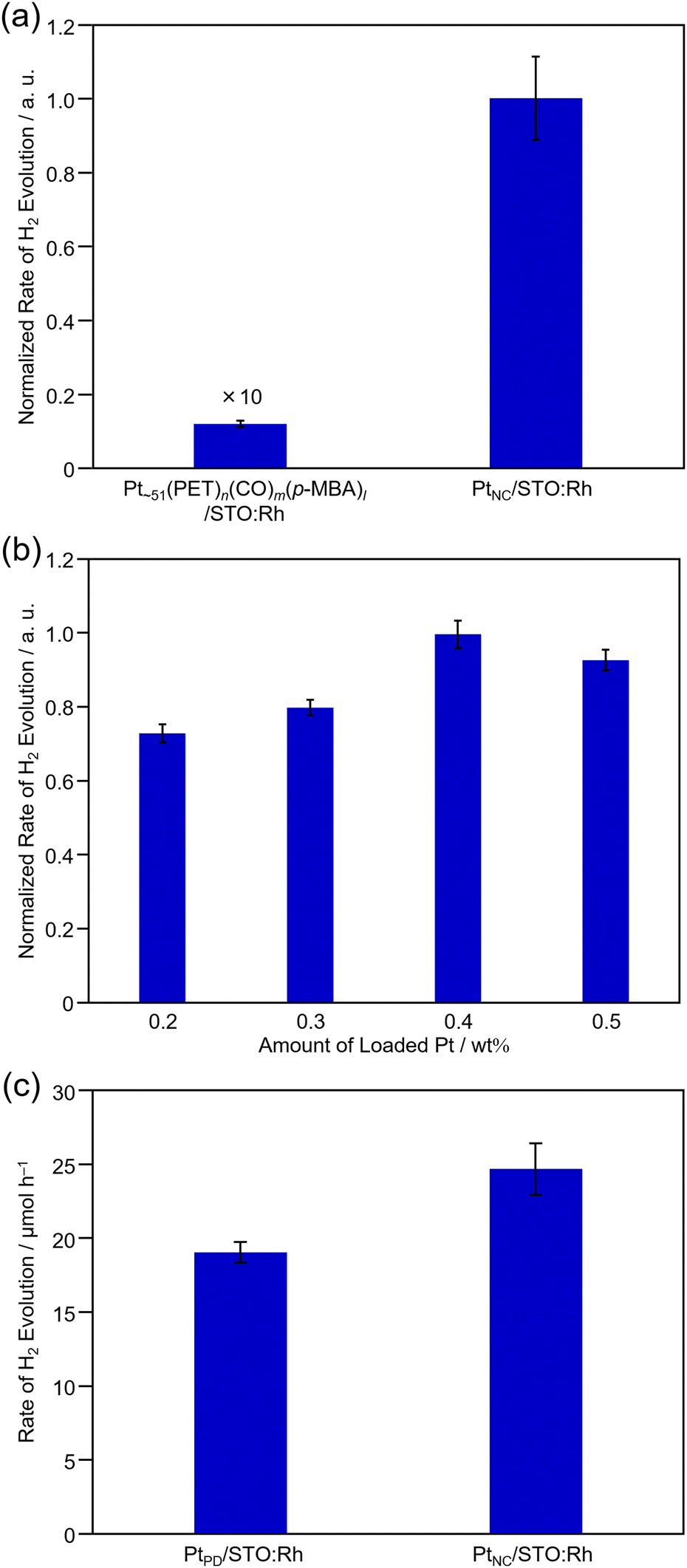 | ||
| Fig. 5 H2-evolution activity of Pt cocatalyst-loaded STO:Rh photocatalysts. (a) Before (Pt∼51(PET)n(CO)m(p-MBA)l/STO:Rh) and after (PtNC/STO:Rh) calcination of Pt NC-loaded STO:Rh and (b) dependence of loading weight of the Pt cocatalyst for PtNC/STO:Rh. (c) Comparison of H2-evolution activity between PtNC/STO:Rh and PtPD/STO:Rh. In (a) and (c), the loading weight of Pt on them was 0.4 wt%. In order to eliminate the effect of the difference in the activity of each photocatalyst, STO:Rh synthesized in the same batch was used in each experiment (a)–(c). | ||
Next, we investigated the effect of the amount of Pt loading on activity (Fig. 5b). The results showed that the maximum activity was obtained at a loading of 0.4 wt% Pt. Comparing the H2-evolution activity of PtNC/STO:Rh and PtPD/STO:Rh when they were both loaded using 0.4 wt% Pt, PtNC/STO:Rh showed 30% higher H2-evolution rate than PtPD/STO:Rh (Fig. 5c and Fig. S14, ESI†). Considering the results of the Pt L3-edge XANES analysis, there is no significant difference in the electronic state of the Pt cocatalyst between PtNC/STO:Rh and PtPD/STO:Rh (Fig. S15, ESI†). This suggests that the difference in activity between PtNC/STO:Rh and PtPD/STO:Rh is mainly due to the increase in the number of active sites for the reaction, which was enabled by decreasing the size of the Pt cocatalyst (Table S4, ESI†). To eliminate the dependence of a hole sacrificial agent, we evaluated H2-evolution activity using FeCl2. The PtNC/STO:Rh shows higher activity than PtPD/STO:Rh and it was indicated that FeCl2 works as a mediator for Z-scheme water splitting (Fig. S16, ESI†).
