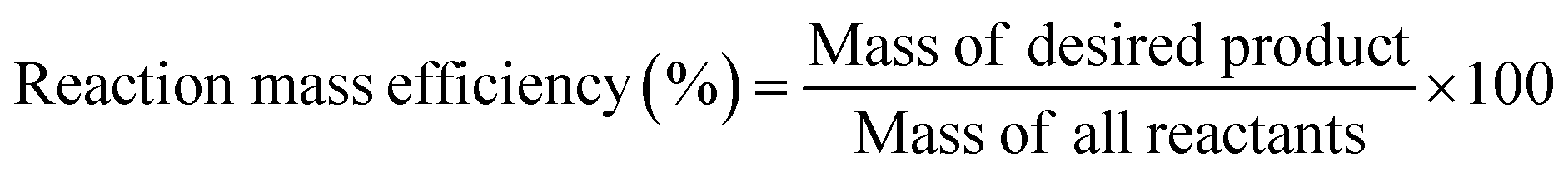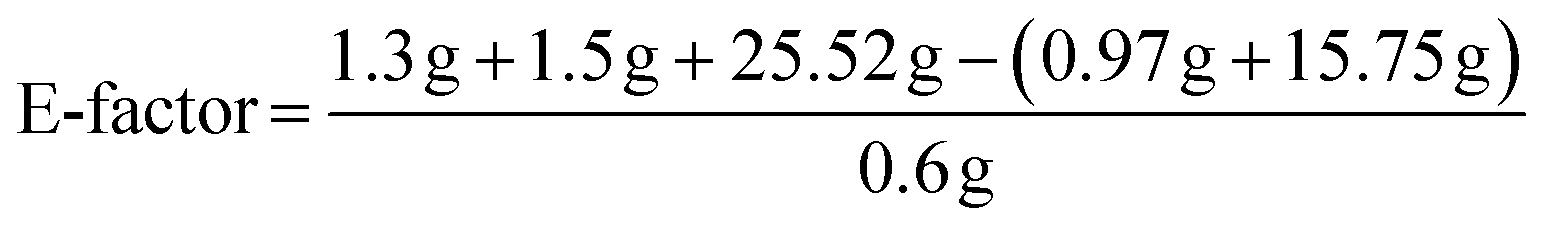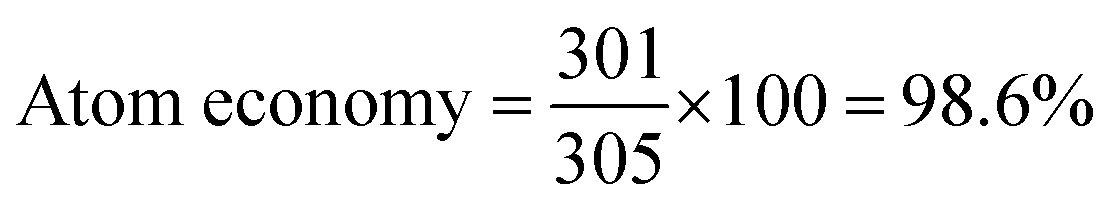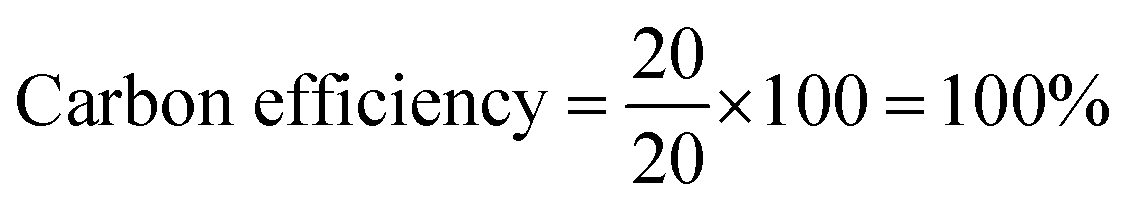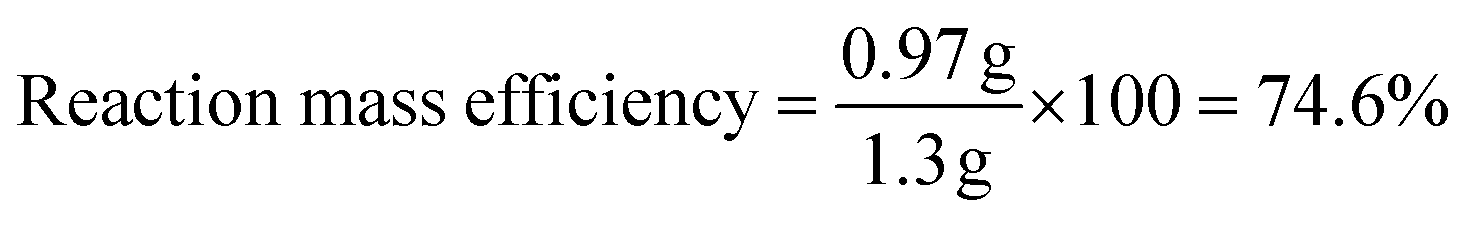DOI:
10.1039/D2RA00585A
(Paper)
RSC Adv., 2022,
12, 8368-8373
Metal-free photoredox-catalyzed direct α-oxygenation of N,N-dibenzylanilines to imides under visible light†
Received
27th January 2022
, Accepted 12th February 2022
First published on 15th March 2022
Abstract
An efficient synthesis of imides using metal-free photoredox-catalyzed direct α-oxygenation of N,N′-disubstituted anilines in the presence of 9-mesityl-10-methylacridinium [Acr+-Mes]BF4 as a photoredox catalyst and molecular oxygen as a green oxidant under visible light was developed. This photochemical approach offered operational simplicity, high atom economy with a low E-factor, and functional group tolerance under mild reaction conditions. Control and quenching experiments confirmed the occurrence of a radical pathway and superoxide radical anion α-oxygenation reactions, and also provided strong evidence for the reductive quenching of [Acr+-Mes]BF4 based on a Stern–Volmer plot, which led to the proposed mechanism of this reaction.
