
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospun TiO2/carbon composite nanofibers as effective (photo)electrodes for removal and transformation of recalcitrant water contaminants†
Ashley Hesterberg
Butzlaff
a,
Madeline
Jensen
a,
Chenxu
Yan
 b,
Abdulsattar
Ghanim
c,
Charles
Werth
b,
David
Cwiertny
*a and
Syed
Mubeen
*c
b,
Abdulsattar
Ghanim
c,
Charles
Werth
b,
David
Cwiertny
*a and
Syed
Mubeen
*c
aDepartment of Civil and Environmental Engineering, University of Iowa, Iowa 52242, USA. E-mail: david-cwiertny@uiowa.edu
bDepartment of Civil, Architectural, and Environmental Engineering, The University of Texas at Austin, 301 E. Dean Keeton St, Stop C1786, Austin, Texas 78712-1173, USA
cDepartment of Chemical and Biochemical Engineering, University of Iowa, Iowa 52242, USA. E-mail: syed-mubeen@uiowa.edu
First published on 4th May 2023
Abstract
Electrochemical (EC) and photoelectrochemical (PEC) water treatment systems are gaining popularity, necessitating new electrode materials that offer reliable performance across diverse application platforms. For applications specifically targeting dilute chemical pollutants (i.e., parts-per-million concentrations or less), beneficial electrode properties include high surface area to overcome kinetic overpotential losses, low electrode areal electrical resistance, and high water permeability with sufficient mechanical strength for use in electroactive membrane-based treatment systems. Here, we used electrospinning to fabricate (photo)electrodes from carbon nanofibers (CNFs) containing titanium dioxide (TiO2) nanoparticles. Optimal CNF/TiO2 composites were electrochemically and photochemically active with a surface area of ∼50 m2 g−1 and electrode areal resistance of 2.66 Ω cm2, values comparable to commercial carbon-based electrode materials (e.g., Kynol Activated Carbon Cloth). Transformation experiments with carbamazepine (CBZ), a recalcitrant organic contaminant, suggest CNF/TiO2 electrodes function dually as sorbents, first binding CBZ prior to oxidation at positive applied potentials. Complete CBZ transformation was observed in both EC (dark) and PEC (UV light; 280 mW cm−2) systems over 90 minutes, with PEC systems exhibiting 1.5-fold higher transformation rates (kobs ∼ 0.18 min−1) at +1.00 V (vs. Ag/AgCl). Composite electrodes also exhibited stability across repeated use, yielding consistent current densities over five experimental cycles (120 min each) of CBZ transformation (0.25 ± 0.03 mA cm−2). Because of their high surface area, electrical conductivity, photoactivity, and electrochemical stability, these electrospun CNF/TiO2 composites represent promising (photo)electrode alternatives for diverse EC and PEC applications.
Environmental significanceAn increasing number of emerging contaminants are present in drinking water supplies, and their risks to human health remain poorly understood. These unregulated contaminants provide unique challenges to traditional water treatment and affect many consumers, including those supplied by private wells. Electrochemical (EC) water treatment holds promise for addressing these challenges, using renewable energy to drive pollutant degradation with minimal chemical input in a decentralized platform. However, high-performance electrode materials remain a critical need for optimal performance, sustainability, and economic feasibility. To meet this need, we herein fabricate a carbon-based, nanoengineered composite electrode material for use in (photo)electrochemical water treatment and demonstrate its performance toward a persistent emerging organic contaminant common to drinking water supplies. |
Introduction
With the growing demand for technologies that can be driven by renewable energy sources, electrochemical (EC) and photoelectrochemical (PEC) systems have the potential to revolutionize water treatment.1–4 EC treatment systems rely on the direct or indirect transfer of electrons between the dissolved pollutant target and the electrode surface under an applied potential.3,5 Building upon EC systems, PEC systems integrate photochemistry to catalyze electrochemical reactions with light energy.6,7 Unlike traditional photocatalytic (PC) systems (Fig. S1a†), PEC systems (Fig. S1b†) provide close contact between the semiconductor and a charge-collecting substrate to promote efficient carrier separation and transport as enabled by an external bias.8–12 Further, by careful choice of (photo)cathode and (photo)anode materials, the entire reaction can be driven using renewable energy with improved efficiencies over a single-semiconductor PC system.10,11,13The working electrode material is perhaps most critical to the performance of EC and PEC treatment systems. In addition to being environmentally (e.g., nontoxic components) and economically responsible, electrode materials must also possess high surface area, electronic conductivity, stability during use, and water/gas permeability to satisfy diverse application platforms and the demand for improved performance over commercial available alternatives.1–3 While PEC electrode materials have been extensively researched for hydrogen and oxygen production, fewer studies have explored suitable electrode materials for water treatment of dissolved chemical contaminants.12,14–18 Indeed, a key practical challenge to implementing EC and PEC configurations in environmental systems is the low pollutant concentrations – typically ranging from parts per trillion (ng L−1) to parts per million (mg L−1) – that induce large kinetic losses at the electrode surface due to mass transport overpotentials. To overcome this and other challenges, work to date has established several favorable electrode properties for EC and PEC water treatment applications including: (i) large interfacial contact between the semiconductor and aqueous phase to decrease kinetic overpotential losses and to maximize light absorption;19–21 (ii) improved carrier collection to minimize recombination;22,23 and (iii) high material strength for durable operation.22–24 Moreover, water permeability is desired to overcome mass transfer limitations, particularly for electrodes acting as reactive (i.e., electroactive) membranes in dead-end or cross-flow filtration systems.25,26
To date, common electrode materials include noble metals, metal oxides (e.g., tungsten, titanium, bismuth, iron) and mixed metal oxides (MMO), graphite, and boron-doped diamond (BDD) electrodes.21,27–29 Integrating a co-catalyst or photocatalyst in these materials can expand applications to PEC systems.29–32 More recent efforts have focused on further developing engineered nanomaterials for use in EC and PEC systems to take advantage of the large surface area to volume ratios, enhanced mass transfer, and improved control of electron flow (i.e., product selectivity).3,32,33 In particular, carbon nanofibers (CNFs) have become a popular choice for self-supported electrodes because the desirable properties of carbon [e.g., (electro)chemical stability, electrocatalytic activity, wide potential window, low cost]22,34 are enhanced in a nanostructured, three-dimensional fiber network that provides improved surface area, pore structure, electrical conductivity, and mechanical flexibility.35,36 Moreover, CNF composites that include secondary components (e.g., a nanoparticle semiconductor) can also be fabricated to introduce properties not realized in carbon-only materials (e.g., photoactivity).28 Although some studies with composite CNF electrodes have demonstrated efficient transformation of environmental pollutants in EC and PEC systems, many prior studies have focused on pollutant targets of limited relevance to emerging water treatment needs (e.g., targeting dyes at high concentrations),28 have been fabricated using toxic materials not suitable for water treatment,27 or employ electrodes and catalysts that may be cost prohibitive (e.g. BDD, platinum group metals).2,3,37
Herein, we fabricated, characterized, and assessed the performance of electrospun CNFs containing titanium dioxide (TiO2) nanoparticles for use as both an electrode and a photoelectrode (in the presence of light) for pollutant transformation. Results from this work demonstrate the potential advantages of the proposed (photo)electrode architecture and design while avoiding complex systems or components (e.g., catalyst recovery, catalyst leaching, additional chemical and energy inputs).38–40 The electrospun CNFs serve as a three-dimensional, low-resistance current collector and provide a high surface area for supporting and exposing embedded TiO2 semiconductor particles for enhanced charge transport. TiO2 – an earth-abundant semiconductor that represents the industry standard for photocatalytic water treatment applications41,42 – was selected due to the extensive literature about its performance as a photocatalyst, particularly for pollutant transformation.14,43–49 While the work builds upon our prior study that utilized CNF/TiO2 composites as a photocatalytic membrane for treating organic micropollutants,47 we herein show for the first time that by carefully controlling synthesis parameters, these materials can also function as both electroactive and photoelectroactive substrates for transforming pollutants recalcitrant to photocatalytic approaches alone.
Using electrospinning, an industrially viable route to large-scale nanofiber fabrication,50 we produced a suite of CNF/TiO2 composites with varying TiO2 levels (30–70 wt% relative to polymer mass) and thicknesses (from 170–520 μm). Through electrochemical characterization using potentiostatic electrochemical impedance spectroscopy (PEIS), we demonstrate that the optimal CNF/TiO2 (photo)electrodes can achieve geometric (areal) resistances (Ω cm2) comparable to commercial activated carbon electrodes (Kynol Activated Carbon Cloth 5092-10; Gun EI Chemical Industry Japan) while demonstrating higher ion mobility by two orders of magnitude (D = 2.70 × 10−8 cm2 s−1). We then evaluated the performance of the CNF/TiO2 composites as electrodes and photoelectrodes in batch water treatment systems targeting the organic micropollutant carbamazepine (CBZ), which is recalcitrant to traditional water treatment approaches.51 The CNF/TiO2 (photo)electrodes demonstrated rates for CBZ transformation that compared favorably to those reported for PC treatment with TiO2 suspensions.52,53 The (photo)electrodes also provided consistent EC performance over multiple reaction cycles with CBZ.
Materials and methods
Reagents
All reagents are listed in the ESI.†Electrode synthesis
To prepare the sol–gels, first, TiO2 and PTA were uniformly dispersed in DMF by ultrasonication for five hours. After sonication, PAN was added to the TiO2 with PTA suspension and thermomixed (2 hours, 60 °C, 700 rpm) to produce a homogeneous sol–gel with no visible settling. All sol–gels were prepared with a PAN concentration of 8 wt% relative to total sol–gel mass (i.e., PAN + DMF + PTA).
To vary the thickness of deposited nonwoven nanofiber mats, the sol–gel volume used during electrospinning was increased. For these materials, sol–gel components were scaled by a factor of 0.5, 1.0, or 1.5 (corresponding to sol–gel DMF volumes of 1.75, 3.5, and 5.25 mL, respectively), where the relative weight percent of each component within the sol–gel was held constant (e.g., PAN concentration of 8 wt% was held constant with volume). Because the electrospun nanofibers were collected on a rotating, grounded collector of constant surface area, CNF mat thickness could be expected to increase with increasing sol–gel volume.
