
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The emission of low pH water from Gulf of Mexico seeps as revealed by δ13C–CO2 and methane oxidation data†
Sydney I.
Louden
 * and
John D.
Kessler
* and
John D.
Kessler
Department of Earth and Environmental Science, University of Rochester, Rochester, NY, USA. E-mail: slouden2@ur.rochester.edu
First published on 9th October 2023
Abstract
Seawater was collected around the MC118 hydrocarbon seep in the Gulf of Mexico and was used previously for a study of aerobic methane (CH4) oxidation. During that experiment, changes in the dissolved concentrations and δ13C isotopes of CH4 and CO2 were recorded. Originally, the CO2 concentrations and isotopes were recognized to qualitatively follow trends supporting the microbial conversion of CH4 to CO2via aerobic oxidation, however, no attempt was made to quantitatively explain this CO2 data. The present study models the δ13C–CO2 changes that occur as a result of CH4 oxidation, accounting for the carbon already present as dissolved inorganic carbon (DIC), DIC added via CH4 oxidation, and the pH of seawater. This study discovers that to accurately model the measured concentration and isotopic data for CO2, the seawater emitted from this seep site must have a pH which is at most between 6.49 and 7.24, and possibly up to 0.43 ± 0.08 pH units lower. These results are corroborated by direct measurements of pH from seeps in the Mediterranean Sea. A first-order extrapolation indicates that while cold seeps in the Gulf of Mexico may be a source of low pH water influencing the carbon dynamics of the deep ocean environment, this influence is likely less than that of current surface ocean acidification caused by the infiltration of atmospheric CO2.
Environmental significanceMethane seeps have been widely studied to understand the amount and fate of released methane, a potent greenhouse gas. However, this study reveals that seeps also emit low pH water, broadening the environmental significance of seafloor seeps to include ocean acidification. Using samples from a Gulf of Mexico seep, measurements and models of aerobic methane oxidation were conducted. These analyses revealed the pH of water emitted from this seep must be between 6.49 and 7.24, much lower than the average ocean pH of 8.1. This low pH water likely has important, localized influences. But a first-order extrapolation suggests the potential rate of deep ocean acidification in this environment is likely lower than the rate of seawater acidification from atmospheric CO2. |
Introduction
Seeps emit methane (CH4) and other hydrocarbons from the seafloor and are concentrated along continental margins. One area with many seeps that has been well documented and studied is the Gulf of Mexico, which contains an estimated 914 natural hydrocarbon seeps.1,2 Both the abundance of hydrocarbons underneath the seafloor and the geology of the basin contribute to the number of seeps found in the Gulf of Mexico.3Methane is emitted from seeps in the form of dissolved CH4 and bubble streams4 which can dissolve into the water column during ascent.5 The rate at which these CH4 bubbles dissolve into the surrounding water depends on a variety of factors including, but not limited to, bubble size and depth of emission6 with substantial amounts of dissolution occurring near the seafloor.7
Once dissolved in the overlying waters, methanotrophic bacteria use CH4 as a carbon and energy source.8 These methanotrophs are present in the water surrounding CH4 seeps and aerobically oxidize CH4via the overall reaction shown in eqn (1).9
| CH4 + 2O2 → CO2 + 2H2O | (1) |
The CO2 produced from aerobic CH4 oxidation then mixes and equilibrates with the dissolved inorganic carbon (DIC) pool already present in the seawater (eqn (2) and (3)),
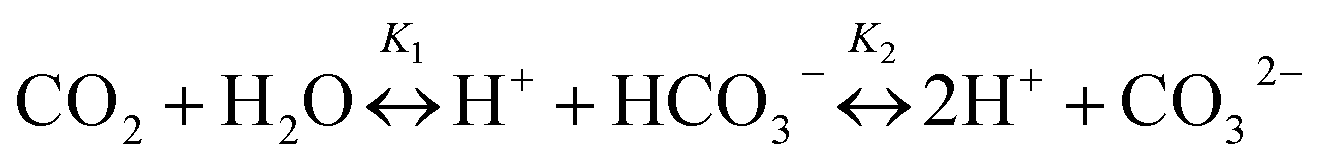 | (2) |
| [DIC] = [CO2] + [HCO3−] + [CO32−] | (3) |
The specific rate of microbial oxidation of CH4 is faster for the light isotopologue (12CH4) than the heavy isotopologue (13CH4), resulting in a kinetic isotope effect and producing measurable changes in the isotopic composition of the residual CH4. Thus, the CO2 produced via CH4 oxidation is enriched in the 12C isotope compared to the residual CH4.10 These isotopic differences change as a function of the extent of oxidation and can be used to confirm that CH4 oxidation is occurring. Additionally, these changes can quantitatively determine the extent of the starting CH4 pool that has been oxidized and have been further used to determine the oxidation rate.11
The investigation presented here began with data measured during an experiment exploring aerobic CH4 oxidation in seawater.9 The experiment measured changes in the dissolved concentrations and δ13C isotopes of CH4 and CO2 in a closed seawater incubation system experiencing significant amounts of aerobic CH4 oxidation.12 The goal of the present study was to develop an isotopic model to quantitatively explain changes in the measured δ13C–CO2 data as a function of the extent of CH4 oxidation, the results of which discovered that seeps emit relatively low pH water alongside CH4. Variables included in this model were the isotopic fractionation associated with aerobic CH4 oxidation, the background concentrations of CO2 and DIC present in the system prior to this CH4 oxidation event, and the added CO2 from CH4 oxidation. This investigation reveals that the model and measurements only agree at pH values significantly below those of the background deep ocean, indicating that seeps are a source of low pH waters to the deep ocean.
