
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogen sorption on microporous carbon/sulfur nanocomposite systems†
Charles D.
Brewster
 a,
Lui R.
Terry
b,
Huan V.
Doan
b,
Sebastien
Rochat
c and
Valeska P.
Ting
*bde
a,
Lui R.
Terry
b,
Huan V.
Doan
b,
Sebastien
Rochat
c and
Valeska P.
Ting
*bde
aDepartment of Aerospace Engineering, University of Bristol, Bristol, BS8 1TR, UK
bDepartment of Mechanical Engineering, University of Bristol, Bristol, BS8 1TR, UK. E-mail: v.ting@bristol.ac.uk
cSchool of Chemistry, University of Bristol, Bristol, BS8 1TR, UK
dCollege of Engineering, Computing and Cybernetics, Australian National University, Canberra, ACT 0200, Australia
eResearch School of Chemistry, Australian National University, Canberra, ACT 0200, Australia
First published on 6th February 2023
Abstract
Encapsulating sulfur in single-walled carbon nanotubes (S@SWCNTs) produces a composite material hitherto unexplored for hydrogen storage. Interactions between sulfur and carbon nanotubes modify the electronic properties of the composite, thus offering methods for improving hydrogen sorption in carbon nanotubes. Here we demonstrate that S@SWCNT composites can provide 35% greater gravimetric excess adsorbed hydrogen per unit specific surface area, and improved hydrogen uptake at lower pressures (<2 MPa), indicating higher enthalpies of adsorption. Through semi-empirical modelling of high-pressure gas sorption isotherms, it was determined that S@SWCNTs can provide 74% higher volumetric hydrogen density compared to an undoped equivalent at 2 MPa and 77 K.
1 Introduction
Poised to replace fossil fuels as a cleaner energy vector, hydrogen (H2) offers several unique properties making it a seemingly ideal successor.1 The potential for a zero-carbon emission, cyclic fuel economy with a relatively uniform global distribution has motivated researchers for several decades. However, the low-density storage of H2 consistently impedes widespread adoption and commercial implementation. Currently, storage and transport methods rely on physical-based manipulation of H2via pressurising (up to 100 MPa), cryogenic cooling (typically between 20 K and 77 K) or a combination of the two to achieve a usable density.2 These techniques though simple, are energy-intensive and raise safety concerns, especially when placed in a mobile setting. Physical adsorption onto a porous scaffold offers an alternative, where molecular H2 is weakly bound and concentrated on the surface of an adsorbent.3,4 The spontaneous adsorption process has been observed to result in a higher density of H2 than that of compressed H2 under similar conditions of temperature and pressure.5–7 Moreover, when the adsorption conditions are reversed (i.e., pressure decreased and/or temperature increased), a complete discharge of the stored H2 can be achieved. Generally, the gravimetric capacity of a physisorption material is dictated by the BET (Brunauer–Emmett–Teller) surface area, approximately 1 wt% of excess H2 per 500 m2 g−1 at 77 K (Chahine's Rule).8–11 However, materials with very high surface areas, such as metal–organic frameworks (MOFs), tend to have poor hydrothermal stabilities and are challenging to synthesise and scale up,12 constraining commercial viability.Carbonaceous scaffolds are promising sorptive materials for meeting the United States Department of Energy targets for H2 storage (ultimately 6.5 wt% including all auxiliary equipment).13 Relatively high stability, low cost and a plethora of derivatives make porous carbons good candidates for practical implementation.14 Furthermore, the high microporosity and low density displayed by these materials offer high gravimetric uptakes, typically ranging between 0.2–5.5 wt% depending on temperature.15 carbon nanotubes (CNTs) (particularly single-walled carbon nanotubes (SWCNTs)) provide an excellent model system for exploring routes for increasing hydrogen storage densities, owing to their simple carbon structure, homogeneous surface and ability to incorporate additional heteroatoms. Single-walled, double-walled and multi-walled carbon nanotubes (SWCNTs, DWCNTs and MWCNTs) are allotropes of carbon that can be conceptualised as rolled graphite/graphene with narrow pseudo-1-dimensional channels on the nanometer scale. Due to their outstanding chemical/thermal stability, high mechanical strength and relatively high surface area (between 300–1300 m2 g−1),16–18 CNTs have attracted much attention in gas sorption and separation. However, compared to other porous carbons, SWCNTs provide relatively low adsorption gravimetric uptakes even under cryogenic conditions; this stems from the relatively weak binding strength of the H2 molecules and the graphitic surface (0.01–0.06 eV).19–21 Improving the CNT–H2 interaction strength through modifying the composition of the material may provide a route to overcoming Chahine's Rule whilst maintaining several of the other desirable properties of CNTs.12,22–25
Modifications to the adsorbent surface through doping or functionalisation may alter the adsorbate–adsorbent interactions through modulating the electronic structure of the surface,26 thus changing the Lennard-Jones interactions between the incoming adsorbate molecules and solid adsorbent.27 Tailoring these interactions may be achieved by adding heteroatoms into the porous structure, replacing specific units of the chemical structure, or adding functional groups such as amines to the surface.28–31 Although not widely reported, sulfur has previously been employed in gas sorption onto microporous carbons and has demonstrated a beneficial effect for H2 storage and carbon dioxide sequestering applications.29,32,33 Activated carbons derived from thiol-containing polymers with micro/mesoscale porosity have shown an admirably high H2 gravimetric uptake of 5.71 wt% at 77 K, and 2 MPa.34 However, these sulfur-doped materials as reported by Sevilla et al. seem to obey Chahine's rule generally.34 This observation implies that the relatively high H2 gravimetric uptake is surface area driven rather than an enhancement due to the thioester groups embedded within the scaffold. This is similar to an observation by Park et al. in gingko leaf derived carbons.35 In contrast, Li et al. report a 22% increase (2.24 wt% to 2.72 wt%) in H2 gravimetric uptake when sulfuric acid was used to catalyse the polymerisation, rather than hydrochloric acid; this resulted in thiophene and sulfone groups forming within the porous carbon.27 This improved performance is despite a noted reduction in surface area compared to benchmark material using hydrochloric acid to catalyse polymerisation. The improvement of H2 gravimetric uptake is attributed to the change in the polarity of the carbon framework. Li et al. further claim that sulfur doping provided the most significant increase in gravimetric uptake to specific surface area ratio among the tested dopants (nitrogen, sulfur and phosphorus).27 Similarly, De Yuso et al. report the addition of sulfur and oxygen-containing groups led to increasing hydrogen capacity at room temperature for hard-templated porous carbons.33 Additionally, density functional theory (DFT) calculations conducted by Mousavipour and Chitsazi showed an enhancement of the binding energies for sulfur-intercalated SWCNTs. They found that the presence of sulfur atoms increased the polarizability of the incoming H2, changing the interactions of the system.36 To further study the role of sulfur for hydrogen sorption in porous carbons, experimental formation and testing of sulfur-doped CNTs needs to be conducted.
In this report, SWCNTs infiltrated with sulfur (S@SWNCTs) are tested for their gravimetric H2 sorption capacities. To the authors' knowledge, S@SWCNTs are yet to be experimentally investigated for their potential in H2 storage applications, though these materials have previously been explored for lithium–sulfur batteries37 and gas-sensing38 applications. SWCNTs offer a good model system to investigate the interactions between the sulfur and carbon sub-systems and how this may modify the surface properties of the sulfur/porous carbon composites, changing the interactions with incoming H2 molecules.36,39 This work provides insight into the role of sulfur doping for improved hydrogen storage in porous carbon structures. Here, the gravimetric sorption capacity of two SWCNTs and their sulfur composite counterparts have been compared and are additionally contrasted against a phenolic-derived activated carbon (TE7).40 The successful synthesis of the composites is confirmed by Raman spectroscopy and spectral features compared to previous literature. Textural and porosity properties are assessed by nitrogen sorption. Furthermore, semi-empirical modelling of the high-pressure H2 sorption isotherms was used to estimate the adsorbate phase H2 density. The maximum excess adsorption of the materials is compared against Chahine's rule and other sulfur-doped materials from the literature.
2 Materials and methodology
2.1 Nanocomposite preparation
Elemental sulfur powder (99.5% purity) was purchased from Alfa Aesar (stock no. 43766.36). 37% trace metal, Primar Plus™ hydrochloric acid, was purchased from Fisher Scientific (product no. 10199832). Nitrogen gas (99.999% purity) and H2 gas Alphagaz™ (>99.999% purity) were purchased from Air Liquide.
