DOI:
10.1039/D2YA00364C
(Paper)
Energy Adv., 2023,
2, 547-555
Efficient N2 electroreduction to ammonia in an isopropanol–PBS electrolyte using NiFe2O4in situ grown on nickel foam†
Received
27th December 2022
, Accepted 2nd March 2023
First published on 3rd March 2023
Abstract
The electrocatalytic nitrogen reduction reaction (ENRR) can produce ammonia under environmental conditions and is considered a sustainable alternative to the traditional Haber–Bosch process. However, the ENRR is limited by the solubility of nitrogen (N2) and the hydrogen evolution reaction (HER) of the aqueous solution. It is found that the solubility of N2 in isopropanol at room temperature and pressure is more than ten times higher than that in an aqueous solution. In addition, K+ can effectively inhibit the HER. Therefore, we use the two-component electrolyte of isopropanol and phosphate buffer solution (PBS) with potassium salt to weaken the influence of low solubility of N2 in an aqueous solution and the competitive HER. In addition, we have in situ grown NiFe2O4 nanoarrays on nickel foam (NiFe2O4/NF) as the electrocatalyst. Finally, a high ammonia yield (1.1 μg h−1 cm−2) is achieved and the faradaic efficiency (FE) exceeds 30% at room temperature and pressure. In the end, a model of N2 adsorption on the active (311) crystal surface of NiFe2O4 was established by means of density functional theory (DFT) calculations, and the activation energy required for the reaction was calculated, proving that the ENRR reported here was an alternate hydrogenation path.
Introduction
Ammonia is a crucial raw material for chemical fertilizers, hydrogen energy carriers and carbon-free fuels in the global economy. To date, the Haber–Bosch process has produced more than 500 million tons of ammonia every year,1,2 but its high-temperature and high-pressure production environment not only consumes about 2% of the world's energy, but also releases more than 900 million tons of carbon dioxide greenhouse gas.3,4 Therefore, it is necessary to develop a truly sustainable alternative technology. Since the first report of electrochemical ammonia synthesis in 1985,5 numerous efforts have been put into this field. Electrochemical nitrogen reduction to ammonia provides an alternative environmentally friendly and sustainable method.6–8 Liu et al. first synthesized an electrocatalyst composed of PC/Sb/SbPO4 for the ENRR at room temperature. In a neutral solution, the catalyst reached a high FE of 34% at −0.1 V (vs. RHE).9 This proves that the ENRR can be carried out at low potentials. Various types of catalysts are used for the ENRR,10–12 including metal oxides,13,14 metal phosphides,15,16 MOFs,17 carbon and nitrogen compounds,18etc., but most of the research is carried out in aqueous solutions,19–21 and the potential required by the ENRR is close to that of the HER from a thermodynamic point of view.22 In addition, N2 molecules have poor solubility in aqueous solution owing to their nonpolarity. At present, the most serious challenge is to select superior catalysts and electrolytes with high N2 solubility to improve the ammonia production rate and the FE of the ENRR. For example, MacFarlane et al. through a reasonable design of an electrode–electrolyte system achieved an FE of the ENRR of 32% and an NH3 yield rate of 2.35 × 10−11 mol s−1 cm−2.23 However, the high cost of ionic liquids and the difficulty in separating ammonia from them make it difficult to be widely used in industry. Compared with an aqueous solution, the solubility of N2 in alcohols increased by more than ten times, and alcohols can better inhibit the occurrence of the HER.24–26 Han et al. used isopropanol as an electrolyte and adjusted the concentration of added sulfuric acid to finally achieve an ammonia production rate of 1.54 × 10−11 mol s−1 cm−2 with an FE of 0.89%.27 In addition, Feng and Hao et al. showed that the presence of K+ can effectively inhibit the adsorption of H+ to the active site of the catalyst in an aqueous solution, thereby inhibiting the occurrence of the HER.28,29 Except for the choice of electrolyte favourable to the ENRR, the selection of catalyst is also crucial. In 2012, Skúlason et al. evaluated the catalytic activity of the ENRR on a series of flat and step transition metal surfaces by assuming that the activation energy is proportional to the free energy difference in each basic step of the ENRR. It can be found from the volcanic map that Fe and Mo are the most active surfaces for ammonia formation.30 As one of the cheapest and richest metals on the Earth, iron-based catalysts are widely used in the field of ENRR.31–33
In this study, isopropanol was used as the electrolyte, and phosphate buffer solution (PBS) was added as a supporting electrolyte to provide protons. In addition, in order to minimize the influence of the binder on the catalytic performance, nickel foam (NF) was selected as the substrate, and NiFe2O4/NF was synthesized in situ and directly used as an electrode for the ENRR. A series of characterization results show that the catalyst does not contain nitrogen, which effectively avoids the possibility of the material itself being reduced to ammonia. As expected, NiFe2O4/NF exhibits outstanding ENRR performance with an NH3 yield rate of 1.1 μg h−1 cm−2 and an FE of 31.4%, which are several times better than those of most previously reported compounds based on non-noble metals. Finally, we revealed the mechanism of ammonia synthesis on the catalyst surface using DFT calculations.
Experimental section
Reagents and chemicals
N2 (99.999%) and Ar (99.999%) were provided by the Beijing Analysis Instrument Factory. Ethanol absolute (CH3CH2OH, Sinopharm), acetone (C3H6O, Sinopharm), potassium hydroxide (99.999%, KOH, Aladdin), sodium hydroxide (NaOH, Sinopharm), potassium phosphate monobasic (KH2PO4, Sinopharm), salicylic acid (99.5%, C7H6O3, Aladdin), sodium nitroferricyanide dihydrate (99.98%, C5FeN6Na2O·2H2O, Aladdin), ammonium chloride (99.99%, NH4Cl, Aladdin), p-dimethylaminobenzaldehyde (98%, (CH3)2NC6H4CHO, Aladdin), ammonium chloride-15N (≥98%, 15NH4Cl, Aladdin), iron nitrate nonahydrate (99.9%, Fe(NO3)3·9H2O, Macklin), hydrazine standard solution (N2H4, Aladdin), sodium citrate dihydrate (≥99%, C6H5Na3O7·2H2O, Macklin), sodium hypochlorite (0.1 mol L−1, NaClO, Macklin), isopropyl alcohol (99.5%, C3H8O, Macklin), and ammonium sulphate (99%, (NH4)2SO4, Aladdin) were used. All materials were used directly without further purification.
Synthesis of the NiFe2O4/NF nanoarrays
Typically, Ni foam (2 × 3 cm2) was immersed in an ultrasound bath of acetone, 6 mol L−1 HCl, deionized water, and ethanol to remove the impurities on its surface and dried in a vacuum oven at 80 °C. 2.5 mmol Fe(NO3)3·9H2O was dissolved in 30 ml of ultrapure water, the obtained homogeneous solution was transferred to a 50 mL Teflon-lined stainless-steel autoclave and the dried Ni foam was placed into an autoclave titled against the wall at a certain angle, which was sealed and maintained at 200 °C for 10 h. After cooling to room temperature, a brown thin film on the Ni foam substrate was formed which was rinsed with deionized water and ethanol 3 times each and dried at 80 °C for 2 h. The synthesis method of NF-200 °C is the same as the above except that Fe(NO3)3 is not added. The as-prepared NiFe2O4/NF was directly used as the working electrode.
