
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative dissolution of lithium and manganese from lithium manganospinel (LiMn2O4): towards climate-smart processes for critical metal recycling†
Rhys A.
Ward
*ab,
Dávid
Kocsis
*abc and
Jay D.
Wadhawan
 *ab
*ab
aDepartment of Chemical Engineering, The University of Hull, Cottingham Road, Kingston-upon-Hull HU6 7RX, UK. E-mail: r.ward4@hull.ac.uk; dkocsis@singletonbirch.co.uk; j.wadhawan@hull.ac.uk
bAura Innovation Centre, Bridgehead Business Park, Meadow Road, Hessle HU13 0GD, UK
cSingleton Birch, Ltd., Melton Ross Quarries, Barnetby, DN38 6AE, North Lincolnshire, UK
First published on 6th April 2023
Abstract
This work reports the oxidative breakdown of the LiMn2O4 structure using alkaline hypochlorite, as a method for recovering both lithium and manganese species. The heterogeneous dissolution rate for this process is monitored through the formation of the permanganate ion. It is found that reaction occurs under activation control, with a barrier that matches that for electron hopping within the spinel. Moreover, this activation barrier is smaller than those observed for typical conventional methods of recovery of lithium or manganese from LiMn2O4; accordingly, the new process is suggested to be climate-smart, despite the low, single-pass, recovery efficiency that results from slow surface kinetics.
The transition to the wide-scale deployment of renewable energy holds a plethora of challenges associated with technical, environmental, societal and governance aspects. Since this clean energy transition is mineral-intensive,1,2 it requires a substantial material footprint, even though the overall amount of greenhouse gas emissions is lower than those associated with the use of fossil fuels.1 Accordingly, many mineral commodities are labelled as “critical” – their economic importance and supply risk both exceed certain thresholds.3–7 However, it is also recognised that supply risk to a particular country is extremely sensitive to the global market conditions, especially when there is little access to indigenous sources of specific minerals at economically viable grades.8–10 For example, the USA is wholly reliant on imports for their manganese supply11 – a key metal chiefly used as a deoxidiser and desulphuriser in steel manufacture,12 and thus impacts on the infrastructure for low-carbon energy technologies (wind, hydroelectric, geothermal power).1,2,4 The current instabilities in Eastern Europe, where Ukraine and Georgia typically contribute ca. 4% of world production (ore or concentrate) of manganese, but hold at least ca. 10% of identified world manganese reserves,13 may cause economic stress in the global manganese supply.14 Likewise, mineral flow analysis indicates that the EU and the UK (notwithstanding current explorations in South-West England15) obtain the majority of their lithium products, required for battery-based energy storage, from China.16,17 This promotes the development of ambitious, industrial recycling programmes, particularly of lithium batteries,18,19 where a number of routes have been proposed for lithium battery cathodes, including pyrometallurgical or hydrometallurgical recovery,20 solvent extraction,21 or electrochemical22 and direct recycling.23 However, environmentally responsible resource recovery and recycling encourages innovation in “climate-smart” pathways, which use lower amounts of energy and/or water than current practices.1,24
One way to achieve climate-smart “urban mining”25 is through the development of chemical leaching processes that require a lower activation energy than alternatives. This requires knowledge of the dissolution kinetics and the identification as to whether leaching operates under activation or mass transfer control. This is important since, if limited by slow mass transfer, the stirring rate of mineral concentrates/powders in the lixiviant will enable faster dissolution; whereas, if the heterogeneous chemical reaction determines the rate of dissolution, the process can capitalise on faster dissolution rates at high temperatures, or smaller particle sizes (larger interfacial areas). It follows that monitoring the kinetics with temperature empowers the determination of the activation energy of the dissolution: this is typically ca. 8–17 kJ mol−1 for diffusion control, and greater than 42 kJ mol−1 for chemical control.26
In this communication, following our earlier work on the redox removal of iron and manganese impurities in slaked lime using alkaline hypochlorite,27 we report a redox method for breaking down the framework of lithium managnospinel, LiMn2O4 – a material used as a lithium-ion battery cathode.19,23 This enables the solubilisation of both lithium and manganese as a pathway for total recovery, rather than merely recovery of one species, with the remainder being used for direct recycling. We focus on the initial dissolution kinetics, since our previous work has illustrated how such initial kinetics can be maintained in a “cascading” mineral processing flowsheet.27 Moreover, this enables the unravelling of the activation energy required for the dissolution – for reactions under activation control, the lower this is, compared with conventional, reductive acidic leaching28 of LiMn2O4, the more climate-smart the recovery process becomes, assuming all other variables (such as leaching time) remain constant.
