
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Which insights can gas diffusion electrode half-cell experiments give into activity trends and transport phenomena of membrane electrode assemblies?†
Nicolai
Schmitt
 a,
Mareike
Schmidt
a,
Jonathan E.
Mueller
b,
Lasse
Schmidt
b,
Michael
Trabold
a,
Katharina
Jeschonek
a and
Bastian J. M.
Etzold
*a
a,
Mareike
Schmidt
a,
Jonathan E.
Mueller
b,
Lasse
Schmidt
b,
Michael
Trabold
a,
Katharina
Jeschonek
a and
Bastian J. M.
Etzold
*a
aTechnical University of Darmstadt, Department of Chemistry, Ernst-Berl-Institut für Technische und Makromolekulare Chemie, 64287 Darmstadt, Germany. E-mail: bastian.etzold@tu-darmstadt.de
bVolkswagen AG, 38436, Wolfsburg, Germany
First published on 4th May 2023
Abstract
Gas diffusion electrode (GDE) half-cell setups were recently presented as a powerful tool to characterize oxygen reduction reaction (ORR) catalyst layers at fuel cell relevant potentials and current densities. In order to pave the way for a broad-based application of the technique, it is essential to assess the comparability of the GDE half-cell technique and real membrane electrode assembly (MEA) measurements. In order to face this concern, we investigate the transferability of trends from GDE half-cell experiments, in which the catalyst layer directly faces the liquid electrolyte, to MEA experiments with (i) an ionic liquid modified and unmodified Pt/C catalyst and (ii) Pt/C catalysts with and without nitrogen modified carbon support at different ionomer to carbon ratios. We show that GDE half-cell experiments can be used to reliably predict trends in catalytic activity for catalyst layers in real MEAs that are related to differences in oxygen mass transport. However, differences in catalytic activity being related to proton accessibility cannot be captured completely due to the differing interphase solid catalyst/liquid electrolyte in GDE testing and solid catalyst/solid electrolyte in MEA testing. In order to account for this, it may be necessary to introduce an ionomer between the catalyst layer and the liquid electrolyte during GDE evaluation, which would, however, dramatically increase the effort required to perform measurements. On the other hand, GDE testing with the catalyst layer being in direct contact with the liquid electrolyte is nevertheless of interest, because it allows for the study of oxygen mass transport properties at application-oriented current densities independent of other transport phenomena.
1. Introduction
Fuel cells converting the chemical energy stored in fuels into electricity are predicted to find increasing application as next-generation power devices due to their high efficiency combined with low emissions. Polymer electrolyte fuel cells (PEMFCs) in particular have received major attention in fuel cell research and development and are currently being commercialized or further developed for many stationary, portable and transportation power applications.1 Furthermore, the optimization of PEMFCs focusses mainly on reducing their cost by maximizing power at minimal precious metal (Pt) content or by avoiding Pt-based materials altogether, and on improvement of the energy conversion efficiency, which is still low compared with batteries. The main focus, therefore, lies on development of improved catalysts layers for the sluggish cathodic oxygen reduction reaction (ORR).2,3 A major obstacle is that catalysts, which are highly active in laboratory-scale test do not automatically maintain their performance when embedded within catalyst layers under industrially relevant conditions. Thus, there are many examples of catalysts which are highly active in fundamental rotating disk electrode (RDE) measurements; however, very few of these have been successfully implemented in membrane electrode assemblies (MEA) operating under industrial relevant conditions.4–6A major reason for the differing performance in RDE and MEA testing is the limited mass transport in RDE experiments resulting in low maximum current densities (max. 6 mA cmgeo−2 at 1600 rpm in RDE vs. up to 3000 mA cmgeo−2 in MEA) and the narrow potential range where catalyst kinetics can be investigated as a result. Additionally, RDE uses a smooth glassy carbon surface with low catalyst loadings (up to 20 μgPt cm−2) resulting in much thinner catalyst layers compared with MEA testing, where higher catalyst loadings (up to 500 μgPt cm−2) on larger gas permeable electrodes are utilized.7–9 Furthermore, in MEA measurements the catalyst layer is in contact with an ionomer membrane, which serves as a solid electrolyte, and with Nafion®, whose addition to the catalyst layer is essential to guarantee sufficient proton transport. In contrast, in RDE evaluation the catalyst is surrounded by liquid electrolyte ensuring good proton transport and Nafion® is mainly added to stabilize the catalyst ink dispersion during electrode preparation, so that a homogenous coating is formed.10,11 Last, but not least, RDE testing is carried out under dynamic potentiostat operation using cyclic voltammetry (CV), while MEA testing is carried out under stationary conditions using potential or current control.12,13
In order to improve the significance of the activity data collected in fundamental research, several approaches for evaluating PEMFC catalysts under conditions that avoid mass transport limitations have been introduced.14–19 Gas diffusions electrode (GDE) half-cells offer a highly promising approach, which avoids mass transport limitations and fulfills the criteria necessary for wide applicability in standard research.14,15,20,21 Mass transport limitations obtained in RDE studies are avoided by distributing the reactant gas directly to the catalyst via a gas diffusion layer (GDL). Most recently, an Inter-lab comparison demonstrated that GDE testing with various, slightly differing setups can deliver data that is both reliable and comparable if standardized measurement protocols are followed.22 While these GDE setups were so far mostly utilized at room temperature and limited to ambient pressure, a setup was recently presented, which allows measurements at temperatures up to 120 °C and pressures of up to 4 bar.23
What is still under debate, is how GDE measurements differ in detail from MEA measurements and what possible limitations the technique has. Despite the temperature difference (mostly room temperature in GDE testing, 80 °C in MEA testing), one important issue is whether GDE half-cell characterization is carried out with or without an ionomer membrane between the catalyst surface and the liquid electrolyte. So far both variants have been utilized in GDE testing and no standard procedure exists, that was agreed on.14,15,24–26 Utilizing a solid electrolyte results in an environment that is much more complex and heterogeneous than the environment in aqueous electrolyte cells. Therefore, comparing both systems can introduce uncertainty regarding interfacial phenomena, reactant solubility, differences in local and bulk pH and transport phenomena such as removal of product water.27 Recently, Ehelebe et al. have shown that degradation of the catalyst layer in long-term durability tests characterizing Pt dissolution is significantly different when the catalyst layer and liquid electrolyte are separated by a membrane.28 As a consequence, employing a membrane as a solid electrolyte in GDE measurements will better mimic the environment in real membrane electrode assemblies. However, in contrast to MEA measurements, it is technically difficult to compress the ionomer membrane and the GDL in liquid half-cells, and thus ensure optimum proton accessibility at the catalyst surface. Furthermore, the production method of the CL and membrane interface plays a critical role, and the resulting interfacial boundary strongly influences the performance at high current densities. Thus, fast, reliable catalyst testing at high current densities is not feasible because the layer/membrane contacting needs to be optimized for each new catalyst, an effort which requires extensive MEA testing. Additionally, measurements employing ionomer membranes require elevated temperatures to provide sufficient proton mobility, which means one must account for water management within the membrane and regulate the humidity of the gases.15 Taken together these differences highlight the dramatic increase in technical complexity and experimental effort that is required to utilize an ionomer membrane as solid electrolyte in GDE experiments. Therefore, it should first be clarified, what GDE half-cell measurements without ionomer membranes provide and especially, how well-suited they are for predicting trends in catalyst activity at high current densities.
In order to address this concern, we compare GDE data collected at room temperature without utilization of an ionomer membrane (direct contact of the catalyst layer to the liquid electrolyte) with MEA data collected at 80 °C. As a test cases we evaluate two catalyst modifications that are the subject of current scientific discussion in both GDE and MEA setups. The catalyst modifications of interest are: (i) an ionic liquid modified and unmodified Pt/C catalyst and (ii) Pt/C catalyst with and without nitrogen modified carbon support at different ionomer to carbon (I/C) ratios.
The state of the art on both modifications is briefly summarized as follows.