Experimental
Sample collection
The full details describing sample collection and analysis can be found in Chan et al.9 In brief, samples were collected in the Gulf of Mexico at site MC118 (28°51′N, 88°29.5′W) during a research expedition from 12–17 April 2015 onboard the E/V Nautilus. Samples were collected at depths of 794 and 888 m using the Suspended-Particle Rosette sampler13 mounted to the remotely operated vehicle (ROV) Hercules. Water samples were taken just above the seafloor in water visibly impacted by CH4 bubbles. Other hydrocarbons, including oil, were present as well.9The collected water samples were incubated at near in situ temperatures using a mesocosm incubation system developed by Chan et al.14 The mesocosm incubation system was connected to a dissolved gas analysis system which measured the concentrations and stable isotopes of CH4 and CO2 throughout the incubation period. Samples for DNA analysis and cell counts were isolated periodically during incubation to characterize the microbial communities and ensure methanotrophic bacteria were present.9
Isotope modeling procedure
Since this experiment incubated seawater samples in a closed vessel, the isotopic fractionation caused by CH4 oxidation was modeled following closed system or Rayleigh fractionation equations.12 We note that other studies of CH4 oxidation in open natural seawater environments still displayed isotopic fractionation following closed system kinetics.11,15 The closed nature of the incubation vessel meant that any CO2 produced from CH4 oxidation accumulated in the vessel. Thus, to model the δ13C–CO2 data, an accumulated product model was incorporated (eqn (4)).16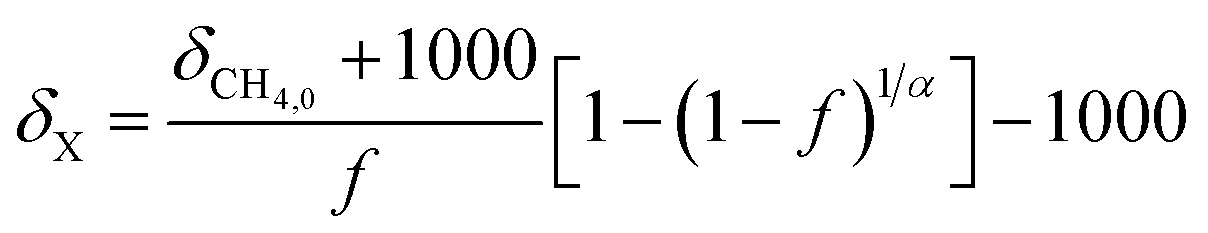 | (4) |
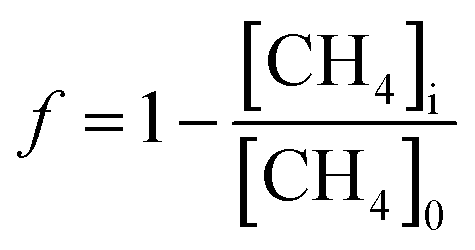 | (5) |
While δX represents the isotopic values of the accumulated CO2 produced during this CH4 oxidation experiment, this CO2 is added to a large pool of CO2 and DIC initially present in the seawater sample. Thus, to accurately model the measured δ13C–CO2 data, a weighted isotopic average was used (eqn (6)).
