
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rigidly linked dinuclear platinum(II) complexes showing intense, excimer-like, near-infrared luminescence†
Piotr
Pander
 *abc,
Melissa T.
Walden
d,
Rebecca J.
Salthouse
d,
Amit
Sil
d,
Dmitry S.
Yufit
d,
Fernando B.
Dias
*c and
J. A. Gareth
Williams
*d
*abc,
Melissa T.
Walden
d,
Rebecca J.
Salthouse
d,
Amit
Sil
d,
Dmitry S.
Yufit
d,
Fernando B.
Dias
*c and
J. A. Gareth
Williams
*d
aFaculty of Chemistry, Silesian University of Technology, Strzody 9, 44-100 Gliwice, Poland
bCentre for Organic and Nanohybrid Electronics, Silesian University of Technology, Konarskiego 22B, 44-100 Gliwice, Poland
cDepartment of Physics, Durham University, Durham, DH1 3LE, UK
dDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 25th October 2023
Abstract
Many luminescent platinum(II) complexes undergo face-to-face interactions between neighbouring molecules, leading to bimolecular excited states that may emit at lower energy (dimers and/or excimers). Detailed photophysical studies are reported on dinuclear complexes, in which two NCN-coordinated Pt(II) units are covalently linked by a xanthene such that intramolecular formation of such dimeric or excimeric states is possible. These complexes display strong excimer-like photoluminescence at low concentrations where their monometallic analogues do not. However, a striking difference emerges between complexes where the Pt(NCN) units are directly connected to the xanthene through the tridentate ligand (denoted Class a) and a new class of compounds reported here (Class b) in which the attachment is through a monodentate acetylide ligand. The former require a substantial geometrical rearrangement to move the metal centres of the Pt(NCN) units to a distance short enough to form excimer-like states. The latter require only a small deformation. Consequently, Class a compounds display negligible excimer-like emission in solid films, as the rigid environment hinders the requisite geometric rearrangement. Class b complexes, in contrast, display strong excimer-like emission in film, even at very low loadings. The new dinuclear molecular architecture may thus offer new opportunities in the quest for efficient NIR-emitting devices.
1. Introduction
The development of molecules that emit deep-red light and/or near-infrared radiation (NIR) attracts interest due to the properties of such low-energy, long-wavelength light that are important for numerous applications.1,2 For instance, in bioimaging, chemosensing, and light-activated medical treatments such as photodynamic therapy (PDT), light in this region of the spectrum is highly desirable in order to allow deeper tissue penetration.3–7 As the Rayleigh scattering of light is inversely proportional to the fourth power of the wavelength, NIR is less scattered than visible light. Moreover, biological tissue has a “window of transparency” in the range 700–1400 nm, due to the absorption of light by endogenous molecules being minimal in this region. As such, NIR has enormous potential for applications in medical therapies and diagnosis using light. The incorporation of NIR emitters into organic light-emitting diodes (OLEDs) for biomedical purposes is particularly attractive, as such technology offers energy-efficient, light, thin and flexible devices that could be used widely in the treatment of disease and in user-device interfaces.8–11However, deep-red and NIR emitters are usually plagued by intrinsically low luminescence efficiency. As the energy gap between the emissive excited state and the ground state decreases – evidently a prerequisite for the generation of long-wavelength light – non-radiative decay to the ground state is rapidly exacerbated through the stronger coupling of the excited state to high-energy vibrational levels of the ground state.12–15 Examples of deep-red/NIR emitters do exist, including pure fluorescent dyes,16–18 thermally activated delayed fluorescence (TADF) emitters,11 and phosphorescent Pt(II) and Ir(III) metal complexes.19–28 Nevertheless, molecular materials with strong luminescence efficiency in this region remain scarce: novel strategies are desirable if new, more efficient NIR emitters are to be developed.
Excimer- and aggregate-forming molecules offer an interesting alternative strategy for obtaining long wavelength emission, since excimers emit at significantly lower energy than the corresponding unimolecular excited state.29–31 Among NIR emitters, heavy-metal complexes that form excimers are particularly intriguing for application in OLEDs. Not only do they allow the generation of low energy emission with high efficiency, but also the presence of the metal centres promotes spin–orbit coupling that accelerates the radiative decay of formally forbidden phosphorescence, which is crucial to avoid polaron and triplet–triplet luminescence quenching.32–36 Excimer-forming Pt(II) complexes are attracting more and more interest as potential deep-red or NIR emitters for OLEDs.21,37–45
It is not always clear, however, whether the red-shifted emission of these complexes is due to a pure excimer state, or due to interactions occurring in the ground state (i.e. aggregation to form dimers or higher oligomers), or indeed to a combination of both. Recent studies that combine experimental and theoretical data have revealed a more complicated picture than might be expected.39,46–49 Further insight into this question is needed, over a more diverse range of molecular structures, in order to better identify and define the targets for the design of novel, efficient NIR emitters based on such molecular materials.