Modifying a Pt/C catalyst with Ionic liquids (ILs) allows us to manipulate the microenvironment of the active site, liquid electrolyte and gaseous reactant. The concept of modifying a solid catalyst with ionic liquid was first presented in 2007 by Kernchen et al. for a heterogeneous catalyzed gas phase reactions and the term “SCILL” (supported catalyst with ionic liquid layer) was invented for this kind of catalyst modification.29 Snyder et al. were the first to bring IL modifications to electrocatalysis and used a similar approach to improve the ORR activity of a PtNi/C catalyst in fundamental RDE studies.30 These promising results were later validated by Etzold et al., who applied the SCILL concepts to carbon supported Pt and Pt-alloy catalysts, utilizing many different kinds of ILs. However, these studies also showed that the amount of IL needs to be balanced very accurately, since excess IL added can result in mass transport limitations, most likely caused by their influence on oxygen diffusivity, and even be observed during low current density RDE testing.31–34 So far, the extraordinary effect of ILs obtained in these RDE studies could rarely be transferred to high current density MEA testing and there are few examples for successful implementation of IL-modified Pt/C catalysts into real MEA applications.35–37 Therefore, this material was chosen as a first example to assess the capability of GDE testing to predict and to distinguish catalyst layer activity trends in the low and the high current density regime.
N-doping of the Pt/C carbon support has been proposed to improve the ionomer-catalyst interaction in PEMFC catalyst layers by introducing nitrogen groups, which develop coulombic interactions with –SO3− groups in the ionomer, resulting in more homogeneous ionomer distribution over the catalyst layer.38–41 According to this amine groups introduced on the carbon surface can react with sulfonic acid groups of the ionomer resulting in positively charged –NH3+ groups leading to strong coulombic attraction between carbon support and ionomer side chains.42,43 Orfanidi et al. and Ott et al. used ammonolysis of Vulcan XC 72R and Ketjenblack EC-300J carbon support, respectively, to apply this concept.39,40 In both studies improved performance in MEA testing was observed and was attributed to improved ionomer distribution after N-doping, resulting in improved proton accessibility to the Pt active sites, and also to the formation of highly accessible pores, optimizing oxygen mass transport. While it is known that adding sufficient amount of ionomer added to the catalyst layer is crucial to ensure sufficient proton conductivity in MEAs, this may be different for GDE testing, since the catalyst layer is in direct contact with the liquid electrolyte. It is not known so far, whether this can result in important differences in catalyst evaluation between the two techniques. In order to further elucidate the effect of N-doping on the ionomer interaction, a variation of the I/C ratio on both unmodified and N-doped Pt/C will be carried out in the present work.
2. Experimental
2.1 Synthesis of ionic liquid modified Pt/C catalyst
IL modified samples were prepared by coating Pt/C catalyst (HiSPEC®3000, 20 wt% Pt) with the ionic liquid [BMIM][beti]. In a typical synthesis procedure 30 mg of catalyst were mixed with 10 mL of isopropyl alcohol containing a certain amount of the IL in a round bottom flask. The mixture was stirred for 30 min at room temperature followed by 30 min of ultrasonication. Then the solvent was slowly removed from the solids by rotary evaporation under low vacuum (120 mbar, 40 °C). After total removal of isopropyl alcohol, the pressure was then further decreased to 10 mbar to ensure full intrusion of IL into the catalyst pores. Varying amounts of IL in isopropyl alcohol solution were used to achieve proportions of IL in the final catalyst of 5, 15 and 20 wt%, respectively.2.2 Nitrogen modification of carbon support
N modification was carried out by using a procedure outlined in literature.44 Therefore, 350 mg of carbon (Ketjenblack® EC300-J) was introduced into a 250 mL three-neck flask together with 12.7 mL of acetic anhydride (99%, Acros Organics). The mixture was placed in an ice bath and 5.6 mL of nitric acid (65 wt%, Acros Organics) was slowly added while stirring. The resulting mixture was stirred on ice for another 5 hours and then for 19 hours at room temperature. Afterwards, the solids were filtered and washed with distilled water until pH neutrality and then dried in a vacuum oven at 70 °C for 12 h. Finally, in order to reduce the amount of acidic oxygen-containing groups, the modified carbon was subjected to a thermal treatment in a tubular furnace (Gero F-A-40-200/13) in N2-atmosphere at a temperature of 800 °C for 2 hours, resulting in the final N-modified carbon support.The carbon support was analyzed before and after N modification with N2 Physisorption and elemental analysis. Details can be found in the ESI.†
2.3 Deposition of Pt on carbon supports
Deposition of platinum on Ketjenblack® EC300-J and on the N-doped carbon was carried out via wet impregnation aiming for a theoretical loading of 40 wt% Pt on carbon. Therefore, 140 mg of Chloroplatinic acid hexahydrate (99.9%, abcr Chemie) was dissolved in 1 mL of Ethanol and then mixed thoroughly with 100 mg of the respective carbon support. In order to remove the solvent, the sample was thereafter dried at 60 °C in a vacuum oven for 12 h.After impregnation of the carbon support with the precursor solution, a gas phase reduction was carried out in a horizontal tubular furnace (Gero F-A-40-200/13). To do this the impregnated sample was transferred into a ceramic bowl (50 × 35 × 12 mm) and positioned in the isothermal zone of the furnace. Afterwards, the sample was heated to 250 °C in a nitrogen gas flow of 10 LN h−1 with a heating ramp of 2.5 K min−1. For reduction of the Pt precursor, the temperature was held for three hours in a mixed gas stream of 7 LN h−1 N2 and 3 LN h−1 H2. Finally, the furnace was cooled down to room temperature under a gas flow of 10 LN h−1 N2 and the resulting Pt/C catalyst was collected and weighed.
The synthesized Pt/C catalysts were analyzed regarding their Pt content via ICP-OES. Details can be found in the ESI.†
2.4 RDE characterization
RDE characterization was carried out on all ionic liquid modified samples and the unmodified HiSPEC®3000 catalyst. The RDE measurements were performed on a Ivium Multichannel Potentiostat (Octostat 5000), which is controlled by IviumSoft software. As a reference electrode, a leak-free double-junction Ag/AgCl electrode (Aldrich) was used. A Pt wire (PINE) served as counter electrode. All potentials reported in this work were calibrated against a reversible hydrogen electrode (RHE) using hydrogen evolution-oxidation reaction on a Pt electrode. For each catalyst sample, two RDE tips were prepared and tested for reproducibility purposes, in order to give the standard deviation between two individual measurements. Further details on the RDE measurements are summarized in the ESI.†2.5 GDE characterization
GDE evaluation was carried out in an automated setup and using a commercial GDE half-cell (Flexcell® PTFE, Gaskatel GmbH) operated at room temperature in 2 M HClO4 as electrolyte. For GDE preparation the respective catalyst was deposited on the gas diffusion media (Sigracet 25 BC, SGL Carbon) by using a drop-casting approach, resulting in a catalyst loading of 100 μgPt cm−2. Detailed information on the coating technique and the measurement procedure is given in our recent publications26,45 and in the ESI.† For the investigation of HiSPEC® 3000 catalyst and the IL modified samples, the I/C ratio was set to 0.5. For the Pt/Ketjenblack catalyst and the N-modified samples, varying I/C ratios of 0.1, 0.5 and 1.7 were investigated. For each catalyst sample, two GDEs were prepared and tested for reproducibility purposes.2.6 MEA characterization