Fig. S2† shows all the different electrospinning solution formulations used in this study. Hereafter, each material will be identified as “XX g PAN/XX wt% TiO2” to specify the TiO2 composition and polymer mass, respectively, as determined by the sol–gel composition. A lab-scale, custom-designed rig described in our earlier work was used for electrospinning.54 The detailed electrospinning procedure and parameters are provided in the ESI.† A photograph of the electrospinning rig is shown in Fig. S3.†
Nanofiber characterization
Energy-dispersive X-ray spectroscopy (EDS) was performed in tangent with SEM (IXRF Systems; 15 kV) to provide the elemental mapping of the optimal fiber composite. Transmission electron microscopy (TEM; Hitachi HT7800) images were also collected for the optimal fiber composite to examine the internal fiber structure and distribution of TiO2 nanoparticles within the fibers. For TEM analysis, samples were suspended in acetonitrile overnight and drop-cast onto carbon-coated TEM grids.
Specific surface area (SBET) and pore volume measurements were conducted under nitrogen using a surface area and pore size analyzer (Quantachrome Nova 4200e). Methods to quantify specific surface area and pore volume are provided in the ESI.† TiO2 crystal structure (anatase, rutile) as a function of carbonization temperature was identified by powder X-ray diffraction (XRD; Bruker D-5000 q – q diffractometer). Similarly, the graphitic character of CNFs fabricated at different carbonization temperatures was identified with Raman spectroscopy (Renishaw), which indicates the extent of graphitic molecular structures using the relative D-band (∼1350 cm−1) and G-band (∼1580 cm−1) intensities.60,61
Bulk (macroscale) tensile strength (Young's modulus, kg mm−2) was measured with a universal test machine equipped with grips and a 50 N load cell (Lloyds Instruments LF-Plus). For tensile testing, samples were cut into coupons with a dog-bone template (35 mm in length, 4 mm in width) and separated uniaxially at a constant rate of 3.5 mm min−1, following the guidelines established in ASTM D882-18.62
Diffusive processes may govern the limiting current and kinetics in an electrochemical system by restricting ion transport to and from the electrode surface (i.e., mass transfer effects). To quantify mass transfer effects for composite electrodes with different TiO2 loadings and carbonization temperatures (450, 1000 °C), the diffusion coefficient (D) was determined by a data-based method,63 which utilizes information provided by PEIS data as represented by the Randles circuit. The details to and equations for this method are provided in the ESI.† In brief, real impedance (Z′) values from PEIS data in the low-frequency range (<10 Hz), where diffusive processes are relevant, were used to determine the Warburg coefficient (σw) (eqn (S3)–(S5)†). Once σw was obtained, the diffusion coefficient was calculated (eqn (S6)†).
Contaminant transformation experiments
Three different systems were examined for transformation studies: photochemical (PC), electrochemical (EC), and photoelectrochemical (PEC). Before all transformation experiments, staircase cyclic voltammetry (SCV) was conducted with CBZ to probe electron transfer processes and to examine the faradaic current available under experimental conditions. SCV was selected to eliminate any current contribution from capacitive (non-faradaic) behavior of the composites. SCV was obtained from −0.70 to +1.55 V vs. Ag/AgCl with a 50 mV step size every 30 seconds. This potential range was selected to capture the positive currents resulting from CBZ oxidation while maintaining reasonable operating conditions for EC and PEC systems.Before the start of each contaminant transformation experiment, the CNF/TiO2 composite (SAgeo = 11.5 cm2) was placed in 10 μM CBZ in 0.5 M phosphate buffer solution (PBS) at circumneutral pH overnight in the dark to reach sorption equilibrium with CBZ. The initial CBZ concentration for transformation experiments (C0 = 10 μM) was greater than typical concentrations in environmental systems (μg L−1 concentrations or less), but this initial concentration was selected to provide accurate analytical detection for a complete mass balance resulting from each experimental step (sorption, transformation, and extraction). After the end of this sorption period, a constant potential was applied (+0.25 to 1.00 V vs. Ag/AgCl) (Biologic VSP-300 Potentiostat) to composites used for EC and PEC experiments. For PEC experiments, the composite was exposed to UV/visible irradiation using a 1000 W xenon arc lamp (Newport Corporation) with a 225 nm cut-on filter (10CGA-225, Newport; 280 mW cm−2) in addition to the constant applied potential. We note that for PEC and EC experiments at all applied potentials, the average geometric current density (Jgeo) excluded the high capacitive current at potential onset (≤300 s). Finally, for PC transformation experiments, only UV/vis radiation (without a constant potential) was applied to the composite after the initial sorption period.
Aqueous samples were taken periodically over a 90 minute transformation period (700 μL per sample) to quantify the dissolved CBZ concentration via high-performance liquid chromatography (HPLC-DAD, Agilent 1100 Series). Additional HPLC method details are provided in the ESI.† At the end of the 90 minute transformation period, CNF/TiO2 composites were removed from the batch system and placed in organic solvent (methanol or acetonitrile) for 24 hours to extract any CBZ remaining on the composite surface or within the fiber matrix. Both methanol and acetonitrile achieved near-complete recovery in controls. After 24 hours, the extraction solvent was sampled and analyzed via HPLC to determine the amount of CBZ mass that remained bound to the CNF/TiO2 at the end of the 90 minute transformation period. This analytical approach provides a complete mass balance for CBZ in our system, from which the mass of CBZ transformed could be determined: (transformed mass) = (initial aqueous mass) − (final aqueous mass) − (sorbed mass).
As a measure of electrode reactivity toward CBZ, we also estimated the pseudo-first-order rate constant for CBZ transformation in EC and PEC systems. This was accomplished by assuming exponential decay of CBZ over the first 15 minutes of applied potential (+1.00 V vs. Ag/AgCl) with or without light. This shortened transformation period allowed for sufficient degradation of CBZ to reasonably estimate a reaction rate constant while also having sufficient CBZ mass remaining in solution and sorbed to the CNF/TiO2 for reliable quantification.
The stability and sustained performance of the CNF/TiO2 composite were also assessed across five reaction cycles with CBZ (i.e., overnight sorption period followed by EC transformation at +1.00 V vs. Ag/AgCl for 120 minutes). After each transformation period, the solution was discarded, and a new solution volume (C0 = 10 μM CBZ) was added to start the sorption period of the next cycle. After the final transformation period, the composite electrode was removed from solution and placed in organic solvent for 24 hours to extract any CBZ remaining on the electrode surface (as previously described for all other transformation experiments).
Results and discussion
Electrode properties
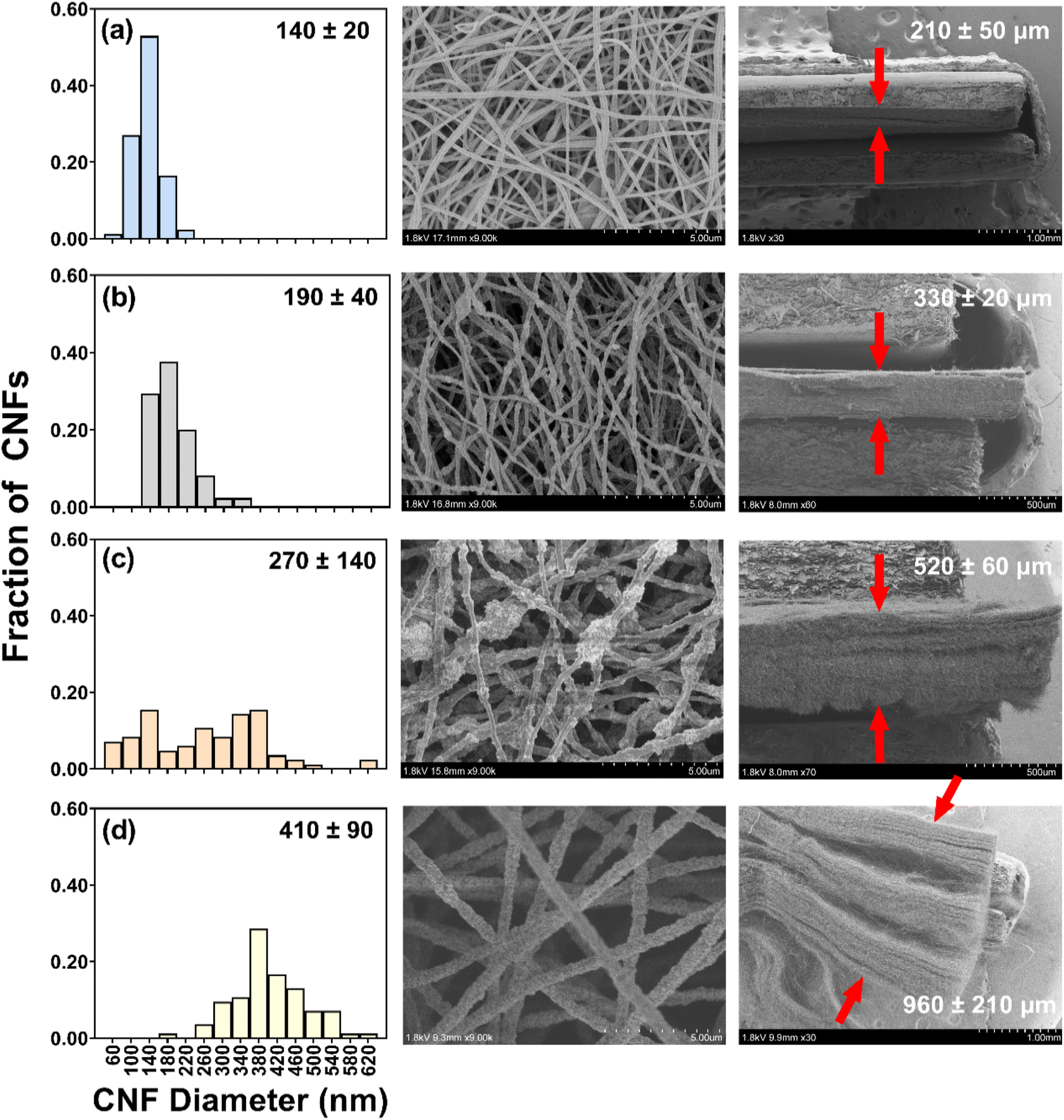 | ||
| Fig. 1 SEM images of CNF/TiO2 composites fabricated with 0.45 g PAN (corresponding to 1.5× sol–gel scaling) and carbonized at 450 °C as a function of TiO2 wt%: (a) 0, (b) 30, (c) 50, and (d) 70. Values in histograms represent mean fiber diameter and standard deviation (n = 85). Values in cross-sectional images represent mean thickness and standard deviation (n = 22). Distance between arrows identifies the electrode thickness as measured. | ||
The SEM images revealed that fiber diameter increased with the addition of TiO2, increasing nearly three-fold from CNFs without TiO2 (140 ± 20 nm) to CNFs with 70 wt% TiO2 (410 ± 90 nm). This increase in diameter is likely due to changes in sol–gel properties in response to increasing TiO2 loading; for example, sol–gel viscosity and conductivity are known to influence the diameter of fibers produced via electrospinning.54,64,65
We note that CNFs with 50 wt% TiO2 exhibited the widest distribution in measured diameters, as evidenced by their considerably larger standard deviation (270 ± 140 nm). We attribute this to some regions along the 50 wt% CNF fibers retaining the diameter of pure CNFs while other areas had TiO2 aggregates that increased diameter. Indeed, the presence of these aggregates was easily discerned in TEM images, shown in Fig. 2 for 0.45 g of PAN/50 wt% TiO2 composite fibers. Despite the presence of these aggregates, SEM-EDS analysis suggests that TiO2 is distributed throughout the fiber matrix (Fig. S7†), thus we do not expect the presence of these aggregates will adversely influence the (photo)electrochemical performance of these materials.
 | ||
| Fig. 2 TEM images of CNF/TiO2 composites with 50 wt% TiO2. | ||
The specific surface area of the CNF/TiO2 composites, as previously determined from N2 adsorption–desorption isotherms,47 increased with TiO2 loading. Composites containing TiO2 at 33, 50, and 80 wt% possessed specific surface areas of 13, 25, and 50 m2 g−1, respectively. This is presumably due to the higher specific surface area of the embedded TiO2 nanoparticles, which become more abundant on the composite surface at higher loadings.