Finally, we attempted Z-scheme water splitting using PtNC/STO:Rh. In this experiment, PtNC/STO:Rh was used as the HEP, BiVO4 as the OEP, and Fe2+ or Fe3+ as a mediator.9,19,51,52 First, the water-splitting activity obtained using Fe3+ is shown in Fig. S17 (ESI†). In this case, O2 was produced in excess and stoichiometric H2 and O2 evolution was not observed (H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 ratio = 1.65 at 16 h light irradiation). This suggests that excess Fe3+ prevented H2 evolution in PtNC/STO:Rh. Next, we evaluated the water-splitting activity using Fe2+ as a mediator (Fig. 6). After a slight induction period, stoichiometric H2 and O2 evolution was observed (H2:O2 ratio = 1.99 at 16 h light irradiation). In this case, stable gas evolution was observed even after 16 h of light irradiation, indicating that PtNC/STO:Rh is highly stable. Thus, when PtNC/STO:Rh is used for the HEP, stoichiometric Z-scheme water splitting can stably proceed when Fe2+ is used as a mediator. It has been reported that the Fe3+ chemical species generated by the addition of FeCl3 in sulfuric acid solutions adsorbs on the surface of the Pt cocatalyst, preventing (1) the formation of dissociative adsorbed H2 on the Pt cocatalyst, (2) the reverse reaction from H2 to H2O, and (3) minor reactions in which Fe3+ is reduced by H2 (Fig. S18, ESI†).19,52 Although there was no significant difference between the electronic structures of PtNC and PtPD, because the number of Pt atoms constituting the PtNC and PtPD cocatalysts is different, it is assumed that there is a difference in the geometric structure of the Pt cocatalysts. The adsorption of Fe3+ chemical species on the surface of the PtNC is likely more inhibited than that on the PtPD because of the difference in the geometric structure. Therefore, using PtNC/STO:Rh as the HEP might decrease the production of H2 when the mediator is Fe3+. Unfortunately, no significant improvement in H2 production was observed with PtNC/STO:Rh-BiVO4 compared with PtPD/STO:Rh-BiVO4 in an overall water splitting using Fe2+ due to probably the high reactivity of PtNC/STO:Rh in terms of the side reactions and reverse reactions in the overall water-splitting reaction. Therefore, it is necessary to search for conditions for Z-scheme water-splitting suitable for the evaluation of the PtNC/STO:Rh-BiVO4 as well as in the case of the PtPD/STO:Rh-BiVO4.19
:O2 ratio = 1.65 at 16 h light irradiation). This suggests that excess Fe3+ prevented H2 evolution in PtNC/STO:Rh. Next, we evaluated the water-splitting activity using Fe2+ as a mediator (Fig. 6). After a slight induction period, stoichiometric H2 and O2 evolution was observed (H2:O2 ratio = 1.99 at 16 h light irradiation). In this case, stable gas evolution was observed even after 16 h of light irradiation, indicating that PtNC/STO:Rh is highly stable. Thus, when PtNC/STO:Rh is used for the HEP, stoichiometric Z-scheme water splitting