Introduction
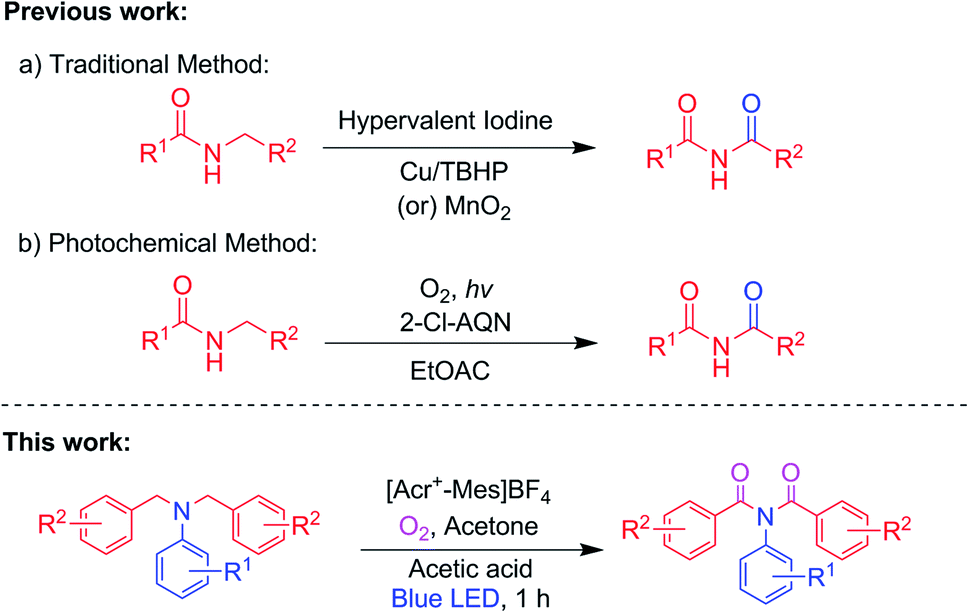
Imides are versatile building blocks in many organic syntheses6 and also have many potential important applications in pharmaceuticals,1,2 agrochemicals,3 material science,4 and organic photovoltaics.5 Because of the importance of imides, various strategies have been established6–9 for synthesizing them, including the Mumm rearrangement of isoimides,10–12 oxidative acylation reactions,13 transition metal carbonylative coupling reactions, C–H/N–H coupling reactions, three-component coupling reactions,14–17 etc. Of these strategies, the direct α-oxygenation of amines has brought special attention to the scientific community due to the easy availability of amines and the environmental friendliness of the approach.18 Direct α-oxygenation of N-alkyl amides to imides using hypervalent iodine, Cu/TBHP and MnO2 (Scheme 1a) has previously been reported13,19–21 – but has been found to require the use of transition metals and a stoichiometric amount of oxidants, and to produce a very large amount of by-products with a high E-factor. Therefore, the use of clean oxidants is essential to minimize the quantity and toxicity of the released waste. In this regard, thus the use of molecular oxygen as a simple, green and environment friendly oxidant has fulfilled the requirements with high atom economy, low E-factor and producing water as a by-product.22
 |
| | Scheme 1 Various strategies involving α-oxygenation reactions of amides and amines for synthesizing imides. | |
In recent years, visible-light-mediated metal-free photocatalyzed oxygenation reactions using molecular oxygen as a green oxidant have become increasingly important in many organic syntheses,23 due to the ability of these reactions to activate O2 into highly reactive oxygen species such as singlet oxygen (1O2) and superoxide radical anion (O2˙−) using energy transfer (ET) or photoinduced electron transfer (PET) processes. One such work published in 2018 by S. Das et al. demonstrated visible-light-mediated metal-free photoredox-catalyzed α-oxygenation of N-benzyl amine by O2 as an oxidant, and this strategy has also been applied to the synthesis of phthalimides from isoindoline.24 In 2014, Akichika and co-workers reported 2-chloroanthraquinone-catalyzed aerobic photo-oxidations of diacylamines (Scheme 1b).25 Recently, our group established visible-light-induced metal-involved and metal-free photocatalyzed organic transformations deploying molecular oxygen as a green oxidant.26 Also, our group has published work on the α-oxidation of N,N-dibenzylanilines to amides via oxidative C–N cleavage as a prominent product in the presence of rose bengal as a photocatalyst and O2.26d However, to the best of our knowledge, direct α-oxygenation of amines to imides has not yet been reported. Hence, we set out to work on and achieved a visible-light-induced α-oxygenation of N,N-dibenzylanilines to imides as prominent products in the presence of [Acr+-Mes]BF4 as a photocatalyst and O2 (Scheme 1).
Experimental section
General considerations
The acquired 1H and 13C NMR spectra were recorded in CDCl3 using a Bruker 300 MHz NMR spectrometer with TMS as an internal standard (see ESI†). Mass spectra were recorded using a Xevo G2S Q-TOF spectrometer (see ESI†). The light source for photochemical reactions was a Kessil 456 nm-wavelength blue light-emitting diode (LED) (see ESI Fig. S1†) (model number: KSPR160L-456-EU). LEDs producing light of different wavelengths including 370 nm, 390 nm, 467 nm and 527 nm were purchased from Kessil and were also used. Reaction tubes made of borosilicate glass were used as reaction vessels. The distance between the light source and the reaction vessel was 8 cm. TLC was performed using Merck pre-coated TLC plates (Merck 60 F254) and the results were visualized using UV light. Column chromatographic separation was carried out with silica gel (100–200 mesh). Reagents and solvents were purified as per standard procedures and used.