For CNF/TiO2 composites electrospun with the same total sol–gel volume and PAN mass, cross-sectional SEM images revealed a notable increase in the mat thickness with increasing TiO2 loading (210 ± 50 μm without TiO2 to 520 ± 60 μm with 50 wt% TiO2; approximately a 2.5-fold increase; see Fig. 1). This trend in mat thickness can primarily be attributed to the observed increase in individual fiber diameter with increasing TiO2 content, which is expected to influence the packing density of nanofibers within the three-dimensional fiber network. It is also possible that the increasing content of TiO2 introduces more electrostatic interactions between the hydroxylated oxide surfaces, resulting in some charge repulsion between fibers that also increases mat thickness. In fact, the nonwoven CNF mats exhibited less cohesiveness at the highest TiO2 loading considered (70 wt%), where mats separated into distinct layers upon cross-sectioning, and henceforth were not prioritized for extensive electrochemical characterization.
For mats with the same TiO2 content, the thickness of the deposited nanofiber layer also varied with the total sol–gel volume and PAN mass, as anticipated. Cross-sectional images are shown in Fig. S8† for composites with 50 wt% TiO2 for sol–gels containing 0.15, 0.30, and 0.45 g of PAN (corresponding to 1.75, 3.75, and 5.25 mL DMF). Because the surface area of the grounded collector was the same in all cases, the thickness of the composite nanofiber layer increased with PAN mass from 170 (±30 μm) (thinnest) to 520 (±30 μm) (thickest).
As a more quantitative measure of material strength through the carbonization process, macroscale tensile strength testing for 0.45 g PAN/50 wt% TiO2 carbonized at 1000 °C provided Young's modulus of 3.8 (±1.0) MPa. This modulus is within the range of values previously provided from a systematic investigation on the strength of nonwoven PAN-based carbon mats fabricated via electrospinning.69 However, we note that this modulus does not exceed the tensile strength of some commercial carbon fibers.70
Surface area and pore size characterization of the synthesized 0.45 g PAN/50 wt% TiO2 composites carbonized at 450 and 1000 °C was performed using adsorption–desorption N2 isotherms (Fig. S11†). From 450 to 1000 °C, the surface area (SBET) increased by 1.75-fold and total pore volume (Vtot) increased by 2.5-fold. Composites carbonized at 1000 °C exhibit greater cumulative pore volume and more pore volume at larger pore sizes. Fig. S12† exhibits a larger mesopore as observed via SEM for 0.45 g PAN/50 wt% TiO2/1000 °C. A detailed discussion on the pore distribution and the corresponding values (see Table S2) are presented in the ESI.†
XRD confirmed that the anatase crystal structure of TiO2 remained dominant at low carbonization temperatures (≤750 °C), and the rutile phase became dominant after carbonization at 1000 °C (26% anatase and 74% rutile) (Fig. 3a). Although the rutile crystal structure is known to have excellent electron transfer, the anatase crystal structure – which is thermodynamically unstable at higher temperatures – possesses greater photoactivity for applications in photocatalysis and photoelectrochemistry.71 This crystal structure transition near 1000 °C also agrees with previous work demonstrating a similar shift for TiO2 nanopowder processed sequentially under air and in vacuum at different temperatures.72
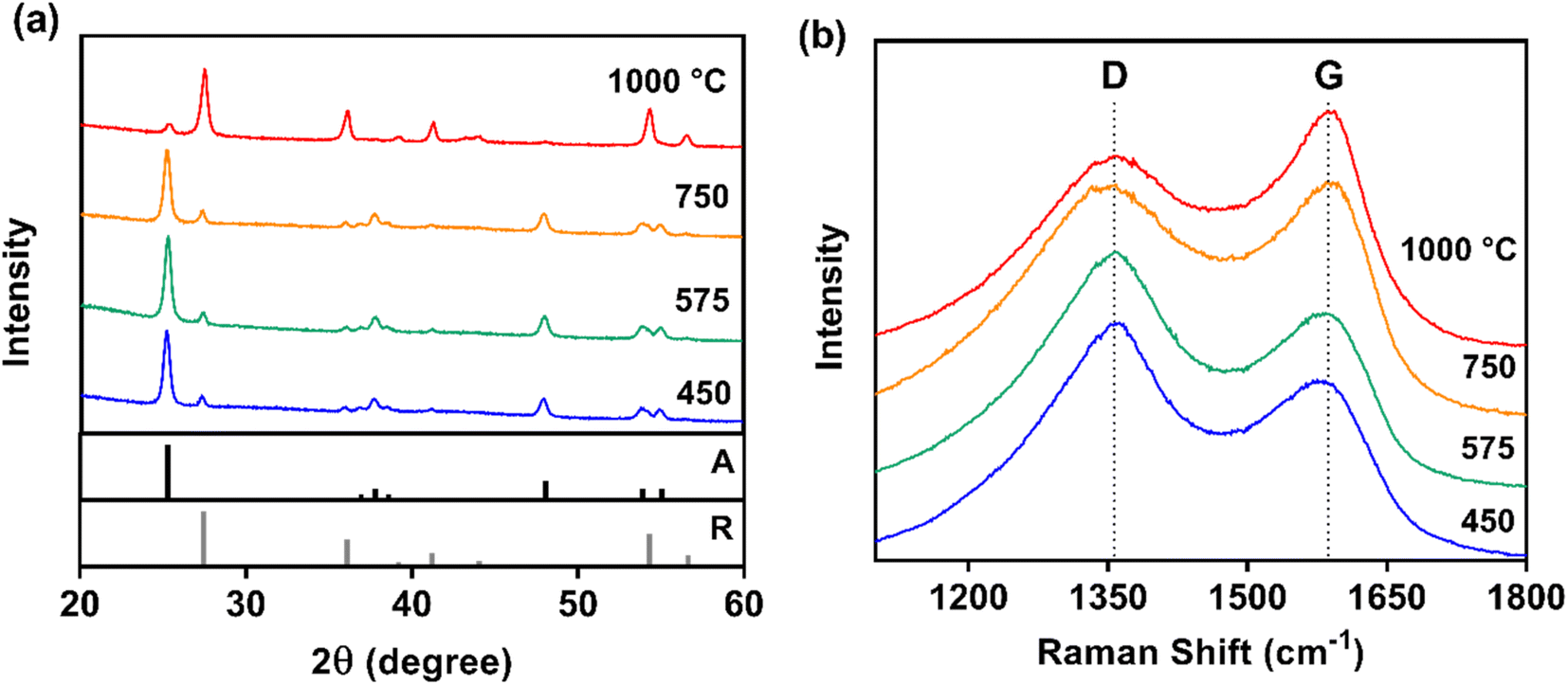 | ||
| Fig. 3 (a) XRD patterns for 0.45 g PAN/50 wt% TiO2 composites as a function of carbonization temperature, with reference diffraction patterns also shown for anatase (A) and rutile (R). (b) Corresponding Raman spectra for these composites, with the amorphous (D-band) and graphitic (G-band) carbon bands indicated by dashed vertical lines. | ||
Because the XRD peaks corresponding to graphite (2θ = 26, 43, and 53°) were overwhelmed by the peaks for TiO2, Raman spectroscopy was used to examine the CNF carbon crystal structure (Fig. 3b), confirming a similar transition with temperature. Graphitic character (R-value) and in-plane crystalline size (La) increased with carbonization temperature (Table S3†). The R-value (graphitic character) increased most notably from 575 to 750 °C, where composites carbonized at 750 °C exhibited markedly different G-band and D-band signatures compared to lower temperatures. Increased G-band intensity indicated ordered (graphitic) carbon became the dominant phase for carbonization temperatures of 750 °C and above. At the highest carbonization temperature (1000 °C), composites had the greatest graphitic character (R-value = 0.81) and the largest in-plane crystalline size (0.54 nm). Graphitic materials with larger in-plane crystallite size are more desirable for electrochemical applications because they exhibit lower electrical resistance as it facilitates carrier hopping between crystallites.61 This trend in carbon structure agrees with the prevailing theory that in-plane crystallite size can be controlled with heat treatment temperature and duration.61