Lithium manganospinel (LiMn2O4) has a normal spinel structure (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with a cubic close-packed oxide lattice, with manganese in the half of the octahedral positions, and lithium in an eighth of the tetrahedral holes.29–33 The manganese is present as both MnIII and MnIV, with the former species (high-spin d4) undergoing a co-operative Jahn–Teller distortion, yielding a tetragonal elongation at low temperature, that is noticeable through a first-order phase change around ambient temperature,32 and which also occurs on increasing lithiation.29–31 In operation as a battery cathode,33 since LiMn2O4 carries DC current through MnIII/MnIV electron hopping,31,32 the expansion and contraction in the lattice, through the Jahn–Teller distortion, can lead to stress-induced cracking, particularly at high current drain.33 Furthermore, it is thought34 that MnIII disproportionates to MnII and MnIV, which gives rise to the well-known capacity-fade (“aging”) of this battery material,35 as the MnII species dissolves in the electrolyte.36 This dissolution process is controlled by surface kinetics, and is faster for smaller particles.37 It occurs with an activation energy of ca. 70 kJ mol−1 (for LiMn2O4 dissolution into a 50
m) with a cubic close-packed oxide lattice, with manganese in the half of the octahedral positions, and lithium in an eighth of the tetrahedral holes.29–33 The manganese is present as both MnIII and MnIV, with the former species (high-spin d4) undergoing a co-operative Jahn–Teller distortion, yielding a tetragonal elongation at low temperature, that is noticeable through a first-order phase change around ambient temperature,32 and which also occurs on increasing lithiation.29–31 In operation as a battery cathode,33 since LiMn2O4 carries DC current through MnIII/MnIV electron hopping,31,32 the expansion and contraction in the lattice, through the Jahn–Teller distortion, can lead to stress-induced cracking, particularly at high current drain.33 Furthermore, it is thought34 that MnIII disproportionates to MnII and MnIV, which gives rise to the well-known capacity-fade (“aging”) of this battery material,35 as the MnII species dissolves in the electrolyte.36 This dissolution process is controlled by surface kinetics, and is faster for smaller particles.37 It occurs with an activation energy of ca. 70 kJ mol−1 (for LiMn2O4 dissolution into a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol% solution of ethylene carbonate and dimethylene carbonate containing 1.0 M LiPF6).37 This activation energy is larger than that required for Mn2+ dissolution using SO2 to reduce MnOOH (16–65 kJ mol−1) under ambient conditions,38 but comparable with those required for dissolution from manganese oxides by sulfuric acid:39 68 ± 2 kJ mol−1 (MnO), 80 ± 2 kJ mol−1 (Mn3O4), 86 ± 2 kJ mol−1 (Mn2O3) and 90 ± 2 kJ mol−1 (MnO2). It is noteworthy that LeBlanc and Fogler40 observed that manganese dissolution from this oxide series using mineral haloacids is more rapid, as the overall manganese oxidation state is lowered, with MnO dissolution having sufficiently low activation energies (ca. 18–20 kJ mol−1) for the chemical dissolution to be mass-transfer controlled, with reductive dissolution of the higher oxidation states requiring activation energies of between ca. 18–97 kJ mol−1. Furthermore, for Mn3O4, the activation energy for manganese dissolution as MnO4− determined by our previous work using alkaline hypochlorite27 is ca. 40% smaller than that reported for sulfuric acid above. Given, the Pourbaix diagram for LiMn2O4 indicates28 its stability above ca. pH 12 (at 90 °C), this encourages the examination of the possibility of manganese (and therefore lithium) dissolution from LiMn2O4 under similar conditions.
:50 vol% solution of ethylene carbonate and dimethylene carbonate containing 1.0 M LiPF6).37 This activation energy is larger than that required for Mn2+ dissolution using SO2 to reduce MnOOH (16–65 kJ mol−1) under ambient conditions,38 but comparable with those required for dissolution from manganese oxides by sulfuric acid:39 68 ± 2 kJ mol−1 (MnO), 80 ± 2 kJ mol−1 (Mn3O4), 86 ± 2 kJ mol−1 (Mn2O3) and 90 ± 2 kJ mol−1 (MnO2). It is noteworthy that LeBlanc and Fogler40 observed that manganese dissolution from this oxide series using mineral haloacids is more rapid, as the overall manganese oxidation state is lowered, with MnO dissolution having sufficiently low activation energies (ca. 18–20 kJ mol−1) for the chemical dissolution to be mass-transfer controlled, with reductive dissolution of the higher oxidation states requiring activation energies of between ca. 18–97 kJ mol−1. Furthermore, for Mn3O4, the activation energy for manganese dissolution as MnO4− determined by our previous work using alkaline hypochlorite27 is ca. 40% smaller than that reported for sulfuric acid above. Given, the Pourbaix diagram for LiMn2O4 indicates28 its stability above ca. pH 12 (at 90 °C), this encourages the examination of the possibility of manganese (and therefore lithium) dissolution from LiMn2O4 under similar conditions.
Incubation of LiMn2O4 (325 mesh, corresponding to particles of diameter ca. 44 μm) with alkaline hypochlorite (8%) at ambient temperature overnight furnished a pale pink supernatant, with a UV-visible absorption spectrum indicating the formation of permanganate (for experimental details, see the ESI,† ESI1). As discussed in ESI2 (ESI†), this is consistent with the following overall process, where de-lithiation occurs with the breakdown of the manganese oxide framework.