For MEA fabrication, GDE cathodes with a size of 5 cm2 were prepared by spray coating using a robot assisted ultrasound nozzle spraying station that includes a CNC table (High-Z S-400, CNC step), an ultrasonic atomizer spray nozzle (Sonozap, Sonaer Ultrasonics) and a syringe pump for catalyst ink feeding. The catalyst ink composition is identical to the one used for half-cell experiments, but contains a higher concentration of solids (8 mg catalyst per 5 mL of solution). During spray coating, the gas diffusion layers (Sigracet 25 BC, SGL Carbon) were heated to 125 °C on a heating plate and the ink feed rate and spraying time were adapted to aim catalyst loadings of 200 μgPt cm−2 on the cathode. After spray coating, the obtained cathode GDEs were weighed to determine the exact catalyst loading and were then hot-pressed together with Nafion™ NR211 membrane (thickness = 25 μm) and a commercial Pt/C GDE (0.2 mgPt cm−2, 20% Pt on Vulcan, FuelCellsEtc) as anode at a temperature of 125 °C and a pressure in relation to the electrode area of 96 bar (LaboPress P200S-VAK, Vogt Labormaschinen GmbH), to obtain the final membrane electrode assemblies. Fuel cell experiments were carried out at 80 °C and a relative humidity (RH) of 17 and 100%, respectively, without backpressure on a Scribner Model 850e (Scribner Associates) under power-optimized conditions with H2 (0.2 L min−1) and O2 (0.2 L min−1). The MEAs were pre-treated with a break-in procedure consisting of repetitive potential steps at OCV, 0.3 V and 0.6 V respectively until a stable fuel cell performance was observed. The polarization data was measured galvanostatically from OCV to a total maximum current of 15 A with 10 measurement points per decade. For iR-correction, the cell resistance at every recorded polarization data point was measured automatically by the test station via current interrupt. For MEA characterization of HiSPEC® 3000 catalyst and the IL modified samples, the I/C ratio was set to 0.5. For the Pt/Ketjenblack catalyst and the N-modified samples, varying I/C ratios of 0.1, 0.5 and 1.7 were investigated. For each catalyst sample, two MEAs were prepared and tested for reproducibility purposes.3. Results
3.1 ORR performance of IL-modified Pt/C in RDE, GDE and MEA
We first performed a comparative study of the influence of modifying a commercial Pt/C catalyst (HiSPEC®3000, 20 wt% Pt) with varying amounts of the ionic liquid [BMIM][beti] in RDE, GDE and MEA. Fig. 1 compares ORR activity of the different catalyst materials obtained in RDE, GDE (both measured at room temperature) and MEA (80 °C, 100%RH) experiments. The corresponding ORR polarization curves for the RDE and the GDE measurements can be found in Fig. S1 in the ESI.†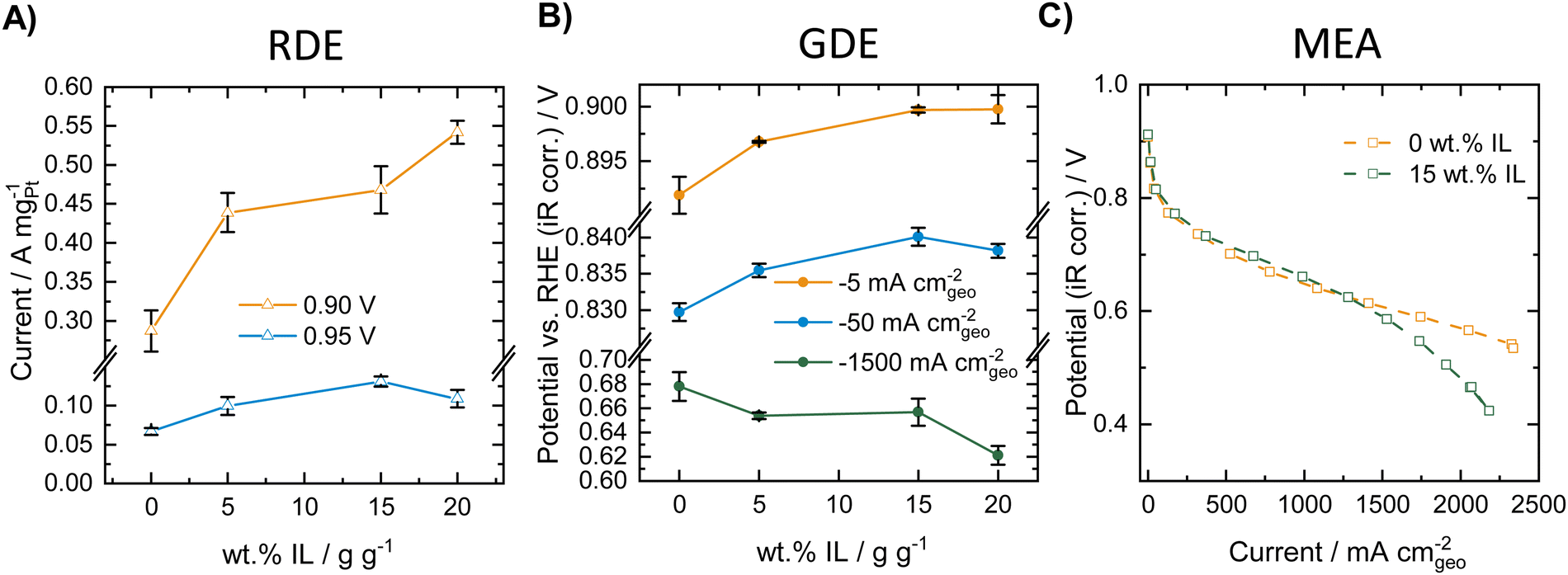 | ||
| Fig. 1 ORR activity of Pt/C catalyst modified with [BMIM][beti]. (A) Mass specific ORR activity, depending on the amount of IL obtained in RDE measurements at room temperature in oxygen saturated 0.1 M HClO4 and 1600 rpm at a catalyst loading of 20 μgPt cm−2. (B) ORR potentials at different current densities depending on the amount of IL obtained in GDE measurements at room temperature in oxygen atmosphere in 2 M HClO4 at a catalyst loading of 100 μgPt cm−2. (C) ORR polarization curve of unmodified Pt/C and Pt/C modified with 15 wt% of IL measured in a MEA at 80 °C and 100%RH in oxygen atmosphere (1 atm) at a catalyst loading of 100 μgPt cm−2. | ||
As can be seen in Fig. 1A, the mass specific activity extracted from the RDE polarization curves increases with increasing amount of ionic liquid both at potentials of 0.90 and 0.95 V vs. RHE. This indicates a strong activity-boosting effect of the IL on the intrinsic ORR activity of the Pt/C catalyst and is well in line with earlier RDE studies.30–36,46,47Fig. 1B shows ORR activity data obtained for the different IL modified samples in the GDE half-cell setup and presents the measured ORR potential at different geometric current densities of −5, −50 and −1500 mA cmgeo−2. The results indicate that at −5 and −50 mA cmgeo−2, the ORR potential increases with increasing amount of IL added to the Pt/C catalyst. Thus, similarly to the RDE results, an activity boosting effect of the IL is obtained in the low current density regime in the GDE setup. In the high current density regime, however, a reverse trend is visible and the measured potential decreases with increasing amount of IL added, indicating a decreasing ORR activity. MEA polarization curves were recorded for the unmodified catalyst and the 15 wt% IL modified sample. Fig. 1C shows the polarization curves obtained in oxygen atmosphere at 80 °C and 100%RH. As can be seen, at low and intermediate current densities, a slight increase in ORR activity is obtained after IL-modification. However, in the high current density regime a worsening of the performance is obtained after IL modification and the corresponding curve shows a strong loss of activity at high current densities. Thus, in the case of IL-modified Pt/C catalyst, similar trends can be observed in GDE and in MEA measurements. The GDE half-cell experiments could therefore very well describe the behavior of the catalysts in the different current regimes, while only considering the RDE data would result in erroneous conclusions. GDE and MEA experiments were also carried out in synthetic air (20% O2 in N2), which is presented in Fig. S2 (ESI†). Herein, identical trends could be observed compared to the measurements in pure oxygen and both setups show an increase in activity at low and intermediate current densities, while in the high current density regime, the IL modification introduces mass transport limitations.
3.2 ORR performance of N-modified Pt/C in GDE and MEA at different ionomer to carbon ratio
In the second example for comparison of ORR activity data collected in GDE and in MEA measurements, a self-synthesized Pt/C catalyst with and without nitrogen modification of the Ketjenblack carbon support was investigated. Elemental analysis of the carbon support before and after the nitrogen modification revealed successful N-doping with a N content of 0.23 at% (see Table S4, ESI†). Physisorption analysis shows that the modification treatment also results in a slight increase in the surface area as determined by application of both a BET analysis and a QSDFT model, as well as a minor increase in the specific pore volume (see Table S3, ESI†).After deposition of Pt on the untreated and on the N-doped carbon support, the resulting Pt/C and Pt/N-C catalysts were analyzed both in the GDE half-cell and in MEA measurements. Thereby, different I/C ratios of 0.1, 0.5 and 1.7 were investigated for the respective catalyst sample. GDE evaluation was carried out at room temperature in oxygen atmosphere, MEA evaluation was carried out at 80 °C both at 17 and 100%RH in oxygen atmosphere. For direct comparison of GDE and MEA data, the GDE data was corrected for the differing reaction conditions (temperature + partial pressure of oxygen reactant). To do this a kinetic and a thermodynamic correction of the GDE data was carried out following14 (for details see ESI†). Furthermore, from the obtained polarization curves, the mass specific activity was calculated, since the catalyst loading varied slightly between the different measurements (see Table S5, ESI†). Fig. 2 shows the resulting mass specific ORR polarization curves of the N-doped and undoped catalysts in the GDE half-cell after correcting the data and in the MEA at 100%RH. GDE polarization curves before and after correction of the data, as well as MEA polarization curves showing the geometric current density are presented in Fig. S3 and S4 (ESI†).