2.2 Characterisation methods
| Ne = 100(ρa − ρb)Vpθ | (1) |
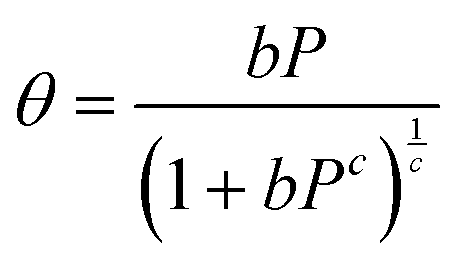 | (2) |
3 Results and discussion
3.1 Sulfur content and conformation
The sulfur content of the materials was estimated through the mass loss at 600 °C, determined via TGA. S@SWCNT-121 and S@SWCNT-777 and S@TE7 contain 9.1 wt%, 6.4 wt% and 23 wt% sulfur, respectively (see ESI,† Fig. S2). A direct comparison between the pristine materials and the sulfur encapsulating composites is challenging due to differences in the thermal history; the composite materials were additionally heated to 600 °C during the vapour deposition. However, it is assumed any mass loss event observed below 600 °C in the pristine carbon samples has already occurred during the vapour deposition process and that the addition of sulfur does not induce any significant instabilities in the material. Therefore, all the mass loss observed up to 600 °C in the sulfur composite is the evaporation of sulfur. The sulfur content of S@SWCNT-121 and S@SWCNT-777 is similar to values of S@SWCNT samples produced in a similar procedure by Nascimento et al. using thermal post-treatment to remove external sulfur following an initial 48 h exposure to sulfur vapour.49 Sulfur contents obtained from TGA differ from those determined via XPS, which reveals sulfur contents of S@SWCNT-121, S@SWCNT-777 and S@TE7 containing 14.7 wt%, 9.3 wt% and 14.4 wt%, respectively, to a maximal probable depth of approximately 10 nm (see ESI,† Table S1). Differences in the quantity of sulfur determined may be due to variation between batches (XPS was conducted on samples produced following testing) or may stem from heterogeneity of sulfur concentration throughout the sample volume. Despite differences in acquisition, the values for TGA are generally trusted to be representative of the composites as they represent a bulk measurement and were conducted on exact samples used for hydrogen sorption.Fitting of the S 2p peak was used to collect information about the speciation of sulfur within the samples. However, fitting of the S 2p is complicated by spin–orbit splitting originating from the angular momentum of the p-orbital (see Fig. 1). Plausible chemical sources of the peaks determined through fitting are given in the summary table (see ESI,† Table S2). The peaks for both S@SWCNT-121 and S@SWCNT-777 at around 163.7 eV in this work (see Fig. 1) are comparable to that reported by Fujimori et al. for S–S bonding modes in S@SWCNT and S@DWCNT and suggest the presence of strained polysulfide chains.42 Furthermore, the existence of polysulfide motifs is suggested by low binding energy modes of 162.4 and 162.3 eV for S@SWCNT-121 and S@SWCNT-777, respectively, which are in good agreement with that of terminal sulfur (162.3 (± 0.1) eV) and central sulfur (163.9 (± 0.1) eV) as determined by Fantauzzi et al. in a lithium sulfide sample.42,50 These peaks may coincide with metal sulfide peaks (such as FeS2, CoS2, MoS2). However, only small quantities of molybdenum were detected in S@SWCNT-777 within the probed volume (see ESI,† Table S1). S@TE7 shows a number of new sulfur modes compared to w-TE7 (see ESI,† Fig. S6) within the S 2p spectra, likely indicating the formation of thiophene, C–Sx–C species and additionally oxidised sulfur species at higher binding energies (see Fig. 1(c)).
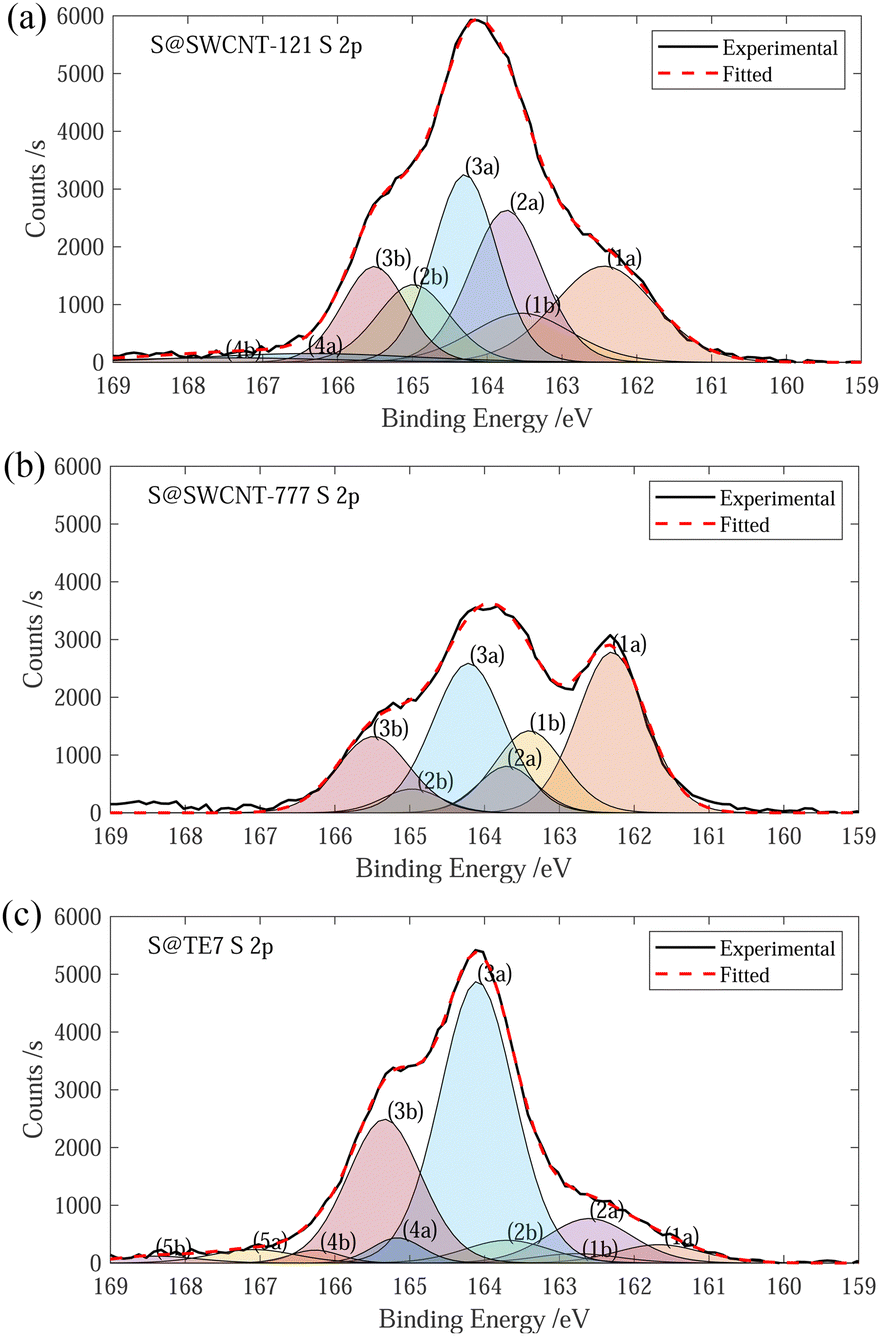 | ||
Fig. 1 (a) Experimental and fitted S 2p XPS spectra for S@SWCNT-121. (b) Experimental and fitted S 2p XPS spectra for S@SWCNT-777. (c) Experimental and fitted S 2p XPS spectra for S@TE7. Peaks denoted “a and “b” show the S 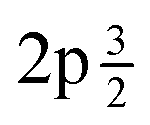 and S and S 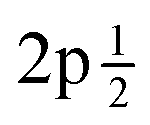 , respectively, as a result of spin–orbital coupling and are separated by a binding energy of 1.16 eV. , respectively, as a result of spin–orbital coupling and are separated by a binding energy of 1.16 eV. | ||