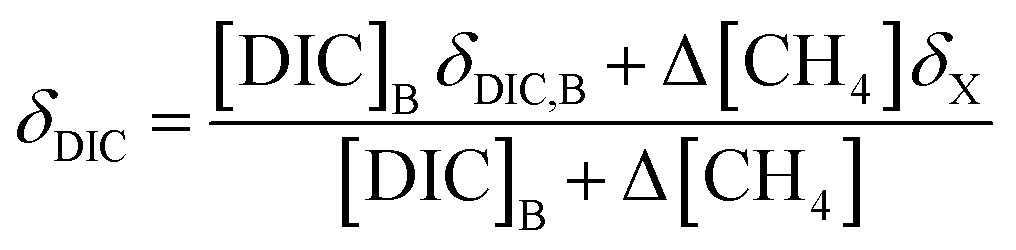 | (6) |
[DIC]B was determined based on the dissolved CO2 concentration measurements and the pH of the seawater sample. Combining eqn (3) with the equilibrium relationships of K1 and K2 (eqn (2)) produces an equation for [DIC]B as a function of CO2 concentration and pH (eqn (7)),
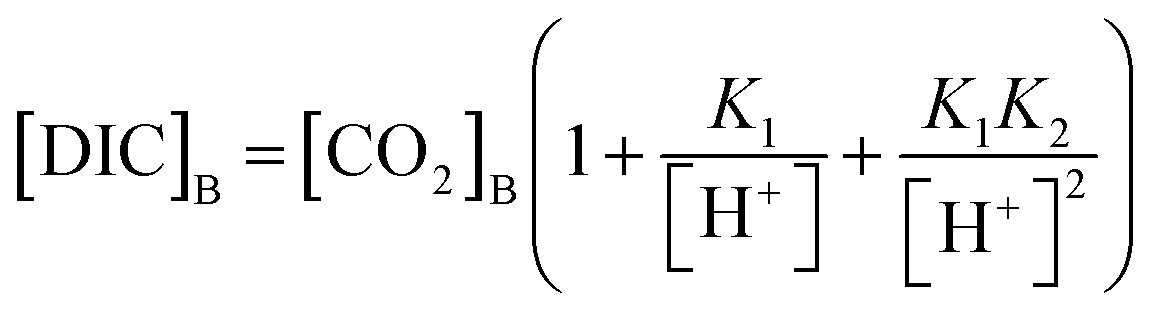 | (7) |
This model calculates the isotopic value of DIC in the system as a mixture of the background DIC and the DIC added from CH4 oxidation (eqn (6)). However, since values of δCO2 were measured rather than δDIC, the modeled values of δDIC (eqn (6)) must be reverted to values of δCO2 for comparison with the data. This is accomplished by subtracting the corresponding isotopic offset value calculated previously.
For each incubation experiment, the pH values incorporated into these calculations were varied until the residuals between the modeled values of δCO2 and the measured data were minimized.
Results and discussion
Data and results
The modeled and measured values of δCO2 for the four incubation experiments are shown in Fig. 1. Incubation experiments S2 and S3 show the best agreement between the modeled and the measured values, especially during more rapid CH4 oxidation, characterized by a large drop in δ13C–CO2. In