| 2LiMn2O4(s) + 7ClO−(aq) + 2OH−(aq) → 2Li+(aq) + 4MnO4−(aq) + 7Cl− + H2O(l) | (A) |
At pH 14, the standard electrode potential for the ClO−/Cl− redox couple is +0.89 V vs. the standard hydrogen electrode (SHE).41 The oxidative de-lithiation of LiMn2O4, (eqn (B)) has been reported to require similar potentials in lithium-based supporting electrolytes:42–45
| LiMn2O4(s) → xLi+(aq) + xe− + Li1−xMn2O4(s) | (B) |
However, this reaction requires much higher potentials in sodium hydroxide electrolytes.46 Accordingly, as indicated in ESI2 (ESI†), the following reaction sequence is likely to occur,
| 2LiMn2O4(s) + xClO−(aq) + xH2O(l) → 2Li1−xMn2O4(s) + 2xLi+(aq) + xCl− + 2xOH−(aq) | (C) |
| Li1−xMn2O4(s) + ½(5 − x)ClO−(aq) + (3 + x)OH−(aq) → (1 − x)Li+(aq) + 2MnO42−(aq) + ½(5 − x)Cl−(aq) + ½(3 + x)H2O(l) | (D) |
| 2MnO42−(aq) + ClO−(aq) + H2O(l) → 2MnO4−(aq) + Cl−(aq) + 2OH−(aq) | (E) |
As mentioned earlier, whilst the dissolution of most manganese oxides typically occurs under the control of surface kinetics, this is not always the case.40 For surface redox reaction-controlled dissolution, a simple cubic law is typically followed.50–53 However, the dissolution rate of irreversible chemical reactions occurring under mixed control by mass transfer and surface kinetics is given by:
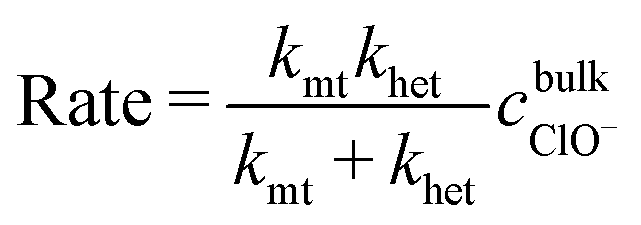 | (1) |
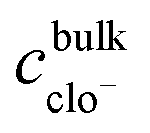 , and is formally derived in ESI3 (ESI†). In eqn (1), both mass transfer and rate coefficients have units of length per unit time, so that the dissolution rate is equivalent to the flux (amount per unit time per unit area). The area involved is the interfacial area, and can be estimated through the total geometrical area of the particles (S). Assuming monodisperse spherical particles of radius r0 (22 μm), we may approximate the total geometrical area as being the area of a single particle multiplied by the number of particles present. The latter is the ratio of the volume of the particles used to that corresponding to a single sphere, where the former is given by the mass of particles used (m0) normalised by the bulk density of LiMn2O4 (ρ = 4.29 g cm−3).54 Thus,
, and is formally derived in ESI3 (ESI†). In eqn (1), both mass transfer and rate coefficients have units of length per unit time, so that the dissolution rate is equivalent to the flux (amount per unit time per unit area). The area involved is the interfacial area, and can be estimated through the total geometrical area of the particles (S). Assuming monodisperse spherical particles of radius r0 (22 μm), we may approximate the total geometrical area as being the area of a single particle multiplied by the number of particles present. The latter is the ratio of the volume of the particles used to that corresponding to a single sphere, where the former is given by the mass of particles used (m0) normalised by the bulk density of LiMn2O4 (ρ = 4.29 g cm−3).54 Thus,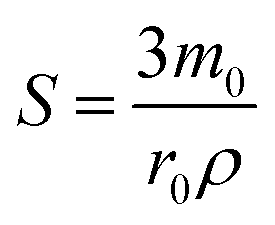 | (2) |
If the mass transport coefficient, kmt, can be estimated, the heterogeneous reaction rate constant can be determined through monitoring the permanganate concentration change in the solution.