General experimental procedure
General procedure A for the synthesis of N,N-dibenzylanilines29. To a mixture of anilines (10 mmol) and K2CO3 (20 mmol) in DMF, benzylbromide (30 mmol) was added dropwise. The reaction was stirred at room temperature overnight. Water was added to the mixture and extracted with ethyl acetate twice. The resulting combined organic phase was washed with brine and dried over Na2SO4. The resulting concentrated residue was purified by performing column chromatography over silica gel using EtOAc/hexanes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) as eluent to furnish the corresponding N,N-dibenzylanilines.
:9) as eluent to furnish the corresponding N,N-dibenzylanilines.
Procedure B for the preparation of imides. The reaction tube was charged with N,N-dibenzylanilines (0.36 mmol), acetic acid (1.8 mmol), and [Acr+-Mes]BF4 (5 mol%) in acetone (8 mL). The resulting reaction mixture was stirred in the presence of O2 (balloon) under blue light for 1 h. After the reaction was completed, the solvent was evaporated under reduced pressure. The resulting crude product was purified by performing column chromatography using EtOAc/hexanes as eluent to furnish the corresponding imide compounds (see ESI† for NMR data of the products).
Results and discussion
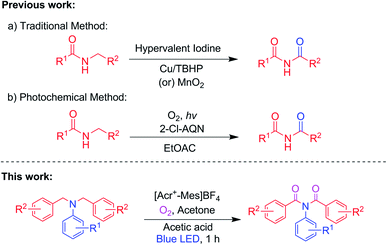
To examine the feasibility of the proposed method, N,N-dibenzylaniline 1a was selected as a model substrate for optimizing the reaction conditions as shown in Table 1. The model substrate 1a was irradiated with blue LED456nm light under O2 in acetone for 35 h to afford the N-benzoyl-N-phenylbenzamide 2a and phenylbenzamide 2a′ in 20% and 32% yields, respectively. The yield of product 2a was reduced when the reactions were carried out in, respectively, DMSO, toluene, and ACN instead of acetone (Table 1, entries 2–4). There was no significant improvement in the yields on the screening of other light sources. Namely LEDs producing light of wavelengths of, respectively, 370 nm, 390 nm, 467 nm, and 525 nm (Table 1, entries 5–8). The use of photoredox catalysts such as methylene blue, eosin Y and [Acr+-Mes]BF4 was screened for further optimization (Table 1, entries 9–11). Gratifyingly, the regioselective product 2a was obtained as a predominant product with a 41% yield in [Acr+-Mes]BF4 as a photocatalyst (Table 1, entry 11). Interestingly, the best yield of 2a, which was 71%, was obtained when carrying out the reaction for 1 hour with the addition of 1.8 mmol of acetic acid as an additive (Table 1, entry 12). Further screening of other acids such as H2SO4 and PTSA led in each case to a lower yield of product 2a (Table 1, entries 13 and 14). Control experiments indicated that [Acr+-Mes]BF4, acetic acid, and molecular oxygen were all needed to furnish imides (Table 1, entries 15–17).