S2 and S3, a period of positive isotopic values is followed by a rapid change to negative isotopic values. This follows the expected values where δ13C–CO2 is positive before more rapid CH4 oxidation begins and quickly becomes negative during more rapid CH4 oxidation. Incubation experiments S1 and S4 also show agreement between the data and the model, however, these samples follow a slightly different trend from S2 and S3. S1 and S4 both begin with negative isotopic values and display a steadier decline.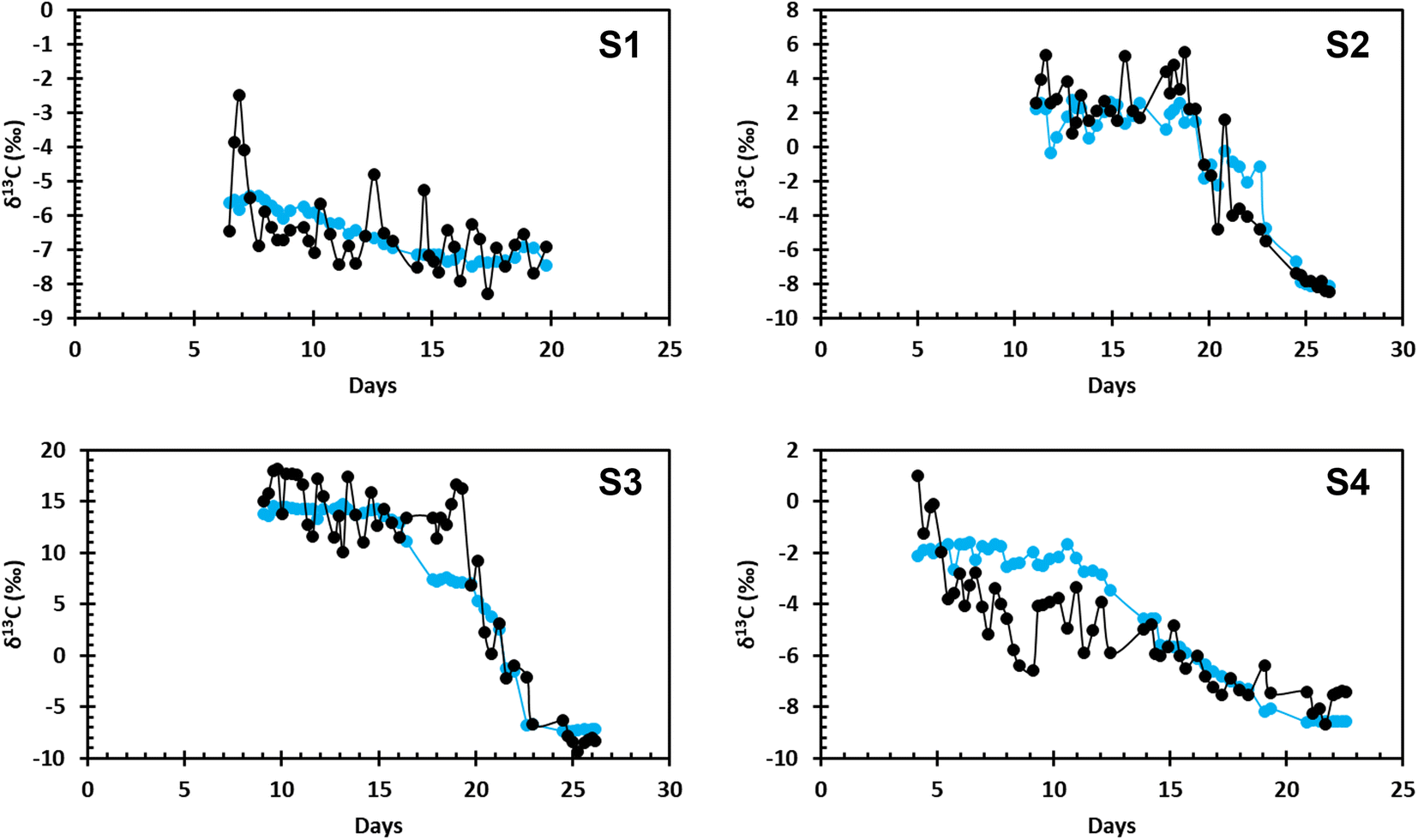 | ||
| Fig. 1 The modeled δ13C–CO2 data (blue) compared to the measured values of δ13C–CO2 (black) over the incubation time for all four samples collected at MC118. | ||
The pH values required to produce the isotopic models shown in Fig. 1 represent the pH at the beginning of the incubation experiment. The pH at the end of the incubation experiment was calculated using eqn (8).
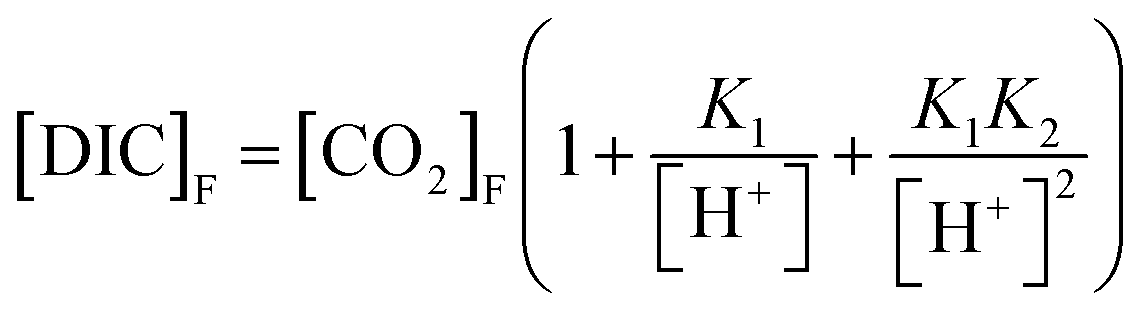 | (8) |
K 1 and K2 are the same as in eqn (7). [CO2]F is [CO2] at the end of the incubation experiment, and [DIC]F is [DIC]B plus the change in CH4, assuming all CH4 removed was added to DIC via CH4 oxidation. The pH values calculated at the beginning and end of each incubation experiment are shown in Table 1.