Electrochemical measurements
All electrochemical measurements were performed using a CH Instruments 760E workstation and a sealed traditional three-electrode electrolysis chamber (Fig. S1, ESI†) under ambient conditions. NiFe2O4/NF (1 cm2), a piece of Pt sheet (1 cm2) and a saturated calomel electrode ((SCE) filled with saturated KCl solution) were used as the working electrode (WE), counter electrode (CE) and reference electrode (RE), respectively. The measurements were performed in a system of three electrodes in a 2-propanol and 0.1 M PBS (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 1:1) double electrolyte system (N2 saturated). Prior to the ENRR, the electrolytic chamber was purged with Ar and N2 for at least 30 min, respectively. During electrolysis, N2 was delivered into the electrolytic cell by N2 gas bubbling at a flow rate of 30 standard cubic centimeters per minute (sccm), and the electrolyte was agitated with a stirring bar at a rate of 500 rpm. As the electrolysis started, N2 was supplied at a rate of 30 sccm, and the whole electrolysis process was carried out under normal temperature and pressure. Linear sweep voltammetry (LSV) was performed at a scanning rate of 5 mV s−1. The ENRR process was achieved by potentiostatic tests at different potentials for 2 h under the condition that N2 was maintained. The electrochemical active surface areas (ECSA) of NiFe2O4/NF and NF were estimated by electrochemical double-layer capacitance (Cdl) measurements using cyclic voltammetry (CV). The detailed steps are as follows: a three-electrode system was immersed in 1 mol L−1 KOH solution, the WE was NiFe2O4/NF or NF, the CE was Pt, and Hg/HgO (1 mol L−1 KOH) was used as the RE.34 The range of 0–0.1 V (vs. RHE), the non-faradaic area, was selected to measure the Cdl value, which is determined by the slope obtained by fitting the current density at 0.5 V against the scan rates. Electrochemical impedance spectroscopy (EIS) was carried out in the frequency range of 105 Hz to 0.1 Hz, the amplitude was 10 mV, and the anode potential selects the open circuit potential. Two possible products in the solution phase, NH3 and N2H4, were determined using spectrophotometric methods at the end of each electrolysis. After electrolytic reduction of the electrolyte, NH3 was quantified after electrolytic reduction of the electrolyte using the calibration curve established using the indophenol blue method,35 and no N2H4 was detected within the detection limit of the spectrophotometric method developed by Watt and Chrisp.36
:v = 1:1) double electrolyte system (N2 saturated). Prior to the ENRR, the electrolytic chamber was purged with Ar and N2 for at least 30 min, respectively. During electrolysis, N2 was delivered into the electrolytic cell by N2 gas bubbling at a flow rate of 30 standard cubic centimeters per minute (sccm), and the electrolyte was agitated with a stirring bar at a rate of 500 rpm. As the electrolysis started, N2 was supplied at a rate of 30 sccm, and the whole electrolysis process was carried out under normal temperature and pressure. Linear sweep voltammetry (LSV) was performed at a scanning rate of 5 mV s−1. The ENRR process was achieved by potentiostatic tests at different potentials for 2 h under the condition that N2 was maintained. The electrochemical active surface areas (ECSA) of NiFe2O4/NF and NF were estimated by electrochemical double-layer capacitance (Cdl) measurements using cyclic voltammetry (CV). The detailed steps are as follows: a three-electrode system was immersed in 1 mol L−1 KOH solution, the WE was NiFe2O4/NF or NF, the CE was Pt, and Hg/HgO (1 mol L−1 KOH) was used as the RE.34 The range of 0–0.1 V (vs. RHE), the non-faradaic area, was selected to measure the Cdl value, which is determined by the slope obtained by fitting the current density at 0.5 V against the scan rates. Electrochemical impedance spectroscopy (EIS) was carried out in the frequency range of 105 Hz to 0.1 Hz, the amplitude was 10 mV, and the anode potential selects the open circuit potential. Two possible products in the solution phase, NH3 and N2H4, were determined using spectrophotometric methods at the end of each electrolysis. After electrolytic reduction of the electrolyte, NH3 was quantified after electrolytic reduction of the electrolyte using the calibration curve established using the indophenol blue method,35 and no N2H4 was detected within the detection limit of the spectrophotometric method developed by Watt and Chrisp.36
Characterization
Microstructures of the NiFe2O4/NF nanoarrays were characterized with an FEI TF20 transmission electron microscope and a Gemini 300 scanning electron microscope (Carl Zeiss AG). The compositions of the samples were examined using an ESCALAB 250Xi high-resolution X-ray photoelectron spectroscope (Thermo Fisher Scientific) and by transmission electron microscopy (TEM) mapping on a Gemini 300 scanning electron microscope equipped with an Ultim MAX energy dispersive spectroscopy system. Raman spectra were obtained using a LabRAM HP Evolution via a Raman microscope with an excitation wavelength of 514 nm. The X-ray diffraction (XRD) data were recorded using a Smartlab (9) X-ray diffractometer with Cu Kα radiation at a generator current of 40 mA and a generator voltage of 40 kV (Rigaku Corporation). Ultraviolet-visible (UV-vis) absorbance spectra were recorded on a SHIMADZU UV-2550 spectrophotometer. A fully automatic gas solubility detector was used to measure N2 solubility (Ou Shi Sheng (Beijing) Technology Co., Ltd).
DFT calculations
We have employed the Vienna Ab Initio Package (VASP) to perform the DFT calculations within the generalized gradient approximation (GGA) in the PBE formulation.37–39 We have chosen the projected augmented wave (PAW) potentials to describe the ionic cores and take valence electrons into account using a plane wave basis set with a kinetic energy cutoff of 450 eV.40,41 Partial occupancies of the Kohn–Sham orbitals were allowed using the Methfessel–Paxton smearing method and a width of 0.10 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. Geometry optimization was considered convergent when the residual forces were smaller than 0.05 eV Å−1. During structural optimization, a 2 × 2 × 1 k-point grid in the Brillouin zone was used for k-point sampling, and the bottom two atomic layers were fixed while the top two were allowed to relax. Finally, the adsorption energies (Eads) were calculated as
where Ead/sub, Ead, and Esub are the total energies of the optimized adsorbate/substrate system, the adsorbate in the structure, and the clean substrate, respectively. The free energy was calculated using the equation:
where G, Eads, ZPE and TSG are the free energy, total energy from DFT calculations, zero-point energy and entropic contributions, respectively.
Results and discussion
Measurement of N2 solubility
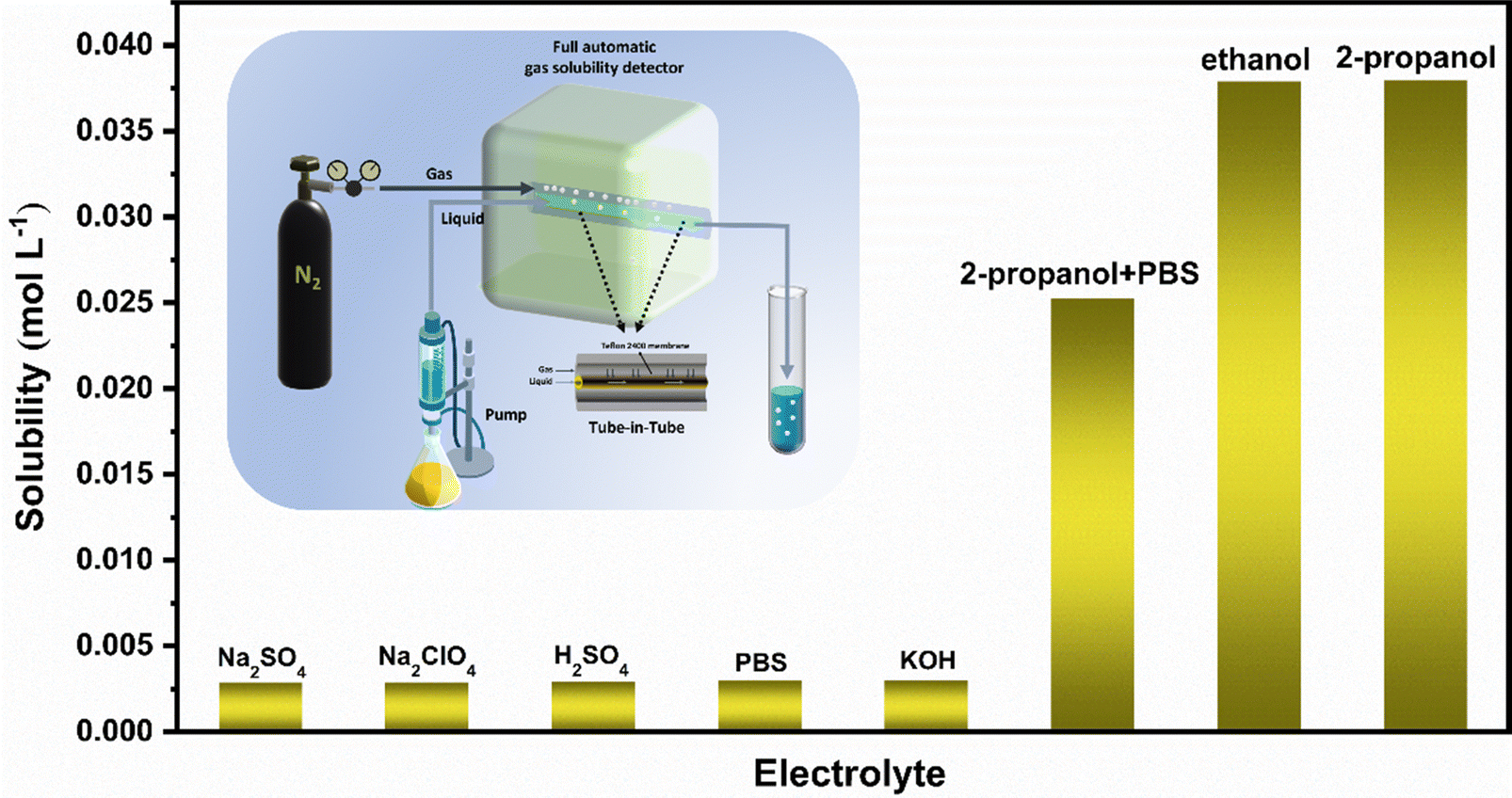
We use an automatic gas solubility detector to measure the solubility of N2 in different electrolytes at normal temperature and pressure. The principle is based on the design of a tube-in-tube reactor constructed using semi-permeable polytetrafluoroethylene AF-2400 tubes, which can rapidly saturate the liquid without direct contact with the gas and liquid (see the ESI† for specific test principle).42 The reactor can quickly measure the solubility of the gas in liquid in situ by balancing the steady flow of gas and liquid flowing into the reactor system within 2–5 minutes (Fig. 1). Finally, we measured that the solubility of N2 in isopropanol under ambient conditions is about 0.038 mol (Fig. 1), which is more than ten times that in an aqueous solution, which provides enough raw materials for the ENRR.