Table 1 Optimization of the α-oxygenation of N,N-dibenzylaniline (1a) to N-benzoyl-N-phenylbenzamide (2a)a
|
| Entry |
Light source (nm) |
PC |
Additive |
Solvent |
Yieldsb (%) |
| 2a |
2a′ |
| Reaction conditions: for each experiment, a mixture of 1a (0.36 mmol), additive (1.8 mmol), and PC (5 mol%) in 8 mL of solvent was irradiated with blue LED light under an O2 atmosphere at rt. Isolated yields. Reaction was performed under air. Reaction was performed under N2. Reaction was performed in the dark. Abbreviations: n.r. = no reaction, PC = photocatalyst, MB = methylene blue, [Acr+-Mes]BF4 = 9-mesityl-10-methylacridinium tetrafluoroborate, AcOH = acetic acid, and PTSA = p-toluenesulfonic acid. |
| 1 |
456 (44 W) |
— |
— |
Acetone |
20 |
32 |
| 2 |
456 (44 W) |
— |
— |
ACN |
10 |
25 |
| 3 |
456 (44 W) |
— |
— |
Toluene |
0 |
0 |
| 4 |
456 (44 W) |
— |
— |
DMSO |
0 |
0 |
| 5 |
370 (43 W) |
— |
— |
Acetone |
15 |
15 |
| 6 |
390 (52 W) |
— |
— |
Acetone |
15 |
22 |
| 7 |
467 (44 W) |
— |
— |
Acetone |
Trace |
15 |
| 8 |
525 (44 W) |
— |
— |
Acetone |
0 |
10 |
| 9 |
456 (44 W) |
MB |
— |
Acetone |
Trace |
35 |
| 10 |
456 (44 W) |
Eosin Y |
— |
Acetone |
9 |
23 |
| 11 |
456 (44 W) |
[Acr+-Mes]BF4 |
— |
Acetone |
41 |
8 |
| 12 |
456 (44 W) |
[Acr+-Mes]BF4 |
AcOH |
Acetone |
75 |
0 |
| 13 |
456 (44 W) |
[Acr+-Mes]BF4 |
H2SO4 |
Acetone |
0 |
0 |
| 14 |
456 (44 W) |
[Acr+-Mes]BF4 |
PTSA |
Acetone |
0 |
0 |
| 15c |
456 (44 W) |
[Acr+-Mes]BF4 |
AcOH |
Acetone |
50 |
0 |
| 16d |
456 (44 W) |
[Acr+-Mes]BF4 |
AcOH |
Acetone |
n.r. |
n.r. |
| 17e |
|
[Acr+-Mes]BF4 |
AcOH |
Acetone |
n.r. |
n.r. |
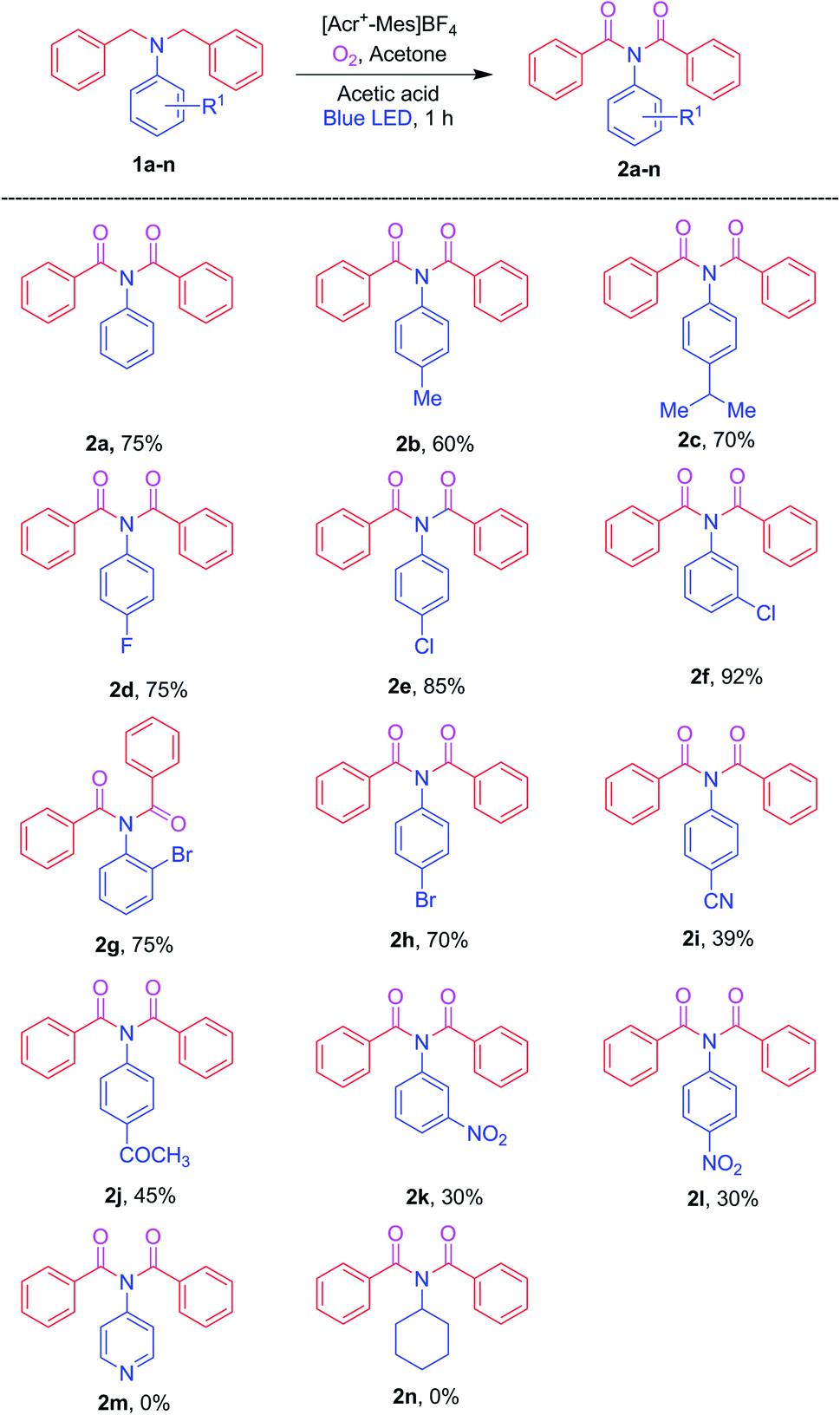
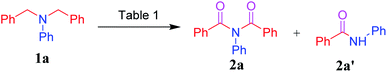
Having optimized the reaction conditions, we investigated the N,N-dibenzylaniline substrate scope, and Scheme 2 summarizes the findings. The R1 substituents including H, 4-CH3 and 4-CH(CH3)2 on anilines 1a–c reacted smoothly to give the corresponding imides 2a–c in good yields. Reaction of the 4-F, 4-Cl, 3-Cl, 2-Br, and 4-Br halogen substituents on the anilines, namely 1d–h, under optimized conditions afforded the imides 2d–h in 70–92% yields. Furthermore, the 4-CN, 4-COCH3, 3-NO2, and 4-NO2 electron-withdrawing groups on anilines 1i–l afforded the corresponding products 2i–l, but did so in lower yields. And heteroaryl amine 1m and aliphatic amine 1n failed to produce the respective products 2m and 2n under the optimized conditions.