Excimers or aggregates require elevated concentrations of the emitter in order to be formed at a proportion sufficient for them to contribute significantly to the photo- or electroluminescence spectrum.50 But, increasing the concentration is not necessarily viable in many applications such as OLEDs, since it is not common for excimer-forming complexes to simultaneously exhibit sufficiently good charge-transport properties. Develay and Williams51 proposed the use of “tweezer” complexes such as 1a (Fig. 1) in which two square-planar, cyclometallated Pt(II) units are covalently linked by a xanthene bridge in a geometry that, despite a Pt⋯Pt distance >4 Å, allows face-to-face interaction and the formation of an “intramolecular excimer”. Low-energy emission was indeed observed in dilute solution, similar to that of the excimer formed by the corresponding mononuclear Pt(II) complex at elevated concentrations. Using an identical structural motif, Wasielewski and co-workers have recently demonstrated a similar behaviour of perylene excimers.52 The term “intramolecular excimer” is, of course, rather unsatisfactory, given that an “excimer” strictly refers to bimolecular species that result from the interaction between two separate molecules under diffusion control. However, it is used here to indicate a process whereby those two “separate” molecular units are linked within a single molecule, with the excimer-like emission thought to arise from the interaction between them in the excited state, without there necessarily being such interaction in the ground state.
 | ||
| Fig. 1 Top: Dinuclear complexes of Class a, in which the Pt(NCN) units are connected to the xanthene bridge through a C–C bond to the tridentate ligand. Middle: Dinuclear complexes of Class b, featuring an acetylide link between the Pt(NCN) units and the xanthene. Bottom: The corresponding parent mononuclear complexes used as models. | ||
In the present work, we describe in-depth photophysical studies on 1a, and on a CF3-substituted analogue 2a (Fig. 1) that displays longer-wavelength emission towards 800 nm. We also consider a new, alternative structure type (b) in which the Pt units are linked to the xanthene bridge via a monodentate acetylide (1b, 2b, 3b) rather than through the tridentate ligand. The studies reveal a complex picture for the generation of long-wavelength photoluminescence in these tweezer-like complexes, with the new class of complexes emitting from states involving ground-state metallophilic interactions, in contrast to more clear-cut excimer-like states in the first class.
2. Results and discussion
2.1. Target compounds and synthetic strategies
Platinum(II) complexes of the NCN-coordinating ligand 1,3-dipyridylbenzene (dpyb) and its derivatives have been widely investigated. Typically, they are not only highly luminescent with solution quantum yields >0.5, but they also form brightly emissive excimers.53–55 The archetypal example is Pt(dpyb)Cl (denoted 1 in Fig. 1).29,56 The excimers formed by these molecules may feature π–π interactions and do not necessarily involve the d–d interactions that lead to the well-known aggregation of Pt(II) complexes with diimine ligands like Pt(bpy)Cl2.57 The behaviour is thus rather different from that of isomeric complexes of NNC ligands based on 2-phenylbipyridine (phbpy), where MMLCT excited states account for the appearance of additional low-energy bands in the emission spectrum (MMLCT = metal–metal bond to ligand charge transfer, i.e., with a Pt–Pt interaction pre-existing in the ground state).58–60The xanthene-bridged dinuclear complex 1a previously communicated51 (Fig. 1) links two Pt(dpyb)Cl units to the relatively rigid xanthene support, through a C–C (aryl–aryl) bond from the central phenyl of the dpyb unit. A similar strategy has been explored to investigate rigidly linked Pt(ONNO) structures,61 as well as metal-free organic systems,52,62,63 including donor–acceptor, through-space, charge-transfer emitters that emit through thermally activated delayed fluorescence (TADF).64 In the present work, we have also prepared an analogue of 1a that features CF3 substituents in the 4-position of the pyridine rings, denoted 2a. The objective here was to explore whether 2a would display excimer-like emission displaced further into the NIR, as has been observed previously for the excimers formed by the parent mononuclear complex 2 compared to 1.49,65 The synthesis of such compounds is, however, arduous and does not lend itself readily to structural variation. A potentially more attractive, alternative way to link Pt(NCN) units is by metathesis of the monodentate chloro ligand to, for instance, an acetylide appended to the linker backbone. Such a strategy has been widely used in the design of multinuclear terpyridyl-based Pt(II) complexes.66,67 In the case of Pt(NCN)X complexes, it is known that the photophysical properties are generally not greatly influenced by the identity of X (with the exception of thiolates, X = RS−),68 and indeed acetylide derivatives of the form Pt(NCN)(–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ar) retain emission properties broadly similar to those of the parent 1.69
C–Ar) retain emission properties broadly similar to those of the parent 1.69
We therefore targeted such xanthene-bis-acetylide-linked dinuclear complexes, which we will refer to as “Class b” (as opposed to “Class a” for 1a and 2a). Compounds 1b and 2b were selected, as they comprise the same Pt(NCN) units as 1a and 2a, allowing for direct comparison of the properties. Compound 3b was also deemed to be of possible interest in the context of low-energy emission, owing to the red-shifted nature of the unimolecular emission of its mononuclear parent 3.49,70
The key intermediate required for the synthesis of the class b compounds is the xanthene bis-acetylide, 2,7-di-tert-butyl-4,5-diethynyl-4,5-9,9-dimethyl-xanthene, which is readily obtainable from the commercial dibromo derivative (see Fig. S2.1 and accompanying synthetic details in the ESI†).71 The reaction of this material with complex 1 (2 equiv.) in the presence of base gave the desired product 1b under mild conditions (see ESI†). The same procedure was likewise applied successfully to the preparation of 2b and 3b, simply by using 2 and 3 in place of 1. The divergent appeal of this methodology for potentially accessing a range of systems with different tridentate ligands is thus clear.