can stably proceed when Fe2+ is used as a mediator. It has been reported that the Fe3+ chemical species generated by the addition of FeCl3 in sulfuric acid solutions adsorbs on the surface of the Pt cocatalyst, preventing (1) the formation of dissociative adsorbed H2 on the Pt cocatalyst, (2) the reverse reaction from H2 to H2O, and (3) minor reactions in which Fe3+ is reduced by H2 (Fig. S18, ESI†).19,52 Although there was no significant difference between the electronic structures of PtNC and PtPD, because the number of Pt atoms constituting the PtNC and PtPD cocatalysts is different, it is assumed that there is a difference in the geometric structure of the Pt cocatalysts. The adsorption of Fe3+ chemical species on the surface of the PtNC is likely more inhibited than that on the PtPD because of the difference in the geometric structure. Therefore, using PtNC/STO:Rh as the HEP might decrease the production of H2 when the mediator is Fe3+. Unfortunately, no significant improvement in H2 production was observed with PtNC/STO:Rh-BiVO4 compared with PtPD/STO:Rh-BiVO4 in an overall water splitting using Fe2+ due to probably the high reactivity of PtNC/STO:Rh in terms of the side reactions and reverse reactions in the overall water-splitting reaction. Therefore, it is necessary to search for conditions for Z-scheme water-splitting suitable for the evaluation of the PtNC/STO:Rh-BiVO4 as well as in the case of the PtPD/STO:Rh-BiVO4.19
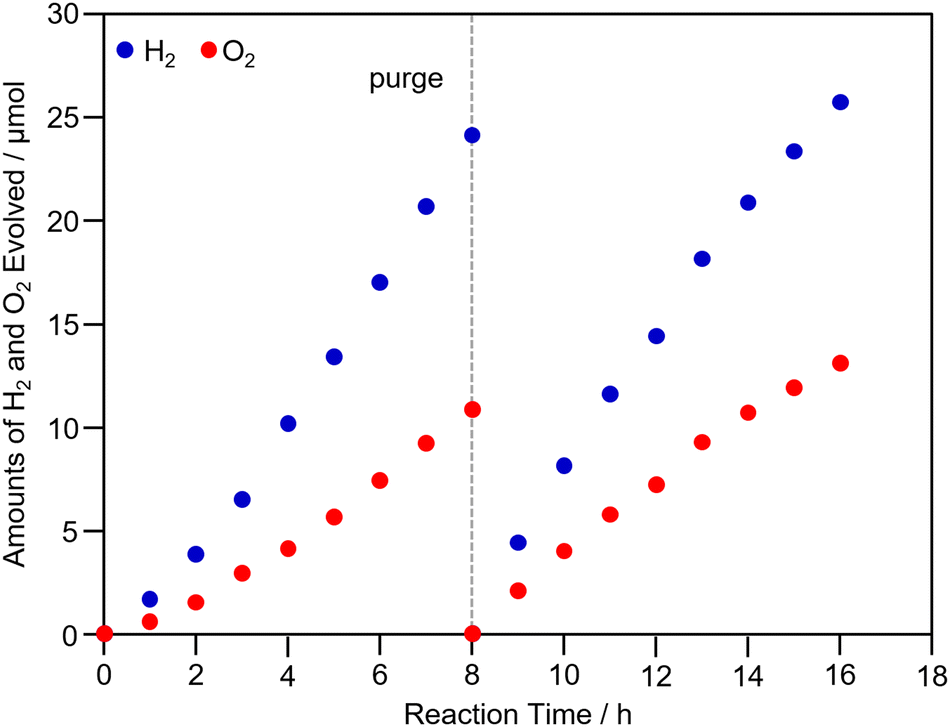 | ||
| Fig. 6 Time course of water-splitting activity for the PtNC/STO:Rh-BiVO4 composite Z-scheme system using FeCl2 as a mediator. Light source: a 300 W Xe lamp with a long-pass filter (>∼410 nm), catalyst: 100 mg for each catalyst, reactant solution: 120 mL of 2 mmol L−1 FeCl2 in H2SO4 aq. (pH 2.4), reaction cell: a top-window reaction cell. | ||
Conclusions
In this study, we loaded Pt cocatalysts with a fine particle size of 1.5 nm onto STO:Rh, which is a promising visible-light-driven photocatalyst, using pre-synthesized hydrophilic Pt∼51 NCs as a precursor. The novel PtNC/STO:Rh-light photocatalyst exhibited 30% higher H2-evolution activity than PtPD/STO:Rh. This improvement was mainly attributed to the increase in active sites when using finer Pt particles. Furthermore, by using the appropriate mediator, the Z-scheme water splitting proceeded stoichiometrically using PtNC/STO:Rh as the HEP. Although STO:Rh has been frequently used as the HEP in Z-scheme water splitting, the reaction on STO:Rh is the rate-limiting step in most cases. The method developed in this report for increasing the activity of STO:Rh may lead to further improvements in the performance of novel visible-light-driven Z-scheme water-splitting systems.Author contributions