After sulfur encapsulation, the Raman spectra (see Fig. 2) for all materials differ between the pristine and washed samples. New intense Raman peaks are observable at 320 cm−1 and 395 cm−1 within S@SWCNT-777, and 323 cm−1 and 394 cm−1 for S@SWCNT-121; these may provide insight into the conformation of the confined sulfur species. Previous reports have explored the origins of these doublet peaks located around 320 cm−1 and 400 cm−1, in detail.49,51 Nascimento et al. predict that a strong doublet peak would be observed in a SWCNT of the chirality (7,6) and suggest these peaks arise from splitting from degenerate stretching modes of a polymeric sulfur chain due to quantum confinement effects enforced by the surrounding CNT.49 Geometry optimisation of similar diameter SWCNTs suggests a helical conformation of the sulfur parallel to the tube wall is the most energetically favourable conformation.49 Overtones of the sulfur modes are clearly observable centered around 710 cm−1 and 716 cm−1 for S@SWCNT-121 and S@SWCNT-777, respectively.49
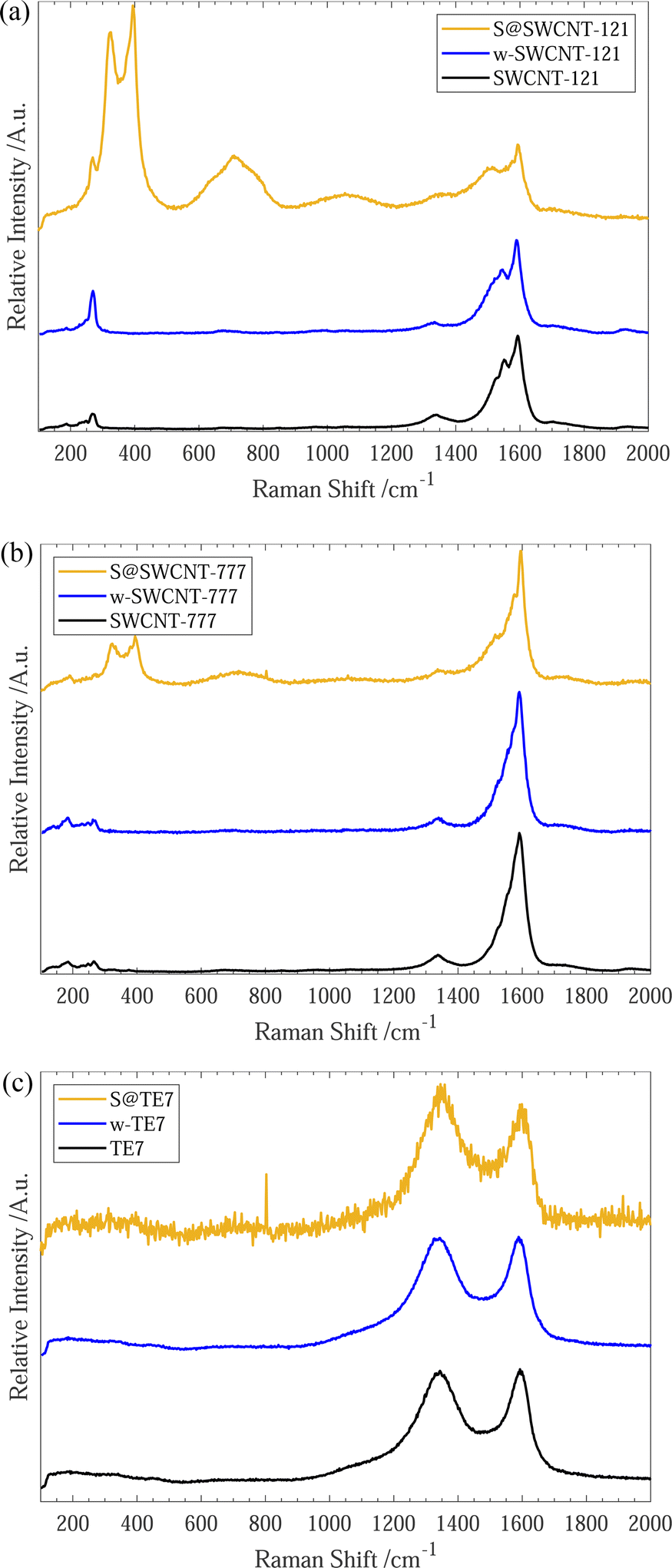 | ||
| Fig. 2 (a) Raman spectra for SWCNT-121, w-SWCNT-121 and S@SWCNT-121, 532 nm excitation wavelength. (b) Raman spectra for SWCNT-777, w-SWCNT-777 and S@SWCNT-777, 532 nm excitation wavelength. (c) Raman spectra for TE7, w-TE7 and S@TE7, 532 nm excitation wavelength. All spectra are normalised to the intensity of the G-band for comparison. Each plot shows the as-purchased (black), washed (blue) and their sulfur composites (yellow). | ||
We conclude that the conformation of sulfur within the S@SWCNT composites is likely helical polysulfide stabilised through confinement effects induced by the surrounding CNT. Furthermore, Raman responses of linear, double linear or zig-zag polymeric sulfur, initially reported by Fujimori et al.,42 do not display the distinctive peaks at 320 cm−1 and 400 cm−1; instead, they show a shift in the radial breathing modes (RBM) (located between 100–350 cm−1), which we do not observe.
Generally, all materials exhibit an increase in the ID/IG ratio with sulfur encapsulation (see Table 1), indicating a relative reduction in the sp2 hybridised bonds52 or increase in defective sites along the SWCNT walls, following sulfur encapsulation, possibly suggesting the occurrence of a reaction between the sulfur and sp2 graphitic material. Under the experimental conditions, the graphitic plates within the carbon material could form C–Sx–C bridges initiated by the diradical sulfur species present in the vapour mixture.51,53,54 Evidence of a sulfur/carbon reaction occurring is given by the emergence of an infrared spectroscopy peak centered around 508 cm−1 (see ESI,† Fig. S7), where a series of alkyl/allyl/aryl disulfide and polysulfide responses are expected to occur55 and is further corroborated by the peaks labelled 3a/3b and 4a/4b within the XPS spectra (see Fig. 1(c)). This is plausible for all samples. However, it is more likely for the S@TE7 sample, where no notable new Raman peaks are present in the spectra for S@TE7, compared to the pristine and washed samples—indicating that no polymeric sulfur species are present at a detectable depth within the sample, on the order of 100 nm (for 488 nm excitation wavelengths).56,57
| ω G/cm−1 | ω D/cm−1 | I D/IG | |
|---|---|---|---|
| SWCNT-121 | 1593 | 1339 | 0.1728 |
| w-SWCNT-121 | 1589 | 1333 | 0.1430 |
| S@SWCNT-121 | 1593 | 1359 | 0.4910 |
| SWCNT-777 | 1590 | 1337 | 0.1359 |