 |
| | Scheme 2 Testing of various R1 substituents on N,N-dibenzylanilines for the imide-producing reaction. a Reaction conditions: for each experiment, a mixture of an aniline (1a–n, 0.36 mmol), AcOH (1.8 mmol), and [Acr+-Mes]BF4 (5 mol%) in 8 mL acetone was irradiated with blue LED456nm light under O2 at rt for 1 h. Yields were determined after purification. | |
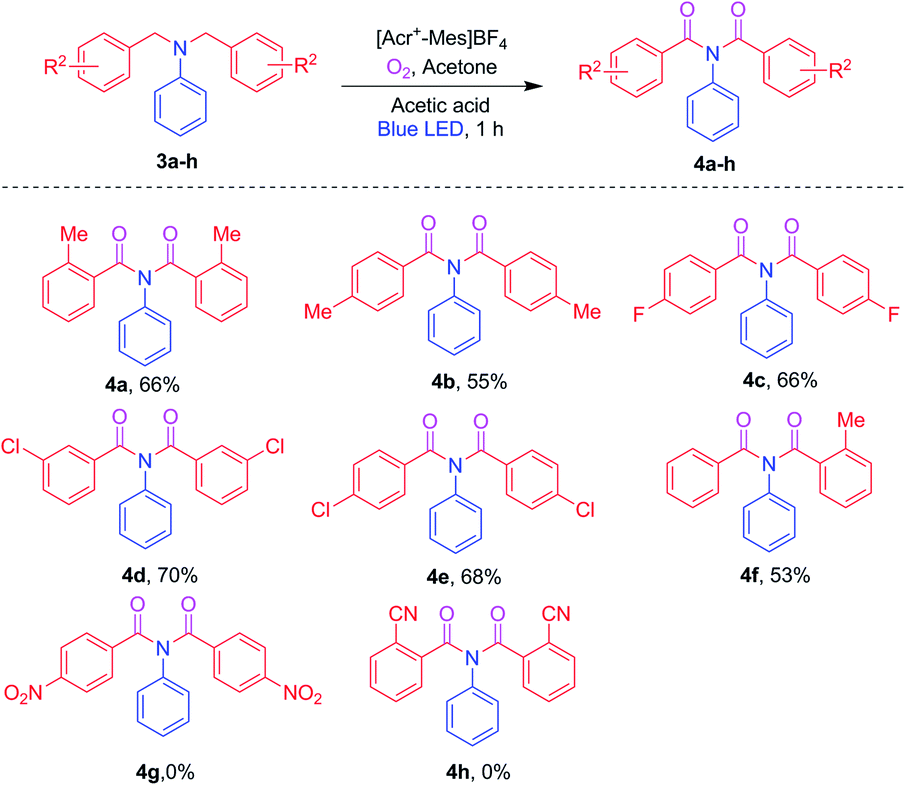
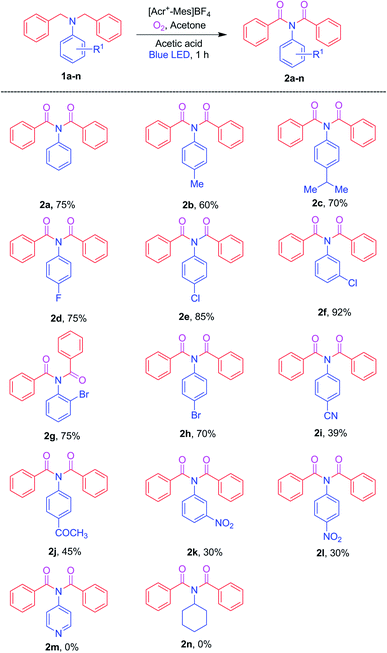
Furthermore, to extend the substrate scope, the R2 substituent on the benzyl aromatic ring of the N,N-dibenzylaniline was investigated as shown in Scheme 3. Various such substituents (2-Me, 4-Me, 4-F, 3-Cl and 4-Cl) on N,N-dibenzylamines 3a–e were reacted under optimized conditions to afford the corresponding imides 4a–e in 53–70% yields, respectively. Interestingly, the unsymmetrical N,N-dibenzylaniline 3f also afforded the corresponding product, namely 4f, in good yield. In contrast, the electron-withdrawing substituents 4-NO2 and 2-CN on N,N-dibenzylamines 3g and 3h failed to produce imides 4g and 4h.
| E-factor = 11.6 kg waste/1 kg product |
 |
| | Scheme 3 Testing of various R2 substituents on N,N-dibenzylanilines for the imide-producing reaction. a Reaction conditions: for each experiment, a mixture of an aniline (3a–h, 0.36 mmol), AcOH (1.8 mmol), and [Acr+-Mes]BF4 (5 mol%) in 8 mL of acetone was irradiated with blue LED456nm light under O2 at rt for 1 h. Yields were determined after purification. | |
To determine the efficiency of this visible-light-mediated α-oxygenation reaction, a scaled-up reaction was performed with 1a (1.3 g, 5 mmol) and AcOH (1.5 g, 25 mmol) under optimized conditions, and afforded the corresponding N-benzoyl-N-phenylbenzamide 2a (0.97 g, 68%) (Scheme 4). Furthermore, we evaluated green chemistry metrics27 for the synthesis of imide 2a. This evaluation showed an E-factor of 11.6, a high atom economy of 98.6%, atom efficiency of 67%, carbon efficiency of 100%, and reaction mass efficiency of 74.6% (Table 2).
 |
| | Scheme 4 Gram-scale synthesis of imide 2a. | |
Table 2 Evaluation of green chemistry metrics for the synthesis of 2a
| Reactant |
1a |
1.3 g |
5 mmol |
FW 273.38 |
| Acid |
AcOH |
1.5 g |
25 mmol |
FW 60.05 |
| Solvent |
Acetone |
25.51 |
— |
— |
| Recycled solvent |
Acetone |
15.75 |
— |
— |
| Product |
2a |
0.97 g |
3.22 mmol |
FW 301.35 |
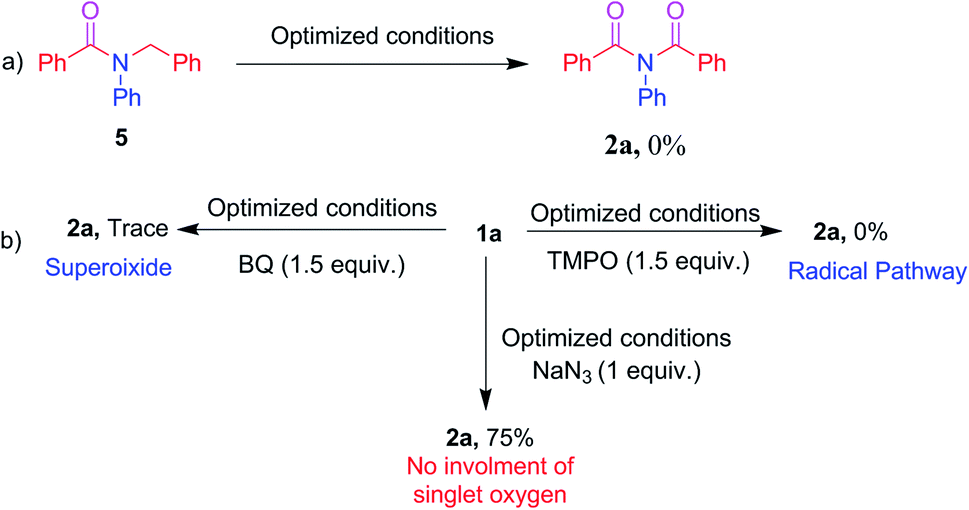
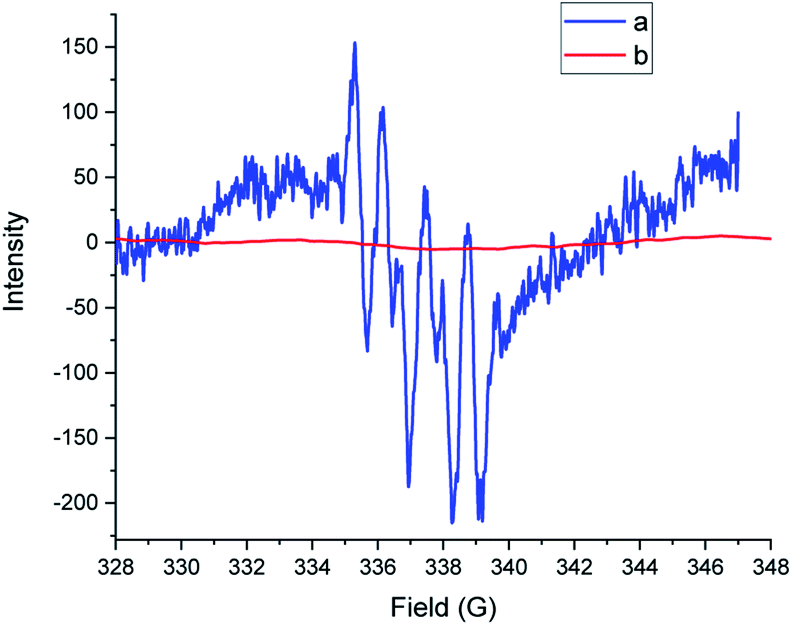