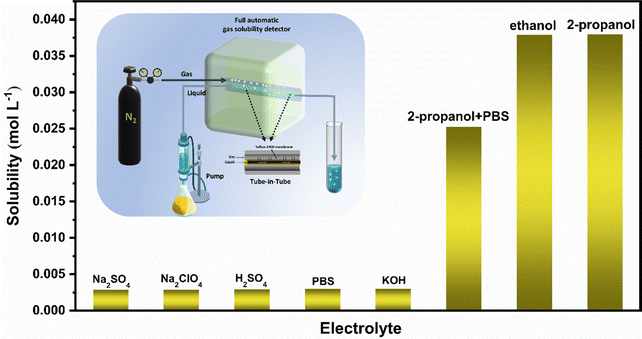 |
| | Fig. 1 Solubility of N2 in different solutions (the illustration shows a schematic diagram of the automatic gas solubility detector). | |
Synthesis and structural characterization of NiFe2O4/NF
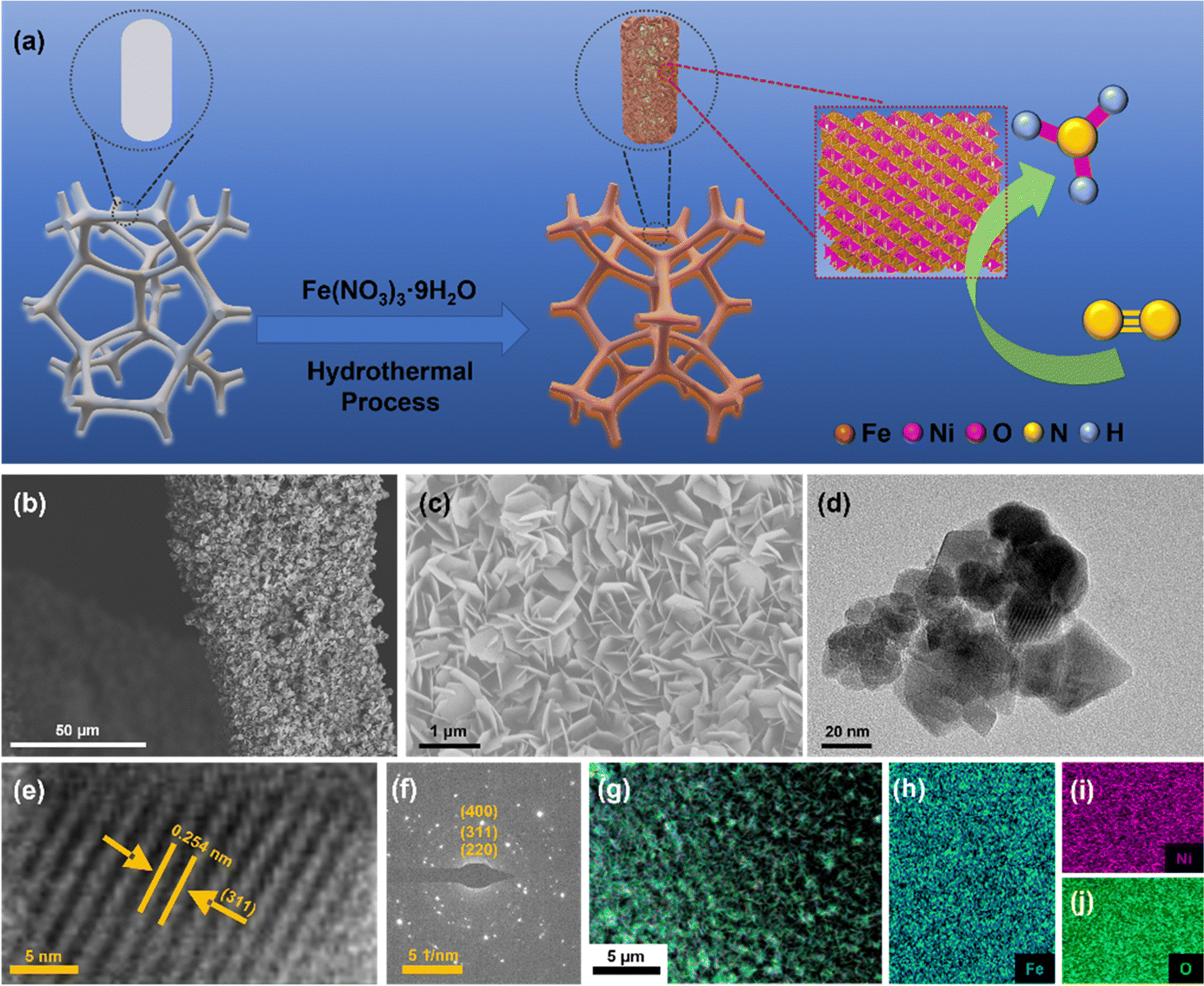
NiFe2O4/NF were synthesized using a hydrothermal method. The synthesis process of NiFe2O4/NF is shown in Fig. 2a. Typically, according to the previous synthesis method,43 in order to eliminate the errors caused by the possible introduction of the nitrogen element contained in the substances to the experiment, 2.5 mmol Fe(NO3)3·9H2O was dissolved in 30 mL of ultrapure water and dissolved by ultrasound. The resultant solution was transferred into a 50 mL Teflon-lined stainless-steel autoclave, and a piece of clean Ni foam (2 × 3 cm2) was then put in the above solution. The autoclave was heated at 200 °C for 9 h and naturally cooled down to room temperature. After synthesis, the resulting Ni foam coated with NiFe2O4 was washed with deionized water. Then the material was allowed to dry in an oven for 2 hours at 80 °C and directly used as a cathode.