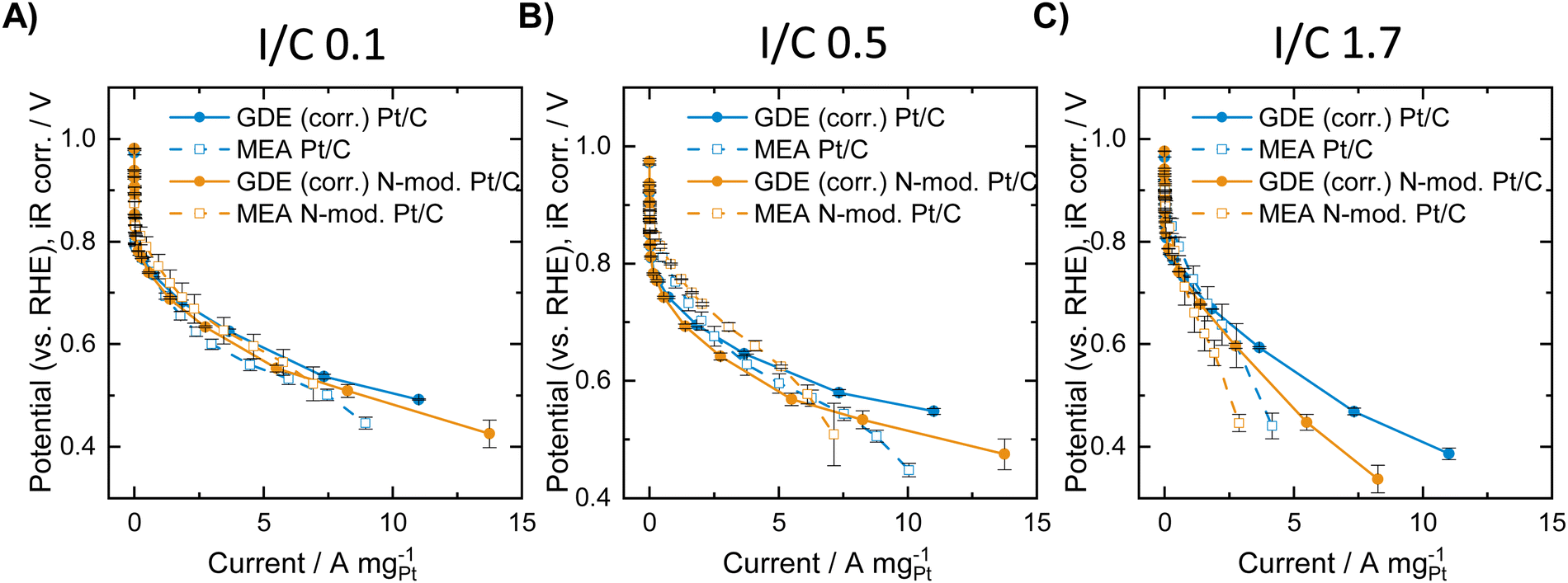 | ||
| Fig. 2 Mass specific ORR polarization curves of the unmodified and N-doped Pt/C catalysts obtained in MEA measurements at 80 °C and 100%RH in oxygen atmosphere (1 atm) and in the GDE half-cell (data collected at room temperature in 2 M HClO4 in oxygen atmosphere and thereafter thermodynamically and kinetically corrected for MEA conditions). (A) I/C ratio = 0.1 (B) I/C ratio = 0.5 (C) I/C ratio = 1.7. | ||
Fig. 2A shows the obtained polarization curves with and without N-modification at an I/C ratio of 0.1. The results indicate a slight improvement of the polarization curve obtained in the MEA after N-modification in the low and intermediate mass specific current regime, and similar activity compared to the unmodified catalyst at high mass specific currents. The GDE half-cell experiments generally deliver very similar mass specific activity curves compared to MEA testing after the applied correction, although the correction can only be seen as rough estimation, since it does not cover any mass transport and MEA effects such as hydrogen crossover and resistance of the ionomer membrane. Furthermore, differences in local pH due to the differing environment solid catalyst/solid ionomer in MEA and solid catalyst/liquid electrolyte in GDE testing are not considered. A detailed look at the GDE results for an I/C ratio of 0.1, however, indicates a reverse trend compared with MEA testing with the unmodified catalyst exhibiting better performance over the entire current range. The results obtained for an I/C ratio of 0.5 in Fig. 2B reveal identical trends with an improvement after N-modification in the MEA and a worsening of the performance in the GDE. Direct comparison of the curves generally reveals the following: in the ohmic regime, MEA activity is better compared to GDE activity, while in the mass-transport limiting regime higher activity is obtained in the GDE half-cell. This is well in line with results earlier presented by Ehelebe et al.14 for a commercial Pt/C catalyst. Fig. 2C shows the obtained mass specific polarization curves at an I/C ratio of 1.7. In this case GDE and MEA data shows the same trend with a better performance of the unmodified catalyst in the high mass specific current regime. For further analysis of the obtained data, voltage losses were determined when lowering or increasing the I/C ratio compared to an I/C ratio of 0.5. Therefore, for the respective unmodified or N-doped material, the polarization curves obtained at an I/C ratio of 0.1 and 1.7, respectively, were subtracted from the ones obtained at I/C 0.5. The resulting voltage loss curves are presented in Fig. 3.