Accordingly, oxidative leaching experiments were undertaken through refluxing 80 mL of alkaline hypochlorite (≤2% alkalinity, ≥8% available chlorine), with a fixed mass of LiMn2O4 (m0 = 0.1 g or 1.0 g) at a known temperature, T, in the range 50 < T/°C < 100, for a constant time period (1 h), see ESI1 (ESI†). Our previous work,27 suggests that this time period is sufficient for the initial rate to be monitored. Fig. 1 illustrates spectrophotometric analysis of an aliquot of the reaction solution taken after 60 min. It is clear that in all cases, MnO4− is present, at concentrations in the range 30–170 μM, based on Beer–Lambert analysis through comparison with external MnO4− calibration (λmax = 525 nm; ε = 2085 L mol−1 cm−1) over the range 25–560 μM.
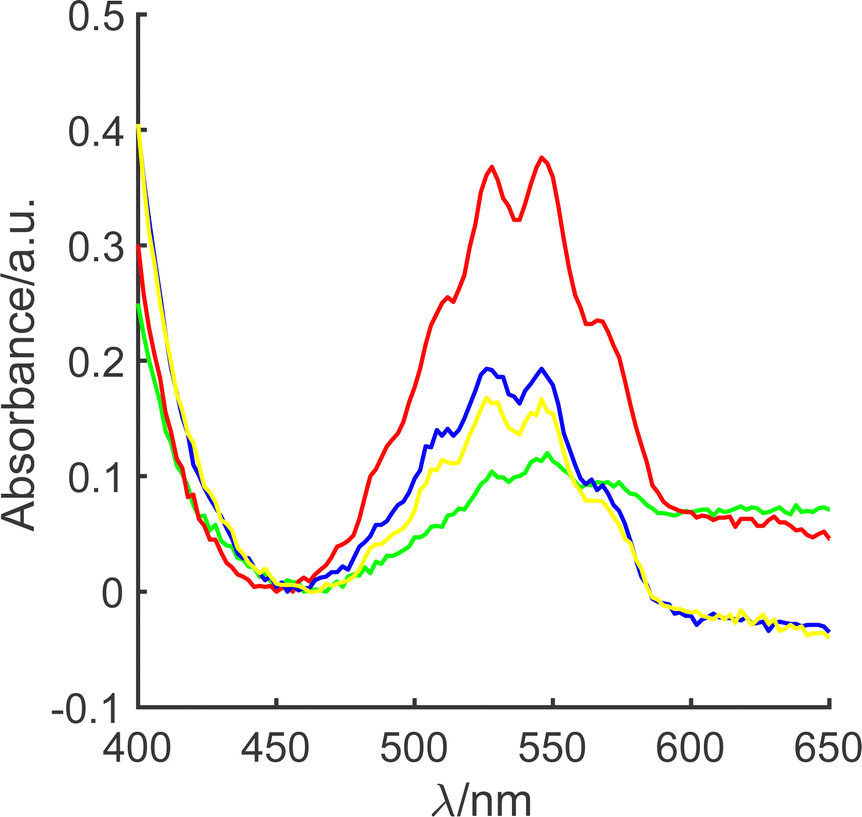 | ||
| Fig. 1 Visible absorption spectrum of the permanganate product obtained from 1.0 g (red, blue and yellow) or 0.1 g (green) LiMn2O4 heated with 80 mL of alkaline hypochlorite solution after one hour at 54 °C (yellow), 72 °C (blue) or 95 °C (red and green). | ||
The rate of LiMn2O4 dissolution is then given by,
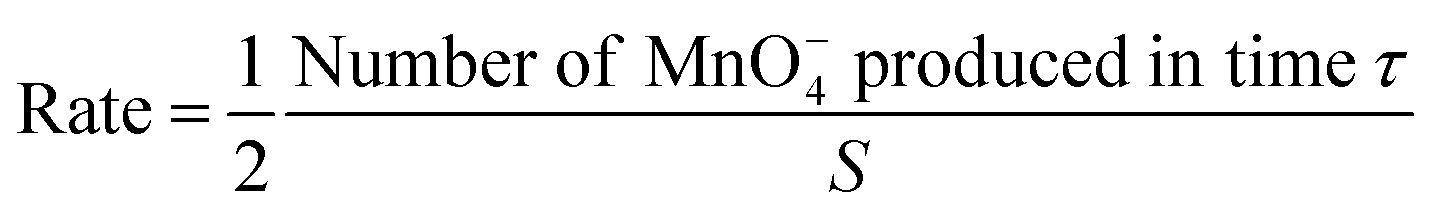 | (3) |
:2 stoichiometry of LiMn2O4: MnO4− in eqn (A). Thus, combining eqn (1)–(3) affords: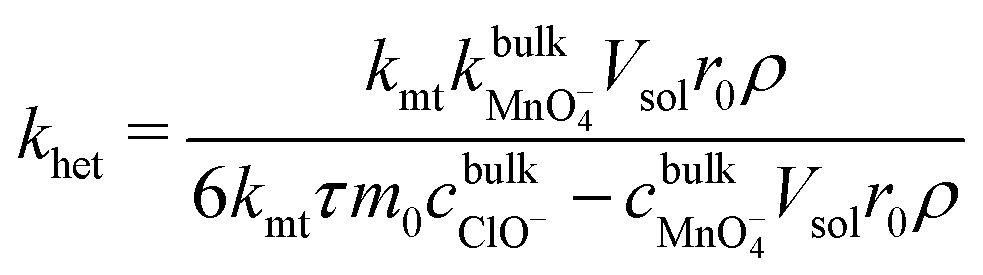 | (4) |
As demonstrated in ESI4 (ESI†), the mass transfer coefficient (kmt) is on the order of 10−4 m s−1 and demonstrates temperature dependence with activation energy of ca. 16 kJ mol−1. In contrast, the mixed activation/mass transfer constant in eqn (1) is on the order of 10−11 m s−1, which is substantially smaller than the mass transfer coefficient. This dissolutive flux thus corresponds to pure kinetics, with no contribution from mass transfer.55 Accordingly, using eqn (4), the heterogeneous rate constant for the dissolution can be estimated. This is presented in Fig. 2, in the form of an Arrhenius plot. It is clear that in the temperature range studied, the true surface chemical rate constant (khet) is independent of the starting mass of LiMn2O4.
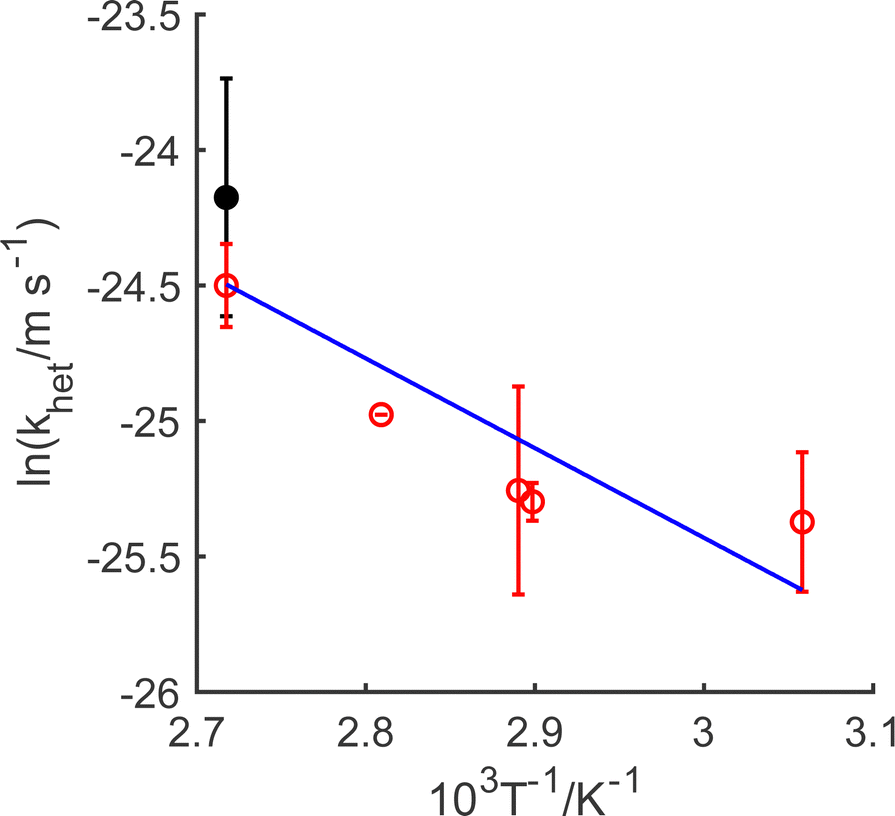 | ||
| Fig. 2 Arrhenius plots illustrating the temperature dependence of the rate constant for oxidative dissolution. Open red circles correspond to 1.0 g LiMn2O4; closed black circles correspond to 0.1 g LiMn2O4. Mean data plotted, with maximum and minimum limits given through the error bars for measurements made in duplicate. | ||
Moreover, the rate constant increases with temperature, as expected; this dependency enables the estimation of the activation energy for the oxidative dissolution of 22 kJ mol−1 (0.23 eV). This is significantly smaller than required by other treatments,36,38,40 and is for slow activation kinetics and not mass transfer in solution. Since the activation energy for electronic conductivity in LiMn2O4 semiconductors through electron hopping (small polaron) between MnIII and MnIV sites has been given as between56–58 0.16–0.34 eV, this indicates that it is transport within the solid of MnIII to the surface that rate limits the dissolution. This is consistent with reaction (C) being the slow step.