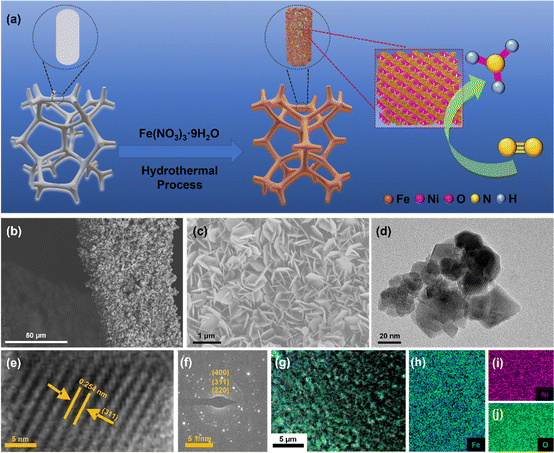 |
| | Fig. 2 (a) Scheme of in situ growth of NiFe2O4 nanoarrays on the nickel foam substrate using a hydrothermal method. (b and c) SEM images of NiFe2O4/NF. (d) TEM images of NiFe2O4 scraped from NF. (e) HRTEM image. (f) The SAED pattern of NiFe2O4. (g–j) EDX elemental mapping images of NiFe2O4/NF. | |
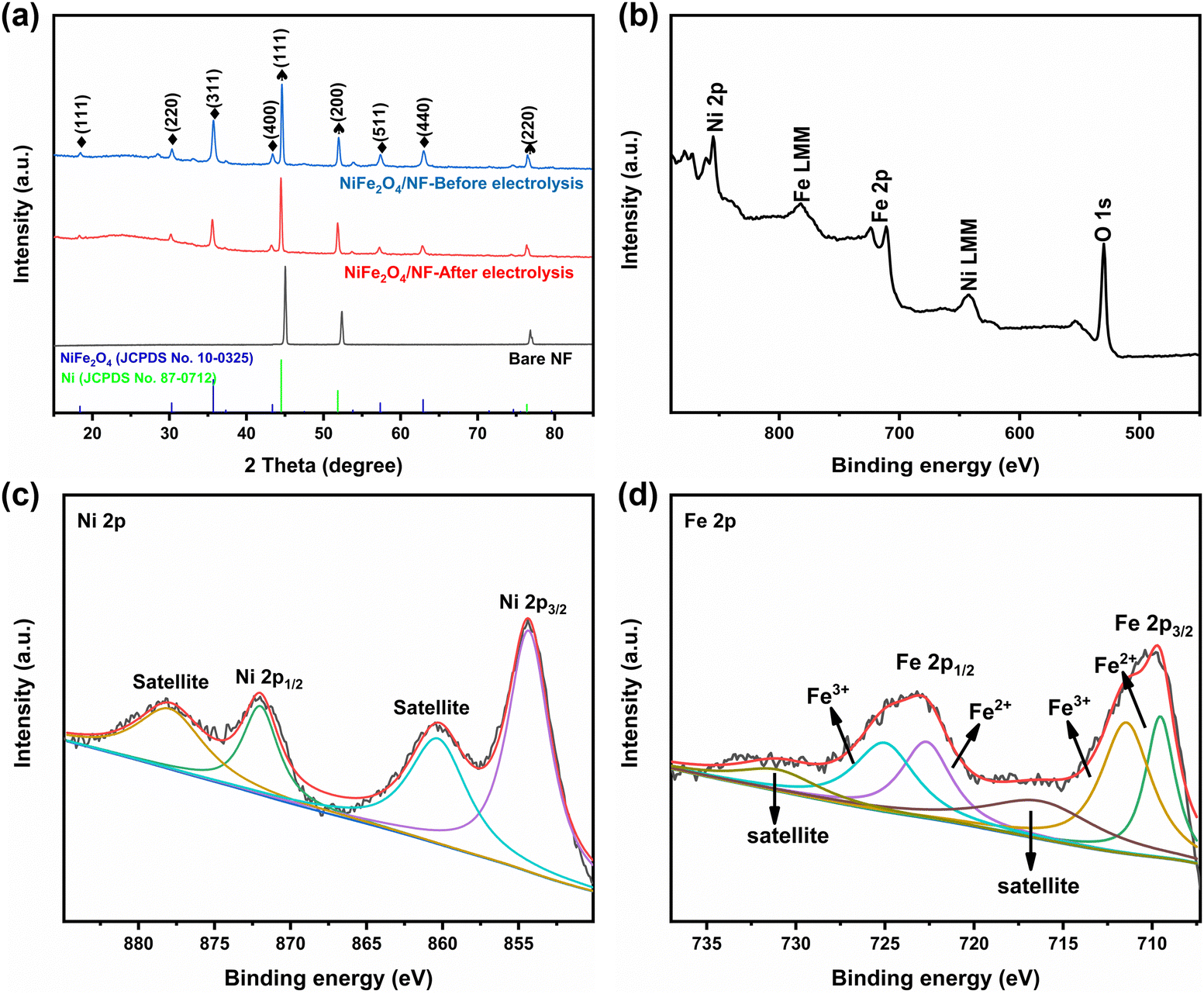
The morphologies and structures of the as-obtained samples were characterized by field emission scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). As seen from the SEM images (Fig. S2 and S3, ESI†), the NiFe2O4 nanosheets are uniformly grown on an NF substrate. In addition, we verified the magnetism of the powder scraped from the surface of nickel foam with a magnet (Fig. S3c, ESI†). Fig. 2b and c show that NiFe2O4 has an apparent hexagonal nanosheet structure with a length of 1 μm. The TEM image in Fig. 2d further proves that NiFe2O4 has a clear hexagonal structure. The HRTEM image (Fig. 2e) was taken from the nanosheets, revealing a well-resolved lattice fringe with an inter-planar distance of 0.254 nm indexed to the (311) plane of NiFe2O4. The selected area electron diffraction (SAED) pattern (Fig. 2f) displays discrete spots indexed to the (220), (311) and (400) planes of the NiFe2O4 phase. The energy-dispersive X-ray (EDX) elemental mapping images (Fig. 2g–j) indicate the uniform distribution of Fe, Ni and O elements evenly dispersed on NiFe2O4/NF. Furthermore, the energy-dispersive X-ray spectroscopy (EDS) confirmed the presence of Ni and Fe with an atomic ratio of about 1:2 (Fig. S4, ESI†). X-ray diffraction phase analysis (XRD) is an effective technique for the phase identification of crystalline materials. The XRD patterns of NiFe2O4/NF before and after the reaction are shown in Fig. 3a. From that, distinct diffraction peaks at 2θ = 18.4°, 30.3°, 35.7°, 43.3°, 57.4° and 62.8° were indexed to the (111), (220), (311), (400), (511) and (440) lattice planes of NiFe2O4 (JCPDS No. 10-0325), respectively. In addition, three diffraction peaks at 45.2°, 52.6° and 76.5° are assigned to Ni foam (JCPDS No. 87-0712). In order to further understand the oxidation state and oxygen vacancy of the synthesized samples, X-ray photoelectron spectroscopy (XPS) measurements were carried out. Fig. 3b shows the XPS survey spectra of NiFe2O4, demonstrating the presence of Fe, Ni, and O in the NiFe2O4 nanosheets, which is in accordance with the EDS elemental mapping results. Fig. 3c shows the core level spectra for Ni at binding energies of 872.3 eV and 853.9 eV, which correspond to Ni 2p3/2 and Ni 2p1/2, respectively, and the other two broad peaks at 860.4 and 878.2 eV correspond to the satellite peak.44 The distance between the two main peaks is 18.4 eV, which is also consistent with the previous report.45 NiFe2O4 is the typical inverse spinel (B(AB)O4).46 It is well known that upon switching between cations A and B, there may be obvious cation disorder in the spinel structure, which usually leads to the distribution of cations between ortho-spinel and anti-spinel.47 As shown in Fig. 3d, Fe 2p splits into Fe 2p3/2 and Fe 2p1/2, where the XPS peaks are situated at 711 eV and 724 eV, respectively. Fe 2p3/2 and Fe 2p1/2 of Fe3+ are present at 711.5 eV and 725.1 eV, respectively, and Fe 2p3/2 and Fe 2p1/2 of Fe2+ are present at 709.5 eV and 722.7 eV, respectively. In addition, there are two satellite peaks at 717 and 731 eV. The XPS results of Fe 2p show that Fe2+ and Fe3+ cations coexist on the surface due to the positive charge loss caused by cation switching between cations and vacancies at different positions.48