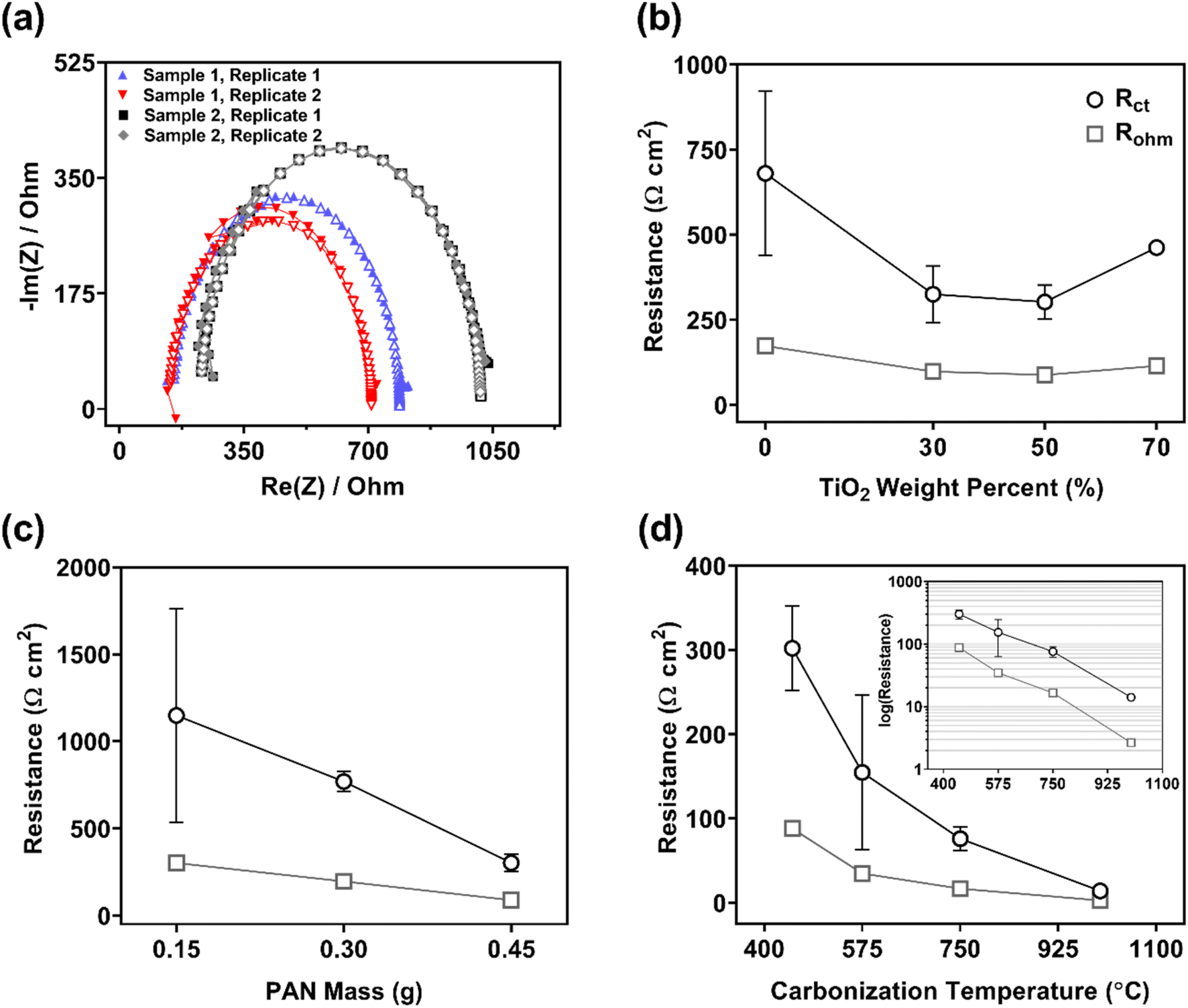 | ||
| Fig. 4 (a) Typical Nyquist plots obtained from PEIS data (▲) for 0.45 g PAN/50 wt% TiO2 composites and corresponding fit (△) by the Simple Randles circuit (see Fig. S5†). The data demonstrate consistency between replicates and batches. (b) Charge transfer resistance (Rct) and ohmic resistance (Rohm) for 0.45 g PAN/TiO2 composites. Standard deviation for 70 wt% composite is small relative to scale. (c) Rct for 50 wt% TiO2 composites with varying PAN mass (0.15, 0.30, and 0.45 g). (d) Rct and Rohm for 0.45 g PAN/50 wt% TiO2 electrodes carbonized at 450, 575, 750, and 1000 °C. PEIS parameters are listed in the ESI.† | ||
An important aspect of this work was demonstrating the reproducible synthesis of composites based on their electrochemical performance metrics. Accordingly, Fig. 4a shows PEIS results obtained for two sample materials of 0.45 g PAN/50 wt% TiO2. Note, in all cases, samples were synthesized from different sol–gels prepared on different days. The agreement in data – across samples and between replicates (i.e., using different sections from the same sample) – demonstrated the ability of our synthesis approach to consistently produce electrode materials with reproducible electrochemical performance. Indeed, in Table S4,† we report average Rct and Rtot values (with standard deviations) from two replicates per sample from samples fabricated over a six-month period. Greater deviation for 0 wt% TiO2 electrodes and for the thinnest electrodes is attributed to their lower material strength. These more fragile electrodes could not be reliably tested because they often broke.
We observed the lowest resistance for 30 and 50 wt% TiO2 composites, with Rct = 330 (±80) and 300 (±50) Ω cm2, respectively. Although composites with 30 and 50 wt% TiO2 exhibited comparable areal resistance, 50 wt% TiO2 composites were preferred over 30 wt% TiO2 composites because they also exhibited less variability in performance between replicates and samples (based on standard deviation of reported performance metrics). Composites with 70 wt% TiO2 demonstrated higher resistance (Rct = 460 ± 10 Ω cm2) than 30 and 50 wt% TiO2, which may be due to the lower structural integrity at this high TiO2 loading (see cross-sectional images in Fig. 1). Ultimately, composites with 50 wt% TiO2 were selected as the optimal composite CNF electrode and used for all subsequent analysis and experiments.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :74%R). Although we anticipate that graphitic character may further develop at temperatures ≥1000 °C, this behavior was not investigated herein because composites are likely to become increasingly brittle and prone to breaking.
:74%R). Although we anticipate that graphitic character may further develop at temperatures ≥1000 °C, this behavior was not investigated herein because composites are likely to become increasingly brittle and prone to breaking.
Contaminant transformation
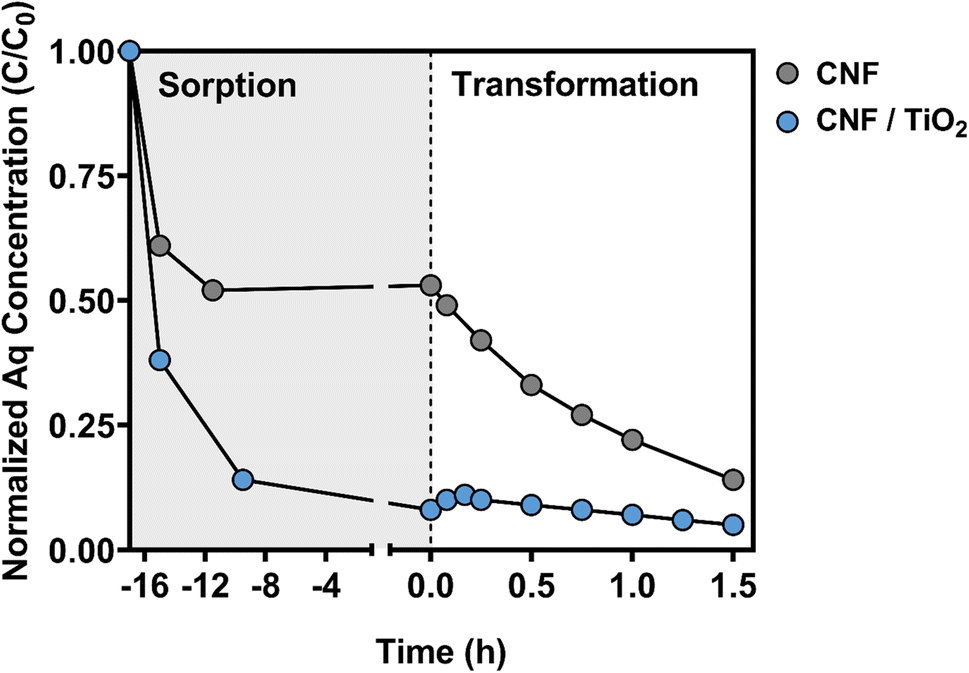 | ||
| Fig. 5 Normalized aqueous (Aq) carbamazepine (CBZ) concentration (C/C0; C0 = 10 μM CBZ) over time during the sorption and electrochemical (EC) transformation periods. CBZ sorption was conducted in the dark, without an applied potential. The EC transformation was conducted at +1.00 V (vs. Ag/AgCl). Data are shown for the optimal composite (0.45 g PAN/50 wt% TiO2/1000 °C) and for CNFs without TiO2 carbonized at 1000 °C. | ||
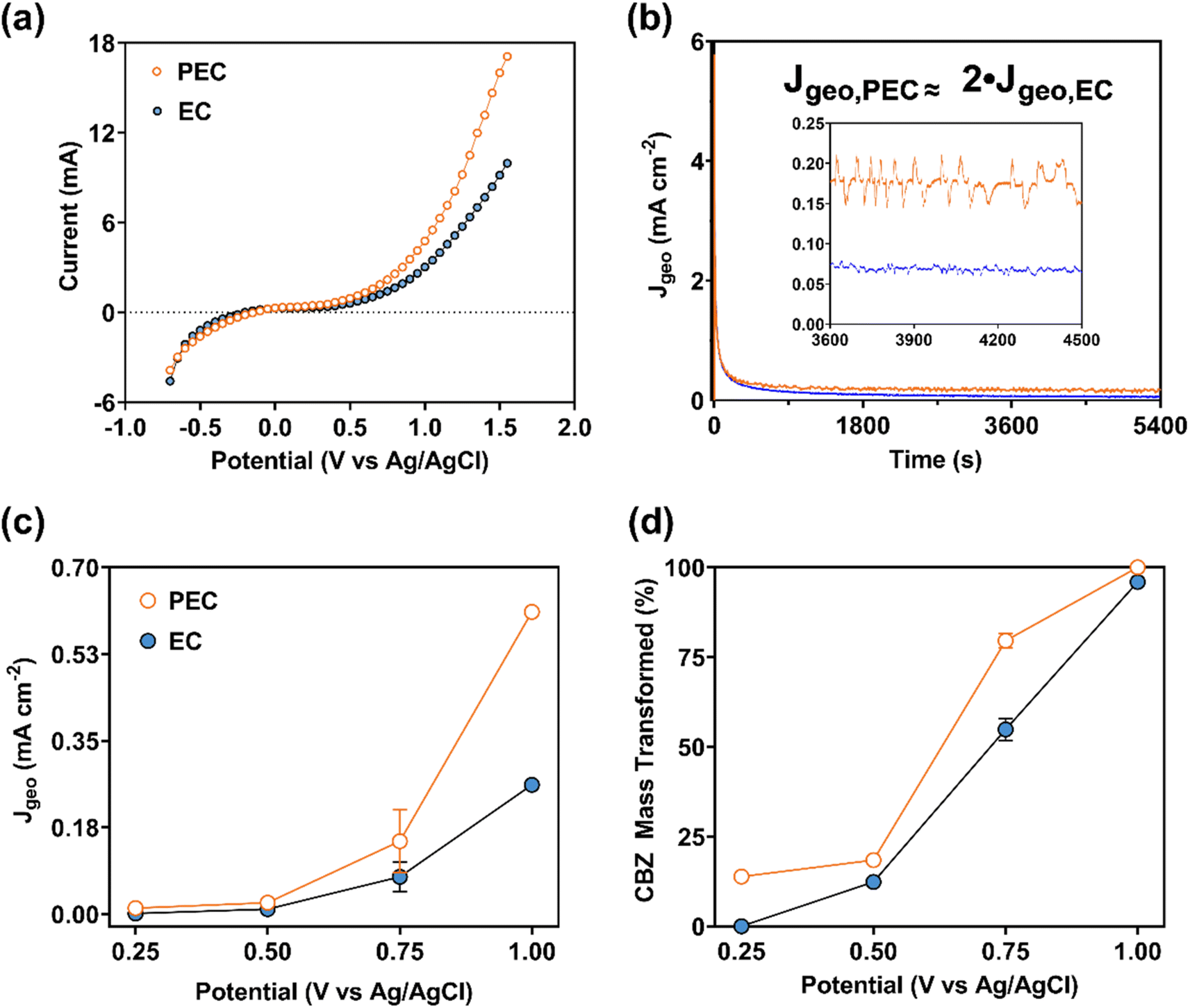 | ||
| Fig. 6 For the optimal (0.45 g PAN/50 wt% TiO2/1000 °C) CNF/TiO2 composite: (a) average faradaic current resulting from staircase cyclic voltammetry (SCV; 50 mV, 30 s, −0.7 to 1.5 V) with and without light; (b) representative current density–time profiles for photoelectrochemical (PEC) and electrochemical (EC) transformation at +0.75 V vs. Ag/AgCl; (c) average faradaic current density during PEC and EC transformation (300–5400 s); and (d) CBZ mass transformed (%) during PEC and EC transformation (0–5400 s). Parameters: C0 = 10 μM CBZ, 0.5 M phosphate buffer at pH 7.4. | ||
In the CNF electrode system, ∼50% of the available CBZ sorbed to the electrode over this initial sorption period (see Fig. 5). This degree of CBZ uptake is far greater than that observed in our previous study using similar composites for photocatalytic water treatment.47 However, CNFs and their TiO2 composites in our previous work were carbonized at a lower temperature (typically 450 °C), and thus would be expected to exhibit less graphitic character than the composites investigated herein. As CBZ is a moderately hydrophobic chemical (with a logarithmic octanol–water partition coefficient of 2.45),78 the greater extent of CBZ sorption observed herein can likely be attributed to the greater hydrophobicity of CNFs generated at higher carbonization temperatures.