| w-SWCNT-777 | 1591 | 1339 | 0.1087 |
| S@SWCNT-777 | 1595 | 1339 | 0.1400 |
| TE7 | 1593 | 1345 | 0.9880 |
| w-TE7 | 1589 | 1342 | 0.9920 |
| S@TE7 | 1599 | 1344 | 1.1546 |
3.2 Surface and porosity measurements
After sulfur encapsulation, it is evident from the nitrogen sorption isotherms (see Fig. 3) that there is a loss of adsorption at low pressure (P/P0 < 0.05) for all samples indicating a loss in observable microporosity, as expected. This is apparent in the pore size distribution (see Fig. 4) where the cumulative pore volume shows a reduced total volume in the microporous range (<2 nm). S@SWCNT-121 demonstrates large reductions in pore volume within the sub-nanometer range (typically desirable for H2 physisorption), where sulfur likely occupies deep sites between CNTs and the narrow endohedral channels. However, S@SWCNT-777 does not show the same reduction in pore volume. We hypothesise the larger diameters and lower sulfur loading of the SWCNTs present in S@SWCNT-777 still allow ingress of N2 molecules to facilitate quantification of these pores. S@TE7 shows considerable loss in volume within the sub-nanometer pores (see Fig. 4(c)) with the emergence of larger micropores between 1–2 nm, this may indicate an expansion of the porous network via oxidation reaction with the sulfur vapour. The reduction in total micropore volume (μVp) is corroborated using the Dubinin–Radushkevich method (see Table 2). Reductions of 13%, 23% and 22% in μVp were found for S@SWCNT-121, S@SWCNT-777 and S@TE7, respectively, compared to the washed materials.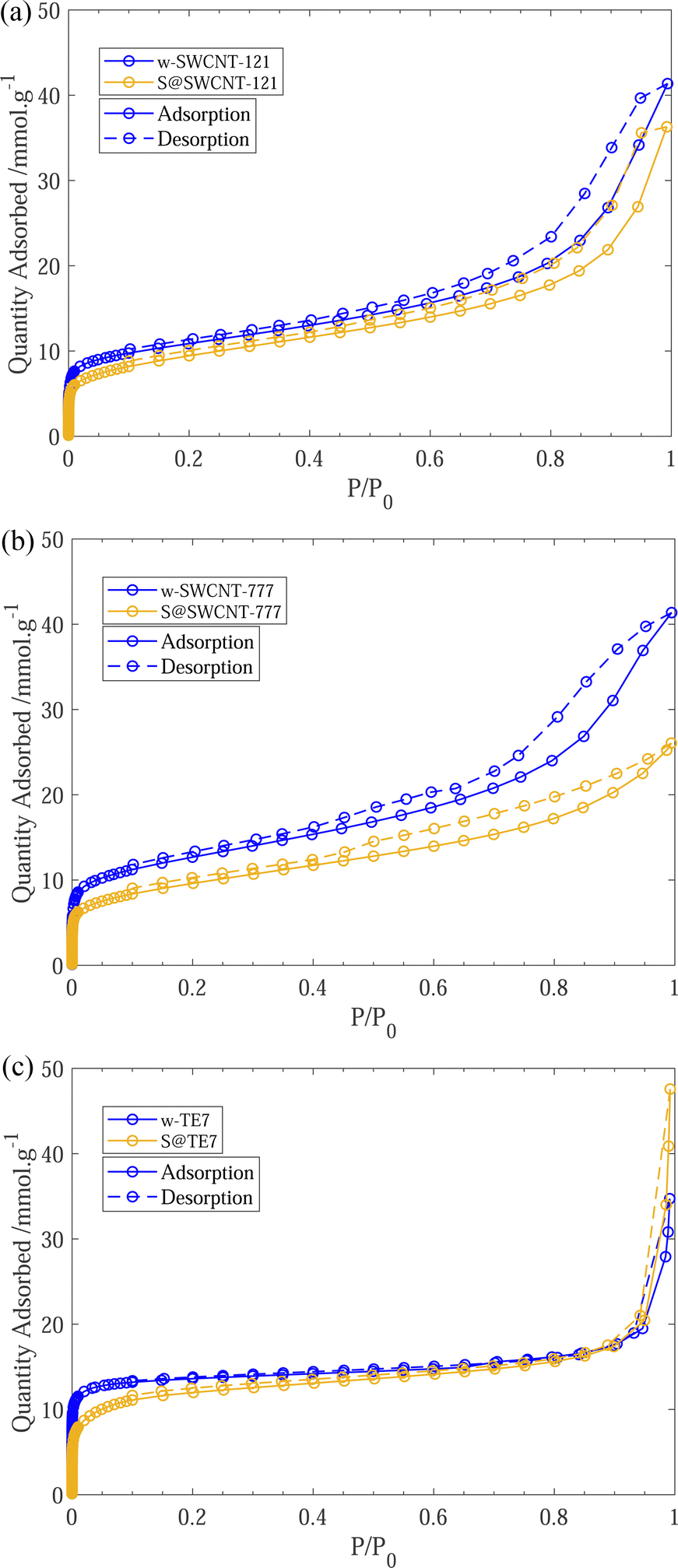 | ||
| Fig. 3 (a) 77 K N2 sorption isotherms for w-SWCNT-121 and S@SWCNT-121. (c) 77 K N2 sorption isotherms for w-SWCNT-777 and S@SWCNT-777. (e) 77 K N2 sorption isotherms for w-TE7 and S@TE7. Each plot shows adsorption (solid line) and desorption (dashed line) for the washed (blue) and sulfur composite (yellow). | ||
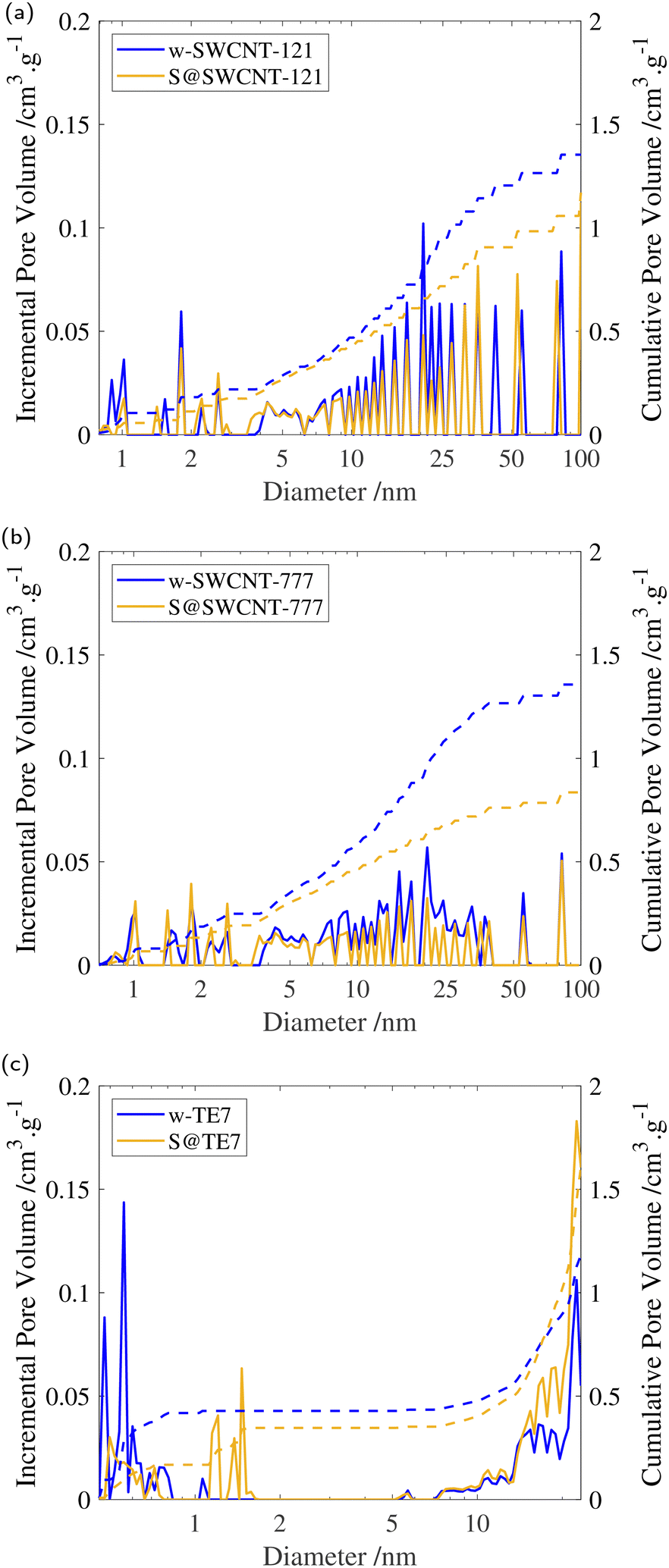 | ||
| Fig. 4 (a) Incremental and cumulative pore volume for w-SWCNT-121 and S@SWCNT-121 determined via NLDFT using cylindrical pore model. (b) Incremental and cumulative pore volume for w-SWCNT-777 and S@SWCNT-777 determined via NLDFT using cylindrical pore model. (c) Incremental and cumulative pore volume for w-TE7 and S@TE7 determined via NLDFT using slit pore model. All plots show the incremental (solid line) and cumulative (dashed line) pore volume for the washed (blue) and sulfur composite (yellow). | ||
| SSA/m2 g−1 | V p/cm3 g−1 | μV p/cm−3 g−1 | |
|---|---|---|---|
| a Standard deviation values taken form the fitting of the BET equation. | |||
| w-SWCNT-121 | 869 ± 2 | 1.19 | 0.31 |
| S@SWCNT-121 | 752 ± 3 | 0.94 | 0.27 |
| w-SWCNT-777 | 1014 ± 2 | 1.28 | 0.36 |
| S@SWCNT-777 | 764 ± 2 | 0.78 | 0.28 |
| w-TE7 | 1200 ± 2 | 0.68 | 0.46 |
| S@TE7 | 1008 ± 6 | 0.71 | 0.36 |
Additional reduction of the total pore volume within the mesoporous range (2–50 nm) is clear for both SWCNTs following sulfur encapsulation (see Table 2). Changes may result from exposure to molten sulfur during the synthesis procedure where changes in the SWCNT packing may have occurred.58 S@SWCNT-777 shows a dramatic 39% reduction in total pore volume. Comparatively, S@SWCNT-121 shows only a 21% decrease in total pore volume. However, S@TE7 displays a +4.4% change in total pore volume, and shows an increase in the proportion of mesoporosity.
SSA for all sulfur composite materials decrease compared to the washed samples (see Table 2). This observation was expected as many of the pores are now occupied by the sulfur or sulfur is occluding pore entrances which can no longer be accessed by the N2 molecules.