To acquire mechanistic insights, several control and quenching experiments were performed as shown in Scheme 5. The reaction performed with N-benzyl-N-phenylbenzamide 5 under optimized conditions afforded a 0% yield of 2a (Scheme 5a), implying that the reaction did not involve formation of reactive intermediates of N-benzyl-N-phenylbenzamide 5. The quenching experiments, shown in Scheme 5b, were specifically carried out to understand the involvement of reactive oxygen species and the mechanism of the α-oxygenation reaction.28 Under optimized conditions, reactions of 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO), benzoquinone (BQ) and sodium azide (NaN3) afforded, respectively, 0%, trace, and 75% yields of 2a. These results clearly indicated the involvement of a radical as well as superoxide radical anion in the photocatalytic α-oxygenation reaction and it also indicated no involvement of singlet oxygen in the reaction pathway. Further confirmation of the involvement of superoxide radical anion using EPR spectroscopy is shown in Fig. 1. An ESR spectrum was recorded after a solution of 5,5-dimethyl-pyrroline-N-oxide (DMPO) (0.02 mL), N,N-dibenzylaniline 1a (0.36 mmol), AcOH (1.8 mmol), and Acr+-Mes (5 mol%) in acetone (8 mL) under an O2 atmosphere was irradiated with blue LED456nm light for 5 minutes. Six strong signals characteristic of DMPO–OOH were observed. In the absence of light, no signal was observed when the reaction with DMPO was conducted (Fig. 1). These results clearly confirmed that the α-oxygenation reaction proceeded via the superoxide radical anion pathway.
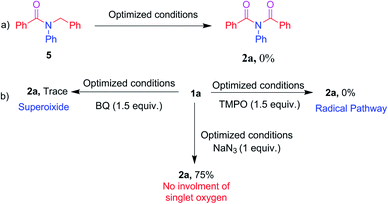 |
| | Scheme 5 Control and quenching experiments. | |
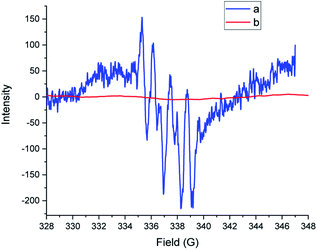 |
| | Fig. 1 Electron spin resonance (ESR) spectra of a solution of DMPO (20 μL) with 1a (0.36 mmol), AcOH (1.8 mmol), and [Acr+-Mes]BF4 (5 mol%) in 8 mL acetone under O2 at rt and (a) with blue LED456nm irradiation for 5 minutes and (b) without blue LED456nm irradiation. | |
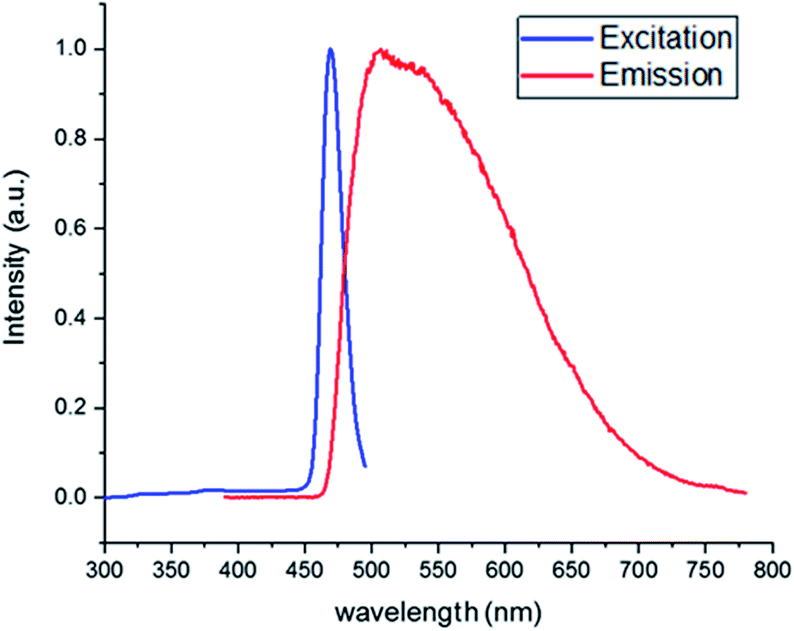
To understand the photocatalytic reaction mechanism of the [Acr+-Mes]BF4 photocatalyst, absorption and emission spectra were recorded to perform a Stern–Volmer plot analysis. Initially, the excitation and emission of [Acr+-Mes]BF4 were recorded, using a photoluminescence spectrometer, to identify the maximum absorption and emission (Fig. 2). As shown in this figure, the excitation maximum and the emission maximum were at, respectively, 444 nm and 504 nm, which were used for further quenching experiments.