2.2. Molecular structures in the solid state
The identities of the dinuclear compounds 1a, 1b, and 3b have been confirmed by X-ray crystallography (Fig. 2 and ESI,† Section S3). The Pt(NCN) units are twisted relative to the plane of the xanthene in each of the dinuclear structures. In 1a, the torsion angles between the planar fragments are 44.5(5) and 43.8(5)°. The intramolecular Pt⋯Pt distance of 4.704(1) Å is clearly too long for significant ground-state interaction between the metal centres to be present. The structure of 1b shows torsion angles in the similar range of 42.31(1)–57.47(1)° (two independent molecules) between the planes of the Pt(NCN) units and the xanthene, with intramolecular Pt⋯Pt distances of 4.5787(2) and 4.6607(2) Å. The Pt⋯Pt distance between nearest metal atoms of adjacent independent molecules is 4.9617(3) Å, whilst the next shortest intermolecular Pt⋯Pt distance is >8.5 Å. Crystals of 3b revealed a shorter intramolecular Pt⋯Pt distance of 3.9757(7) Å and slightly larger torsion angles of the Pt(NCN) planes relative to the xanthene: 65.6(9) and 49.5(8)° (Fig. 2). It is noticeable that the acetylide bonds “point inwards” in this case, rather than outwards in 1b, leading to the closer separation of the Pt atoms in 3b. Nevertheless, the intramolecular π–π distances are actually quite similar in each case, with the values for the acetylide systems being only marginally shorter: 1a = 3.572(8) Å; 1b = 3.439(2) Å; 3b = 3.443(9) Å. | ||
| Fig. 2 The molecular structures of 1a (top), 1b (bottom left) and 3b (bottom right) in crystals of the complexes. Further details of crystallography are provided in the ESI.† | ||
2.3. Steady-state absorption and emission spectra in solutions
The absorption and emission spectra of 1a, 2a, 1b, 2b, and 3b in dilute solution in CH2Cl2 (10−5 M) are shown in Fig. 3, together with spectra of the mononuclear chloro complexes 1, 2, and 3 for comparison. The absorption spectra of the Class a dinuclear complexes display a similar absorption onset to their corresponding mononuclear parents 1 and 2 (Fig. 3, left). However, for the Class b complexes (1b–3b), it can be seen that the low-energy absorption bands extend out significantly further towards the red than the respective models 1–3 (Fig. 3, right). This is not attributable simply to the replacement of the monodentate Cl ligand by an acetylide, since earlier work reveals no such change in mononuclear Pt(NCN)(–CC–Ar) complexes. The divergent behaviour of the two classes of molecules thus suggests that they may have a different electronic ground-state structure. A possible explanation for the extended absorption to lower energy in 1b–3b could be the presence of additional absorption bands originating from intramolecular, metallophilic Pt–Pt interactions in the ground state.39,49 We discuss this phenomenon further in the computational section below.
 | ||
| Fig. 3 Absorption and photoluminescence spectra of Class a dinuclear complexes 1a and 2a (left), and Class b complexes 1b, 2b, and 3b (right) in deoxygenated CH2Cl2 solution at 10−5 M (T = 295 K, λex = 365 nm), together with the spectra of the parent mononuclear complexes 1–3 in the same panel in each case, recorded at the same concentration of 10−5 M. | ||
At the low concentration of 10−5 M at which the emission spectra of Fig. 3 are recorded, there is very little excimer contribution to the emission spectra of the mononuclear complexes 1–3 (Fig. 3). They display almost exclusively the structured profiles associated with unimolecular emission.49 In contrast, at the same concentration, the emission spectra of all of the dinuclear complexes show two sets of bands. The higher-energy set match those of the associated monometallic complexes, suggesting that this emission arises from non-interacting Pt(NCN) units, and we shall refer to it as the “monomer” band in the subsequent discussion. It is accompanied by a broad, structureless band of comparable or greater intensity at longer wavelength (λmax in the region 690–800 nm). This emission band closely resembles the excimer band exhibited by the corresponding mononuclear complexes at higher concentrations,49,72 and is thus attributed to an excimer-like excited state, tentatively one that spans two Pt(NCN) units within the same molecule (as opposed to two such units in two separate molecules). We shall therefore refer to it as the “excimer-like” or “red” band to reflect the ambiguity in its origin at this stage. It is, however, crucial to note the difference in concentration required for the excimer-like band to contribute to the luminescence. To observe such a substantial contribution from the excimer of 1 requires a concentration >3 × 10−4 M (Fig. 4), i.e., some 30 times higher than that at which the spectra of 1a or 1b are recorded in Fig. 3.