3.3 H2 sorption
Supercritical high-pressure H2 sorption isotherms were used to determine the excess gravimetric uptakes of the porous samples (see Fig. 5). w-SWCNT-121 and w-TE7 show relatively high maximum excess H2 gravimetric uptakes at 77 K and surpass the forecasted value of 1 wt% per 500 m2 g−1, where w-SWCNT-121 and w-TE7 adsorbed 1.99 ± 0.03 wt% and 3.02 ± 0.03 wt%, respectively. In comparison, w-SWCNT-777 falls short of Chahine's rule and only adsorbs 1.67± 0.02 wt% of H2. This may be attributed to the more significant proportion of ultra-micropores around the claimed optimum pore size of approximately 0.7 nm in the w-SWCNT-121 and w-TE7 (see Table 2), where increased overlap of electrostatic potentials from opposing walls generates stronger binding sites,5,25 thus, providing more desirable adsorption sites and allowing for more significant H2 densification.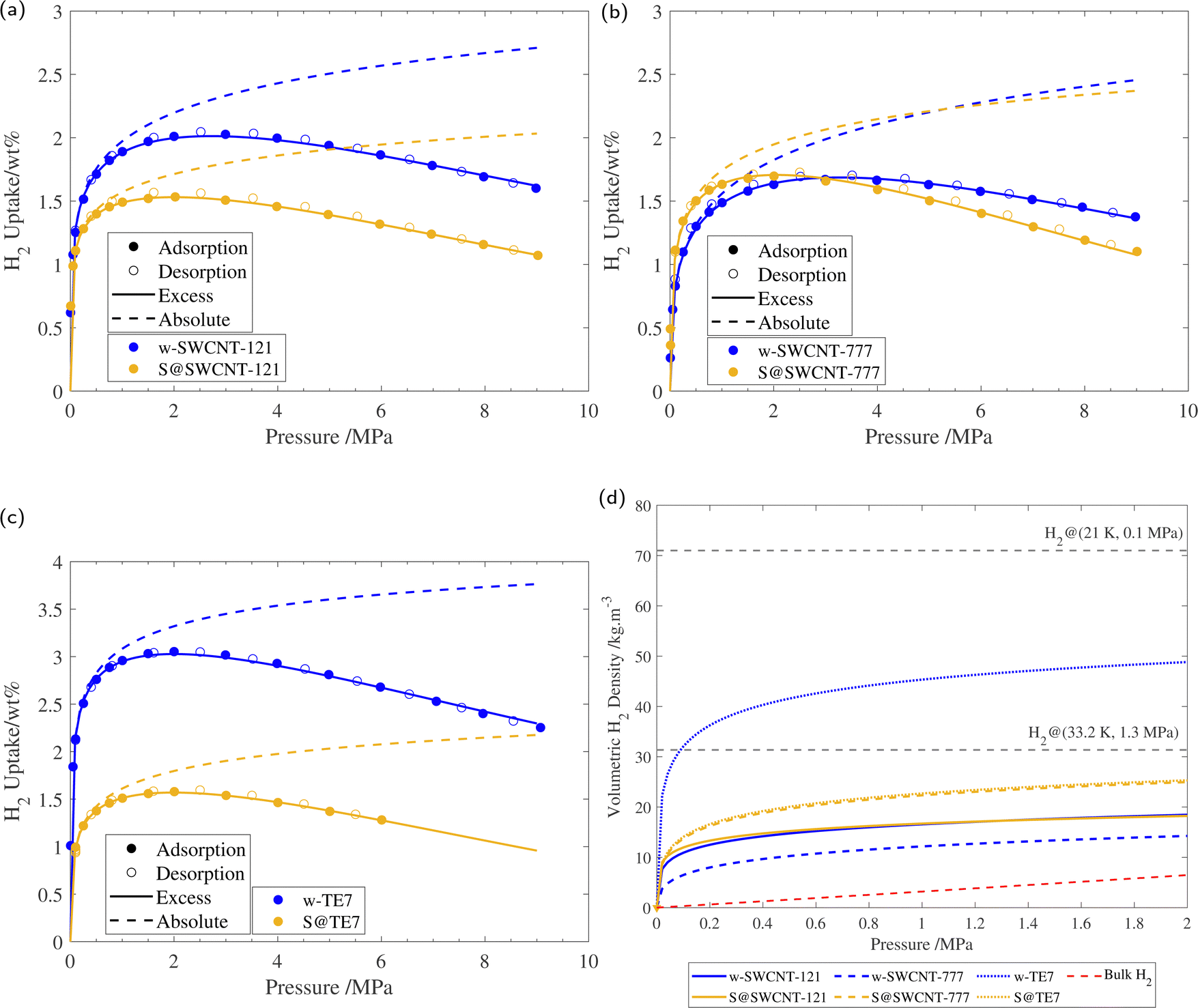 | ||
| Fig. 5 (a) 77 K H2 sorption isotherm of w-SWCNT-121 and S@SWCNT-121, with the respective excess isotherm determined through fitting and absolute isotherm, calculated using eqn (3). (b) 77 K H2 sorption isotherm of w-SWCNT-777 and S@SWCNT-777, with the respective excess isotherm determined through fitting and absolute isotherm, calculated using eqn (3). (c) 77 K H2 sorption isotherm of w-TE7 and S@TE7, with the respective excess isotherm determined through fitting and absolute isotherm, calculated using eqn (3). (d) Volumetric hydrogen density versus pressure up to 2 MPa for w-SWCNT-121 (blue solid line), S@SWCNT-121 (yellow solid line), w-SWCNT-777 (blue dashed line), S@SWCNT-777 (yellow dashed line), w-TE7 (blue dotted line) and S@TE7 (yellow dotted line), calculated using eqn (4). NIST bulk hydrogen data at 77 K (red dashed line), Liquid hydrogen density (20.25 K, 0.1 MPa), and density of hydrogen at its critical point (33.20 K, 1.3 MPa) are shown for reference. Each isotherm shows the result of the 3rd repeat run, all isotherms can be found in the ESI† (see Fig. S8). | ||
Following sulfur encapsulation, both S@SWCNT-121 and S@TE7 show a reduction in maximum excess H2 sorption by 32% and 50%, at 77 K, respectively. The decrease in the adsorbed quantity may originate from a reduction in available pore sizes around 0.7 nm shown by the N2 sorption experiments. The higher sulfur loading may further exaggerate this reduction in adsorbed hydrogen within these samples, particularly in w-TE7, which displayed the largest sulfur content. In contrast, S@SWCNT-777 shows comparable maximum gravimetric excess sorption uptakes to the w-SWCNT-777 sample; this is made more remarkable due to the reduction in SSA induced by the presence of sulfur. This surprising enhancement to the H2 per unit surface area (+33%) could originate from an increase in the enthalpy of adsorption. An enhancement to the enthalpy of adsorption is empirically suggested by the steeper initial H2 gravimetric uptake within the lower pressure regime; S@SWCNT-777 reaches the maximum excess gravimetric uptake at a lower pressure compared to w-SWCNT-777, excess sorption maxima are 2.0 MPa and 3.2 MPa, respectively. However, this effect is limited to the low-pressure regime where direct adsorbent–adsorbate interactions are prominent. At higher pressures, the H2 uptake of S@SWCNT-777 can be seen to crossover with w-SWCNT-777 to a lower value. Here multi-layer formation in mesopores begins where substrate–adsorbate interactions are negligible. At this point, the quantity of hydrogen is then dictated by the total available pore volume remaining, which is reduced in the S@SWCNT-777 sample, resulting in the reduction in excess gravimetric uptake at higher pressures.59 Mousavipour and Chitsazi computationally predicted increased binding energy (analogous to the enthalpy of adsorption) for H2 on sulfur-decorated SWCNTs. Sulfur atoms decorated on the external surface increase the polarizability of the incoming H2, thus increasing its interaction strength with the surface.36,60 Recently, Sedelnikova et al. demonstrated modification to the electronic surface where there is an electron accumulation on the external surface of (5,5) chirality CNTs (diameter approx. 0.73 nm) due to the encapsulation.39 Higher enthalpies of adsorption are common in chemisorptive materials where chemical bonding occurs. However, the cryogenic conditions and lack of a desorption hysteresis suggest little to no chemical bonding between H2 and adsorbent; therefore, this enhancement is deemed physisorption purely. From the PSD of S@SWCNT-777 and w-SWCNT-777, little to no reduction to the pore volume is observed in the ultra-microporous range (see Fig. 4), showing a synergistic effect between the high densification due to these sorption sites and modifications to the surface properties of the material.
A summary of other literature examples of sulfur-doped porous materials for hydrogen sorption at 77 K is given in the ESI† (see Table S3). Improved gravimetric uptakes are comparable to the improvements reported by Li et al. for sulfuric acid catalysed templated carbons, where the best performing material reported by Li et al. adsorbed approximately 59% more excess H2 per surface area compared to a hydrochloric acid catalysed reference material (see ESI,† Table S3). Despite similar SSA to the sulfur-containing materials described by Li et al. and Xia et al., the S@SWCNTs adsorb less H2. It is well known that slit-pore materials generally perform better than cylindrical-pore materials for H2 sorption and that cylindrical pores only demonstrate better performance at low pressures due to capillary condensation of H2.61,62 This, coupled with potential pore blockages likely explains the difference between the samples reported here and other sulfur-doped carbons.