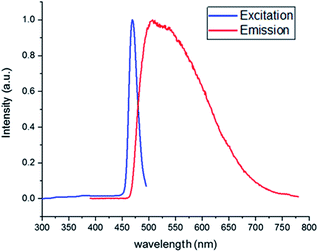 |
| | Fig. 2 Absorption-emission spectrum of [Acr+-Mes]BF4 in acetone. | |
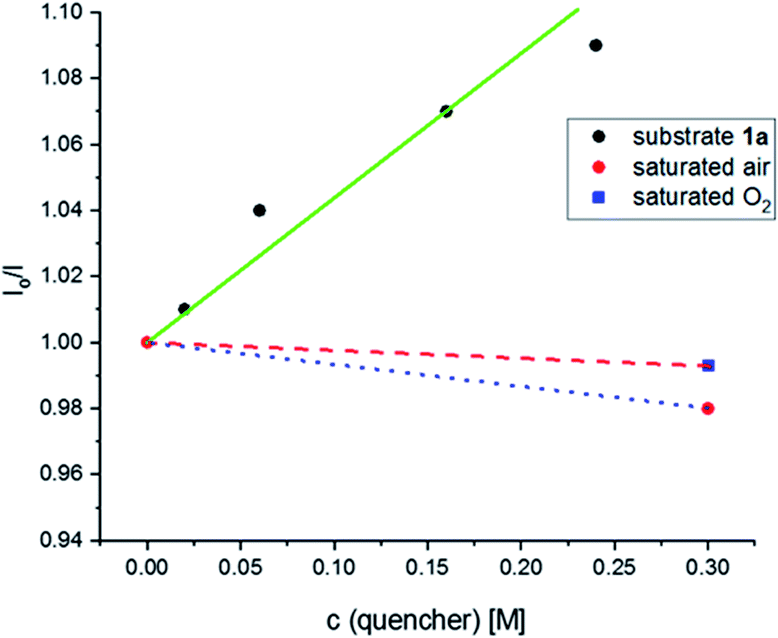
Initially, the emission of the photocatalyst was recorded under an N2 atmosphere and the resulting intensity was taken as I0. Then, the effect of concentration of substrate 1a was examined. A solution with saturated air and another with molecular oxygen were also investigated. As shown in Fig. 3, the emission decreases as the concentration concentration of substrate 1a and on the concentration of saturated air and oxygen showed only a negligible change in emissions.
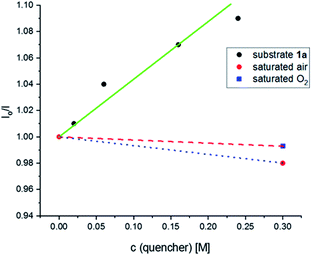 |
| | Fig. 3 Stern–Volmer plot for oxygenation of N,N-dibenzylaniline 1a. | |
Stern–Volmer quenching studies were performed to understand the catalytic cycle in the reaction mechanism (Fig. 3). The excited state of the Acr˙-Mes˙+ photocatalyst was quenched by applying N,N-dibenzylaniline 1a. The corresponding Stern–Volmer plot showed that there was a significant decrease in light emission intensity as the concentration of substrate 1a was increased. In contrast, there was essentially no such change in the light emission intensity when either saturated air or molecular oxygen was added. These results clearly showed that the N,N-dibenzylaniline 1a underwent reductive quenching with Acr˙-Mes˙+ in the catalytic cycle.
Based on the above experiments and literature reports,24,26a a possible reaction mechanism was sketched out, and is shown in Scheme 6. Initially, according to this mechanism, Acr+-Mes was excited upon irradiation of visible light to form Acr˙-Mes˙+. Then, Acr˙-Mes˙+ underwent a single electron transfer process with 1a to generate nitrogen radical cation A and Acr˙-Mes, and Acr˙-Mes was oxidized by O2 via a single electron transfer process to generate a superoxide radical anion radical (O2˙−) and it regenerated Acr+-Mes. Then, the radical cation A lost H+ to generate α-amino radical B. At that point, the hydroperoxide intermediate C was produced as a result of the nucleophilic attack on the amino radical B by in situ-generated O2˙− and H+. Similarly, hydroperoxide intermediate C was converted to dihydroperoxide D in the catalytic cycle in the presence of blue LED light, followed by the nucleophilic addition of in situ-generated O2˙− and H+. Finally, according to the proposed mechanism, imide 2a was obtained from dihydroperoxide D as a result of the elimination of H2O.
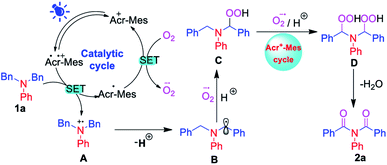 |
| | Scheme 6 A plausible mechanism of α-oxygenation for the synthesis of imides. | |
Conclusions
To summarize, we developed a visible-light-mediated direct α-oxygenation of N,N-dibenzylanilines to form imides in the presence of [Acr+-Mes]BF4 as a photocatalyst and molecular oxygen as a green oxidant. The α-oxygenation reaction method was operationally simple, and showed high atom economy and a low E-factor under mild conditions. Control and quenching experiments were also conducted and indicated the reaction mechanism to involve a radical pathway and superoxide radical anion, and also provided strong evidence for the reductive quenching of [Acr+-Mes]BF4 according to the acquired Stern–Volmer plot.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors RA and NN thank DST-SERB for a research grant (vide Grant No. EMEQ/2018/001129), and DST-FIST for providing NMR and HRMS facilities to the Department of Organic Chemistry. NN acknowledges DST-SERB for financial support.