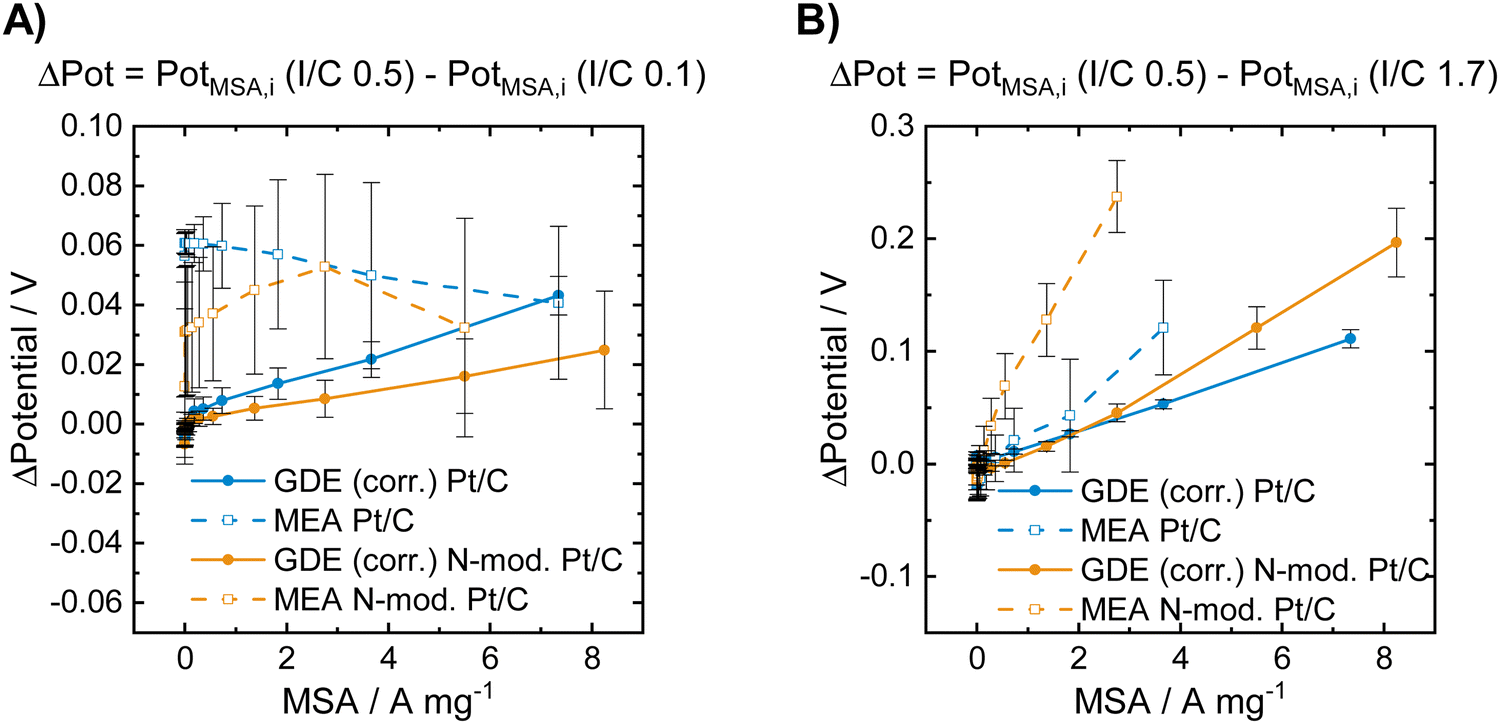 | ||
| Fig. 3 Voltage losses obtained for the unmodified and for the N-doped Pt/C catalysts in GDE and in MEA characterization at low and high I/C ratio compared to an I/C ratio of 0.5. The data presented in (A) was obtained by subtracting the polarization curve of the respective catalyst in the respective cell type shown in Fig. 2B by the one shown in Fig. 2A. The data presented in (B) was obtained by subtracting the polarization curve of the respective catalyst in the respective cell type shown in Fig. 2B by the one shown in Fig. 2C. | ||
Fig. 3A shows that a lowering of the I/C ratio results in significant activity losses in the MEA in the low current regime. These losses are not visible in the GDE half-cell, where at low mass specific currents no performance loss compared to an I/C ratio of 0.5 is obtained for both unmodified and N-doped catalyst. In Fig. 3B the obtained potential losses after increasing the I/C ratio to 1.7 is shown. In this case no activity losses are obtained for the materials in both GDE and MEA in the low current regime. At higher currents, however, strong activity losses and identical trends are observed in both cell types, while the absolute numbers differ between GDE and MEA. These activity losses might be attributed to increased oxygen mass transport limitations at higher currents for excess ionomer content and the resulting diffusion barrier through the thick ionomer film.48
MEA mesaurements carried out under dry conditions at 17%RH with both unmodified and N-doped Pt/C are presented in Fig. S5 and S6 (ESI†). In this case N-doping results in a dramatic increase in performance for all I/C ratios. Since proton transport is limiting under dry conditions for non optimized ionomer coverage,49 this lets us assume that N modification of the carbon support could strongly improve dry proton accessibility of the Pt particles in the catalyst, as was previously shown by Ott et al.40
3.3 Which insights can gas diffusion electrode half-cell experiments give into activity trends and transport phenomena of membrane electrode assemblies?
The two model materials investigated in this study present the following outcome: in the case of IL-modified and unmodified Pt/C catalyst identical trends are observed in GDE and in MEA, with an increase in activity after IL modification at low and intermediate current densities and a lower activity compared to the pristine catalyst in the high current density regime. In the case of Pt/C catalyst with and without N-doped carbon support identical trends are observed at an I/C ratio of 1.7, with a decrease in activity after N-doping in the high current density regime. For lower I/C ratio of 0.1 and 0.5, however, only MEA evaluation showed an improvement after N-doping, while a reverse trend is observed in the GDE half-cell. Based on these observations, we subsequently want to consider the circumstances, under which GDE half-cell experiments with the catalyst layer in direct contact with the liquid electrolyte, can correctly predict trends for real MEAs. To do this, we now analyze each case, to determine which transport mechanism (proton accessibility or oxygen mass transport) limits the MEA performance of unmodified catalyst compared to modified catalyst, or vice versa.In the case of the ionic liquid modified catalyst, the activity boosting effect observed in RDE studies was mostly attributed to higher oxygen solubility in the IL compared to the liquid electrolyte and suppressed adsorption of non-reactive oxygenated species.30,32,33,50 Recent studies, however, point out that the improved proton conductivity in the catalyst layer after IL modification due to the higher ionic conductivity of ILs compared to water or liquid electrolyte is responsible for improved ORR kinetics.37,51,52 According to this, ILs fill micropores and smaller mesopores of high surface area carbons, which are not accessible by the ionomer due to size exclusion effects, and can thus improve proton accessibility to Pt active sites within these pores.37,53 Based on this, we exclude the hypothesis that the decrease in MEA performance of IL-modified Pt/C compared to unmodified Pt/C in the mass transport limiting regime is attributed to proton transport limitations of the IL-modified catalyst. More likely, pores filled with IL can affect oxygen transport to the Pt active sites within these pores, resulting in oxygen transport limitations at high current densities, where oxygen consumption is at a high level (see Fig. 4). This behavior would correspond well to the GDE half-cell measurement.
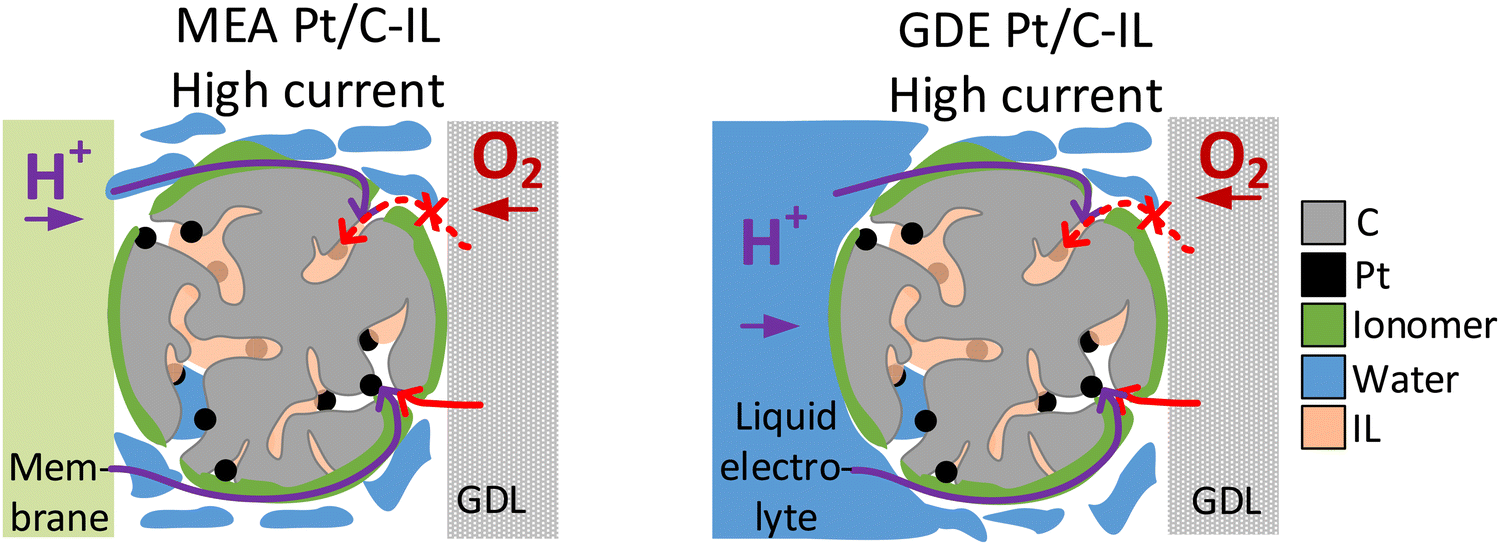 | ||
| Fig. 4 Schematic illustration of oxygen transport being affected by the ionic liquid modification of Pt/C in the high current density regime in MEA (left) and GDE (right). | ||