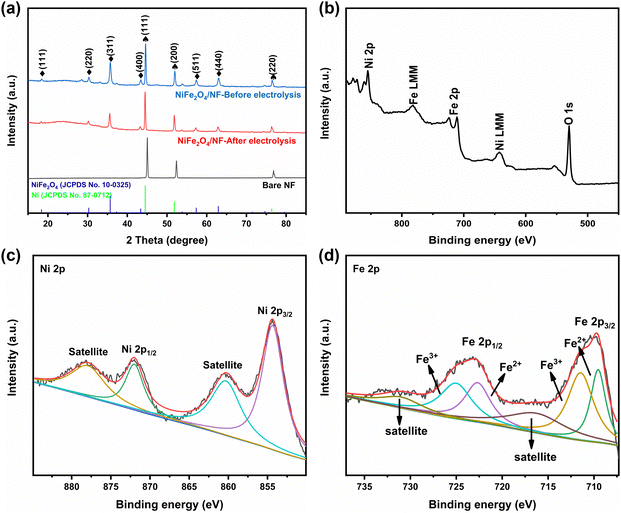 |
| | Fig. 3 (a) XRD patterns of NiFe2O4/NF, (b) XPS survey of NiFe2O4/NF, and high-resolution XPS of (c) Ni 2p and (d) Fe 2p. | |
Raman spectroscopy further confirmed that the mixed Ni/Fe oxide material synthesized by the hydrothermal technology was indeed NiFe2O4. As shown in Fig. S5 (ESI†), the Raman spectra of powder scraped from nickel foam before and after electrolysis are very similar to those of commercial NiFe2O4. The characteristic Raman phonon modes of NiFe2O4 were observed at approximately 683 cm−1 (A1g), 316 cm−1 (Eg), 535 and 471 cm−1 (T2g), which are in accordance with the earlier report.49 The A1g mode is due to the symmetric stretching vibration of the oxygen atom with the Fe and Ni atoms at the tetrahedral position. Eg and T2g are caused by the symmetric bending vibration of oxygen relative to metal ions and the asymmetric bending of oxygen relative to the tetrahedral and octahedral cations, respectively.50,51
Electrocatalytic ammonia synthesis
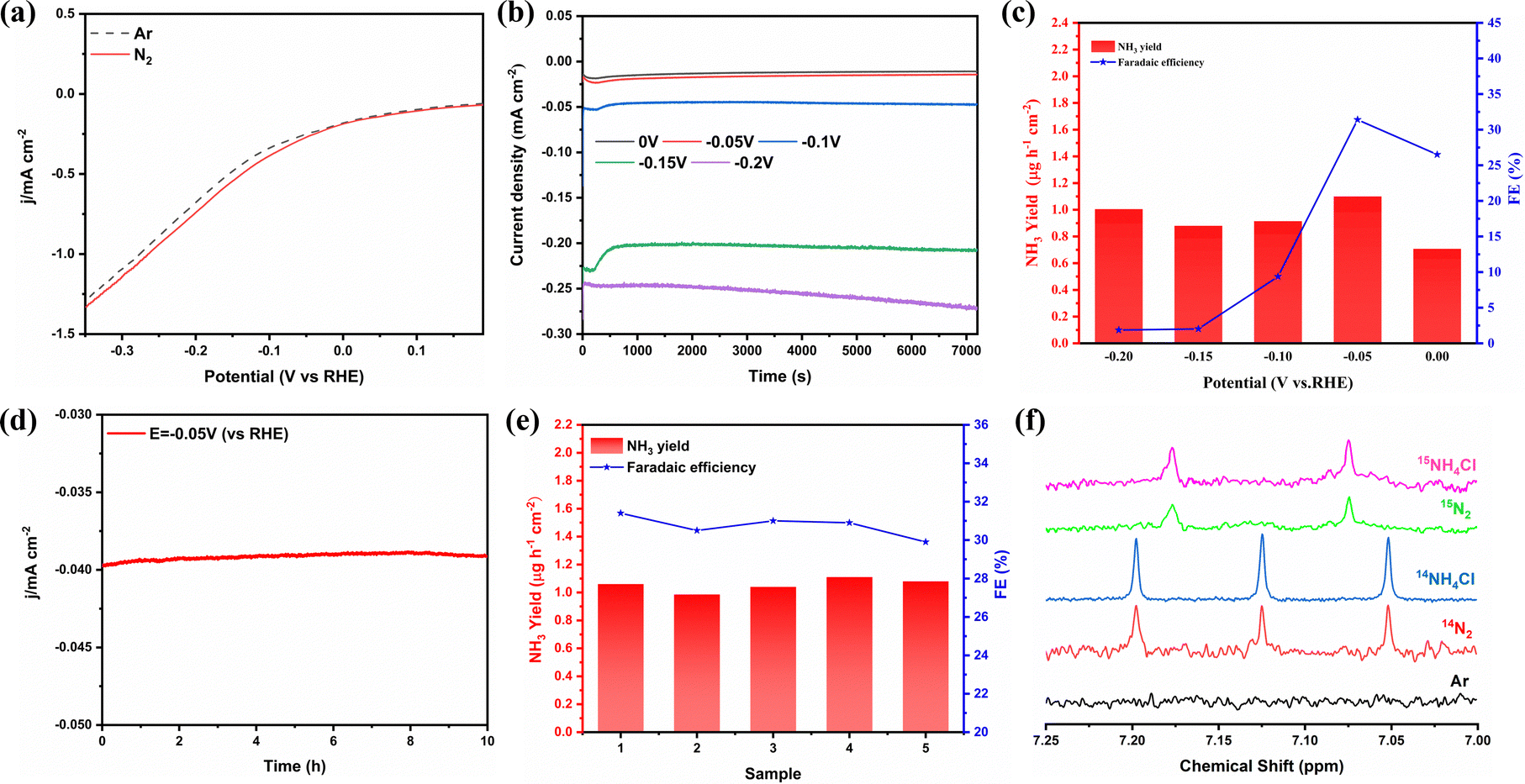
The electrochemical N2 reduction test was carried out in 2-propanol and 0.1 mol L−1 PBS, which was purged with high-purity Ar for 30 min prior to the test. As shown in Fig. S6 (ESI†), NiFe2O4/NF was directly used as the working electrode. During the ENRR process, high-purity N2 (99.999%) gas was bubbled into the electrolyte, which further reacts with electrons on the surface of NiFe2O4 to produce NH3. All potentials are reported on the saturated calomel electrode (SCE) scale, and without any correction. In order to evaluate the electrocatalytic activity of NiFe2O4/NF for the ENRR, linear sweep voltammetry (LSV) curves were initially measured under an N2 or Ar atmosphere (Fig. 4a). Compared with Ar-purging, when N2 is introduced, the LSV curve moves negatively at about 0 V, and the trend is more obvious with the increase of the working electrode voltage, which indicates that ammonia is produced on the cathode surface. To investigate the ENRR performance of the catalyst, the electrocatalyst was measured by chronoamperometry at different voltages (0, −0.05, −0.1, −0.15, and −0.2 V vs. RHE) for 2 h under the condition of N2 saturation (Fig. 4b). The concentration of NH3 generated by electrocatalysis and the possible by-product N2H4 were measured using the indophenol blue method and the ammonia selective electrode method, respectively (Fig. S7–S9, ESI†). After electrolysis for 2 h, the possible products (NH3 and N2H4) obtained in the electrolyte are determined using an ultraviolet-visible spectrophotometer (UV-vis) according to the indophenol blue method and the Watt–Chrisp method, respectively (Fig. S10, ESI†). In addition, to confirm the nitrogen source of the NH3 product, we also used the isotope labeling method to detect the product ammonia. As shown in Fig. 4f, the 15NH4+ or 14NH4+ signal can be observed clearly under the 15N2 atmosphere or the 14N2 atmosphere, when 15N2 is used as the feed gas, new double coupling peaks are generated, and the electrolyte after electrolysis was tested to correspond to them, which proves that the product ammonia is actually generated by electrocatalysis. Fig. 4c shows the ammonia yields and FE of NiFe2O4/NF under different cathodic potentials. Both the NH3 yield and FE increase with the increase of the negative potential until −0.05 V, and then they gradually decrease, which is due to the competitive selectivity of the HER. The maximum value of NH3 yield is 1.1 μg h−1 cm−2 and FE is 31.4%, respectively, and these results are superior to most reported ENRR electrocatalysts (see Table S1 for details, ESI†).