T. K. and Y. N. conceived the research and designed the experiments. T. K. and Y. N. designed the synthesis and photocatalytic tests. D. Y., T. T., D. H., and Y. S. performed the synthesis, characterization, and photocatalytic activity. T. K. and Y. N. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant number 20H02698, 20H02552), Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion” (grant number 18H05178 and 20H05115), Scientific Research on Innovative Areas “Hydrogenomics” (grant number 21H00027) and Joint Usage/Research Center for Catalysis (Proposal 22AY0056 and 23AY0189). Funding from the Takahashi Industrial and Economic Research Foundation, the Yazaki Memorial Foundation for Science and Technology, the Ogasawara Foundation for the Promotion of Science and Engineering, the Kao Foundation for Arts and Sciences, the Sasakawa Scientific Research Grant from the Japan Science Society, TEPCO Memorial Foundation Research Grant (Basic Research), Advanced Technology Institute Research Grants 2022 and the Kumagai Foundation for Science and Technology is gratefully acknowledged.References
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825–2850 RSC.
- T. Hisatomi, K. Takanabe and K. Domen, Catal. Lett., 2015, 145, 95–108 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- S. Kawasaki, K. Akagi, K. Nakatsuji, S. Yamamoto, I. Matsuda, Y. Harada, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, C. Sakai, H. Niwa, M. Oshima, K. Iwashina and A. Kudo, J. Phys. Chem. C, 2012, 116, 24445–24448 CrossRef CAS.
- B. Modak and S. K. Ghosh, J. Phys. Chem. B, 2015, 119, 11089–11098 CrossRef CAS PubMed.
- D. H. K. Murthy, H. Matsuzaki, Q. Wang, Y. Suzuki, K. Seki, T. Hisatomi, T. Yamada, A. Kudo, K. Domen and A. Furube, Sustainable Energy Fuels, 2019, 3, 208–218 RSC.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 RSC.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CrossRef CAS.
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS.
- Q. Wang, Y. Li, T. Hisatomi, M. Nakabayashi, N. Shibata, J. Kubota and K. Domen, J. Catal., 2015, 328, 308–315 CrossRef CAS.
- Q. Wang, T. Hisatomi, Y. Suzuki, Z. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675–1683 CrossRef CAS PubMed.
- B. Moss, Q. Wang, K. T. Butler, R. Grau-Crespo, S. Selim, A. Regoutz, T. Hisatomi, R. Godin, D. J. Payne, A. Kafizas, K. Domen, L. Steier and J. R. Durrant, Nat. Mater., 2021, 20, 511–517 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- H. Kato, Y. Sasaki, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef CAS.
- Q. Wang, S. Okunaka, H. Tokudome, T. Hisatomi, M. Nakabayashi, N. Shibata, T. Yamada and K. Domen, Joule, 2018, 2, 2667–2680 CrossRef CAS.
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 150–151, 187–196 CrossRef CAS.
- S. S. K. Ma, K. Maeda, T. Hisatomi, M. Tabata, A. Kudo and K. Domen, Chem. – Eur. J., 2013, 19, 7480–7486 CrossRef CAS PubMed.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS PubMed.
- K. Ogawa, A. Nakada, H. Suzuki, O. Tomita, M. Higashi, A. Saeki, H. Kageyama and R. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 5642–5650 CrossRef CAS PubMed.