 | ||
| Fig. 4 The photoluminescence spectra of the dinuclear complexes in deoxygenated CH2Cl2 at 10−5 M and 295 K, as in Fig. 3, but in this case shown superimposed on the excimer emission spectra of the respective monometallic analogues, obtained at a much higher concentration of 3 × 10−4 M. | ||
It is clear, then, that both the Class b and – apparently to an even greater extent – Class a diplatinum(II) complexes show an overwhelmingly greater ability to form excimer-like states than their mononuclear counterparts. Such an observation strongly points towards intramolecular interaction of the Pt(NCN) units. Yet, the molecular structures in the solid state (Section 2.2) revealed intramolecular Pt⋯Pt distances of around 4–5 Å, which are clearly longer than the optimal distance of about 3.5 Å expected for such interactions.49 Some form of geometry rearrangement must be necessary in solution to account for the observed long wavelength luminescence in both classes, as well as for the low-energy MMLCT states featuring in the absorption spectra of the Class b compounds (Fig. 3).
2.4. Calculations
We used density functional theory (DFT) and time-dependent DFT (TD-DFT) methods implemented in Orca 4.2.173,74 to model the behaviour of complexes 1a, 2a and 1b–3b in the ground and excited states. Due to the complexity of the studied structures, the BP86 functional with a def2-SVP basis set was used for geometry calculations. We also used dispersion correction to account for the long-range interaction of the adjacent Pt(NCN) units. Excited-state energies and molecular orbitals were calculated using a range-separated, hybrid functional CAM-B3LYP with the same basis set. The model systems were considered as adopting two principal geometries: (1) with a large Pt⋯Pt distance, leading to negligible metallophilic interactions; (2) with short (i.e., ∼3 Å) Pt⋯Pt contacts. For the purpose of this section, we will name them monomer (S0 or T1) and dimer (S0 or T1), respectively, to highlight the interaction – or lack thereof – between the metal centres. Due to the complexity introduced by the relatively flexible acetylide linkers in 1b–3b complexes, we were not able to obtain some of the geometries for 2b and 3b reliably. We discuss the results for 1a and 1b in the main text, while the results pertaining to the remaining molecules are presented in the ESI† (Fig. S4.1–S4.6).The Class a complexes 1a and 2a adopt two ground state geometry minima: S0-monomer and S0-dimer (Fig. 5 for 1a and Fig. S4.1 for 2a, ESI†). The S0-monomer geometry resembles that of the X-ray structures (Fig. 2) with Pt⋯Pt distances >4.4 Å. Due to the relative rigidity of the Class a structure, the only possible way to bring metal centres of the two Pt(NCN) units to a distance of ∼3 Å is by bending the xanthene unit at the central C–O axis to give the S0-dimer structures. Formation of the bent conformer of xanthene has been reported earlier,75 while similar bent conformers have been postulated or observed experimentally in analogous systems, such as in phenothiazine derivatives.76
 | ||
| Fig. 5 Optimised ground (S0) and triplet excited state (T1) geometries of 1a. | ||
Regardless of the initial structure (S0-monomer or S0-dimer) used for optimisation of the T1 excited state, both of the Class a complexes adopt only one minimal triplet state geometry T1-dimer, which is analogous to that of the S0-dimer and displays the same bent conformation of the xanthene unit. From the short Pt⋯Pt distance of 2.88 Å in the T1-dimer structure of 1a, we conclude that this state is responsible for the intense excimer-like PL at ∼690 nm. It is clear from this analysis that complexes 1a and 2a must undergo significant reorganisation, by adopting a bent conformation of the xanthene linker, in order to form the excimer-like excited state.
Unlike the Class a complexes, those of Class b (1b–3b) form (under the approximation of the computational method used) only one type of ground state, S0-dimer, but two geometrically distinct T1 excited states: T1-monomer and T1-dimer, as shown for 1b in Fig. 6. The S0-dimer geometry shows displaced Pt(NCN) units with the Pt⋯Pt distances approaching ∼3 Å, but importantly with a flat rather than bent xanthene conformation. The S0-dimer geometry of 1b can be achieved by a relatively subtle displacement of the Pt(NCN) units by deformation of the flexible acetylide linkers, rather than through the bending of the xanthene bridge that appears to be necessary in 1a. The related, excited-state T1-dimer geometry responsible for the excimer-like luminescence of complexes 1b–3b is similar to that of S0-dimer, but with a slightly shorter Pt⋯Pt distance. The T1-monomer geometry resembles that of the X-ray structures (Fig. 2), with Pt⋯Pt distances >4 Å. We attribute this structure to the one which is responsible for the monomer PL of complexes 1b–3b.
 | ||
| Fig. 6 Optimised ground-state (S0) and triplet excited-state (T1) geometries of 1b. | ||
It is worth noting that, although the geometries of the S0-monomer of 1b and T1-monomer geometry of 1a could not be obtained definitively, that is most likely due to these structures having very shallow energy minima. Such structures are nevertheless expected to be present and to contribute to the experimental data, and the T1-monomer structure is most likely responsible for the residual monomer PL observed in 1a and 2a.