During the sorption period, CBZ uptake was faster and occurred to a far greater extent (∼90%) on CNF/TiO2 composites carbonized at 1000 °C relative to CNFs without TiO2 (see Fig. 5). Additional studies with CNF/TiO2 composites confirmed that the extent of CBZ sorption increased with carbonization temperature (Fig. S13†), consistent with expectations from the behavior of CNFs. Compared to the CNF electrode, the increased sorption capacity of the CNF/TiO2 composites is notable because the surface hydroxyl groups of the oxide should produce a more polar surface that hinders hydrophobic partitioning of CBZ. It is likely that the increased CBZ uptake is due in part to the greater degree of surface area available on CNF/TiO2 composites.47 It is also possible that there are other, more specific binding interactions (e.g., H-bonding, van der Waals forces) that are promoted by the surface hydroxyl groups on TiO2 that can also contribute to CBZ uptake. Regardless, at the start of the transformation period, nearly all of the CBZ mass is bound to the electrode for CNF/TiO2 composites, and only a small fraction (C/C0 < 0.1) remains available in the aqueous phase.
During the EC transformation period, evidence suggests that degradation of both aqueous and sorbed CBZ occurs with CNF and CNF/TiO2 electrodes. In the CNF system, which started with roughly equal portions of CBZ in the aqueous and sorbed phase, a near exponential decrease in aqueous CBZ was observed over time upon application of +1.00 V to the electrode. In contrast, slightly different behavior was observed for the aqueous phase concentration of CBZ in the CNF/TiO2 composite system, where CBZ concentration initially, albeit briefly, increased before slowly decaying away over time. We attribute the slight increase to electrostatic repulsion from the positively charged electrode surface in response to the applied potential, which in turn promoted the release of the previously bound CBZ.
At the conclusion of the 90 minute EC transformation period, a small portion of CBZ was detected in methanol extracts of the CNF electrode, indicating that some CBZ remained bound to the electrode at the conclusion of the reaction (approximately 10% of the initial concentration). With roughly 15% of the initial CBZ concentration also remaining in the aqueous phase at the conclusion of the transformation period, evidence suggests that the remaining 75% of CBZ mass was degraded via electrochemical oxidation on the CNFs over the 90 minute period. For CNF/TiO2 electrodes, CBZ was not detected in methanol extracts and only ∼5% remained in aqueous solution, suggesting that there was near-complete degradation of CBZ using the CNF/TiO2 composites. Thus, evidence suggests that CNF/TiO2 composite electrodes were able to achieve a greater extent of CBZ electrochemical oxidation relative to unmodified CNFs over the same transformation period.
During the EC transformation period, the aqueous CBZ concentration varied over time, as in Fig. 5, but its behavior depended on system conditions (i.e., applied potential, light versus dark). More discussion is provided in the ESI,† but there were three general trends in aqueous CBZ over time in these systems: (i) at low applied potential, very low aqueous CBZ concentrations remained essentially constant over the transformation period; (ii) at intermediate applied potentials, aqueous CBZ increased over time consistent with release of sorbed CBZ from the electrode; and (iii) at higher potentials, aqueous CBZ often exhibited an initial, very brief increase before decreasing over time, consistent with degradation via electrochemical oxidation. Notably, aqueous CBZ transformation was more prominent in PEC systems, which exhibited decreases in aqueous concentration over time at lower applied potentials than in EC systems. For example, in PEC systems at +1.00 V, aqueous CBZ was not detected after the first 15 minutes of the transformation period; notably, aqueous CBZ was detectable over the entire EC transformation period at +1.00 V. With light only (no applied potential) for 90 minutes, no CBZ degradation (photolysis) was observed (C/C0 ≈ 1.0).
The electrochemical performance for the CNF/TiO2 electrode during EC and PEC transformation is summarized in Fig. 6. Collectively, results in Fig. 6 are consistent with the (photo)electrochemical transformation of CBZ in both the EC and PEC systems. For example, SCV (Fig. 6a) clearly captured the faradaic current of the CNF/TiO2 composites in the EC system with CBZ, also illustrating the relevant applied potential range where faradaic current increased when the composite was exposed to light in the PEC system. Faradaic current in the PEC system began to deviate from the current in the EC system at approximately +0.25 V; for example, at +1.55 V, the irradiated CNF/TiO2 composite produced a current that was ∼70% greater than in the dark EC system.
Greater activity in PEC systems relative to EC systems is also supported by measurements of the geometric current density (Jgeovs. t), as shown in Fig. 6b for transformation experiments at +0.75 V. Higher current density was consistently observed in PEC systems relative to EC systems (Jgeo,PEC ≈ 2Jgeo,EC), illustrating the ability of UV-light to enhance CBZ transformation at a given potential. PEC current density was greater than EC current density at several of the applied potentials investigated (+0.50, 0.75, and 1.00 V; see Fig. 6c), with the greatest difference observed at +1.00 V. Consistent with SCV results, a minimal difference in PEC and EC current was observed at +0.25 V.
Based on a mass balance of CBZ in experimental systems, we were able to calculate the amount of CBZ transformed (%) at the end of the 90 minute transformation period as a function of applied potential in EC and PEC systems (Fig. 6d). The extent of CBZ transformation increased with applied potential, and for a given potential PEC consistently transformed more total CBZ mass than EC, as anticipated from the greater current density in the PEC system (see Fig. 6c). Near complete CBZ transformation was observed at the conclusion of the 90 minute transformation period at +1.0 V in both EC and PEC systems, but it is likely that all of the CBZ in the PEC system was consumed prior to 90 minutes. Accordingly, we estimated the rate constant (kobs) for the transformation of CBZ mass in EC and PEC systems at +1.0 V by quantifying the mass remaining in both the aqueous and sorbed phase after 15 minutes of reaction. As with other advanced oxidation processes, the CBZ oxidation reaction was assumed as a pseudo-first-order process [i.e., ln(Ct/C0) = −kobst],79 allowing the determination of a single-point kobs value based on the change in total CBZ mass over this 15 minute interval. Based on these estimated kobs values at +1.00 V, the rate of CBZ transformation was roughly 1.5-fold faster in the PEC [kobs(PEC) = 0.18 min−1] system relative to the EC system [kobs(EC) = 0.12 min−1], corresponding to half-lives of 3.8 and 5.8 minutes, respectively. Applying this same approach to the CNF electrode in Fig. 5, we obtained a rate constant for total CBZ transformation of 0.015 min−1 (corresponding to a half-life of roughly 45 minutes), which is roughly one order of magnitude less than the kobs values measured for CNF/TiO2 composites.