3.4 Estimation of hydrogen densities
Additional information can be extracted from the H2 isotherms through implementing eqn (1). Semi-empirical modelling is a powerful tool to determine properties that would otherwise present significant challenges or costs to directly determine, such as adsorbate density, where the low atomic cross-section of H2 makes it a challenging target for most experimental techniques. Excess isotherms shown in Fig. 5 reveal information regarding the quantity of H2 as a direct result of the attractive electrostatic potential of the surface material. However, by adding the contribution of bulk H2 within the Vp, the absolute quantity of H2 can be estimated using the following equation (see eqn (3)).| Na = Ne + ρbVp | (3) |
Following the introduction of sulfur to the porous system, there is a consistent decrease in the maximum absolute H2 gravimetric uptake (see Fig. 5). This observation can be attributed to the change in the morphology (SSA and Vp) of the porous materials, simply no longer being able to accommodate large quantities of H2 at higher bulk densities. This translates to a lower H2 density at pore saturation (see Table 3). To obtain an estimate of volumetric density during pore filling (i.e. θ < 1), Na(P) was simply divided by Vp (see eqn (4)).
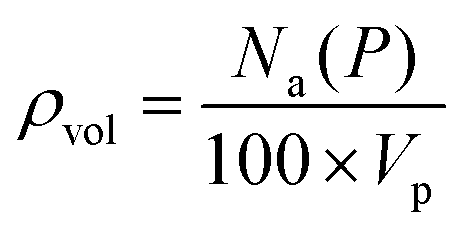 | (4) |
| N maxe/wt% |
N
(2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MPa)a/wt% MPa)a/wt% |
N
(θ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) =1)a/wt% =1)a/wt% |
ρ
(2MPa)a/kg m−3 |
ρ
(θ=1)a/kg m−3 |
|
|---|---|---|---|---|---|
| a Maximum measured excess gravimetric uptake (average of all repeat measurements). b Absolute gravimetric hydrogen uptake at 2 MPa determined from fitting using eqn (1) (average of all repeat measurements). c Absolute gravimetric uptake at pore saturation determined from fitting using eqn (1) (average of all repeat measurements). d Volumetric hydrogen density determined using eqn (4) (average of all repeat measurements). e Volumetric hydrogen density at pore saturation using eqn (4) (average of all repeat measurements). | |||||
| w-SWCNT-121 | 1.99 ± 0.03 | 2.16 ± 0.02 | 8.22 ± 0.19 | 18.19 ± 0.21 | 69.05 ± 1.61 |
| S@SWCNT-121 | 1.35 ± 0.11 | 1.66 ± 0.21 | 5.40 ± 0.52 | 17.62 ± 2.21 | 57.49 ± 5.58 |
| w-SWCNT-777 | 1.67 ± 0.02 | 1.82 ± 0.01 | 8.22 ± 0.06 | 14.24 ± 0.11 | 64.29 ± 0.48 |
| S@SWCNT-777 | 1.70 ± 0.03 | 1.93 ± 0.03 | 4.17 ± 0.12 | 24.76 ± 0.33 | 53.52 ± 1.65 |
| S2@SWCNT-777 | 0.66 ± 0.04 | 0.84 ± 0.02 | 1.72 ± 0.34 | 15.25 ± 0.37 | 31.38 ± 6.14 |
| w-TE7 | 3.02 ± 0.03 | 3.29 ± 0.02 | 4.93 ± 0.04 | 48.37 ± 0.32 | 72.49 ± 0.61 |
| S@TE7 | 1.52 ± 0.04 | 1.74 ± 0.05 | 3.50 ± 0.10 | 24.57 ± 0.67 | 49.26 ± 1.39 |
At lower pressures (<2 MPa), ρvol of both S@SWCNT-121 and S@SWCNT-777 is greater than in SWCNTs without sulfur (see Fig. 5(d)). This suggests that in both SWCNTs, there is a stronger attractive electrostatic potential as a result of the interactions between the sulfur and the SWCNT surface. The effect is more pronounced in S@SWCNT-777, likely owing to the preservation of the PSD within the sub-nanometer range, providing additional attraction. To test the effect of sulfur loading and microporosity, a second batch of S@SWCNT-777 (S2@SWCNT-777) was prepared with greater sulfur content and highly diminished total micropore volume (see ESI,† Fig. S9). The sulfur content of S2@SWCNT-777 was determined to be 19 wt% from TGA, and a Dubinin–Radushkevich μVp of 0.16 cm3 g−1 was calculated. Much of the loss in microporosity originates from the reduction of total pore volume in the ultra-microporous range (<0.7 nm). S2@SWCNT-777 shows a severely reduced hydrogen density both at pore saturation and 2 MPa by 32% and 49.6%, respectively. S2@SWCNT-777 demonstrates that, while sulfur can improve adsorbate density, microporosity has a larger impact on densification. Many of the stronger binding sites for SWCNTs are located in exohedral grooves between adjacent CNTs.63 Due to the greater sulfur loading, it is expected more exohedral sites within the bundle will be occupied by the sulfur; this prevents hydrogen from experiencing the improved electrostatic potential provided by the confined sulfur species.
4 Conclusion
Non-metal doping of existing porous materials offers an exciting and sustainable route to improved H2 storage and densification. We report the increased H2 surface packing and H2 densities within sulfur vapour infiltrated SWCNTs at pressures up to 2 MPa. The enhanced properties are proposed to stem from the retention of ultra-micropore volume and modifications to the polarizability of the material surface through electrostatic interactions between the CNT surface and the encapsulated sulfur species, which produced greater densities of hydrogen. However, maintaining the ultra-micropores (approx. 0.7 nm) is imperative to achieving high densities of H2. Limiting the location of sulfur to the endohedral cavity of the SWCNTs while preserving high ultra-microporosity may provide the greatest enhancement to hydrogen densification. This is illustrated particularly by a comparative activated carbon sample (TE7 and S@TE7) and a second higher sulfur-loaded SWCNT-777, where the inclusion of the sulfur resulted in significant changes to the PSD, reducing the strong densification contributions from the desirable ultra-microporous sites. Future work will further explore the changes in the properties of the dense hydrogen adsorbate phase and identify the preferable adsorbate sites in these composites through neutron scattering and Raman spectroscopy, amongst other complementary techniques. Furthermore, experimental validation of increased enthalpy of adsorption may be provided through the application of the Clausius–Clapeyron method using additional adsorption isotherms at a range of temperatures.Author contributions
CDB performed the experiments, data analysis and drafted the manuscript. LRT contributed to methodology, data analysis, conceptualisation, supervision, and reviewing & editing of the manuscript. HVD contributed through the methodology and data analysis. SR contributed to data analysis, project supervision and reviewing & editing of the manuscript. VPT contributed to project conceptualisation, data analysis, supervision and reviewing & editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Engineering and Physical Science Research Council through the EPSRC Centre for Doctoral Training in Composites Science, Engineering and Manufacturing [EP/S021728/1] and an EPSRC Research Fellowship for VPT [EP/R01650X/1]. HVD thanks the Royal Society of Chemistry for the Research Fund grant [R20-8172]. HVD acknowledges support from the UK Engineering and Physical Sciences Research Council [EP/T517872/1]. SR acknowledges the support of the University of Bristol Dean's Fellowship. XPS experiments were performed at the Cardiff hub of the EPSRC National Facility for X-ray Photoelectron Spectroscopy (‘HarwellXPS’), operated by Cardiff University and UCL under contract No. PR16195. Special thanks to Dr John Day (University of Bristol) for conducting the Raman Spectroscopy, Dr Mi Tian (University of Exeter) for consultation on this project, Dr Prasanth Prabhakaran (Teeside University) for training on the Hiden Isochema XEMIS-001 and Mr Duncan Tarling (University of Bristol) for manufacturing the H-shaped quartz ampule.Notes and references
- S. Bouckaert, A. F. Pales, C. McGlade, U. Remme, B. Wanner, L. Varro, D. D’Ambrosio and T. Spencer, Net Zero by 2050, IEA, IEA, Paris, 2021 Search PubMed.
- R. Moradi and K. M. Groth, Hydrogen storage and delivery: Review of the state of the art technologies and risk and reliability analysis, Int. J. Hydrogen Energy, 2019, 44(23), 12254–12269, DOI:10.1016/j.ijhydene.2019.03.041.