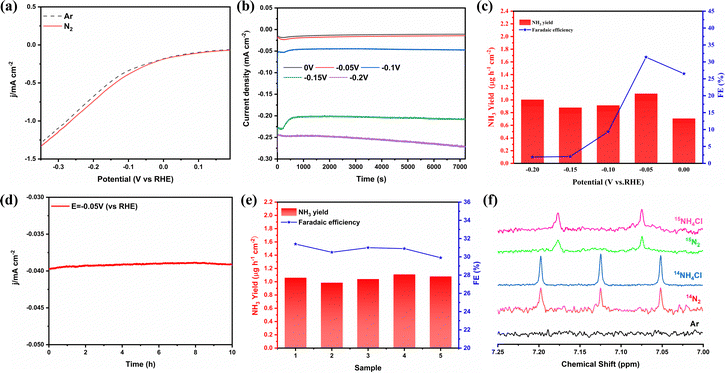 |
| | Fig. 4 (a) LSV curves in the N2 and Ar-saturated electrolyte. (b) Chronoamperometry curves recorded on NiFe2O4/NF at different potentials. (c) NH3 yield rates and faradaic efficiencies of NiFe2O4/NF at each given potential. (d) Long time-dependent current density curve for NiFe2O4/NF at −0.05 V. (e) Cycling test for 5 times at −0.05 V vs. RHE. (f) 1H NMR spectra (700 MHz) of 15NH4+ and 14NH4+ produced from the ENRR reaction using 15N2 and 14N2 as the N2 source, respectively. | |
We performed a series of comparative tests to verify the role of isopropanol and NiFe2O4 in the ENRR. In order to verify the effect of isopropanol, the ENRR experiment was carried out with pure PBS as the electrolyte. As shown in Fig. S11 (ESI†), the results showed that the ammonia production efficiency in pure PBS solution is only 0.27 μg h−1 cm−2, and the FE is 1.9%. In order to explore the role of NiFe2O4, bare NF is used as the electrode, and the ammonia production rate is only 0.3 μg h−1 cm−2 and the FE is 7.4% under the same potential (−0.05 V vs. RHE) and other conditions are not changed (Fig. S12a, ESI†), both of which are far lower compared to the effect of doping iron. In addition, to further determine whether nickel foam has a catalytic effect on the NRR, NF-200 °C is synthesized using the same method without adding Fe(NO3)3. As shown in Fig. S13 (ESI†), SEM showed that nickel foam remained stable at high temperatures, and the XRD results proved that there is no NiO formation. The results of the ENRR experiment proved that nickel foam treated in a high-temperature aqueous solution showed no obvious difference in the electrocatalytic synthesis of ammonia from bare NF. Furthermore, potentiostatic tests were carried out on the two electrodes under the conditions of Ar and N2, respectively. Fig. S12b (ESI†) shows the ultraviolet-visible absorption spectrum of the electrolyte tested with the indophenol blue method, which proves that the product ammonia is generated through electrolytic N2.
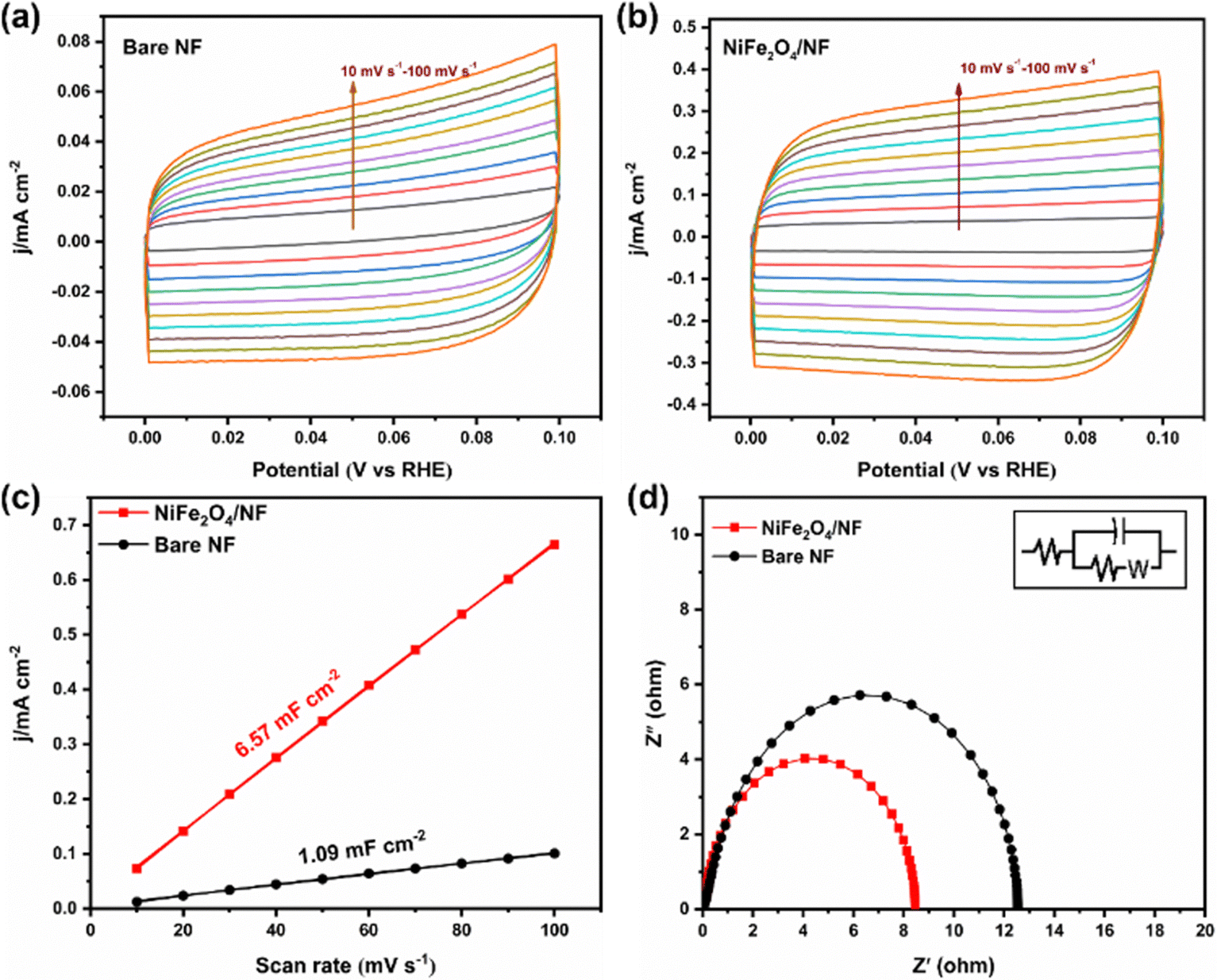
Stability is an important index to evaluate the catalyst, so we carried out a 10 h potentiostatic experiment and took samples every two hours for UV testing. As shown in Fig. 4d, the electrode was tested continuously at −0.05 V potential for 10 h, and the current density remained in a stable range, indicating that the catalyst on the electrode surface did not change significantly. SEM characterization of the electrode after electrolysis showed that it still maintained the structure of the nano-sheet array (Fig. S14, ESI†). In addition, the XRD results showed that its crystal form did not change (Fig. S15, ESI†). Fig. 4e displays that the NH3 yield and FE have hardly changed during 5 times of ENRR tests. Furthermore, cyclic voltammetry was used to measure the electric double-layer capacitance of the electrode materials and then compare the electrochemically active surface areas. According to the formula ECSA = Cdl/Cs, it can be found that the Cdl of NiFe2O4/NF is much larger than that of bare NF (Fig. 5a–c). This indicated that the ECSA increased significantly after iron loading, providing more active sites. Electrochemical impedance spectroscopy (Fig. 5d) also proves that the resistance is significantly reduced when NiFe2O4 is loaded on nickel foam, which is conducive to electron transfer on the electrode surface. More importantly, we used a PBS buffer solution prepared using KH2PO4 and NaH2PO4 as the control group to verify that K+ plays a crucial role in inhibiting the HER.27 As shown in Fig. S16a (ESI†), when K+ is replaced with Na+, it can be seen that the current of the two electrodes increases significantly during the ENRR process, which indicates that K+ is more selective to the ENRR at the same potential. After K+ is replaced with Na+, ammonia production and FE also decrease significantly (Fig. S16b, ESI†).