The rate constants for CNF/TiO2 composites in EC and PEC systems compare favorably to those previously reported for CBZ transformation with other advanced water treatment processes.79 These rate constants also meet or surpass the rate constants for photocatalysis as obtained with metal-augmented carbon catalysts and electrospun TiO2 fibers.52,53 Moreover, these rate constants also meet or exceed those reported for EC oxidation as obtained at lower supporting electrolyte concentrations with commercial electrodes.80,81 These comparison rates, and corresponding experimental parameters, are presented in Table S6.† Unlike the CNF/TiO2 composites presented herein, other nanostructured TiO2 electrodes exhibited little to no CBZ degradation for EC conditions (bias only) in the presence of real secondary effluent wastewater spiked with CBZ (10 μg L−1).82
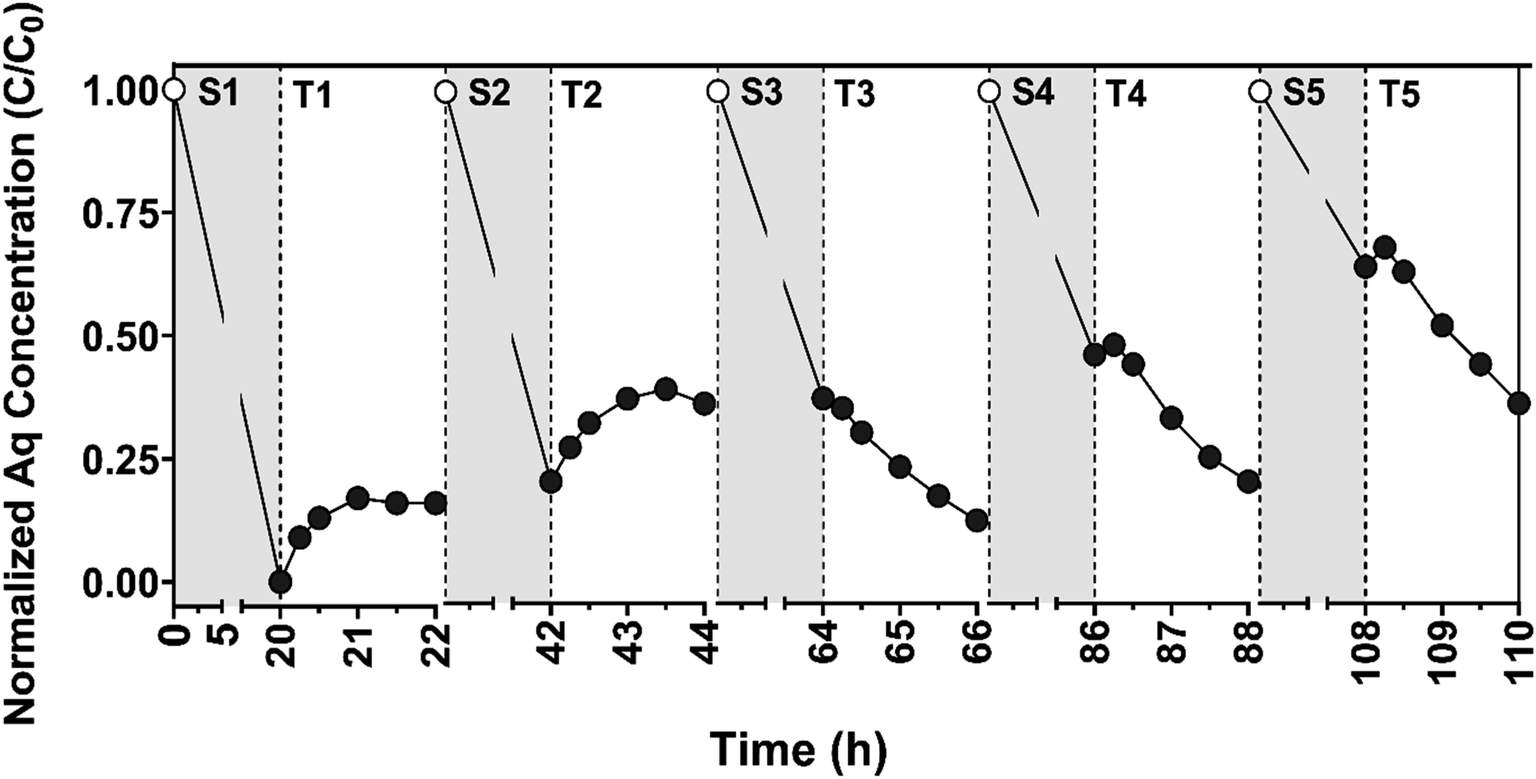 | ||
| Fig. 7 Normalized aqueous (Aq) CBZ concentration (C/C0; C0 = 10 μM CBZ) observed during the sorption (gray) and transformation (white) periods during cycling experiments to test the long-term stability and reactivity of CNF/TiO2 composites (0.45 g PAN/50 wt% TiO2/1000 °C) across five cycles. Electrochemical (EC) transformation occurred at +1.0 V vs. Ag/AgCl. Lines do not represent model fits and are only shown to help illustrate trends in data. | ||
Despite the loss in sorption capacity, it does not appear that the electrochemical performance of the electrode was diminished over the five reaction cycles. In fact, measured current densities (Jgeo) were almost constant across all trials, with an average value of 0.25 (±0.03) mA cm−2 across the 5 cycles. The trends in aqueous CBZ concentration over time during the transformation period, particularly in later cycles, also suggest sustained electrode performance. In the first two cycles, CBZ was released from the surface upon the application of +1.00 V, as was observed in Fig. 5 and S14† for EC systems and suggesting that most of the CBZ transformation during these cycles resulted from oxidation of CBZ previously sorbed to the electrode surface. In later cycles (three through five), however, when electrode sorption capacity decreased, the near-equivalent rate of aqueous CBZ decay over time (corresponding to a kobs of ∼0.008 min−1 in each cycle, assuming exponential decay), suggests a comparable transformation of CBZ for these systems.
Based on the results of these cycling experiments, we conceptualize the CNF/TiO2 composite electrodes as having at least two distinct surface sites. One surface site (or family of sites) is primarily responsible for sorption of CBZ, and likely consists primarily of the graphitic carbon generated during carbonization. A second surface site (or family of sites) appears primarily responsible for charge transfer, and these presumably consist of the embedded TiO2 surfaces. We propose distinct sites for sorption and charge transfer because sorption capacity markedly decreased across the five reaction cycles while current density, a measure of charge transfer, was effectively unchanged. Because the majority of CBZ in the system is sorbed to the electrode during early transformation cycles, surface-bound CBZ must be loosely held and able to migrate between sorption and charge transfer sites to allow for the extent of transformation that we observed. Accordingly, and because CBZ was not detected in extracts of the electrode at the conclusion of the five reaction cycles, we speculate that some CBZ transformation products must be more irreversibly bound to sorption sites such that they are occupied and no longer available for CBZ uptake. We emphasize that the loss of sorption capacity does not appear to be detrimental to electrode performance during applications in water treatment; in the later cycles, based off the decay in CBZ concentration over time in aqueous solution, the electrode continued to achieve rate constants for CBZ degradation that are comparable to those reported for some UV-based treatment of CBZ.79
Conclusion
Herein, we demonstrate that electrospun CNF/TiO2 composites, when optimized for their physical and (photo)chemical properties, can function as promising (photo)electrodes for advanced water treatment applications. Electrospinning provides a reproducible, single-batch synthesis approach for producing electroactive nanomaterial composites with tunable performance properties. Using this synthesis approach, we were able to enhance the performance of standard CNFs by easily integrating photoactivity (through the addition of TiO2 nanoparticles) and tailoring conductivity (through carbonization temperature) without need for more extensive material post-processing or surface modification (e.g., doping, etching, physical vapor deposition). CNF/TiO2 composites produced via electrospinning hold the advantages of easily controlled TiO2 loading, bulk chemical structure, and diffusivity (which is important for applications targeting contaminants at low environmental concentrations), while also being able to maintain high surface area and mechanical integrity after heat treatment. Detailed structural and electrochemical characterizations found that the optimized CNF/TiO2 (photo)electrodes exhibited high surface area (50 m2 g−1) and low geometric (areal) resistance (2.66 Ω cm2). Notably, we also demonstrate that these CNF/TiO2 composites can be successfully applied as a free-standing, binder-free electrode to transform a recalcitrant water contaminant (CBZ), which may enable the use of such systems for renewably driven water treatment. Indeed, a benefit of this composite electrode material is its ability to outperform more traditional CNF electrodes when used in either electrochemical or photoelectrochemical treatment configurations.There are several other practical advantages of these composite (photo)electrodes relative to other commercially available electrodes and electrode synthesis approaches. In producing these materials, we avoided the use of expensive, unsustainable, and/or rare materials, such as noble metals that are commonly integrated via nanoparticles, coatings, or thin films to augment electrode performance. Our electrodes add to the existing body of research demonstrating that nanoparticles (e.g., TiO2) commonly used in dispersions that are difficult to apply at water treatment scale can maintain performance in EC and PEC systems when immobilized into conductive platforms. Further, as with commercial carbon electrodes, the CNF (photo)electrode material can be fabricated with sufficient strength to withstand routine handling (e.g., bend, twist; see Fig. S9†) and versatile deployment across a range of reactor configurations.
There remain several opportunities to further validate and improve upon the performance of electrode materials. From the material performance demonstrated here, an EC or PEC system equipped with the CNF (photo)electrodes would be well-suited as a final polishing step for drinking water to remove micropollutant mixtures, where competition and fouling from other dissolved co-solutes would be minimal. Moreover, electrode performance can likely be improved further if used as an electroactive membrane in a flow-through system designed to alleviate mass transfer limitations; our previous efforts with the photoactive CNF/TiO2 composites demonstrated the ability to remove the majority of recalcitrant organic micropollutants in a single pass during dead end filtration at fluxes comparable to that required for microfiltration (540 L m−2 h−1).47 While promising, better removals efficiencies toward a wider range of contaminants should be achievable using an analogous photoelectroactive membrane. Therefore, future work will need to assess the performance of CNF/TiO2 composites in reactive filtration systems at comparable fluxes under EC and PEC conditions and toward a broader suite of recalcitrant organic micropollutants. Using such a configuration, it may also be possible to alleviate the loss of electrode sorption capacity over time (see Fig. 7) through periodic regeneration or washing steps (e.g., with acid, base or an organic solvent).
Conflicts of interest
There are no conflicts to declare.References
- J. Radjenovic and D. L. Sedlak, Challenges and Opportunities for Electrochemical Processes as Next-Generation Technologies for the Treatment of Contaminated Water, Environ. Sci. Technol., 2015, 49, 11292–11302 CrossRef CAS PubMed.
- B. P. Chaplin, The Prospect of Electrochemical Technologies Advancing Worldwide Water Treatment, Acc. Chem. Res., 2019, 52, 596–604 CrossRef CAS PubMed.
- S. Garcia-Segura, X. Qu, P. J. J. Alvarez, B. P. Chaplin, W. Chen, J. C. Crittenden, Y. Feng, G. Gao, Z. He, C.-H. Hou, X. Hu, G. Jiang, J.-H. Kim, J. Li, Q. Li, J. Ma, J. Ma, A. B. Nienhauser, J. Niu, B. Pan, X. Quan, F. Ronzani, D. Villagran, T. D. Waite, W. S. Walker, C. Wang, M. S. Wong and P. Westerhoff, Opportunities for nanotechnology to enhance electrochemical treatment of pollutants in potable water and industrial wastewater – a perspective, Environ. Sci.: Nano, 2020, 7, 2178–2194 RSC.
- N. Singh and B. R. Goldsmith, Role of Electrocatalysis in the Remediation of Water Pollutants, ACS Catal., 2020, 10, 3365–3371 CrossRef CAS.
- C. A. Martínez-Huitle and M. Panizza, Electrochemical oxidation of organic pollutants for wastewater treatment, Curr. Opin. Electrochem., 2018, 11, 62–71 CrossRef.
- C. A. Martínez-Huitle and E. Brillas, Decontamination of wastewaters containing synthetic organic dyes by electrochemical methods: A general review, Appl. Catal., B, 2009, 87, 105–145 CrossRef.