Notes and references
- S. M. Capitosti, T. P. Hansen and M. L. Brown, Org. Lett., 2003, 5, 2865–2867 CrossRef CAS PubMed.
- E. P. Balskus and E. N. Jacobsen, J. Am. Chem. Soc., 2006, 128, 6810–6812 CrossRef CAS PubMed.
- C. Takayama, O. Kirino, Y. Hisada and A. Fujinami, Agric. Biol. Chem., 1987, 51, 1547–1552 CAS.
- Z. Cheng, X. Zhu, E. T. Kang and K. G. Neoh, Macromolecules, 2006, 39, 1660–1663 CrossRef CAS.
- A. M. Higgins and R. A. L. Jones, Nature, 2000, 404, 476–478 CrossRef CAS PubMed.
- J. Sperry, Synth, 2011, 2011, 3569–3580 CrossRef.
- M. B. Andrus, W. Li and R. F. Keyes, Tetrahedron Lett., 1998, 39, 5465–5468 CrossRef CAS.
- L. Xu, S. Zhang and M. L. Trudell, Chem. Commun., 2004, 1668–1669 RSC.
- C. S. Digwal, U. Yadav, P. V. S. Ramya, S. Sana, B. Swain and A. Kamal, J. Org. Chem., 2017, 82, 7332–7345 CrossRef CAS PubMed.
- J. S. P. Schwarz, J. Org. Chem., 1972, 37, 2906–2908 CrossRef CAS.
- K. Brady and A. F. Hegarty, J. Chem. Soc., Perkin Trans. 2, 1980, 121–126 RSC.
- O. Mumm, H. Hesse and H. Volquartz, Ber. Dtsch. Chem. Ges., 1915, 48, 379–391 CrossRef CAS.
- K. C. Nicolaou and C. J. N. Mathison, Angew. Chem., 2005, 117, 6146–6151 CrossRef.
- K. Kataoka, K. Wachi, X. Jin, K. Suzuki, Y. Sasano, Y. Iwabuchi, J.-y. Hasegawa, N. Mizuno and K. Yamaguchi, Chem. Sci., 2018, 9, 4756–4768 RSC.
- A. Schnyder and A. F. Indolese, J. Org. Chem., 2002, 67, 594–597 CrossRef CAS PubMed.
- H. Q. Li, K. W. Dong, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10239–10243 CrossRef CAS PubMed.
- Y. Li, K. Dong, F. Zhu, Z. Wang and X.-F. Wu, Angew. Chem., Int. Ed., 2016, 55, 7227–7230 CrossRef CAS PubMed.
- P. Nagaraaj and V. Vijayakumar, Org. Chem. Front., 2019, 6, 2570–2599 RSC.
- H. Yu and Y. Zhang, Eur. J. Org. Chem., 2015, 2015, 1824–1828 CrossRef CAS.
- H. Yu, Y. Chen and Y. Zhang, Chin. J. Chem., 2015, 33, 531–534 CrossRef CAS.
- S. Biswas, H. S. Khanna, Q. A. Nizami, D. R. Caldwell, K. T. Cavanaugh, A. R. Howell, S. Raman, S. L. Suib and P. Nandi, Sci. Rep., 2018, 8, 13649–13656 CrossRef PubMed.
- V. P. Charpe, A. Ragupathi, A. Sagadevan and K. C. Hwang, Green Chem., 2021, 23, 5024–5030 RSC.
-
(a) Y. Zhang, W. Schilling, D. Riemer and S. Das, Nat. Protoc., 2020, 15, 822–839 CrossRef CAS PubMed;
(b) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823–1829 CrossRef CAS PubMed.
- Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659–6664 CrossRef CAS.
- I. Itoh, Y. Matsusaki, A. Fujiya, N. Tada, T. Miura and A. Itoh, Tetrahedron Lett., 2014, 55, 3160–3162 CrossRef CAS.
-
(a) M. Bhargava Reddy, N. Neerathilingam and R. Anandhan, Org. Chem. Front., 2021, 8, 87–93 RSC;
(b) M. Bhargava Reddy, K. Prasanth and R. Anandhan, Org. Biomol. Chem., 2020, 18, 9601–9605 RSC;
(c) M. Bhargava Reddy and R. Anandhan, Chem. Commun., 2020, 56, 3781–3784 RSC;
(d) N. Neerathilingam, M. Bhargava Reddy and R. Anandhan, J. Org. Chem., 2021, 86, 15117–15127 CrossRef CAS PubMed;
(e) K. Prasanth, M. Bhargava Reddy and R. Anandhan, Asian J. Org. Chem., 2022, 11, e202100590 CrossRef.
- A. Sagadevan, P.-C. Lyu and K. C. Hwang, Green Chem., 2016, 18, 4526–4530 RSC.
-
(a) Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898–2910 CrossRef CAS PubMed;
(b) W. Schilling, Y. Zhang, D. Riemer and S. Das, Chem.–Eur. J., 2020, 26, 390–395 CrossRef CAS PubMed;
(c) J. Kollmann, Y. Zhang, W. Schilling, T. Zhang, D. Riemer and S. Das, Green Chem., 2019, 21, 1916–1920 RSC;
(d) Y. Zhang, N. Hatami, N. S. Lange, E. Ronge, W. Schilling, C. Jooss and S. Das, Green Chem., 2020, 22, 4516–4522 RSC.
- X. Ling, Y. Xiong, R. Huang, X. Zhang, S. Zhang and C. Chen, J. Org. Chem., 2013, 78, 5218–5226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data for all new compounds. See DOI: 10.1039/d2ra00585a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*