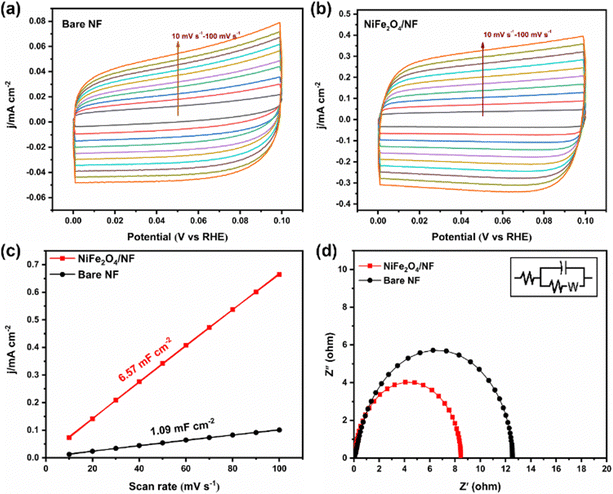 |
| | Fig. 5 CV curves of (a) bare NF and (b) NiFe2O4/NF measured at different scan rates of 10–100 mV s−1. (c) The dependence of current density on the potential scan rate for bare NF and NiFe2O4/NF. (d) EIS plots of bare NF and NiFe2O4/NF. | |
DFT calculations
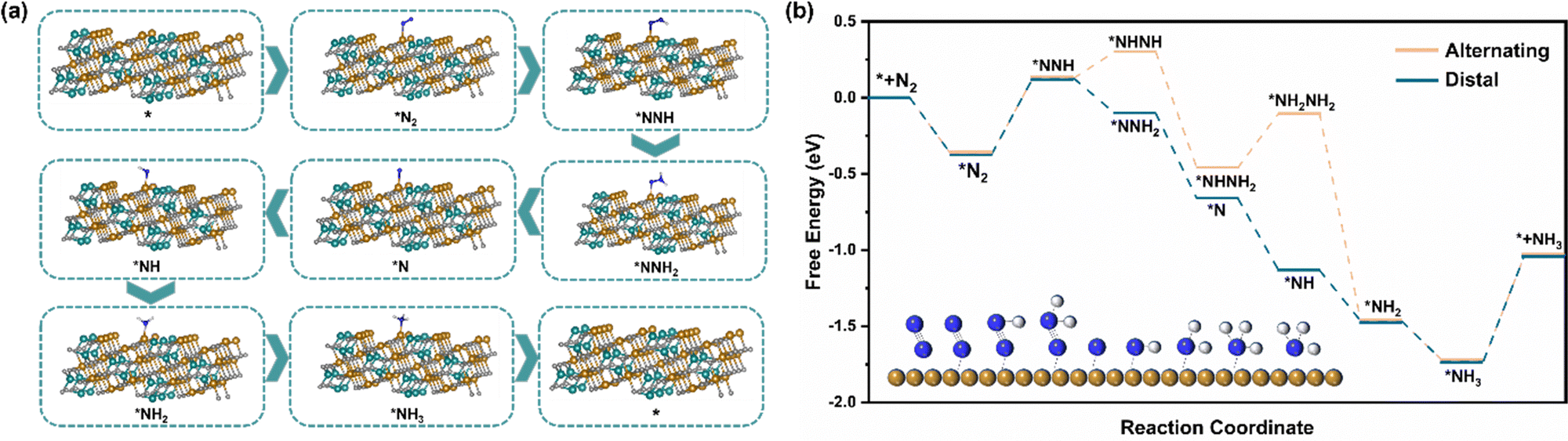
Different from the dissociation path followed by the Haber–Bosch process at high temperatures, under environmental conditions, the ENRR is more inclined to the association path, in which N2 molecules are gradually reduced and protonated by electrons and protons.52 There are two mechanisms underlying the association pathway, the distal pathway and the alternative pathway. Theoretically, the distal route is more advantageous because the alternative route is not only expected to have a higher energy barrier but also tends to generate the by-product N2H4.28 The XRD and HRTEM results show that the exposed surface of NiFe2O4 is mainly the (311) crystal plane, so we have constructed two paths from the atomic point of view to further understand the ENRR mechanism (Fig. 6a and Fig. S17, ESI†). Furthermore, we carried out DFT calculations to describe the thermodynamics of the distal and alternative path of N2 reduction and calculated the adsorption free energy of intermediate products on the NiFe2O4 (311) surfaces (Fig. 6b). It is well known that the most likely rate-determining step in the ENRR pathway is the destruction protonation of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (*N2 → *NNH),53 and the calculation shows that the two paths have the same ΔG(*N2 → *NNH) = 0.497 eV. However, considering that the total energy required for the steps (*NNH → *NHNH) and (*NHNH2 → *NH2NH2) in the alternative mechanism is 0.53 eV, which is much higher than that of a distal path in the remote hydrogenation mechanism, this step is easy to generate N2H4. From the experimental results that N2H4 was not detected, combined with the calculation results, it is obvious that the distal pathway is more suitable for our ENRR system.
N (*N2 → *NNH),53 and the calculation shows that the two paths have the same ΔG(*N2 → *NNH) = 0.497 eV. However, considering that the total energy required for the steps (*NNH → *NHNH) and (*NHNH2 → *NH2NH2) in the alternative mechanism is 0.53 eV, which is much higher than that of a distal path in the remote hydrogenation mechanism, this step is easy to generate N2H4. From the experimental results that N2H4 was not detected, combined with the calculation results, it is obvious that the distal pathway is more suitable for our ENRR system.
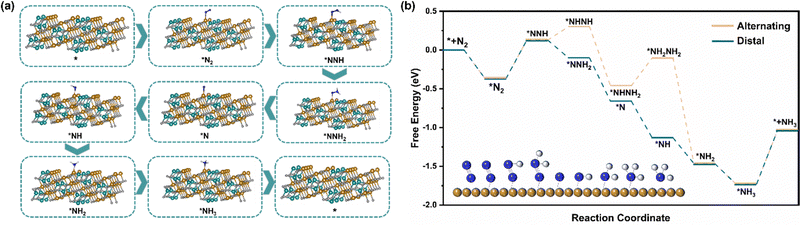 |
| | Fig. 6 (a) Schematic diagram of the atomic structure of the distal hydrogenation ENRR mechanism (blue, brown, cyan, white and gray balls represent N, Fe, Ni, H and O, respectively.) (b) The calculated free energy diagram for the electrocatalytic ENRR processes occurring on the NiFe2O4 (311) surfaces (the illustration shows the reaction mechanism of conversion of N2 to NH3 in the distal pathway; blue, brown and gray balls represent N, Fe and H, respectively). | |
Conclusions
In summary, we found that the solubility of N2 in isopropanol was much higher than that in an aqueous solution, which is conducive to the adsorption of N2 molecules on the catalyst surface in the ENRR. In addition, K+ can effectively inhibit the HER. We used the isopropanol and PBS two-component electrolyte and NiFe2O4/NF as the electrode to conduct the ENRR under ambient conditions and finally obtained a higher ammonia yield and FE. When doped with iron, the productivity and efficiency are much higher than those of nickel foam, which further proves that the existence of iron is crucial. The model of N2 adsorption on the (311) crystal surface of NiFe2O4 was constructed using DFT calculations, the free energy changes of two different paths were calculated, and the distal pathway was finally determined. Our method represents an efficient, clean and cheap synthetic ammonia route and provides a new idea for artificial nitrogen fixation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Science Foundation of China (No. 22178360), the Natural Science Foundation of Hebei Province (No. B2021208006), and the 2021 Industrial Technology Basic Public Service Platform Project-Public Service Platform for Emission Peak and Carbon Neutral of Key Raw Material Industries (No. 2021-H029-1-1).