In the case of N-doped Pt/C catalyst, we see an improvement in the MEA performance after N modification for I/C ratios of 0.1 and 0.5, which is pronounced in the low and intermediate current regime. Ott et al.40 have shown for the same carbon support and a similar nitrogen modification procedure that at an I/C ratio of 0.66, N-doping results in a significant improvement of the dry proton accessibility due to more homogenous ionomer coverage. Thus, for the unmodified catalyst, the lower performance we observed might be related to inhomogeneous distribution of ionomer and result from uncovered Pt active sites on the carbon surface (see Fig. S7, ESI†) that suffer from proton transport losses. This is also supported by the fact, that the activity boosting effect introduced by N-doping is much more pronounced under dry conditions. These trends in activity linked to proton accessibility cannot be covered in GDE half-cell measurements with the catalyst layer being surrounded by liquid electrolyte. This also becomes obvious looking at the results in Fig. 3A, which show voltage losses at an I/C ratio of 0.1 compared to an I/C ratio of 0.5. In the low current regime, these voltage losses are pronounced in the MEA measurements, while no losses are observed during GDE evaluation. Thus, the liquid electrolyte surrounding the catalyst in the GDE half-cell can also ensure good proton accessibility for unideal ionomer coverage (see Fig. 5). This phenomenon is also well known in RDE evaluation, where measurements without ionomer are standard to avoid poisoning of the Pt surface by ionomer.54,55 Additionally, similar phenomena were also recently described by Lin et al. using a floating electrode (FE) setup.57 The FE technique generally follows the same principle as the herein presented GDE approach with the catalyst being in contact with the liquid electrolyte and the gaseous reactant being delivered through a porous hydrophobic gas diffusion media, but uses much thinner catalyst layers (catalyst loading <10 μgPt cm−2) and a different substrate (Au coated polymer membranes with hydrophobic coating).18 Lin et al. have compared commercial Pt/C catalyst at different I/C ratio and also without addition of any ionomer. It could be shown that the ionomer free catalyst layer exhibits similar activity compared to an I/C ratio of 1 at lower overpotential and even higher activity in the high current density regime, while the maximum geometric current densities were lower compared to this study (<300 mA cmgeo−2vs. 2500 mA cmgeo−2). In our study, in difference, improved performance in the high current density regime was found at an I/C ratio of 0.5 compared to 0.1. This might be linked to the much thicker catalyst layers utilized in this study compared to the FE approach. While for the ultrathin catalyst layers utilized in FE, also at a higher reaction rate good proton accessibility might be guaranteed by the surrounding electrolyte, in the thicker catalyst layers utilized in GDE, the ionomer might play a bigger role for proton transport and thus give a more realistic picture of trends for real MEAs.
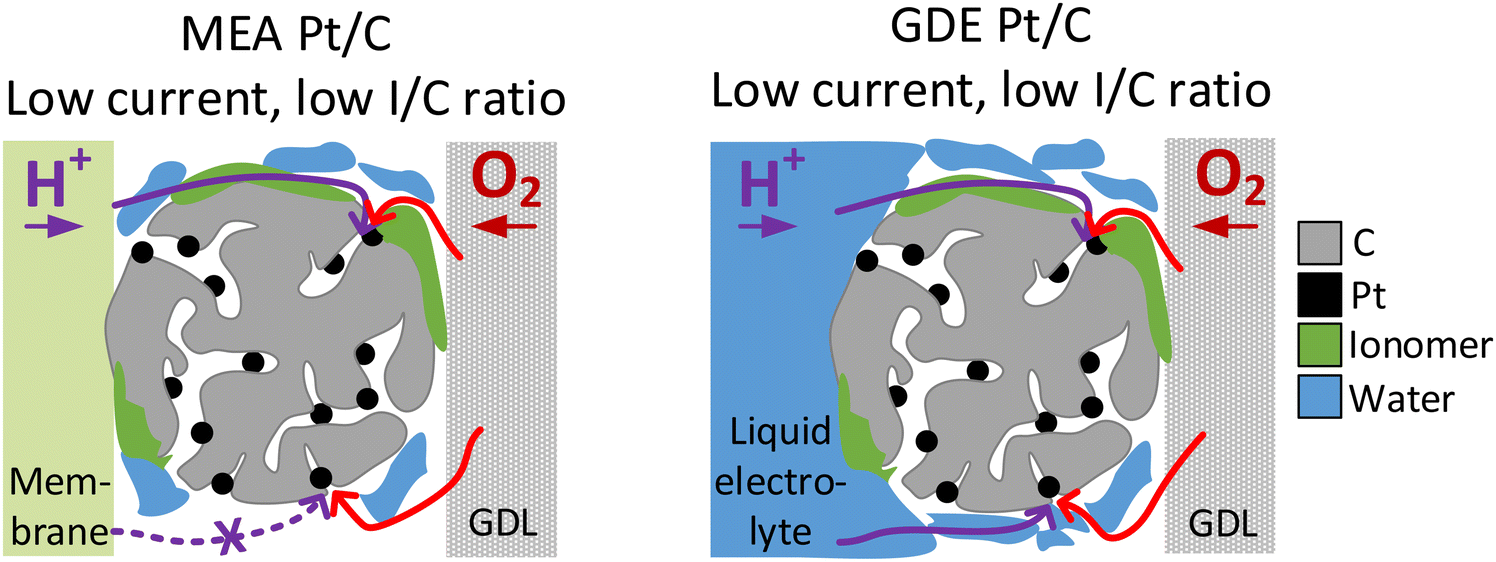 | ||
| Fig. 5 Comparison of proton accessibility of Pt active sites in MEA catalyst layers (left) and GDE catalyst layers (right) at low current densities and for low I/C ratio and/or unideal ionomer coverage. | ||
The presented observations, however, also imply that the catalyst layer is always flooded with electrolyte to a certain extent during GDE evaluation and raises the question whether this will affect oxygen transport. Indeed, we see better performance in the MEA compared with the GDE in Fig. 2 for all catalyst samples in the ohmic current regime. This observation could be linked to slower oxygen mass transport in the GDE due to partial flooding of the catalyst layer by the electrolyte. At even higher current densities this behavior is no longer observed and the GDE performance becomes better compared with the MEA performance for all investigated materials. This is connected with another interesting phenomenon: in MEA experiments it is well known that flooding the gas diffusion layer on the cathode side hinders gas transport and is thus responsible for the observed limiting current density.56 In contrast, in GDE experiments the water produced does not need to be transported through the cathodic gas diffusion electrode but will more likely dilute the aqueous acidic electrolyte instead. This is also supported by the fact, that no flooding of the GDLs is visually observed during GDE testing. This might explain how flooding the channels in the gas diffusion layer is prevented and the resulting improved performance of GDE over MEA in the mass transport limiting current regime (see Fig. 6). Besides differences in water removal pathways, this behaviour might also be linked to the differing measurement times in both GDE and MEA. For MEA polarization curves, the holding time per point was 60 s and a higher number of measurement points was applied in the high current density regime compared to GDE. In GDE, the number of high current density measurement points, as well as the holding time at these points (5 s) was kept low to prevent heating of the electrolyte. Thus, the amount of excess water produced in the MEA is also much higher, which could also result in excess flooding compared to the GDE. Similar behaviour and conclusions were recently also presented by Jackson et al. comparing MEA data of Pt/C with measurements carried out in a FE setup with ultrathin catalyst layers.59 Interestingly, in this study this behaviour in the MEA mass-transport limitation regime is thus also observed for much thicker catalyst layers compared to the FE and close to industrial application. The study by Jackson et al. generally has seen an improved performance of FE over MEA in the whole current regime, and does not show the behaviour seen in this study in the ohmic regime with a superior performance of MEA over GDE. This difference observed between GDE and FE is most likely linked to the differences in the catalyst layer thickness and partial flooding of the thicker catalyst layer in the GDE, as discussed above.