- H. Ibach, Physics of Surfaces and Interfaces, Springer, Berlin, Heidelberg, 2006 Search PubMed.
- L. Liu, X.-B. Luo, L. Ding and S.-L. Luo, in Nanomaterials for the Removal of Pollutants and Resource Reutilization, ed. L. Liu, X.-B. Luo, L. Ding and S.-L. Luo, Elsevier, Amsterdam, 2019, ch. 4, pp. 83–147 Search PubMed.
- V. P. Ting, A. J. Ramirez-Cuesta, N. Bimbo, J. E. Sharpe, A. Noguera-Diaz, V. Presser, S. Rudic and T. J. Mays, Direct evidence for solid-like hydrogen in a nanoporous carbon hydrogen storage material at supercritical temperatures, ACS Nano, 2015, 9(8), 8249–8254, DOI:10.1021/acsnano.5b02623.
- N. C. Gallego, L. He, D. Saha, C. I. Contescu and Y. B. Melnichenko, Hydrogen confinement in carbon nanopores: extreme densification at ambient temperature, J. Am. Chem. Soc., 2011, 133(35), 13794–13797, DOI:10.1021/ja202432x.
- R. Chahine and T. Bose, Low-pressure adsorption storage of hydrogen, Int. J. Hydrogen Energy, 1994, 19(2), 161–164, DOI:10.1016/0360-3199(94)90121-X.
- P. Bénard and R. Chahine, Storage of hydrogen by physisorption on carbon and nanostructured materials, Scr. Mater., 2007, 56(10), 803–808, DOI:10.1016/j.scriptamat.2007.01.008.
- S. S. Samantaray, S. T. Putnam and N. P. Stadie, Volumetrics of Hydrogen Storage by Physical Adsorption, Inorganics, 2021, 9, 45, DOI:10.3390/inorganics9060045.
- G. Sdanghi, R. L. Canevesi, A. Celzard, M. Thommes and V. Fierro, Characterization of carbon materials for hydrogen storage and compression, C, 2020, 6(3), 46, DOI:10.3390/c6030046.
- E. Boateng and A. Chen, Recent advances in nanomaterial-based solid-state hydrogen storage, Mater. Today Adv., 2020, 6, 100022, DOI:10.1016/j.mtadv.2019.100022.
- S. P. Shet, S. S. Priya, K. Sudhakar and M. Tahir, A review on current trends in potential use of metal-organic framework for hydrogen storage, Int. J. Hydrogen Energy, 2021, 46(21), 11782–11803, DOI:10.1016/j.ijhydene.2021.01.020.
- U. S. Department of Energy, Target explanation document: onboard hydrogen storage for light-duty fuel cell vehicles, https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_targets_onboard_hydro_storage_explanation.pdf, (accessed August 24 2022).
- T. Kopac, Hydrogen storage characteristics of bio-based porous carbons of different origin: A comparative review, Int. J. Energy Res., 2021, 45, 20497–20523, DOI:10.1002/er.7130.
- Y. Xia, Z. Yang and Y. Zhu, Porous carbon-based materials for hydrogen storage: advancement and challenges, J. Mater. Chem. A, 2013, 1(33), 9365–9381, 10.1039/c3ta10583k.
- A. Peigney, C. Laurent, E. Flahaut, R. Bacsa and A. Rousset, Specific surface area of carbon nanotubes and bundles of carbon nanotubes, Carbon, 2001, 39(4), 507–514, DOI:10.1016/S0008-6223(00)00155-X.
- R. Bacsa, C. Laurent, R. Morishima, H. Suzuki and M. Le Lay, Hydrogen Storage in High Surface Area Carbon Nanotubes Produced by Catalytic Chemical Vapor Deposition, J. Phys. Chem. B, 2004, 108(34), 12718–12723, DOI:10.1021/jp0312621.
- M. E. Birch, T. A. Ruda-Eberenz, M. Chai, R. Andrews and R. L. Hatfield, Properties that influence the specific surface areas of carbon nanotubes and nanofibers, Ann. Occup. Hyg., 2013, 57(9), 1148–1166, DOI:10.1093/annhyg/met042.
- G. E. Froudakis, Hydrogen storage in nanotubes & nanostructures, Mater. Today, 2011, 14(7–8), 324–328, DOI:10.1016/S1369-7021(11)70162-6.
- S. Seenithurai, R. K. Pandyan, S. V. Kumar, C. Saranya and M. Mahendran, Al-decorated carbon nanotube as the molecular hydrogen storage medium, Int. J. Hydrogen Energy, 2014, 39(23), 11990–11998, DOI:10.1016/j.ijhydene.2014.05.184.
- V. Tozzini and V. Pellegrini, Prospects for hydrogen storage in graphene, Phys. Chem. Chem. Phys., 2013, 15, 80–89, 10.1039/C2CP42538F.
- M. Hirscher, V. A. Yartys, M. Baricco, J. B. von Colbe, D. Blanchard, R. C. Bowman Jr, D. P. Broom, C. E. Buckley, F. Chang and P. Chen, Materials for hydrogen-based energy storage – past, recent progress and future outlook, J. Alloys Compd., 2020, 827, 153548, DOI:10.1016/j.jallcom.2019.153548.
- D. P. Broom, C. Webb, G. S. Fanourgakis, G. E. Froudakis, P. N. Trikalitis and M. Hirscher, Concepts for improving hydrogen storage in nanoporous materials, Int. J. Hydrogen Energy, 2019, 44(15), 7768–7779, DOI:10.1016/j.ijhydene.2019.01.224.
- I. Rossetti, G. Ramis, A. Gallo and A. Di Michele, Hydrogen storage over metal-doped activated carbon, Int. J. Hydrogen Energy, 2015, 40(24), 7609–7616, DOI:10.1016/j.ijhydene.2015.04.064.
- L. R. Terry, S. Rols, M. Tian, I. Da Silva, S. J. Bending and V. P. Ting, Manipulation of the crystalline phase diagram of hydrogen through nanoscale confinement effects in porous carbons, Nanoscale, 2022, 14, 7250–7261, 10.1039/D2NR00587E.
- I. Cabria, M. López and J. Alonso, Enhancement of hydrogen physisorption on graphene and carbon nanotubes by Li doping, J. Chem. Phys., 2005, 123(20), 204721, DOI:10.1063/1.2125727.
- D. Li, W. Li, J. Shi and F. Xin, Influence of doping nitrogen, sulfur, and phosphorus on activated carbons for gas adsorption of H2, CH4 and CO2, RSC Adv., 2016, 6(55), 50138–50143, 10.1039/C6RA06620H.
- M. Murialdo, N. J. Weadock, Y. Liu, C. C. Ahn, S. E. Baker, K. Landskron and B. Fultz, High-Pressure Hydrogen Adsorption on a Porous Electron-Rich Covalent Organonitridic Framework, ACS Omega, 2019, 4(1), 444–448, DOI:10.1021/acsomega.8b03206.
- J. P. Paraknowitsch and A. Thomas, Doping carbons beyond nitrogen: an overview of advanced heteroatom doped carbons with boron, sulphur and phosphorus for energy applications, Energy Environ. Sci., 2013, 6(10), 2839–2855, 10.1039/C3EE41444B.
- L. S. Blankenship, N. Balahmar and R. Mokaya, Oxygen-rich microporous carbons with exceptional hydrogen storage capacity, Nat. Commun., 2017, 8, 1–12, DOI:10.1038/s41467-021-26590-4.
- M. I. M. Kusdhany, Z. Ma, A. Mufundirwa, H.-W. Li, K. Sasaki, A. Hayashi and S. M. Lyth, Hydrogen and carbon dioxide uptake on scalable and inexpensive microporous carbon foams, Microporous Mesoporous Mater., 2022, 343, 112141, DOI:10.1016/j.micromeso.2022.112141.
- Y. Xia, Y. Zhu and Y. Tang, Preparation of sulfur-doped microporous carbons for the storage of hydrogen and carbon dioxide, Carbon, 2012, 50(15), 5543–5553, DOI:10.1016/j.carbon.2012.07.044.
- A. M. de Yuso, M. De Fina, C. Nita, P. Fioux, J. Parmentier and C. M. Ghimbeu, Synthesis of sulfur-doped porous carbons by soft and hard templating processes for CO2 and H2 adsorption, Microporous Mesoporous Mater., 2017, 243, 135–146, DOI:10.1016/j.micromeso.2016.12.001.
- M. Sevilla, A. Fuertes and R. Mokaya, Preparation and hydrogen storage capacity of highly porous activated carbon materials derived from polythiophene, Int. J. Hydrogen Energy, 2011, 36(24), 15658–15663, DOI:10.1016/j.ijhydene.2011.09.032.