| Sample | Estimated pH range |
|---|---|
| MC118-S1 | 7.13–7.24 |
| MC118-S2 | 6.67 |
| MC118-S3 | 6.49–6.62 |
| MC118-S4 | 6.69–6.84 |
The average difference between final and initial pH values for all four incubation experiments is 0.1 ± 0.07 pH units. Overall, S1 is the most basic, and S3 is the most acidic. S2 shows no change in pH over the duration of the incubation experiment, while S4 shows the largest change. Data from all four incubation experiments suggests a pH considerably lower than the average ocean pH of 8.1.22
Disagreement between the modeled and measured data can be attributed to the natural variation in the measured data, differences in initial CH4 concentration, oxidation of other carbon compounds in the system, and/or analytical uncertainties. The variability in the measured data is not as present in the modeled values due to the averaging of initial data before it was used in the model. Each of the four incubations had different starting concentrations of CH4. In S2 and S3, initial CH4 concentrations were approximately 150 μM; in S1 and S4, initial CH4 concentrations were approximately 50 μM. Methane oxidation rates were higher in S2 and S3 compared to S1 and S4. Higher initial CH4 concentrations and faster oxidation rates indicate CH4 oxidation was contributing more CO2 to the DIC pool than in experiments S1 and S4. Oxidation of seep-derived dissolved organic carbon (DOC) would also add CO2 to the DIC pool in these experiments.23 Other geologic hydrocarbons were present in the incubations, which was visible at the sample collection site. Sequencing of the 16S rRNA gene at the end of each of the four incubations showed the presence of many species of hydrocarbon oxidizing bacteria, with CH4 oxidizing bacteria making up a relatively low percentage of the microbial community.9
Discussion
A pH measurement was not taken at the sampling site of this dataset, however, there is strong evidence that the water directly above the CH4 seep is relatively acidic. Isotope and concentration data suggest that the pH of the sample site is between 6.49 and 7.24, if not lower. One assumption made in modeling the isotope data was that all CH4 removed during the incubation experiment was converted to CO2. Based on eqn (1), two moles of O2 should be removed for every mole of CH4. Dissolved O2 (DO) data from Chan et al. indicate a DO to CH4 ratio less than 2 to 1.9 Chan et al. hypothesize that this discrepancy is due to the formation of biomass that has not been fully oxidized to CO2 by the end of the incubation experiment.9 Thus, if we assume that only half of the CH4 is fully oxidized to CO2 while the rest remains as biomass at the conclusion of this experiment, an assumption supported by the DO to CH4 ratio,9 the estimated pH of the water emitted from this seep is approximately 0.43 ± 0.08 pH units lower.The pH of fluids emitted from hydrothermal vents has been well documented24,25 with the development of in situ measurement techniques,26,27 however, few measurements of pH at CH4 seep sites have been published. The limited published data that exist indicate that water surrounding and coming from cold CH4 seeps has an approximately neutral pH.1,28 Sisma-Ventura et al. report measurements of pH as low as 6.83 in the water column above a hydrocarbon seep in the Southeast Mediterranean Sea with low pH values of 6.8 to 7.4 recorded throughout the water column up to 50 m above the seafloor.28
The data and conclusions of Sisma-Ventura et al. support the findings of this study.28 The measurement of a pH of 6.83 in the water above a hydrocarbon seep falls within the calculated pH range for the MC118 seep. The lower pH values calculated for MC118 could be attributed to the fact that water samples were collected directly above the seep emission via ROV whereas Sisma-Ventura et al. collected water up to 50 m above the seep using a carousel of Nisken bottles.28
Sisma-Ventura et al. conclude that the low pH values measured above hydrocarbon seeps are a result of the oxidation of CH4 and other hydrocarbons to CO2 which is added to the DIC pool of the background bottom water, lowering the pH.28,29 Sisma-Ventura et al. hypothesize that hydrocarbon seeps have a substantial impact on bottom water chemistry.28 The results of the present study support this conclusion and suggest that CH4 seeps are a source of low pH water to the deep Gulf of Mexico.
To estimate the potential impact of this low pH water, the modeled results from this study were extrapolated to the entire Gulf of Mexico to estimate the amount of low pH water emitted and its influence on the total pH of the Gulf of Mexico. For these calculations, the Gulf of Mexico was modeled as a cylinder with a total volume of 2.434 × 1015 m3 and a height equivalent to the average depth, 1615 m.30 It was assumed that all estimated 914 seeps in the Gulf of Mexico2 emit the same low pH water at the same, constant rate. Estimates of water flux from Gulf of Mexico seeps range from 9.4 to 30 mm per year, and seep diameters from which water is emitted are estimated to be between 0.2 and 1.2 km.31,32 Maximum values were chosen for this calculation to prevent against underestimating the potential impact on bottom water pH. Therefore, the lowest pH value determined of 5.91 was used, which assumes half of the oxidized CH4 remained as biomass. It was assumed that the background pH of the deep Gulf of Mexico was 8.1.22 This also maximizes the estimated pH impact as the effect of low pH water emitted from seeps would be reduced in less basic surrounding water. The amount of low pH water emitted by all seeps over the span of one year was then calculated, and the resulting pH changes were determined assuming this low pH seep water impacts (i) the total volume of the Gulf of Mexico, (ii) the bottom 50 m of the Gulf of Mexico, and (iii) the bottom 10 m of the Gulf of Mexico.
Any pH change was negligible assuming the total volume or the bottom 50 m were influenced by low pH water emitted from cold seeps. When we assumed that low pH seep water only impacts the bottom 10 m of the Gulf of Mexico, the pH decreased by 1.38 × 10−4 in one year. For comparison, the average yearly decrease in pH from surface ocean acidification from the infiltration of atmospheric CO2 is 2.00 × 10−3.22 Only if the water flow from all seeps in the Gulf of Mexico was higher by a factor of 10 would a similar decrease in pH be observed as in the surface waters, assuming only the bottom 10 m are impacted by seep water. A hypothetical increase in water flow by a factor of 100 would be necessary for the bottom 50 m to display a similar annual pH decrease to the surface waters. The emission of low pH waters from CH4 seeps impacts the carbon system of the deep ocean, however, the impact on deep ocean pH is likely less than the impact of acidification in the surface ocean.