In their T1-dimer geometry, all the dinuclear complexes feature T1 excited states that are composed of two main excitations: HOMO → LUMO and HOMO−1 → LUMO (1a and 2a) and HOMO → LUMO and HOMO−2 → LUMO (1b–3b). A clear contribution of the dz2(Pt) and pz(Cl) orbitals to the HOMO is observed (1a and 2a), and a similar composition of the HOMO−2 for 1b–3b but with X = acetylide playing the role of a π-donor (Fig. 7). The LUMO is localised mainly on the NCN chelating ligands. This behaviour of the molecular orbitals is in line with that of the analogous mononuclear platinum(II) complexes forming intermolecular 3MMLCT excited states, reported earlier by us.39,49
 | ||
| Fig. 7 Molecular orbital iso surfaces relevant to the T1-dimer excited state of molecules 1a and 1b. | ||
The calculated excited-state energies may be compared with the experimental excimer-like photoluminescence spectra of the diplatinum(II) complexes 1a, 2a and 1b–3b; a clear linear relationship is observed between the two quantities for 1b–3b (Fig. S4.6, ESI†), supporting the validity of the calculated structures. Complexes 1a and 2a are offset by ∼0.3 eV relative to the approximate trendline set by 1b–3b. We attribute the need for this offset to a significant difference in excited-state geometries between the two groups, resulting in relative underestimation of the T1 energy for class a complexes. A similar relationship has recently been presented for the mononuclear complexes 1, 2, and 3.49
2.5. Time-resolved luminescence in solution
Additional insight into the behaviour of the dinuclear complexes comes from time-resolved measurements, which reveal very different behaviour for the photoluminescent decays compared to those of the mononuclear compounds. In the mononuclear complexes 1–3, the lifetimes of the red excimer band mirror that of the unimolecular emission, but the former displays a clear grow-in of intensity from t = 0 (Fig. 8b).49 Such behaviour conforms to the well-established Birks’ scheme for excimer formation under diffusion control, as discussed previously for 1.31,49 In the dinuclear complexes, on the other hand, no clear relationship between the decays of the excimer-like and monomer photoluminescence bands is observed, indicating that the species responsible for each are not kinetically linked. This can be explained by the monomer and excimer-like emissions originating from species with distinct molecular geometries, if the transition between them is slow on the timescale of the excited-state lifetime.It is notable that the lifetimes of the red emission band in all the dinuclear complexes are generally longer than those of their monometallic counterparts. The difference is most evident in the case of 3b and 3, where the lifetime of the excimer-type emission in 3b is five-fold longer than that in 3. This is indicative of suppressed vibrational/rotational quenching of the excited states of the more rigidly organised dinuclear systems compared to the excimer emission of the mononuclear complexes. This observation highlights an important advantage of our dinuclear design that is significant for applications where intense NIR luminescence is required in solution – such as in bioimaging applications.77
In order to probe the kinetic evolution of the different emission bands in more detail, emission decay data have also been acquired on a much shorter timescale. For the mononuclear complexes, following excitation, a very clear increase in the intensity of the red band is observed over >1 μs (see example in Fig. 8(b)),49 pointing to diffusion-controlled excimer formation being responsible for this emission. In contrast, no rise time for the red band in the dinuclear compounds is detectable, at least on a timescale down to around 5 ns approaching the instrument response function (IRF) (Fig. 8(a)). This indicates either that the population of the excimer-like states occurs nearly instantaneously from the initial excited-state population (complexes 1a and 2a, also likely 1b–3b), or that the excited state population in question is created from molecular geometries with pre-existing metallophilic interactions, such as ground state intramolecular dimers (complexes 1b–3b). Regardless of which mechanism is at work, all the dinuclear complexes clearly diverge in their behaviour from that of the respective mononuclear model compounds, indicating that the origin of the red excimer-like band in the former is intramolecular.