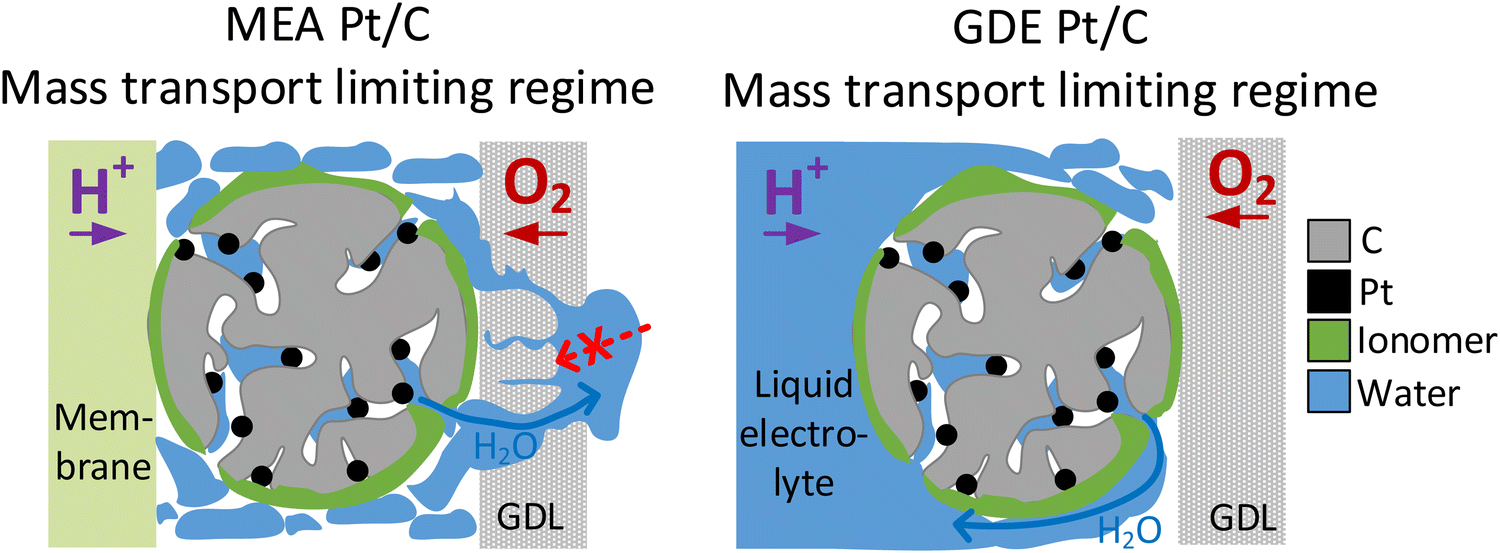 | ||
| Fig. 6 Comparison of water transport in the mass transport limiting regime in MEA catalyst layers (left) and GDE catalyst layers (right). | ||
For the N-doped Pt/C catalyst at an I/C ratio of 1.7, worse performance in the high current density regime and a stronger break-in of the polarization curve compared with the unmodified material was observed during MEA evaluation. Since the previous results indicated an improved proton accessibility after N-modification, these performance losses will be linked to increased oxygen transport losses for the N-doped catalyst, which are well described by GDE testing. Ott et al.58 showed that N modification not only influences the ionomer-carbon support interaction, but can also alter the pore structure of the carbon support. Thus, the harsh conditions during the nitric acid treatment performed in this study may have resulted in a detrimental change in the pore structure within the carbon support. This is not visible under dry conditions and in the case of low ionomer content, where proton transport losses are dominating for the unmodified catalyst. However, at an I/C ratio of 0.5 (in the high current density regime) and 1.7 under wet conditions, where proton transport is sufficient for both unmodified and N-doped catalyst, it becomes evident. This could also explain why the GDE experiments, which guarantee good proton accessibility due to the surrounding liquid electrolyte, display a decrease in performance after N-doping in the ohmic regime, independent of the I/C ratio.
4. Conclusion
In this study we systematically compared ORR data collected in GDE half-cell experiments, where the catalyst layer directly faces the liquid electrolyte, and in real membrane electrode assemblies. The investigated materials were (i) an ionic liquid modified and unmodified Pt/C catalyst and (ii) Pt/C catalyst on unmodified and nitrogen modified carbon supports at various ionomer to carbon ratios. In the case of the IL modified Pt/C catalyst it could be shown that oxygen mass transport is negatively affected by the IL in the high current density regime in MEA experiments, resulting in pronounced mass transport limitations compared to the unmodified catalyst. This behavior was well described in the GDE experiment, where correct trends in MEA activity could be predicted for the respective current regime. In the case of the N-doped Pt/C catalyst, MEA characterization displayed an improvement after N modification under dry conditions and in the case of low ionomer content, where proton transport losses are dominant for the unmodified catalyst. However, at higher I/C ratio and under wet conditions, where proton transport is sufficient for both unmodified and N-doped catalyst, worse performance after N modification is observed in the high current density regime. During GDE evaluation only the latter phenomenon could be captured and the unmodified catalyst showed a superior performance over the N-doped material at any I/C ratio in the ohmic and in the mass transport limiting regime.In conclusion, our results show that GDE half-cell experiments with the catalyst layer in direct contact with the liquid electrolyte are a reliable proxy to describe trends for real MEAs under high current densities in case of differences in catalytic activity being linked to oxygen mass transport. In particular, GDE testing is superior in this regard compared to the wide-spread RDE technique, which cannot capture the high-current density regime and can therefore give misleading results. However, for a reliable description of all transport phenomena in real fuel cells (e.g. proton accessibility of Pt active sites and water transport within the gas diffusion layer), introduction of an ionomer membrane between catalyst layer and liquid electrolyte might be necessary during GDE evaluation. On the other hand, our results also indicate that GDE measurements without the utilization of an ionomer membrane allow for the study of oxygen mass transport properties independent of other transport phenomena, which is typically not possible in a MEA setup. This valuable information will help guide future applications of the GDE technique for the evaluation of fuel cell electrocatalysts.
Author contributions
N. Schmitt: writing – original draft, writing – review & editing, conceptualization, visualization, methodology, investigation M. Schmidt + M. Trabold + K. Jeschonek: writing – review & editing, investigation J. Mueller + L. Schmidt: writing – review & editing, project administration, conceptualization; B. Etzold: writing – review & editing, supervision, conceptualization, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported financially by the Volkswagen AG. The results, opinions and conclusions expressed in this publication are not necessarily those of Volkswagen Aktiengesellschaft.References
- Y. Wang, Y. Pang, H. Xu, A. Martinez and K. S. Chen, Energy Environ. Sci., 2022, 15, 2288–2328 RSC.
- Y. Wang, D. F. R. Diaz, K. S. Chen, Z. Wang and X. C. Adroher, Mater. Today, 2020, 32, 178–203 CrossRef CAS.
- A. Alaswad, A. Omran, J. R. Sodre, T. Wilberforce, G. Pignatelli, M. Dassisti, A. Baroutaji and A. G. Olabi, Energies, 2020, 14, 144 CrossRef.
- B. Han, C. E. Carlton, A. Kongkanand, R. S. Kukreja, B. R. Theobald and L. Gan, Energy Environ. Sci., 2015, 8, 258–266 RSC.
- I. E. L. Stephens, J. Rossmeisl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed.
- A. Ly, T. Asset and P. Atanassov, J. Power Sources, 2020, 478, 228516 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354 CrossRef CAS.
- H. A. Gasteiger and S. G. Yan, J. Power Sources, 2004, 127, 162–171 CrossRef CAS.
- K. Shinozaki, J. W. Zack, S. Pylypenko, B. S. Pivovar and S. S. Kocha, J. Electrochem. Soc., 2015, 162, F1384 CrossRef CAS.
- Y. Garsany, I. L. Singer and K. E. Swider-Lyons, J. Electroanal. Chem., 2011, 662, 396–406 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- Y. Garsany, J. Ge, J. St-Pierre, R. Rocheleau and K. E. Swider-Lyons, J. Electrochem. Soc., 2014, 161, F628 CrossRef CAS.
- K. Ehelebe, D. Seeberger, M. T. Y. Paul, S. Thiele, K. J. J. Mayrhofer and S. Cherevko, J. Electrochem. Soc., 2019, 166, F1259 CrossRef CAS.
- M. Inaba, A. W. Jensen, G. W. Sievers, M. Escudero-Escribano, A. Zana and M. Arenz, Energy Environ. Sci., 2018, 11, 988–994 RSC.