Conclusions
This study quantitatively interpreted the concentration and δ13C content of CO2 measured during incubation experiments exploring aerobic CH4 oxidation. The water and CH4 used in this experiment were collected immediately above a Gulf of Mexico CH4 seep site via ROV.9,12 A model was developed to match the measured changes in δ13C–CO2 during aerobic CH4 oxidation. This model had to account for the inorganic carbon already in the system in the form of DIC as well as the CO2 added from CH4 oxidation.The model reveals the importance of pH in determining the impact of added CO2 on background DIC. This model and analysis suggest that the pH of water emitted at the MC118 CH4 seep site is between 6.49 and 7.24 – and possibly up to 0.43 ± 0.08 pH units lower – considerably lower than the average ocean pH of 8.1.22 While seeps in the Gulf of Mexico are an important source of CH4, this study proposes that they are also a source of low pH water. Based on a first-order extrapolation of the results from this isotopic model, the influence of this low pH water on the overall bottom water chemistry is relatively small, especially when compared to the rates of surface ocean acidification.
A paradox seemingly raised by these findings is the existence of authigenic carbonate formations, which are characteristic of CH4 seeps,33–36 surrounding an emission point of more acidic water and CH4 bubbles. While we do not have a quantitative explanation for this paradox, we offer two potential avenues for future study. First, while anaerobic oxidation of methane (AOM) is the primary reaction driving authigenic carbonate precipitation,37 organoclastic sulfate reduction (OSR) can also contribute to carbonate precipitation under low pH conditions38 and may be contributing to carbonate precipitation in these environments. Second, beyond the point of emission of the seep itself, local sediment production and consumption of CH4 may control carbonate production.39 A highly localized seep environment would allow for the dual existence of both low pH water coming from the seep and authigenic carbonate formation in the surrounding environment. In addition, a previous modeling study of carbonate formation at Hydrate Ridge also suggested that carbonate can form under relatively acidic conditions with a pH of 6.9 at the sediment–water interface.40
While the regional significance of low pH water emissions from CH4 seeps in the Gulf of Mexico is likely relatively minor in the context of today's environmental change, their impact on local seep environments requires further investigation. More direct measurements of the pH of water emitted from seep sites in the Gulf of Mexico and globally are needed to confirm and expand upon the findings of this study. Even if the global influence of low pH waters from CH4 seeps is relatively minor compared to surface ocean acidification in the anthropocene, the results of this study highlight the need to further characterize the influence of these low pH waters on the surrounding seafloor environment.
Author contributions
S. L. and J. K. designed the study, performed the research, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by grants to J. K. from NSF (OCE-2241873 and OCE-1851402). The authors also acknowledge Eric Chan for his original collection of the data used in this study.Notes and references
- S. B. Joye, The geology and biogeochemistry of hydrocarbon seeps, Annu. Rev. Earth Planet. Sci., 2020, 48, 205–231 CrossRef CAS.
- I. R. MacDonald, O. Garcia-Pineda, A. Beet, S. Daneshgar Asl, L. Feng, G. Graettinger, D. French-McCay, J. Holmes, C. Hu, F. Huffer, I. Leifer, F. Muller-Karger, A. Solow, M. Silva and G. Swayze, Natural and unnatural oil slicks in the Gulf of Mexico, J. Geophys. Res. Oceans, 2015, 120, 8364–8380 CrossRef CAS PubMed.
- C. Fisher, H. Roberts, E. Cordes and B. Bernard, Cold seeps and associated communities of the Gulf of Mexico, Oceanography, 2007, 20, 118–129 CrossRef.
- T. C. Weber, L. Mayer, K. Jerram, J. Beaudoin, Y. Rzhanov and D. Vovalv, Acoustic estimates of methane gas flux from the seabed in a 6000 km2 region in the Northern Gulf of Mexico, Geochem., Geophys., Geosyst., 2014, 15, 1911–1925 CrossRef CAS.
- M. Leonte, B. Wang, S. A. Socolofsky, S. Mau, J. A. Breier and J. D. Kessler, Using carbon isotope fractionation to constrain the extent of methane dissolution into the water column surrounding a natural hydrocarbon gas seep in the Northern Gulf of Mexico, Geochem., Geophys., Geosyst., 2018, 19, 4459–4475 CrossRef CAS.