- Z. Zheng and I. M. C. Lo, Multifunctional photoelectrochemical systems for coupled water treatment and high-value product generation: current status, mechanisms, remaining challenges, and future opportunities, Curr. Opin. Electrochem., 2021, 34, 100711 CrossRef.
- S. Chang, Q. Wang, B. Liu, Y. Sang and H. Liu, Hierarchical TiO2 nanonetwork–porous Ti 3D hybrid photocatalysts for continuous-flow photoelectrodegradation of organic pollutants, Catal. Sci. Technol., 2017, 7, 524–532 RSC.
- R. Song, H. Chi, Q. Ma, D. Li, X. Wang, W. Gao, H. Wang, X. Wang, Z. Li and C. Li, Highly Efficient Degradation of Persistent Pollutants with 3D Nanocone TiO2-Based Photoelectrocatalysis, J. Am. Chem. Soc., 2021, 143, 13664–13674 CrossRef CAS PubMed.
- S. Xiang, Z. Zhang, Z. Wu, L. Sun, P. Radjenovic, H. Ren, C. Lin, Z. Tian and J. Li, 3D Heterostructured Ti-Based Bi2MoO6/Pd/TiO2 Photocatalysts for High-Efficiency Solar Light Driven Photoelectrocatalytic Hydrogen Generation, ACS Appl. Energy Mater., 2019, 2, 558–568 CrossRef CAS.
- G. Wang, X. Xiao, W. Li, Z. Lin, Z. Zhao, C. Chen, C. Wang, Y. Li, X. Huang, L. Miao, C. Jiang, Y. Huang and X. Duan, Significantly Enhanced Visible Light Photoelectrochemical Activity in TiO2 Nanowire Arrays by Nitrogen Implantation, Nano Lett., 2015, 15, 4692–4698 CrossRef CAS PubMed.
- M. S. Koo, K. Cho, J. Yoon and W. Choi, Photoelectrochemical Degradation of Organic Compounds Coupled with Molecular Hydrogen Generation Using Electrochromic TiO2 Nanotube Arrays, Environ. Sci. Technol., 2017, 51, 6590–6598 CrossRef CAS PubMed.
- G. M. Peleyeju, E. H. Umukoro, J. O. Babalola and O. A. Arotiba, Solar-Light-Responsive Titanium-Sheet-Based Carbon Nanoparticles/B-BiVO4/WO3 Photoanode for the Photoelectrocatalytic Degradation of Orange II Dye Water Pollutant, ACS Omega, 2020, 5, 4743–4750 CrossRef CAS PubMed.
- C. Liu, A.-Y. Zhang, Y. Si, D.-N. Pei and H.-Q. Yu, Photochemical Protection of Reactive Sites on Defective TiO2–x Surface for Electrochemical Water Treatment, Environ. Sci. Technol., 2019, 53, 7641–7652 CrossRef CAS PubMed.
- L. Barrera and R. Bala Chandran, Harnessing Photoelectrochemistry for Wastewater Nitrate Treatment Coupled with Resource Recovery, ACS Sustainable Chem. Eng., 2021, 9, 3688–3701 CrossRef CAS.
- J.-S. Yang, W. W.-P. Lai, S. C. Panchangam and A. Y.-C. Lin, Photoelectrochemical degradation of perfluorooctanoic acid (PFOA) with GOP25/FTO anodes: Intermediates and reaction pathways, J. Hazard. Mater., 2020, 391, 122247 CrossRef CAS PubMed.
- C. Wang, M. Sun, Y. Zhao, M. Huo, X. Wang and M. Elimelech, Photo-electrochemical Osmotic System Enables Simultaneous Metal Recovery and Electricity Generation from Wastewater, Environ. Sci. Technol., 2021, 55, 604–613 CrossRef CAS PubMed.
- D.-H. Nam, D. Lee and K.-S. Choi, Electrochemical and photoelectrochemical approaches for the selective removal, recovery, and valorization of chloride ions, Chem. Eng. J., 2021, 404, 126378 CrossRef CAS.
- K. Kinoshita and S. C. Leach, Mass-Transfer Study of Carbon Felt, Flow-Through Electrode, J. Electrochem. Soc., 1993, 129, 6 Search PubMed.
- X. You, Q. Ye and P. Cheng, The Dependence of Mass Transfer Coefficient on the Electrolyte Velocity in Carbon Felt Electrodes: Determination and Validation, J. Electrochem. Soc., 2017, 164, E3386–E3394 CrossRef CAS.
- B. P. Chaplin, Critical review of electrochemical advanced oxidation processes for water treatment applications, Environ. Sci.: Processes Impacts, 2014, 16, 1182–1203 RSC.
- R. McCreery, Advanced Carbon Electrode Materials for Molecular Electrochemistry, Chem. Rev., 2008, 108, 41 CrossRef PubMed.
- X. Chen, G. Chen and P. L. Yue, Anodic oxidation of dyes at novel Ti/B-diamond electrodes, Chem. Eng. Sci., 2003, 58, 995–1001 CrossRef CAS.
- M. Qiao, Y. Zhang, L. Zhai and M. Sun, Corrosion of graphite electrode in electrochemical advanced oxidation processes: Degradation protocol and environmental implication, Chem. Eng. J., 2018, 344, 8 CrossRef.
- M. Sun, X. Wang, L. R. Winter, Y. Zhao, W. Ma, T. Hedtke, J.-H. Kim and M. Elimelech, Electrified Membranes for Water Treatment Applications, ACS ES&T Engg, 2021, 1, 725–752 Search PubMed.
- X. Zhu and D. Jassby, Electroactive Membranes for Water Treatment: Enhanced Treatment Functionalities, Energy Considerations, and Future Challenges, Acc. Chem. Res., 2019, 52, 1177–1186 CrossRef CAS PubMed.
- M. Shestakova and M. Sillanpää, Electrode materials used for electrochemical oxidation of organic compounds in wastewater, Rev. Environ. Sci. Bio/Technol., 2017, 16, 223–238 CrossRef CAS.
- M. J. Nalbandian, S. Kim, H. E. Gonzalez-Ribot, N. V. Myung and D. M. Cwiertny, Recent advances and remaining barriers to the development of electrospun nanofiber and nanofiber composites for point-of-use and point-of-entry water treatment systems, J. Hazard. Mater. Adv., 2022, 8, 100204 CrossRef CAS PubMed.
- W. Wu, Z.-H. Huang and T.-T. Lim, Recent development of mixed metal oxide anodes for electrochemical oxidation of organic pollutants in water, Appl. Catal., A, 2014, 480, 58–78 CrossRef CAS.
- H. Wu, H. L. Tan, C. Y. Toe, J. Scott, L. Wang, R. Amal and Y. H. Ng, Photocatalytic and Photoelectrochemical Systems: Similarities and Differences, Adv. Mater., 2020, 32, 1904717 CrossRef CAS PubMed.
- Y. Han, S. Zhang, H. Zhao, W. Wen, H. Zhang, H. Wang and F. Peng, Photoelectrochemical Characterization of a Robust TiO2/BDD Heterojunction Electrode for Sensing Application in Aqueous Solutions, Langmuir, 2010, 26, 6033–6040 CrossRef CAS PubMed.
- Y. Wang, M. Zu, X. Zhou, H. Lin, F. Peng and S. Zhang, Designing efficient TiO2-based photoelectrocatalysis systems for chemical engineering and sensing, Chem. Eng. J., 2020, 381, 122605 CrossRef CAS.
- Y.-n. Zhang, Q. Niu, X. Gu, N. Yang and G. Zhao, Recent progress on carbon nanomaterials for the electrochemical detection and removal of environmental pollutants, Nanoscale, 2019, 11, 11992–12014 RSC.
- W. Zhang, S. Zhu, R. Luque, S. Han, L. Hu and G. Xu, Recent development of carbon electrode materials and their bioanalytical and environmental applications, Chem. Soc. Rev., 2016, 45, 715–752 RSC.
- S. Peng, L. Li, J. Kong Yoong Lee, L. Tian, M. Srinivasan, S. Adams and S. Ramakrishna, Electrospun carbon nanofibers and their hybrid composites as advanced materials for energy conversion and storage, Nano Energy, 2016, 22, 361–395 CrossRef CAS.
- L.-F. Chen, Y. Feng, H.-W. Liang, Z.-Y. Wu and S.-H. Yu, Macroscopic-Scale Three-Dimensional Carbon Nanofiber Architectures for Electrochemical Energy Storage Devices, Adv. Energy Mater., 2017, 7, 1700826 CrossRef.
- R. Stirling, W. S. Walker, P. Westerhoff and S. Garcia-Segura, Techno-economic analysis to identify key innovations required for electrochemical oxidation as point-of-use treatment systems, Electrochim. Acta, 2020, 338, 135874 CrossRef CAS.
- G. Song, M. Zhou, X. Du, P. Su and J. Guo, Mechanistic Insight into the Heterogeneous Electro-Fenton/Sulfite Process for Ultraefficient Degradation of Pollutants over a Wide pH Range, ACS ES&T Water, 2021, 1, 1637–1647 Search PubMed.
- K. Deng, Y. Gu, T. Gao, Z. Liao, Y. Feng, S. Zhou, Q. Fang, C. Hu and L. Lyu, Carbonized MOF-Coated Zero-Valent Cu Driving an Efficient Dual-Reaction-Center Fenton-like Water Treatment Process through Utilizing Pollutants and Natural Dissolved Oxygen, ACS ES&T Water, 2022, 2, 174–183 Search PubMed.
- S. O. Ganiyu, M. Zhou and C. A. Martínez-Huitle, Heterogeneous electro-Fenton and photoelectro-Fenton processes: A critical review of fundamental principles and application for water/wastewater treatment, Appl. Catal., B, 2018, 235, 103–129 CrossRef CAS.
- S. K. Loeb, P. J. J. Alvarez, J. A. Brame, E. L. Cates, W. Choi, J. Crittenden, D. D. Dionysiou, Q. Li, G. Li-Puma, X. Quan, D. L. Sedlak, T. David Waite, P. Westerhoff and J. H. Kim, The Technology Horizon for Photocatalytic Water Treatment: Sunrise or Sunset?, Environ. Sci. Technol., 2019, 53, 2937–2947 CrossRef CAS PubMed.
- M. A. Lazar, S. Varghese and S. S. Nair, Photocatalytic Water Treatment by Titanium Dioxide: Recent Updates, Catalysts, 2012, 2, 572–601 CrossRef CAS.