- J. Park, N. F. Attia, M. Jung, M. E. Lee, K. Lee, J. Chung and H. Oh, Sustainable nanoporous carbon for CO2, CH4, N2, H2 adsorption and CO2/CH4 and CO2/N2 separation, Energy, 2018, 158, 9–16, DOI:10.1016/j.energy.2018.06.010.
- S. Mousavipour and R. Chitsazi, A theoretical study on the effect of intercalating sulfur atom and doping boron atom on the adsorption of hydrogen molecule on (10,0) single-walled carbon nanotubes, J. Iran. Chem. Soc., 2010, 7, S92–S102, DOI:10.1007/BF03246188.
- C. Fu, M. B. Oviedo, Y. Zhu, A. von Wald Cresce, K. Xu, G. Li, M. E. Itkis, R. C. Haddon, M. Chi and Y. Han, Confined Lithium–Sulfur Reactions in Narrow-Diameter Carbon Nanotubes Reveal Enhanced Electrochemical Reactivity, ACS Nano, 2018, 12(10), 9775–9784, DOI:10.1021/acsnano.7b08778.
- J. Yang, J. Lee, J. Lee and W. Yi, Gas sensing mechanism of sulfur chain-encapsulated single-walled carbon nanotubes, Diamond Relat. Mater., 2019, 97, 107474, DOI:10.1016/j.diamond.2019.107474.
- O. V. Sedelnikova, V. I. Sysoev, O. A. Gurova, Y. P. Ivanov, V. O. Koroteev, R. Arenal, A. A. Makarova, L. G. Bulusheva and A. V. Okotrub, Role of interface interactions in the sensitivity of sulfur-modified single-walled carbon nanotubes for nitrogen dioxide gas sensing, Carbon, 2022, 186, 539–549, DOI:10.1016/j.carbon.2021.10.056.
- A. Hruzewicz-Kołodziejczyk, V. P. Ting, N. Bimbo and T. J. Mays, Improving comparability of hydrogen storage capacities of nanoporous materials, Int. J. Hydrogen Energy, 2012, 37(3), 2728–2736, DOI:10.1016/j.ijhydene.2011.03.001.
- N. Lertrojanachusit, O. Pornsunthorntawee, B. Kitiyanan, J. Chavadej and S. Chavadej, Separation and purification of carbon nanotubes using froth flotation with three sequential pretreatment steps of catalyst oxidation, catalyst removal, and silica dissolution, Asia-Pac. J. Chem. Eng., 2013, 8(6), 830–842, DOI:10.1002/apj.1727.
- T. Fujimori, A. Morelos-Gómez, Z. Zhu, H. Muramatsu, R. Futamura, K. Urita, M. Terrones, T. Hayashi, M. Endo and S. Y. Hong, Conducting linear chains of sulphur inside carbon nanotubes, Nat. Commun., 2013, 4, 1–8, DOI:10.1038/ncomms3162.
- H. Kataura, Y. Maniwa, T. Kodama, K. Kikuchi, K. Hirahara, K. Suenaga, S. Iijima, S. Suzuki, Y. Achiba and W. Krätschmer, High-yield fullerene encapsulation in single-wall carbon nanotubes, Synth. Met., 2001, 121(1–3), 1195–1196, DOI:10.1016/S0379-6779(00)00707-4.
- ISO, ISO 9277:2010 Determination of the specific surface area of solids by gas adsorption-BET method, 2010.
- J. Tóth, Uniform interpretation of gas/solid adsorption, Adv. Colloid Interface Sci., 1995, 55, 1–239, DOI:10.1016/0001-8686(94)00226-3.
- J. E. Sharpe, N. Bimbo, V. P. Ting, A. D. Burrows, D. Jiang and T. J. Mays, Supercritical hydrogen adsorption in nanostructured solids with hydrogen density variation in pores, Adsorption, 2013, 19, 643–652, DOI:10.1007/s10450-013-9487-6.
- J. W. Leachman, R. T. Jacobsen, S. Penoncello and E. W. Lemmon, Fundamental Equations of State for Parahydrogen, Normal Hydrogen, and Orthohydrogen, J. Phys. Chem. Ref. Data, 2009, 38(3), 721–748, DOI:10.1063/1.3160306.
- E. W. Lemmon, I. H. Bell, M. L. Huber and M. O. McLinden, Thermophysical Properties of Fluid Systems, 2022 Search PubMed.
- V. V. Nascimento, W. Q. Neves, R. S. Alencar, G. Li, C. Fu, R. C. Haddon, E. Bekyarova, J. Guo, S. S. Alexandre and R. W. Nunes, Origin of the Giant Enhanced Raman Scattering by Sulfur Chains Encapsulated inside Single-Wall Carbon Nanotubes, ACS Nano, 2021, 15(5), 8574–8582, DOI:10.1021/acsnano.1c00390.
- M. Fantauzzi, B. Elsener, D. Atzei, A. Rigoldi and A. Rossi, Exploiting XPS for the identification of sulfides and polysulfides, RSC Adv., 2015, 5, 75953–75963, 10.1039/C5RA14915K.
- G. Li, C. Fu, M. B. Oviedo, M. Chen, X. Tian, E. Bekyarova, M. E. Itkis, B. M. Wong, J. Guo and R. C. Haddon, Giant Raman Response to the Encapsulation of Sulfur in Narrow Diameter Single-Walled Carbon Nanotubes, J. Am. Chem. Soc., 2016, 138(1), 40–43, DOI:10.1021/jacs.5b10598.
- N. Shimodaira and A. Masui, Raman spectroscopic investigations of activated carbon materials, J. Appl. Phys., 2002, 92(2), 902–909, DOI:10.1063/1.1487434.
- G. Carotenuto, V. Romeo, S. De Nicola and L. Nicolais, Graphite nanoplatelet chemical cross-linking by elemental sulfur, Nanoscale Res. Lett., 2013, 8, 1–6, DOI:10.1186/1556-276X-8-94.
- J. Mao, Y. Wang, J. Zhu, J. Yu and Z. Hu, Thiol functionalized carbon nanotubes: Synthesis by sulfur chemistry and their multi-purpose applications, Appl. Surf. Sci., 2018, 447, 235–243, DOI:10.1016/j.apsusc.2018.03.188.
- B. A. Trofimov, L. M. Sinegovskaya and N. K. Gusarova, Vibrations of the S–S bond in elemental sulfur and organic polysulfides: a structural guide, J. Sulfur Chem., 2009, 30, 518–554, DOI:10.1080/17415990902998579.
- B. Elman, M. Shayegan, M. Dresselhaus, H. Mazurek and G. Dresselhaus, Structural characterization of ion-implanted graphite, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 25(6), 4142, DOI:10.1103/PhysRevB.25.4142.
- R. Escribano, J. Sloan, N. Siddique, N. Sze and T. Dudev, Raman spectroscopy of carbon-containing particles, Vib. Spectrosc., 2001, 26(2), 179–186, DOI:10.1016/S0924-2031(01)00106-0.
- D. Y. Kim, J. H. Kim, M. Li, S. Noda, J. Kim, K.-S. Kim, K. S. Kim and C.-M. Yang, Controllable pore structures of pure and sub-millimeter-long carbon nanotubes, Appl. Surf. Sci., 2021, 566, 150751, DOI:10.1016/j.apsusc.2021.150751.
- N. Bimbo, V. P. Ting, J. E. Sharpe and T. J. Mays, Analysis of optimal conditions for adsorptive hydrogen storage in microporous solids, Colloids Surf., A, 2013, 437, 113–119, DOI:10.1016/j.colsurfa.2012.11.008.
- D. Wang, Y. Shen, Y. Chen, L. Liu and Y. Zhao, Microwave-assistant preparation of N/S co-doped hierarchical porous carbons for hydrogen adsorption, Chem. Eng. J., 2019, 367, 260–268, DOI:10.1016/j.cej.2019.02.150.
- M. Rzepka, P. Lamp and M. De la Casa-Lillo, Physisorption of Hydrogen on Microporous Carbon and Carbon Nanotubes, J. Phys. Chem. B, 1998, 102(52), 10894–10898, DOI:10.1021/jp9829602.
- Q. Wang and J. K. Johnson, Molecular simulation of hydrogen adsorption in single-walled carbon nanotubes and idealized carbon slit pores, J. Chem. Phys., 1999, 110(1), 577–586, DOI:10.1063/1.478114.
- B. Panella and M. Hirscher, Raman studies of hydrogen adsorbed on nanostructured porous materials, Phys. Chem. Chem. Phys., 2008, 10, 2910–2917, 10.1039/B719678D.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00242f |
| This journal is © The Royal Society of Chemistry 2023 |