- D. F. McGinnis, J. Greinert, Y. Artemov, S. E. Beaubien and A. Wüest, Fate of rising methane bubbles in stratified waters: How much methane reaches the atmosphere?, J. Geophys. Res. Oceans, 2006, 111, C09007 CrossRef.
- B. Wang, I. Jun, S. A. Socolofsky, S. F. DiMarco and J. D. Kessler, Dynamics of gas bubbles from a submarine hydrocarbon seep within the hydrate stability zone, Geophys. Res. Lett., 2020, 47, e2020GL089256 CrossRef CAS.
- R. S. Hanson and T. E. Hanson, Methanotrophic bacteria, Microbiol. Rev., 1996, 60, 439–471 CrossRef CAS PubMed.
- E. W. Chan, A. M. Shiller, D. J. Joung, D. L. Valentine, M. C. Redmond, J. A. Breier, S. A. Socolofsky and J. D. Kessler, Investigations of aerobic methane oxidation in two marine seep environments: Part 1 – Chemical Kinetics, J. Geophys. Res. Oceans, 2019, 124, 8852–8868 CrossRef CAS.
- J. F. Barker and P. Fritz, Carbon isotope fractionation during microbial methane oxidation, Nature, 1981, 293, 289–291 CrossRef CAS.
- M. Leonte, J. D. Kessler, M. Y. Kellerman, E. C. Arrington, D. L. Valentine and S. P. Sylva, Rapid rates of aerobic methane oxidation at the feather edge of gas hydrate stability in the waters of Hudson Canyon, US Atlantic Margin, Geochim. Cosmochim. Acta, 2017, 204, 357–387 CrossRef.
- E. W. Chan, A. M. Shiller, D. J. Joung, E. C. Arrington, D. L. Valentine, M. C. Redmond, J. A. Breier, S. A. Socolofsky and J. D. Kessler, Investigations of aerobic methane oxidation in two marine seep environments: Part 2 – Isotopic Kinetics, J. Geophys. Res. Oceans, 2019, 124, 8392–8399 CrossRef CAS.
- J. A. Breier, C. G. Rauch, K. McCartney, B. M. Toner, S. C. Fakra, S. N. White and C. R. German, A suspended-particle rosette multi-sampler for discrete biogeochemical sampling in low-particle-density waters, Deep-Sea Res. I: Oceanogr. Res., 2009, 56, 1579–1589 CrossRef CAS.
- E. W. Chan, J. D. Kessler, A. M. Shiller, D. J. Joung and F. Colombo, Aqueous mesocosm techniques enabling the real-time measurement of the chemical and isotopic kinetics of dissolved methane and carbon dioxide, Environ. Sci. Technol., 2016, 50, 3039–3046 CrossRef CAS PubMed.
- N. J. Grant and M. J. Whiticar, Stable carbon isotope evidence for methane oxidation in plumes above Hydrate Ridge, Cascadia Oregon Margin, Global Biogeochem. Cycles, 2002, 16, 71 Search PubMed.
- J. Hoefs, Stable Isotope Geochemistry, Springer, Berlin, 4th edn, 1997, vol. 1, pp. 5–10 Search PubMed.
- R. E. Zeebe and D. Wolf-Gladrow, CO2 in Seawater Equilibrium, Kinetics, Isotopes, Elsevier, Amsterdam, 2001, vol. 3, pp. 168–182 Search PubMed.
- W. G. Mook, 13C in atmospheric CO2, Neth. J. Sea Res., 1986, 20, 211–223 CrossRef CAS.
- J. Zhang, P. D. Quay and D. O. Wilbur, Carbon isotope fractionation during gas-water exchange and dissolution of CO2, Geochim. Cosmochim. Acta, 1995, 59, 107–114 CrossRef CAS.
- A. G. Dickson and F. J. Millero, A comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media, Deep-Sea Res., Part A, 1987, 34, 1733–1743 CrossRef CAS.
- C. Mehrbach, C. H. Culberson, J. E. Hawley and R. M. Pytkowicx, Measurement of the apparent dissociation constants of carbonic acid in seawater at atmospheric pressure, Limnol. Oceanogr., 1973, 18, 897–907 CrossRef CAS.
- T. Takahashi, S. C. Sutherland, D. W. Chipman, J. G. Goddard, C. Ho, T. Newberger, C. Sweeny and D. R. Munro, Climatological distributions of pH, pCO2, total CO2, alkalinity, and CaCO3 saturation in the global surface ocean, and temporal changes at selected locations, Mar. Chem., 2014, 164, 95–125 CrossRef CAS.
- J. W. Pohlman, J. E. Bauer, W. F. Waite, C. L. Osburn and N. R. Chapman, Methane hydrate-bearing seeps as a source of aged dissolved organic carbon to the oceans, Nat. Geosci., 2010, 4, 37–41 CrossRef.