- S. Franz, E. Falletta, H. Arab, S. Murgolo, M. Bestetti and G. Mascolo, Degradation of Carbamazepine by Photo(electro)catalysis on Nanostructured TiO2 Meshes: Transformation Products and Reaction Pathways, Catalysts, 2020, 10, 169 CrossRef CAS.
- J. Matos, S. Miralles-Cuevas, A. Ruíz-Delgado, I. Oller and S. Malato, Development of TiO2-C photocatalysts for solar treatment of polluted water, Carbon, 2017, 122, 361–373 CrossRef CAS.
- P. Aghasiloo, M. Yousefzadeh, M. Latifi and R. Jose, Highly porous TiO2 nanofibers by humid-electrospinning with enhanced photocatalytic properties, J. Alloys Compd., 2019, 790, 257–265 CrossRef CAS.
- S. Kim, G. Piao, D. S. Han, H. K. Shon and H. Park, Solar desalination coupled with water remediation and molecular hydrogen production: a novel solar water-energy nexus, Energy Environ. Sci., 2018, 11, 344–353 RSC.
- K. E. Greenstein, M. R. Nagorzanski, B. Kelsay, E. M. Verdugo, N. V. Myung, G. F. Parkin and D. M. Cwiertny, Carbon–titanium dioxide (C/TiO2) nanofiber composites for chemical oxidation of emerging organic contaminants in reactive filtration applications, Environ. Sci.: Nano, 2021, 8, 711–722 RSC.
- R. Montenegro-Ayo, J. C. Morales-Gomero, H. Alarcon, A. Corzo, P. Westerhoff and S. Garcia-Segura, Photoelectrocatalytic degradation of 2,4-dichlorophenol in a TiO2 nanotube-coated disc flow reactor, Chemosphere, 2021, 268, 129320 CrossRef CAS PubMed.
- B. Zhang, F. Yang, H. Liu, L. Yan, W. Yang, C. Xu, S. Huang, Q. Li, W. Bao, B. Liu and Y. Li, Assembling Graphene-Encapsulated Pd/TiO2 Nanosphere with Hierarchical Architecture for High-Performance Visible-Light-Assisted Methanol Electro-Oxidation Material, Ind. Eng. Chem. Res., 2019, 58, 19486–19494 CrossRef CAS.
- L. Persano, A. Camposeo, C. Tekmen and D. Pisignano, Industrial Upscaling of Electrospinning and Applications of Polymer Nanofibers: A Review, Macromol. Mater. Eng., 2013, 298, 504–520 CrossRef CAS.
- T. Kosjek, H. R. Andersen, B. Kompare, A. Ledin and E. Heath, Fate of Carbamazepine during Water Treatment, Environ. Sci. Technol., 2009, 43, 6 CrossRef PubMed.
- G. Chen, W. Dong, H. Wang, Z. Zhao, F. Wang, F. Wang and C. Nieto-Delgado, Carbamazepine degradation by visible-light-driven photocatalyst Ag3PO4/GO: Mechanism and pathway, Environ. Sci. Ecotechnology, 2022, 9, 100143 CrossRef CAS PubMed.
- S. K. Maeng, K. Cho, B. Jeong, J. Lee, Y. Lee, C. Lee, K. J. Choi and S. W. Hong, Substrate-immobilized electrospun TiO2 nanofibers for photocatalytic degradation of pharmaceuticals: The effects of pH and dissolved organic matter characteristics, Water Res., 2015, 86, 25–34 CrossRef CAS PubMed.
- K. T. Peter, J. D. Vargo, T. P. Rupasinghe, A. De Jesus, A. V. Tivanski, E. A. Sander, N. V. Myung and D. M. Cwiertny, Synthesis, Optimization, and Performance Demonstration of Electrospun Carbon Nanofiber-Carbon Nanotube Composite Sorbents for Point-of-Use Water Treatment, ACS Appl. Mater. Interfaces, 2016, 8, 11431–11440 CrossRef CAS PubMed.
- Y. Zhang, N. Tajaddod, K. Song and M. L. Minus, Low temperature graphitization of interphase polyacrylonitrile (PAN), Carbon, 2015, 91, 479–493 CrossRef CAS.
- M. Inagaki, Y. Yang and F. Kang, Carbon nanofibers prepared via electrospinning, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, A review of heat treatment on polyacrylonitrile fiber, Polym. Degrad. Stab., 2007, 92, 1421–1432 CrossRef CAS.
- D. Edie, The effect of processing on the structure and properties of carbon fibers, Carbon, 1998, 36, 18 CrossRef.
- N. Yusof and A. F. Ismail, Post spinning and pyrolysis of processes of polyacrylonitrile (PAN)-based carbon fiber and activated carbon fiber: A review, J. Anal. Appl. Pyrolysis, 2012, 93, 13 CrossRef.
- F. Tuinstra and J. L. Koenig, Raman Spectrum of Graphite, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Studying disorder in graphite-based systems by Raman spectroscopy, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- Standard Test Method for Tensile Properties of Thin Plastic Sheeting ASTM International, 2018, ASTM D882-18.
- T. Q. Nguyen and C. Breitkopf, Determination of Diffusion Coefficients Using Impedance Spectroscopy Data, J. Electrochem. Soc., 2018, 165, E826–E831 CrossRef CAS.
- J. M. Deitzel, J. Kleinmeyer, D. Harris and N. C. Beck Tan, The effect of processing variables on the morphology of electrospun nanofibers and textiles, Polymer, 2001, 42, 261–272 CrossRef CAS.
- B. Zhang, F. Kang, J.-M. Tarascon and J.-K. Kim, Recent advances in electrospun carbon nanofibers and their application in electrochemical energy storage, Prog. Mater. Sci., 2016, 76, 319–380 CrossRef CAS.
- H. Kakida and K. Tashiro, Mechanism and Kinetics of Stabilization Reaction of Polyacrylonitrile and Related Copolymers II. Relationships between Iosthermal DSC Thermograms and FT-IR Spectral Changes of Polyacrylonitrile in Comparison with the Case of Acrylonitrile/Methacrylic Acid Copolymer, Polym. J., 1997, 29, 5 Search PubMed.
- A. Oberlin, Carbonization and Graphitization, Carbon, 1984, 22, 21 Search PubMed.
- Phthalic acid and its isomers (isophthalic acid and terephthalic acid) [MAK Value Documentation, 2009], in The MAK-Collection for Occupational Health and Safety, 2012, DOI:10.1002/3527600418.mb8899isme0025.
- B. I. Waisi, S. S. Manickam, N. E. Benes, A. Nijmeijer and J. R. McCutcheon, Activated Carbon Nanofiber Nonwovens: Improving Strength and Surface Area by Tuning Fabrication Procedure, Ind. Eng. Chem. Res., 2019, 58, 4084–4089 CrossRef CAS.
- E. Frank, L. M. Steudle, D. Ingildeev, J. M. Spörl and M. R. Buchmeiser, Carbon Fibers: Precursor Systems, Processing, Structure, and Properties, Angew. Chem., Int. Ed., 2014, 53, 5262–5298 CrossRef CAS PubMed.
- R. I. Bickley, T. Gonzalez-Carreno, T. S. Lees, L. Palmisano and R. J. Tilley, Structural Investigation of Titanium Dioxide Photocatalysts, J. Solid State Chem., 1991, 92, 178–190 CrossRef CAS.
- G. Cerrato, L. Marchese and C. Morterra, Structural and morphological modifications of sintering microcrystalline TiO2: an XRD, HRTEM and FTIR study, Appl. Surf. Sci., 1993, 70/71, 200–205 CrossRef.
- A. Elmouwahidi, E. Bailón-García, J. Castelo-Quibén, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar and F. Carrasco-Marín, Carbon–TiO2 composites as high-performance supercapacitor electrodes: synergistic effect between carbon and metal oxide phases, J. Mater. Chem. A, 2018, 6, 633–644 RSC.
- H. Lu, C. Yang, H. Bao, L. Wang, C. Li and H. Wang, Unusual Improvement of Pseudocapacitance of Nanocomposite Electrodes: Three-Dimensional Amorphous Carbon Frameworks Triggered by TiO2 Nanocrystals, ACS Appl. Mater. Interfaces, 2019, 11, 48039–48053 CrossRef CAS PubMed.
- H. Lin, X. Ji, Q. Chen, Y. Zhou, C. E. Banks and K. Wu, Mesoporous-TiO2 nanoparticles based carbon paste electrodes exhibit enhanced electrochemical sensitivity for phenols, Electrochem. Commun., 2009, 11, 1990–1995 CrossRef CAS.
- L.-C. Jiang and W.-D. Zhang, Electrodeposition of TiO2 Nanoparticles on Multiwalled Carbon Nanotube Arrays for Hydrogen Peroxide Sensing, Electroanalysis, 2009, 21, 988–993 CrossRef CAS.
- A. Ramadoss and S. J. Kim, Improved activity of a graphene–TiO2 hybrid electrode in an electrochemical supercapacitor, Carbon, 2013, 63, 434–445 CrossRef CAS.
- P. Westerhoff, Y. Yoon, S. Snyder and E. Wert, Fate of Endocrine-Disruptor, Pharmaceutical, and Personal Care Product Chemicals during Simulated Drinking Water Treatment Processes, Environ. Sci. Technol., 2005, 39, 6649–6663 CrossRef CAS PubMed.
- C.-m. Dai, X.-f. Zhou, Y.-l. Zhang, Y.-p. Duan, Z.-m. Qiang and T. C. Zhang, Comparative study of the degradation of carbamazepine in water by advanced oxidation processes, Environ. Technol., 2012, 33, 1101–1109 CrossRef CAS PubMed.
- J. D. García-Espinoza, P. Mijaylova-Nacheva and M. Avilés-Flores, Electrochemical carbamazepine degradation: Effect of the generated active chlorine, transformation pathways and toxicity, Chemosphere, 2018, 192, 142–151 CrossRef PubMed.
- C. García-Gómez, P. Drogui, F. Zaviska, B. Seyhi, P. Gortáres-Moroyoqui, G. Buelna, C. Neira-Sáenz, M. Estrada-alvarado and R. G. Ulloa-Mercado, Experimental design methodology applied to electrochemical oxidation of carbamazepine using Ti/PbO2 and Ti/BDD electrodes, J. Electroanal. Chem., 2014, 732, 1–10 CrossRef.
- S. Murgolo, S. Franz, H. Arab, M. Bestetti, E. Falletta and G. Mascolo, Degradation of emerging organic pollutants in wastewater effluents by electrochemical photocatalysis on nanostructured TiO2 meshes, Water Res., 2019, 164, 114920 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00017f |
| This journal is © The Royal Society of Chemistry 2023 |