 | ||
| Fig. 8 Time-correlated single photon counting (TCSPC) traces of the temporal evolution of: (a) the excimer-like band of the dinuclear complexes in dilute CH2Cl2 solution (10−5 M); (b) monomer and excimer bands of the monometallic complex 1 at 10−4 M. | ||
2.6. Steady-state emission spectra in film
The effects of a more rigid environment on the properties of the dinuclear complexes can be studied in polystyrene (PS) dispersions. The behaviour of 1a and 2a in PS is fundamentally different from that of 1b–3b (Fig. 9 and Table 1). The former display negligible contributions of the excimer-like band at a concentration of ∼1% by weight, with a significant long-wavelength band only visible in neat films. This behaviour of 1a and 2a in polymer film is in stark contrast to solution, where significant excimer-like PL was observed (vide supra). In contrast, 1b–3b display dominant excimer-like PL even at 1% loading, with only very small contributions from monomer PL that reduce further at higher concentrations in the PS film. | ||
| Fig. 9 Steady-state photoluminescence spectra of complexes 1a, 2a and 1b–3b in polystyrene film at the % by weight indicated in each legend. | ||
| CH2Cl2 | Polystyrene | ||||||
|---|---|---|---|---|---|---|---|
| Compound | λ max , nm (monomer) | λ max , nm (excimer/dimer) | Φ PL | τ M , /μs | τ E , /μs | τ M /μs | τ E /μs |
| a Emission λmax and observed lifetime τM for the high-energy set of bands, attributed to non-interacting Pt(NCN) units. b Emission λmax and observed lifetime τE for the low-energy band, attributed to an excited state involving interacting Pt(NCN) units. c Total emission quantum yield, measured using [Ru(bpy)3]Cl2(aq) as the standard, for which ΦPL = 0.04. * Negligible excimer luminescence. d τ M is determined as the lifetime at infinite dilution from plots of the observed lifetime as a function of concentration extrapolated to the intercept (see, e.g., ref. 29 for this approach). τE is measured directly for dilute solutions of approximately 10−5 M. In polystyrene, τE and τM were recorded at 1 wt% loading of the diplatinum(II) complex. | |||||||
| 1a | 523 | 690 | 0.25 | 6.7 | 1.42 | 7.0 | * |
| 2a | 544, 586sh | 762 | 0.08 | 6.0 | 0.43 | 4.5 | * |
| 1b | 490, 522, 566sh | 685 | 0.58 | 3.9 | 1.4 | 3 | 1.4 |
| 2b | 534, 573sh | 752 | 0.35 | 6.0 | 0.65 | 2.2 | 0.85 |
| 3b | 593, 642, 699sh | 788 | 0.08 | 3.0 | 0.43 | 2.4 | 0.71 |
From this general picture, one may conclude that the striking contrast between the two classes of complexes is related to the degree of rigidity in the solid-state medium. For complexes 1a and 2a, there exist negligible ground state Pt⋯Pt contacts, as such contacts require the xanthene bridge to remain in a much less favourable bent conformation, as discussed in Section 2.4. The planar xanthene conformation does not allow for significant Pt⋯Pt interaction as the distance (>4 Å) is too large. Evidently, then, the polymer host significantly restricts the conformational change that would lead to the intramolecular excimer-like luminescence. As a result, the strongly concentration-dependent, excimer-like PL must originate from intermolecular short contacts already present in the ground state.