The activation energy for the oxidative, surface-controlled leaching of LiMn2O4 is lower than that reported for the surface kinetics-limited reductive leaching (activation energy of ∼50 kJ mol−1) LiCoO2.59 Both materials are semiconductors at ambient temperature: LiMn2O4 is an n-type semiconductor (band gap of 1.99 eV),58 whilst, LiCoO2 undergoes a semiconductor-to-metallic transition as lithium de-intercalation occurs.60 When a metal is immersed into a redox electrolyte, equilibration of the Fermi level of the metal with that of the redox electrolyte occurs, and heterogeneous electron transfer is dominated by the continuum of electronic energy states in the electrode, where the density of states (the number of states per atom per unit energy) is assumed to be independent of energy, and for which the Fermi–Dirac distribution governs the occupancy probability of a given electronic energy state.61 For oxidative leaching (corrosion) of metals, reduction of the chemical oxidant is an iso-energetic electron transfer into an unoccupied energy state in the solution species. This can occur from any occupied state within the electrode into the (assumed) Gaussian density of states function for the aqueous redox species. In contrast, when a semiconductor is in electronic contact with a redox electrolyte, charge can flow across the interface, which leads to the development of a space-charge region in the semiconductor, in which the band edges are pinned relative to the Fermi level of the solution phase species (assuming the semiconductor is defect-free).61 In the case of LiMn2O4, the space-charge region has been reported to have a width of 44 nm.58 Such band bending phenomena changes the concentrations of electrons and holes at the semiconductor side of the interface, and thus, hole (or electron) transfer can only occur through a narrow distribution of energies within the band continuum, whenever electronic overlap of semiconductor states with the solution Fermi energy occurs. As noted by LeBlanc and Folger,40 the difference between the solution redox couple and the semiconductor band edge can be considered as the activation barrier for the heterogeneous redox process. Under standard conditions, the experimental solution at ca. pH 14 indicates that the valence band edge of LiMn2O4 has a value of ca. 0.78 V vs. SHE,58 the oxidation process is predicted to be under activation control, as observed experimentally.
The manganese leaching efficiency (χ) is calculated from the ratio of the amount of manganese dissolved in solution as permanganate to the amount of manganese in the initial solid powder:28
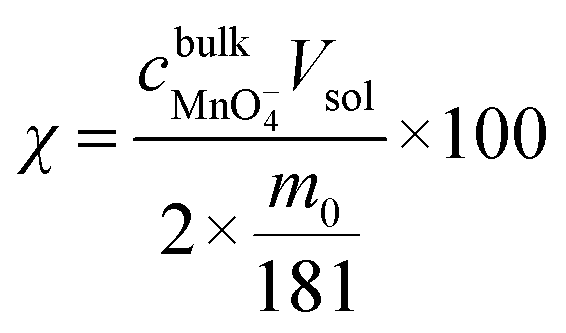 | (5) |
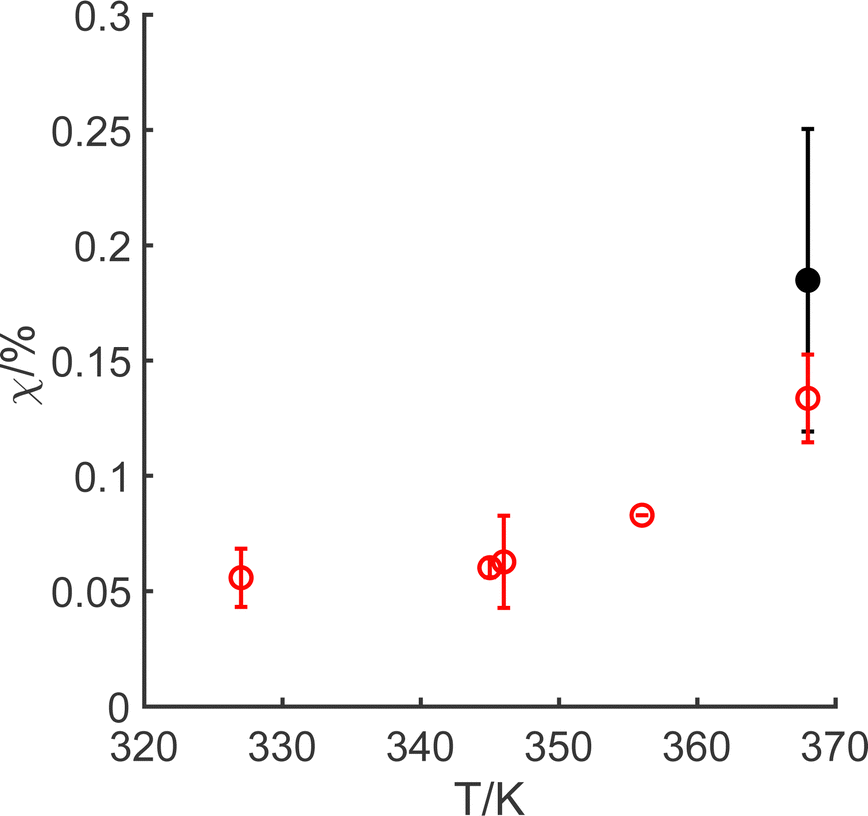 | ||
| Fig. 3 Temperature dependence of the manganese leaching efficiency through oxidative dissolution with alkaline permanganate. Open red circles correspond to 1.0 g LiMn2O4; closed black circles correspond to 0.1 g LiMn2O4. Mean data plotted, with maximum and minimum limits given through the error bars for measurements made in duplicate. | ||
In summary, the alternative oxidative leaching method illustrated in this work requires lower energy expenditure than current methods, owing to the smaller activation energy for the chemical reaction. Whilst only ca. 0.15% of the available manganese has been demonstrated to have dissolved through oxidation under the short time period, this is a climate-smart process. Although we have only studied a small range of solid/water levels (1.25 g L−1 to 12.5 g L−1), since the chemistry is under pure kinetic control by the heterogeneous electron transfer, there is no dependence on the mass of the solid. This suggests that optimisation of this chemistry is possible, and this may enable a reduction in the water stress required for lithium and manganese recovery from LiMn2O4, although the 7 F mol−1 required by the overall reaction, reaction (A), indicates a compromise is necessary as hypochlorite is needed for both the initial and subsequent steps. The chemical system enables dissolution of the manganese framework,62 and therefore allows both lithium ions and permanganate ions to be based in the solution. These two ions are readily separated in subsequent steps, since MnO4− is highly redox-active and can be reduced to MnO2 by, for example, abating a reduced gas species (such as H2S, SO2, NO2, etc.) at an appropriate pH, during industrial symbiotic chemical processes. In line with previous work,27 in taking this forward, a cascading leaching process will need to be developed, to overcome the thermal decomposition of hypochlorite,63 in order enable optimisation of the process conditions.