- B. A. Pinaud, A. Bonakdarpour, L. Daniel, J. Sharman and D. P. Wilkinson, J. Electrochem. Soc., 2017, 164, F321–F327, DOI:10.1149/2.0891704jes.
- C. Zalitis, A. Kucernak, X. Lin and J. Sharman, ACS Catalysis, 2020, 10, 4361–4376 CrossRef CAS.
- C. M. Zalitis, D. Kramer and A. R. Kucernak, Phys. Chem. Chem. Phys., 2013, 15, 4329–4340, 10.1039/C3CP44431G.
- G. K. H. Wiberg, M. Fleige and M. Arenz, Rev. Sci. Instrum., 2015, 86, 24102 CrossRef PubMed.
- J. Schröder, J. Quinson, J. K. Mathiesen, J. J. K. Kirkensgaard, S. Alinejad, V. A. Mints and M. Arenz, J. Electrochem. Soc., 2020, 167, 134515 CrossRef.
- S. Alinejad, M. Inaba, J. Schröder, J. Du, J. Quinson, A. Zana and M. Arenz, J. Phys.: Energy, 2020, 2, 24003 CAS.
- K. Ehelebe, N. Schmitt, G. Sievers, A. W. Jensen, A. Hrnjić, P. Collantes Jiménez, P. Kaiser, M. Geuß, Y.-P. Ku and P. Jovanovič, ACS Energy Lett., 2022, 7, 816–826 CrossRef CAS.
- G. K. H. Wiberg, S. Nösberger and M. Arenz, Curr. Opin. Electrochem., 2022, 101129 CrossRef.
- S. Nösberger, J. Du, J. Quinson, E. Berner, A. Zana, G. K. H. Wiberg and M. Arenz, Electrochem. Sci. Adv., 2021, e2100190 Search PubMed.
- A. Hrnjić, F. Ruiz-Zepeda, M. Gaberšček, M. Bele, L. Suhadolnik, N. Hodnik and P. Jovanovič, J. Electrochem. Soc., 2020, 167, 166501 CrossRef.
- N. Schmitt, M. Schmidt, G. Hübner and B. J. M. Etzold, J. Power Sources, 2022, 539, 231530 CrossRef CAS.
- J. G. Petrovick, G. C. Anderson, D. I. Kushner, N. Danilovic and A. Z. Weber, J. Electrochem. Soc., 2021, 168, 56517 CrossRef CAS.
- K. Ehelebe, J. Knöppel, M. Bierling, B. Mayerhöfer, T. Böhm, N. Kulyk, S. Thiele, K. J. J. Mayrhofer and S. Cherevko, Angew. Chem., 2021, 133, 8964–8970 CrossRef.
- U. Kernchen, B. Etzold, W. Korth and A. Jess, Chem. Eng. Technol., 2007, 30, 985–994 CrossRef CAS.
- J. Snyder, T. Fujita, M. W. Chen and J. Erlebacher, Nat. Mater., 2010, 9, 904–907 CrossRef CAS PubMed.
- G.-R. Zhang, M. Munoz and B. J. M. Etzold, ACS Appl. Mater. Interfaces, 2015, 7, 3562–3570, DOI:10.1021/am5074003.
- G.-R. Zhang, T. Wolker, D. J. S. Sandbeck, M. Munoz, K. J. J. Mayrhofer, S. Cherevko and B. J. M. Etzold, ACS Catalysis, 2018, 8, 8244–8254 CrossRef CAS PubMed.
- G.-R. Zhang, M. Munoz and B. J. M. Etzold, Angew. Chem., Int. Ed., 2016, 55, 2257–2261 CrossRef CAS PubMed.
- M. George, G.-R. Zhang, N. Schmitt, K. Brunnengräber, D. J. S. Sandbeck, K. J. J. Mayrhofer, S. Cherevko and B. J. M. Etzold, ACS Catalysis, 2019, 9, 8682–8692 CrossRef CAS PubMed.
- J. Snyder, K. Livi and J. Erlebacher, Adv. Funct. Mater., 2013, 23, 5494–5501 CrossRef CAS.
- H. Zhang, J. Liang, B. Xia, Y. Li and S. Du, Front. Chem. Sci. Eng., 2019, 13, 695–701 CrossRef CAS.
- A. Avid, J. L. Ochoa, Y. Huang, Y. Liu, P. Atanassov and I. V. Zenyuk, Nat. Commun., 2022, 13, 1–13 Search PubMed.
- R. Arrigo, M. Hävecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden and A. Knop-Gericke, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef CAS PubMed.
- A. Orfanidi, P. Madkikar, H. A. El-Sayed, G. S. Harzer, T. Kratky and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, F418 CrossRef CAS.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hübner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- H. Schmies, E. Hornberger, B. Anke, T. Jurzinsky, H. N. Nong, F. Dionigi, S. Kühl, J. Drnec, M. Lerch and C. Cremers, Chem. Mater., 2018, 30, 7287–7295 CrossRef CAS.
- L.-X. Sun and T. Okada, J. Membr. Sci., 2001, 183, 213–221 CrossRef CAS.
- K. Miyazaki, N. Sugimura, K. Kawakita, T. Abe, K. Nishio, H. Nakanishi, M. Matsuoka and Z. Ogumi, J. Electrochem. Soc., 2010, 157, A1153, DOI:10.1149/1.3483105.
- A. Weiß, M. Munoz, A. Haas, F. Rietzler, H.-P. Steinrück, M. Haumann, P. Wasserscheid and B. J. M. Etzold, ACS Catalysis, 2016, 6, 2280–2286 CrossRef.
- N. Schmitt, M. Schmidt, J. E. Mueller, L. Schmidt and B. J. M. Etzold, Electrochem. Commun., 2022, 141, 107362 CrossRef CAS.
- G.-R. Zhang and B. J. M. Etzold, Adv. Funct. Mater., 2021, 31, 2010977 CrossRef CAS.
- G.-R. Zhang and B. J. M. Etzold, J. Energy Chem., 2016, 25, 199–207 CrossRef.
- R. Alink, R. Singh, P. Schneider, K. Christmann, J. Schall, R. Keding and N. Zamel, Molecules, 2020, 25, 1523 CrossRef CAS PubMed.
- Y. Liu, M. W. Murphy, D. R. Baker, W. Gu, C. Ji, J. Jorne and H. A. Gasteiger, J. Electrochem. Soc., 2009, 156, B970, DOI:10.1149/1.3143965.
- H. Liu, Y. Liu and J. Li, Phys. Chem. Chem. Phys., 2010, 12, 1685–1697 RSC.
- K. Huang, T. Song, O. Morales-Collazo, H. Jia and J. F. Brennecke, J. Electrochem. Soc., 2017, 164, F1448 CrossRef CAS.
- X. Yan, F. Zhang, H. Zhang, H. Tang, M. Pan and P. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 6111–6117 CrossRef CAS PubMed.
- T. Soboleva, X. Zhao, K. Malek, Z. Xie, T. Navessin and S. Holdcroft, ACS Appl. Mater. Interfaces, 2010, 2, 375–384 CrossRef CAS PubMed.
- A. Ohma, K. Fushinobu and K. Okazaki, Electrochim. Acta, 2010, 55, 8829–8838 CrossRef CAS.
- K. Shinozaki, Y. Morimoto, B. S. Pivovar and S. S. Kocha, J. Power Sources, 2016, 325, 745–751 CrossRef CAS.
- R. Anderson, M. Blanco, X. Bi and D. P. Wilkinson, Int. J. Hydrogen Energy, 2012, 37, 16093–16103 CrossRef CAS.
- X. Lin, C. M. Zalitis, J. Sharman and A. Kucernak, ACS Appl. Mater. Interfaces, 2020, 12, 47467–47481, DOI:10.1021/acsami.0c12718.
- S. Ott, F. Du, M. L. Luna, T. A. Dao, S. Selve, B. R. Cuenya, A. Orfanidi and P. Strasser, Appl. Catal., B, 2022, 306, 121118 CrossRef CAS.
- C. Jackson, X. Lin, P. B. J. Levecque and A. R. J. Kucernak, ACS Catal., 2022, 12, 200–211, DOI:10.1021/acscatal.1c03908.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00055a |
| This journal is © The Royal Society of Chemistry 2023 |