The intense excimer-like PL displayed by the complexes 1b–3b shows very little concentration dependence in the 1–100% range of concentrations. Studies were therefore also made at even lower loadings <1% (Fig. 10), to obtain further insight into the origin of their long-wavelength PL. Gradual dilution down to 0.005% does reveal some concentration dependence (Fig. 10), but the clear dominance of the excimer-like PL even at such a low loading of the complex implies that the long-wavelength PL must originate from species involving intramolecular, as well as intermolecular, interactions. These interactions are illustrated schematically in Fig. 11 as A and B respectively, and they are consistent with the observations made in solution, where 1b–3b showed a high propensity for intramolecular Pt⋯Pt interactions. Such behaviour is likely linked to the energy cost being lower to deform the relatively flexible acetylide ancillary ligands and form short metal–metal contacts, than to form the necessary bent conformer of 1a and 2a. This fundamental difference in mechanism underpinning the excimer-like PL between these two classes of dinuclear complexes will clearly render the class b compounds more favourable as potential candidates for NIR phosphors in solid-state applications such as OLEDs.
 | ||
| Fig. 10 Steady-state photoluminescence spectra of complexes 1b–3b in polystyrene film at weight concentrations between 0.005 and 1%, as indicated in the figure legend. | ||
 | ||
| Fig. 11 Possible modes of face-to-face interactions between Pt(NCN) units (represented by the orange ovals) in the dinuclear complexes. | ||
2.7. Time-resolved photoluminescence in films
Further insight into the excimer-like luminescence of 1b–3b in films is obtained through time-resolved PL spectra (Fig. 12; corresponding spectra for class a are shown in Fig. S5.6, ESI†). A relatively fast decay of the dominant, long-wavelength PL of 1a-3b is followed by a significantly slower decay of the monomer emission. Thus, the spectra at short delay times <1 μs are dominated by excimer-like PL, while at >2 μs the monomer emission dominates. Interestingly, however, in the short delay regime <0.5 μs, both the monomer and excimer-like bands are present in the spectrum, and they remain in the same proportion in this timeframe, indicating that they have identical decay lifetimes. Thus, despite most monomer emission originating from isolated molecules with their intrinsic lifetimes, some of it originates from species in equilibrium with the intramolecular dimer. The behaviour of 1b–3b is alike at 80 K (Fig. 12).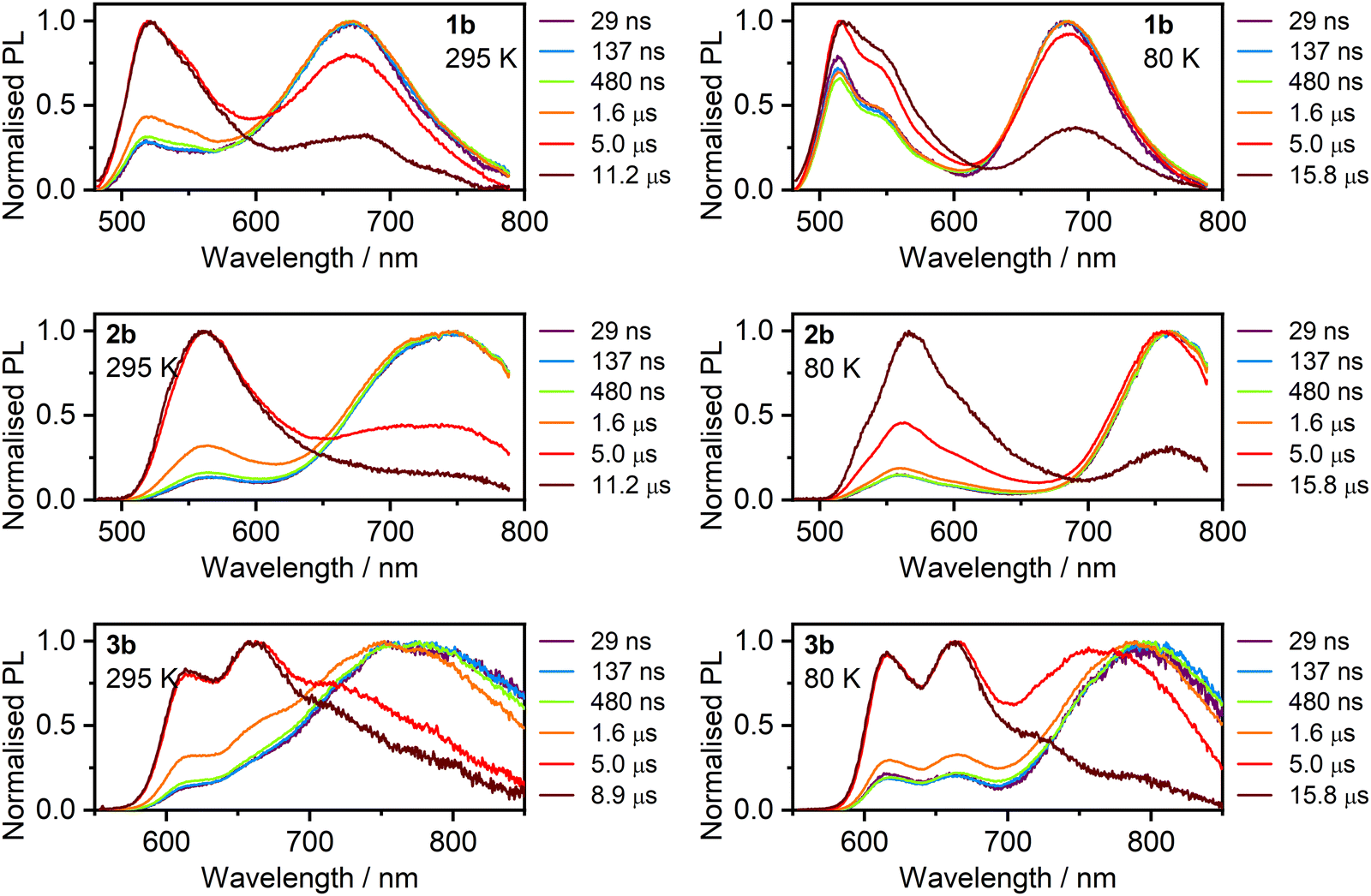 | ||
| Fig. 12 Time-resolved photoluminescence spectra of complexes 1b–3b in polystyrene film at a loading of 1% by weight at 295 K and 80 K. | ||
This general picture of the time-resolved PL of 1b–3b is in line with the conclusions made from the steady state spectra:
(1) The excimer-like and monomer PL components generally originate from different species that are not kinetically linked; and
(2) Excimer-like PL is in a kinetic equilibrium with a weak monomer band.