- H. Kumagai, R. Aoyagi, K. Kato, A. Yamakata, M. Kakihana and H. Kato, ACS Appl. Energy Mater., 2021, 4, 2056–2060 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4317–4318 CrossRef CAS.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 12, 543–544 RSC.
- Z. Wang, Y. Luo, T. Hisatomi, J. J. M. Vequizo, S. Suzuki, S. Chen, M. Nakabayashi, L. Lin, Z. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed.
- L. Qin, G. Si, X. Li and S.-Z. Kang, RSC Adv., 2015, 5, 102593–102598 RSC.
- S. Y. Xiao, Y. Liu, X. F. Wu, L. T. Gan, H. Y. Lin, L. R. Zheng, S. Dai, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2021, 9, 14786–14792 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- T. Kawawaki, Y. Kataoka, S. Ozaki, M. Kawachi, M. Hirata and Y. Negishi, Chem. Commun., 2021, 57, 417–440 RSC.
- T. Kawawaki, Y. Mori, K. Wakamatsu, S. Ozaki, M. Kawachi, S. Hossain and Y. Negishi, J. Mater. Chem. A, 2020, 8, 16081–16113 RSC.
- T. Kawawaki, Y. Negishi and H. Kawasaki, Nanoscale Adv., 2020, 2, 17–36 RSC.
- N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106–109 RSC.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- M. Luo, Y. Hong, W. Yao, C. Huang, Q. Xu and Q. Wu, J. Mater. Chem. A, 2015, 3, 2770–2775 RSC.
- S. Cao, J. Jiang, B. Zhu and J. Yu, Phys. Chem. Chem. Phys., 2016, 18, 19457–19463 RSC.
- M. Luo, P. Lu, W. Yao, C. Huang, Q. Xu, Q. Wu, Y. Kuwahara and H. Yamashita, ACS Appl. Mater. Interfaces, 2016, 8, 20667–20674 CrossRef CAS PubMed.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- W. Kurashige, Y. Mori, S. Ozaki, M. Kawachi, S. Hossain, T. Kawawaki, C. J. Shearer, A. Iwase, G. F. Metha, S. Yamazoe, A. Kudo and Y. Negishi, Angew. Chem., Int. Ed., 2020, 59, 7076–7082 CrossRef CAS PubMed.
- D. Yazaki, T. Kawawaki, D. Hirayama, M. Kawachi, K. Kato, S. Oguchi, Y. Yamaguchi, S. Kikkawa, Y. Ueki, S. Hossain, D. J. Osborn, F. Ozaki, S. Tanaka, J. Yoshinobu, G. F. Metha, S. Yamazoe, A. Kudo, A. Yamakata and Y. Negishi, Small, in press.
- C. M. Pelicano, M. Saruyama, R. Takahata, R. Sato, Y. Kitahama, H. Matsuzaki, T. Yamada, T. Hisatomi, K. Domen and T. Teranishi, Adv. Funct. Mater., 2022, 32, 2202987 CrossRef CAS.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Akinaga, R. Takahata, K. Wakamatsu, Y. Fujiki, M. Kataoka, S. Kikkawa, A. S. Alotabi, S. Hossain, D. J. Osborn, T. Teranishi, G. G. Andersson, G. F. Metha, S. Yamazoe and Y. Negishi, Angew. Chem., Int. Ed., 2021, 60, 21340–21350 CrossRef CAS PubMed.
- T. Kawawaki, N. Shimizu, K. Funai, Y. Mitomi, S. Hossain, S. Kikkawa, D. J. Osborn, S. Yamazoe, G. F. Metha and Y. Negishi, Nanoscale, 2021, 13, 14679–14687 RSC.
- S. E. Creager and C. M. Steiger, Langmuir, 1995, 11, 1852–1854 CrossRef CAS.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513–1519 RSC.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, and experimental, characterization, peak assignments of FT-IR, additional figures, characterization of Pt NCs, additional UV-Vis, XPS, FT-IR and FT-EXAFS spectra, SEM and TEM images, and photocatalytic activity. Correspondence and requests for materials should be addressed to T.K. and Y.N. See DOI: https://doi.org/10.1039/d3ya00159h |
| This journal is © The Royal Society of Chemistry 2023 |