Notes and references
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- C. J. M. Van Der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- C. J. Pickett and J. Talarmin, Nature, 1985, 317, 652–653 CrossRef CAS.
- T. Wu, W. Fan, Y. Zhang and F. Zhang, Mater. Today Phys., 2021, 16, 100310 CrossRef CAS.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- X. Liu, H. Jang, P. Li, J. Wang, Q. Qin, M. G. Kim, G. Li and J. Cho, Angew. Chem., 2019, 131, 13463–13468 CrossRef.
- C. Guo, J. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- J. Liang, Q. Liu, A. A. Alshehri and X. Sun, Nano Res. Energy, 2022, 1, 1–8 Search PubMed.
- Y. Luo, Q. Li, Y. Tian, Y. Liu and K. Chu, J. Mater. Chem. A, 2022, 10, 1742–1749 RSC.
- H. Chen, J. Liang, K. Dong, L. Yue, T. Li, Y. Luo, Z. Feng, N. Li, M. S. Hamdy, A. A. Alshehri, Y. Wang, X. Sun and Q. Liu, Inorg. Chem. Front., 2022, 9, 1514–1519 RSC.
- H. J. Chen, Z. Q. Xu, S. Sun, Y. Luo, Q. Liu, M. S. Hamdy, Z. S. Feng, X. Sun and Y. Wang, Inorg. Chem. Front., 2022, 9, 4608–4613 RSC.
- Z. Du, J. Liang, S. Li, Z. Xu, T. Li, Q. Liu, Y. Luo, F. Zhang, Y. Liu, Q. Kong, X. Shi, B. Tang, A. M. Asiri, B. Li and X. Sun, J. Mater. Chem. A, 2021, 9, 13861–13866 RSC.
- Q. Liu, Y. T. Lin, S. Gu, Z. Q. Chen, L. S. Xie, S. G. Sun, L. C. Zhang, Y. S. Luo, Q. Liu, Y. Lin, S. Gu, Z. Cheng, L. Xie, S. Sun, L. Zhang, Y. Luo, A. A. Alshehn, M. S. Hamdy, Q. Q. Kong, J. H. Wang and X. P. Sun, Nano Res., 2022, 15, 7134–7138 CrossRef CAS.
- Y. Sun, W. Wu, L. Yu, S. Xu, Y. Zhang, L. Yu, B. Xia, S. Ding, M. Li, L. L. Jiang, J. Duan, J. Zhu and S. Chen, Carbon Energy, 2022, 1–12 Search PubMed.
- Q. Li, P. Shen, Y. Tian, X. Li and K. Chu, J. Colloid Interface Sci., 2022, 606, 204–212 CrossRef CAS PubMed.
- D. Yang, T. Chen and Z. Wang, J. Mater. Chem. A, 2017, 5, 18967–18971 RSC.
- M. Liu, S. Yin, T. Ren, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 47458–47464 CrossRef CAS.
- Z. Wang, Z. Dai, S. Wang, H. Zhang, W. Tian, Y. Xu, X. Li, L. Wang and H. Wang, Chem. Eng. J., 2021, 416, 129105 CrossRef CAS.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017, 7, 706–709 CrossRef CAS.
- B. H. R. Suryanto, C. S. M. Kang, D. Wang, C. Xiao, F. Zhou, L. M. Azofra, L. Cavallo, X. Zhang and D. R. Macfarlane, ACS Energy Lett., 2018, 3, 1219–1224 CrossRef CAS.
- K. Ohta, M. Kawamoto, T. Mizuno and D. A. Lowy, J. Appl. Electrochem., 1998, 28, 717–724 CrossRef CAS.
- M. Murugananthan, M. Kumaravel, H. Katsumata, T. Suzuki and S. Kaneco, Int. J. Hydrogen Energy, 2015, 40, 6740–6744 CrossRef CAS.
- F. Köleli and T. Röpke, Appl. Catal., B, 2006, 62, 306–310 CrossRef.
- K. Kim, N. Lee, C.-Y. Yoo, J.-N. Kim, H. C. Yoon and J.-I. Han, J. Electrochem. Soc., 2016, 163, F610–F612 CrossRef CAS.
- L. Hu, A. Khaniya, J. Wang, G. Chen, W. E. Kaden and X. Feng, ACS Catal., 2018, 8, 9312–9319 CrossRef CAS.
- Y. C. Hao, Y. Guo, L. W. Chen, M. Shu, X. Y. Wang, T. A. Bu, W. Y. Gao, N. Zhang, X. Su, X. Feng, J. W. Zhou, B. Wang, C. W. Hu, A. X. Yin, R. Si, Y. W. Zhang and C. H. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- F. Studt, T. Bligaard, J. Rossmeisl, F. Abild-pedersen, T. Vegge, H. Jo and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- J. Liang, Q. Zhou, T. Mou, H. Chen, L. Yue, Y. Luo, Q. Liu, M. S. Hamdy, A. A. Alshehri, F. Gong and X. Sun, Nano Res., 2022, 15, 4008–4013 CrossRef CAS.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS.
- J. Abed, S. Ahmadi, L. Laverdure, A. Abdellah, C. P. O’Brien, K. Cole, P. Sobrinho, D. Sinton, D. Higgins, N. J. Mosey, S. J. Thorpe and E. H. Sargent, Adv. Mater., 2021, 33, 1–11 CrossRef.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- A. Aparna, B. Anupama, G. Vindya and G. D. Rao, Orient. J. Chem., 2010, 26, 1207–1209 CAS.
- G. Kresse and J. Furthmiiller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmu, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16–28 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 11–19 Search PubMed.
- P. E. Bloechl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef CAS PubMed.
- J. Zhang, A. R. Teixeira, H. Zhang and K. F. Jensen, Anal. Chem., 2017, 89, 8524–8530 CrossRef CAS.
- X. Meng, J. Han, L. Lu, G. Qiu, Z. L. Wang and C. Sun, Small, 2019, 15, 1–10 CAS.
- Y. Zhang, L. Ye, M. Zhang, L. Ma and Y. Gong, Appl. Surf. Sci., 2022, 589, 152957 CrossRef CAS.
- X. Zhang, Z. Zhang, S. Sun, Y. Wu, Q. Sun and X. Liu, RSC Adv., 2018, 8, 15222–15228 RSC.
- M. S. Matseke, H. Luo, L. Wen and H. Zheng, J. Phys. Chem. Solids, 2022, 165, 110644 CrossRef CAS.
- W. Y. Ching, S. Di Mo, I. Tanaka and M. Yoshiya, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 641021 Search PubMed.
- S. Anjum, G. H. Jaffari, A. K. Rumaiz, M. S. Rafique and S. I. Shah, J. Phys. D: Appl. Phys., 2010, 43, 265001 CrossRef.
- R. Singh Yadav, J. Havlica, J. Masilko, L. Kalina, J. Wasserbauer, M. Hajdúchová, V. Enev, I. Kuřitka and Z. Kožáková, J. Magn. Magn. Mater., 2015, 394, 439–447 CrossRef CAS.
- M. B. Askari and P. Salarizadeh, Int. J. Hydrogen Energy, 2020, 45, 27482–27491 CrossRef CAS.
- R. Nongjai, S. Khan, K. Asokan, H. Ahmed and I. Khan, J. Appl. Phys., 2012, 112, 084321 CrossRef.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- W. Zang, T. Yang, H. Zou, S. Xi, H. Zhang, X. Liu, Z. Kou, Y. Du, Y. P. Feng, L. Shen, L. Duan, J. Wang and S. J. Pennycook, ACS Catal., 2019, 9, 10166–10173 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Penglei
Cui
b,
Cong
Zhang
a,
Qian
Yang
b and
Jujie
Ren
*a
*b,
Penglei
Cui
b,
Cong
Zhang
a,
Qian
Yang
b and
Jujie
Ren
*a