Conflicts of interest
There are no competing pecuniary interests to declare.Acknowledgements
We thank both Singleton Birch, Ltd., and The University of Hull for funding this work.Notes and references
-
K. Hund, D. La Porta, T. P. Fabregas, T. Laing and J. Drexhage, Minerals for Climate Action: The Mineral Intensity of the Clean Energy Transition, World Bank, Washington, DC, 2020 Search PubMed
.
-
International Energy Agency, The Role of Critical Minerals in Clean Energy Transitions, IEA, Paris, 2021 Search PubMed
-
G. Gun, ed., Critical Metals Handbook, Wiley, Chichester, 2014 Search PubMed
-
National Research Council, Minerals, Critical Minerals, and the US Economy, The National Academies Press, Washington DC, 2008 Search PubMed
- L. Simandl, G. J. Simandl and S. Paradis, Economic geology models V: speciality, critical, battery, magnet and photovoltaic materials – market, facts, projections and implications for exploration and development, Geosci. Canada, 2021, 48, 73 CrossRef
- R. Pell, L. Tijsseling, K. Goodenough, F. Wall, Q. Dehaine, A. Grant, D. Deak, X. Yan and P. Whattoff, Towards sustainable extraction of technology materials through integrated approaches, Nat. Rev. Earth Environ., 2021, 2, 665 CrossRef
- R. Harrington, Mining our green future, Nat. Rev. Mater., 2021, 6, 456 CrossRef
- N. T. Nassar, J. Brainard, A. Gulley, R. Manley, G. Matos, G. Lederer, L. R. Bird, D. Pineault, E. Alonso, J. Gambogi and S. M. Fortier, Evaluating the mineral commodity supply risk of the U.S. manufacturing sector, Sci. Adv., 2020, 6, eaay8647 CrossRef CAS PubMed
- B. K. Sovacol, S. H. Ali, M. Bazilian, B. Radley, B. Nemery, J. Okatz and D. Mulvaney, Sustainable minerals and metals for a low-carbon future: policy co-ordination is needed for global supply chains, Science, 2020, 367, 30 CrossRef PubMed
- See, for example: D. Schrijvers, A. Hool, G. A. Blengini, W.-Q. Chen, J. Dewulf, R. Eggert, L. van Ellen, R. Gauss, J. Goddin, K. Habib, C. Hagelüken, A. Hirohata, M. Hofmann-Amtenbrink, J. Kosmol, M. Le Gleuher, M. Grohol, A. Ku, M.-H. Lee, G. Liu, K. Nansai, P. Nuss, D. Peck, A. Reller, G. Sonnemann, L. Tercero, A. Thorenz and P. A. Wäger, A review of methods and data to determine raw material criticality, Resour., Conserv. Recycl., 2020, 155, 104617 CrossRef
- K. J. Schulz, J. H. DeYoung Jr., R. R. Seal II and D. C. Bradley, ed., Critical Mineral Resources of the United States: Economic and Environmental Geology and Prospects for Future Supply, U. S. Geological Survey Professional Paper 1802, U. S. Geological Survey, Reston, Virginia, 2017.
- See, for example: A. Street and W. Alexander, Metals in the Service of Man, Penguin Books, London, 11th edn, 1998 Search PubMed
- Estimates based on data provided in U.S. Geological Survey, Mineral Commodity Summaries 2022, U.S. Geological Survey, Reston, Virginia, 2022.
- This has been seen recently for the nickel market: following “unprecedented overnight increases in the three month nickel price…owing to the “evolving situation in Russia and Ukraine,” the London Metal Exchange suspended nickel trading on March 8, 2022, with a number of subsequent trading updates in the light of price disruption events given since then, q.v. https://www.lme.com/en/metals/non-ferrous/lme-nickel (accessed March 30, 2022).
- Two companies, British Lithium, Ltd. (https://britishlithium.co.uk, accessed March 13, 2022) and Cornish Lithium (https://cornishlithium.com, accessed March 13, 2022) have been exploring the extraction of lithium from granite-based pegmatites and geothermal brine. For an overview of availability of lithium resources in the UK, see British Geological Survey, Raw Materials for Decarbonisation: The Potential for Lithium in the UK, British Geological Survey, Keyworth, 2020.
- X. Sun, H. Hao, F. Zhao and Z. Liu, Tracing global lithium flow: a trade-linked material flow analysis, Resour., Conserv. Recycl., 2017, 124, 50 CrossRef
- E. Petavratzi and P. Josso, Global Material Flows of Lithium for the Lithium-Ion and Lithium Iron Phosphate Battery Markets, British Geological Survey Open Report, OR/21/055.