- W. E. Seyfried Jr, N. J. Pester, B. M. Tutolo and K. Ding, The Lost City hydrothermal system: Constraints imposed by vent fluid chemistry and reaction path models on subseafloor heat and mass transfer processes, Geochim. Cosmochim. Acta, 2015, 163, 59–79 CrossRef.
- W. E. Seyfried Jr, C. Tan, X. Wang, S. Wu, G. N. Evans, L. A. Coogan, S. F. Mihály and M. D. Lilley, Time series of hydrothermal vent fluid chemistry at Main Endeavour Field, Jan de Fuca Ridge: Remote sampling using the NEPTUNE cabled observatory, Deep-Sea Res. I: Oceanogr. Res., 2022, 186, 103809 CrossRef.
- K. Ding, W. E. Seyfried Jr, Z. Zhang, M. K. Tivey, K. L. Von Damm and A. M. Bradley, The in situ pH of hydrothermal fluids at mid-ocean ridges, Earth Planet. Sci. Lett., 2005, 237, 167–174 CrossRef CAS.
- C. Tan, K. Ding and W. E. Seyfried Jr, Development and Application of a New Mobile pH Calibrator for Real-Time Monitoring of pH in Diffuse Flow Hydrothermal Vent Fluids, Mar. Technol. Soc. J., 2016, 50, 37–47 CrossRef.
- G. Sisma-Ventura, O. M. Bialik, Y. Makovsky, E. Rahav, T. Ozer, M. Kanari, S. Marmen, N. Belkin, T. Guy-Haim, G. Antler, B. Herut and M. Rubin-Blum, Cold seeps alter the near-bottom biogeochemistry in the ultraoligotrophic Southeastern Mediterranean Sea, Deep-Sea Res. I: Oceanogr. Res., 2022, 183, 103744 CrossRef CAS.
- P. Aharon, E. R. Graber and H. H. Roberts, Dissolved carbon and 33-133-133-1 anomalies in the water column caused by hydrocarbon seeps on the northwestern Gulf of Mexico slope, Geo-Mar. Lett., 1992, 12, 33–40 CrossRef CAS.
- R. E. Turner, The Gulf of Mexico Large Marine Ecosystem, ed. H. Kumpf, K. Steidinger and K. Sherman, Blackwell Science, Oxford, 1999, Inputs and outputs of the Gulf of Mexico, pp. 64–73 Search PubMed.
- A. J. Smith, P. B. Flemings and P. M. Fulton, Hydrocarbon flux from natural deepwater Gulf of Mexico vents, Earth Planet. Sci. Lett., 2014, 395, 241–253 CrossRef CAS.
- A. J. Smith, P. B. Flemings, X. Lie and K. Darnell, The evolution of methane vents that pierce the hydrate stability zone in the world's oceans, J. Geophys. Res. Solid Earth, 2014, 119, 6337–6356 CrossRef.
- A. Y. Lein, Authigenic Carbonate Formation in the Ocean, Lithol. Miner. Resour., 2004, 39, 1–30 CrossRef CAS.
- S. Ritger, B. Carson and E. Suess, Methane-derived authigenic carbonates formed by subduction-induced pore-water expulsion along the Oregon/Washington margin, Geol. Soc. Am. Bull., 1987, 98, 147–156 CrossRef CAS.
- C. K. Paull, J. P. Chanton, A. C. Neumann, J. A. Coston, C. S. Martens and W. Showers, Indicators of methane-derived carbonates and chemosynthetic organic carbon deposits: examples from the Florida Escarpment, Palaios, 1992, 7, 361–375 Search PubMed.
- N. G. Prouty, D. Sahy, C. D. Ruppel, E. B. Roark, D. Condon, S. Brooke, S. W. Ross and A. W. J. Demopoulos, Insights into methane dynamics from analysis of authigenic carbonates and chemosynthetic mussels at newly-discovered Atlantic Margin seeps, Earth Planet. Sci. Lett., 2016, 449, 332–334 CrossRef CAS.
- W. S. Reeburgh, Oceanic Methane Biogeochemistry, Chem. Rev., 2007, 107, 486–513 CrossRef CAS PubMed.
- J. Blouet, S. Arndt, P. Imbert and P. Regnier, Are seep carbonates quantitative proxies of CH4 leakage? Modeling the influence of sulfate reduction and anaerobic oxidation of methane on pH and carbonate precipitation, Chem. Geol., 2021, 557, 120254 CrossRef.
- B. B. Bernard, Methane in marine sediments, Deep-Sea Res., Part A, 1979, 26, 429–443 CrossRef CAS.
- R. Luff, K. Wallmann and G. Aloisi, Numerical modeling of carbonate crust formation at cold vent sites: significance for fluid and methane budgets and chemosynthetic biological communities, Earth Planet. Sci. Lett., 2004, 221, 337–353 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00117b |
| This journal is © The Royal Society of Chemistry 2023 |