- P. Meshram, B. D. Pandey and T. R. Mankhand, Extraction of lithium from primary and secondary sources by pre-treatment,
leaching and separation: a comprehensive review, Hydrometallurgy, 2014, 150, 192 CrossRef CAS
- See, for example: L. Li, V. G. Deshmane, M. P. Paranthaman, R. Bhave, B. A. Moyer and S. Harrison, Lithium recovery from aqueous resources and batteries: a brief review, Johnson Matthey Technol. Rev., 2018, 62, 161 CrossRef CAS
- L. Brückner, J. Frank and T. Elwert, Industrial recycling of lithium-ion batteries: a critical review of metallurgical process route, Metals, 2020, 10, 1107 CrossRef
- T. H. Nguyen and M. S. Lee, A review on the separation of lithium ion from leach liquors of primary and secondary resources by solvent extraction with commercial extractants, Processes, 2018, 6, 55 CrossRef
- See, for example: M. J. Lain, Recycling of lithium cells and batteries, J. Power Sources, 2001, 97–98, 736 CrossRef CAS
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Recycling lithium-ion batteries from electric vehicles, Nature, 2019, 575, 75 CrossRef CAS PubMed
- Water-stress is increasingly becoming an important consideration for the mining industry – see, for example: M. B. Revuelta, Whilst the water footprint of critical metal recycling facilities is not yet catalogued, it is important to design recovery processes that minimise water use, Mineral Resources: From Exploration to Sustainability Assessment, Springer Nature, Cham, 2018 Search PubMed
- See, for example: C. Hageluken, Recycling of (critical) metals, in G. Gunn, ed., Critical Metals Handbook, Wiley, Chichester, 2014, p. 41 Search PubMed
- See, for example: K. N. Han and M. C. Fuerstenau, Hydrometallurgy and solution kinetics, in M. C. Fuerstenau and K. N. Han, ed., Principles of Mineral Processing, Society for Mining, Metallurgy and Exploration, Englewood, Colorado, 2003, p. 413 Search PubMed
- D. Kocsis, R. A. Ward, C. R. Meyer, M. Thompson, T. J. Prior, S. M. Kelly, N. S. Lawrence and J. D. Wadhawan, Empowering clean water whilst safeguarding water distribution pipeline integrity: towards manganese- and iron-free lime hydrate for water treatment, Environ. Sci.: Water Res. Technol., 2023, 9, 833 RSC
- See, for example: Z. Li, L. He, Z. Wei Zhao, D. Wang and W. Xu, Recovery of lithium and manganese from scrap LiMn2O4 by slurry electrolysis, ACS Sustainable Chem. Eng., 2019, 7, 16738 CrossRef CAS
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Lithium insertion into manganese spinels, Mater. Res. Bull., 1983, 18, 461 CrossRef CAS
- M. M. Thackeray, Structural considerations of layered and spinel lithiated oxides for lithium-ion batteries, J. Electrochem. Soc., 1995, 142, 2558 CrossRef CAS
- H. Berg, K. Goransson, B. Nolang and J. O. Thomas, Electronic structure and stability of the LixMn2O4 (0 < x < 2) system, J. Mater. Chem., 1999, 9, 2813 RSC
- S. Chitra, P. Kalyani, T. Mohan, M. Massot, S. Ziolkiewicz, R. Gangandharan, M. Eddrief and C. Julien, Physical properties of LiMn2O4 spinel prepared at moderate temperature, Ionics, 1998, 4, 8 CrossRef CAS
- T. Liu, A. Dai, J. Lu, Y. Yuan, Y. Xiao, L. Yu, M. Li, J. Gim, L. Ma, J. Liu, C. Zhan, L. Li, J. Zheng, Y. Ren, T. Wu, R. Shahbazian-Yasser, J. Wen, F. Pan and K. Amine, Correlation between manganese dissolution and dynamic phase stability in spinel-based lithium-ion battery, Nat. Commun., 2019, 10, 4721 CrossRef PubMed
- J. C. Hunter, Preparation of a new crystal form of manganese dioxide: λ-MnO2, J. Solid State Chem., 1981, 39, 142 CrossRef CAS
- D. Larcher, P. Courjal, R. H. Urbina, B. Gerand, A. Blyr, A. du Pasquier and J.-M. Tarascon, Synthesis of MnO2 phases from LiMn2O4 in aqueous acidic media: mechanisms of phase transformations, reactivity and effects of Bi species, J. Electrochem. Soc., 1998, 145, 3392 CrossRef CAS
- Y. Tesfamhret, H. Liu, Z. Chai, E. Berg and R. Younesi, On the manganese dissolution process from LiMn2O4 cathode materials, ChemElectroChem, 2021, 8, 1516 CrossRef CAS
- C. Lu and S. Lin, Dissolution kinetics of spinel lithium manganite and its relation to capacity fading in lithium-ion batteries, J. Mater. Res., 2002, 17, 1476 CrossRef CAS
- W. Zhang and C. Y. Cheng, Manganese metallurgy review part 1: leaching of ores/secondary materials and recovery of electrolytic/chemical manganese dioxide, Hydrometallurgy, 2007, 89, 137 CrossRef CAS
- E. B. Godunov, A. D. Izotov and I. G. Gorichev, Dissolution of manganese oxides of various compositions in sulfuric acid solutions studied by kinetic methods, Inorg. Mater., 2018, 54, 66 CrossRef CAS
- B. S. E. LeBlanc and H. S. Fogler, The role of conduction/valence bands and redox potential in accelerated mineral dissolution, AIChE J., 1986, 32, 1702 CrossRef
-
W. M. Latimer, The Oxidation States of the Elements and their Potentials in Aqueous Solution, Prentice-Hall, New York, 2nd edn, 1952 Search PubMed
- B. Tao, L. C. Yule, E. Daviddi, C. L. Bentley and P. R. Unwin, Correlative electrochemical microscopy of Li-ion (de)intercalation at a series of individual LiMn2O4 particles, Angew. Chem., Int. Ed., 2019, 58, 4606 CrossRef CAS PubMed
- L. Köhler, M. E. Abrishami, V. Roddatis, J. Geppert and M. Risch, Mechanistic parameters of electrocatalytic water oxidation on LiMn2O4 in comparison to natural photosynthesis, ChemSusChem, 2017, 10, 4479 CrossRef PubMed
- T. Ohzuku, M. Kitagawa and T. Hirai, Electrochemistry of manganese dioxide in lithium non-aqueous cell III: X-ray diffractional study on the reduction of spinel-related manganese dioxide, J. Electrochem. Soc., 1990, 137, 769 CrossRef CAS
- E. Hosono, T. Kudo, I. Honma, H. Matsuda and H. Zhou, Synthesis of single crystalline spinel LiMn2O4 nanowires for a lithium-ion battery with high power density, Nano Lett., 2009, 9, 1045 CrossRef CAS PubMed
- M. Baumung, L. Kollenbach, L. Xi and M. Risch, Undesired bulk oxidation of LiMn2O4 increases overpotential of electrocatalytic water oxidation in lithium hydroxide electrolytes, ChemSusChem, 2019, 20, 2981 CAS
- S. K. Ghosh, Diversity in the family of manganese oxides at the nanoscale: from fundamentals to applications, ACS Omega, 2020, 5, 25493 CrossRef CAS PubMed
-
B. Gilbert and G. A. Waychunas, The timescale of mineral redox reactions, in I. A. M. Ahmed and K. A. Hudson-Edwards, ed., Redox-Reactive Minerals: Properties, Reactions and Applications in Natural Systems and Clean Technologies, EMU Notes in Mineralogy, The Mineralogical Society of Great Britain and Ireland, London, 2017, ch. 4, vol. 17, p. 55 Search PubMed
- T. A. Zordan and L. G. Hepler, Thermochemistry and oxidation potentials of manganese and its compounds, Chem. Rev., 1968, 68, 737 CrossRef CAS
- M. G. Segal and R. M. Sellers, Reactions of solid iron(III) oxides with aqueous reducing agents, J. Chem. Soc., Chem. Commun., 1980, 991 RSC
- A. Mills and D. Worsley, Kinetics of redox dissolution of soft-centre particles, J. Chem. Soc., Faraday Trans., 1990, 86, 3405 RSC
- J. D. Wadhawan, R. G. Evans, C. E. Banks, S. J. Wilkins, R. R. France, N. J. Oldham, A. J. Fairbanks, B. Wood, D. J. Walton, U. Schroder and R. G. Compton, Voltammetry of electroactive oil droplets: electrochemically induced ion insertion, expulsion and reaction processes at microdroplets of N,N,N’,N’-tetraalkyl-para-phenylenediamine (TRPD, R = n-butyl, n-hexyl, n-heptyl, n-nonyl), J. Phys. Chem., 2002, 106, 9619 CrossRef CAS
- C. L. Forryan, O. V. Klymenko, C. M. Brennan and R. G. Compton, Reactions at the solid-liquid
interface: surface-controlled dissolution of solid particles: the dissolution of potassium bicarbonate in dimethylformamide, J. Phys. Chem. B, 2005, 109, 2862 CrossRef CAS PubMed
- Bulk crystalline density of LiMn2O4 is reported at the following URL. https://materials.springer.com/isp/crystallographic/docs/sd_1628205 (accessed March 15, 2022).
- See, for example: J. H. Atherton, Mechanism in two-phase reaction systems: coupled mass transfer and chemical reaction, in R. G. Compton and G. Hancock, ed., Research in Chemical Kinetics, Elsevier, Amsterdam, 1994, vol. 2, p. 193 Search PubMed
- S. Chitra, P. Kalyani, T. Mohan, M. Massot, S. Ziolkiewicz, R. Gangandharan, M. Eddriel and C. Julien, Physical properties of LiMn2O4 spinel prepared at moderate temperature, Ionics, 1998, 4, 8 CrossRef CAS
- E. Iguchi, Y. Tokuda, H. Nakatsugawa and F. Munakata, Electrical transport properties in LiMn2O4, Li0.95Mn2O4 and LiMn1.95B0.05O4 (B = Al or Ga) around room temperature, J. Appl. Phys., 2002, 91, 2149 CrossRef CAS
- S. Douafer, H. Lahmar, M. Benamira, G. Rekhila and M. Trari, Physical and photoelectrochemical properties of the spinel LiMn2O4 and its application in photocatalysis, J. Phys. Chem. Solids, 2018, 118, 62 CrossRef CAS
- C. K. Lee and K.-I. Rhee, Reductive leaching of cathodic active materials from lithium-ion battery wastes, Hydrometallurgy, 2003, 68, 5 CrossRef CAS
- See, for example: H. Kang, J. Lee, T. Rodgers, J.-H. Shim and S. Lee, Electrical conductivity of de-lithiated lithium cobalt oxides: conductive atomic force microscopy and density functional theory study, J. Phys. Chem. C, 2019, 123, 17703 CrossRef CAS
- See, for example: H. Gerischer, Charge transfer processes at semiconductor-electrolyte interfaces in connection with problems of catalysis, Surf. Sci., 1969, 18, 97 CrossRef CAS
- This type of approach for lithium-ion recovery has already been used before – see: K. Ooi, Y. Miyai, S. Kathoh, H. Maeda and M. Abe, Lithium-ion insertion/extraction with λ-MnO2 in the aqueous phase, Chem. Lett., 1988, 989 CrossRef CAS
- L. C. Adam and G. Gordon, Hypochlorite ion decomposition: effects of temperature, ionic strength, and chloride ion, Inorg. Chem., 1999, 38, 1299 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ya00196a |
| This journal is © The Royal Society of Chemistry 2023 |