Conclusion (1) indicates that the species that give rise to monomer or excimer-like emissions are ‘locked’, such that they display a limited mobility and ability to adapt different geometries. Therefore, in the PS films, they retain the configurations that give rise solely either to monomer or to excimer-like PL. Conclusion (2) suggests that the species giving rise to the excimer-like luminescence are somewhat kinetically linked to a small population of species emitting monomer PL. For example, only a small fluctuation of the Pt⋯Pt distance will switch between monomer-like and excimer-like excited states, leading to the observed equilibrium. Such conclusions remain consistent with behaviour observed previously for related mononuclear complexes in film.78
3. Conclusions
An in-depth investigation of bimetallic-Pt(II) “sandwich” complexes reveals important insights into the mechanism of their NIR emission. In contrast with the classical mechanism of excimer formation that is observed in monometallic Pt(II) complexes and other excimer-forming molecules, the bimetallic complexes presented here preferentially form intramolecular excimers or ground state dimers. These complexes, therefore, display strong “excimer-like” photoluminescence at concentrations where their monometallic analogues show negligible rates of excimer formation. We demonstrate a striking difference between the known Class a complexes – with the Pt(NCN) units connected to the xanthene linker through the central benzene ring – and the novel Class b complexes, in which the Pt(NCN) units are attached to the xanthene bridge via acetylides. The former require a geometrical rearrangement through formation of a bent xanthene conformer in order to move the metal centres of the Pt(NCN) units to a distance short enough to form excimer-like 3MMLCT excited states. On the other hand, complexes 1b–3b only require a small deformation to form the necessary short Pt⋯Pt contacts, which does not significantly affect the molecule's footprint. As a result, the Class a complexes display negligible intensity of the excimer-like band in solid films, in which environment the geometry rearrangement necessary to form intramolecular 3MMLCT excited states is strongly hindered. Class b complexes, on the other hand, display strong excimer formation in film due to the contributions involving both intra- and intermolecular interactions. We believe that the new dinuclear molecular architecture may offer an exciting opportunity to introduce excimer NIR emission into doped OLEDs, for example, without the necessity to rely on non-doped (i.e., neat) films of the emitter.Experimental
All experimental details are provided in the accompanying ESI†: synthetic details, crystallography, DFT calculations, and photophysical measurements.Author contributions
P. P. – formal analysis, investigation (photophysics and DFT calculations), writing – original draft; M. T. W. – investigation (synthesis and photophysics); R. J. S. – investigation (synthesis and photophysics), writing – original draft; A. S. – investigation (synthesis and crystallisation); D. S. Y. – investigation (X-ray diffraction); F. B. D. – funding acquisition, project administration, writing – review & editing; J. A. G. W. – conceptualization, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank EPSRC (grant ref. EP/S012788/1) and the Samsung GRO program for supporting parts of this work. We also thank the Diamond Light Source for an award of instrument time on Station I19 (MT 16177), enabling the structure of 1b to be obtained, and the support of the instrument scientists there. The computational work made use of the facilities of the Hamilton HPC Service of Durham University.References
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Chem. Soc. Rev., 2020, 49, 765–838 RSC.
- P. Liu, X. Mu, X.-D. Zhang and D. Ming, Bioconjugate Chem., 2020, 31, 260–275 CrossRef CAS PubMed.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC.
- Y. Chen, R. Guan, C. Zhang, J. Huang, L. Ji and H. Chao, Coord. Chem. Rev., 2016, 310, 16–40 CrossRef CAS.
- E. Baggaley, J. A. Weinstein and J. A. G. Williams, in Luminescent and Photoactive Transition Metal Complexes as Biomolecular Probes and Cellular Reagents, ed. K. K.-W. Lo, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp.205–256 Search PubMed.
- L. Huang, Z. Li, Y. Zhao, Y. Zhang, S. Wu, J. Zhao and G. Han, J. Am. Chem. Soc., 2016, 138, 14586–14591 CrossRef CAS PubMed.
- Nat. Inst. for Health & Care Excellence, https://www.nice.org.uk/guidance/mtg6, (accessed 21 February 2021).
- P. Avci, A. Gupta, M. Sadasivam, D. Vecchio, Z. Pam, N. Pam and M. R. Hamblin, Semin. Cutaneous Med. Surg., 2013, 32, 41–52 Search PubMed.
- T. Yamanaka, H. Nakanotani, S. Hara, T. Hirohata and C. Adachi, Appl. Phys. Express, 2017, 10, 074101 CrossRef.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley, 2008 Search PubMed.
- P. Lai and T. S. Teets, Chem. – Eur. J., 2019, 25, 6026–6037 CrossRef CAS PubMed.
- T. M. Stonelake, K. A. Phillips, H. Y. Otaif, Z. C. Edwardson, P. N. Horton, S. J. Coles, J. M. Beames and S. J. A. Pope, Inorg. Chem., 2020, 59, 2266–2277 CrossRef CAS PubMed.
- X. Lian, M.-Y. Wei and Q. Ma, Front. Bioeng. Biotechnol., 2019, 7, 1–16 Search PubMed.
- S. Zhu, B. C. Yung, S. Chandra, G. Niu, A. L. Antaris and X. Chen, Theranostics, 2018, 8, 4141–4151 CrossRef CAS PubMed.
- K. Umezawa, D. Citterio and K. Suzuki, Anal. Sci., 2014, 30, 327–349 CrossRef CAS PubMed.
- M. Ibrahim-Ouali and F. Dumur, Molecules, 2019, 24, 1412 CrossRef CAS PubMed.
- M. Z. Shafikov, P. Pander, A. V. Zaytsev, R. Daniels, R. Martinscroft, F. B. Dias, J. A. G. Williams and V. N. Kozhevnikov, J. Mater. Chem. C, 2021, 9, 127–135 RSC.
- K. Tuong, Ly, R. W. Chen-Cheng, H. W. Lin, Y. J. Shiau, S. H. Liu, P. T. Chou, C. S. Tsao, Y. C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63–68 CrossRef.
- Y. Zhang, Q. Li, M. Cai, J. Xue and J. Qiao, J. Mater. Chem. C, 2020, 8, 8484–8492 RSC.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, Inorg. Chem., 2011, 50, 11446–11460 CrossRef CAS PubMed.
- Y. Zhang, Z. Yin, F. Meng, J. Yu, C. You, S. Yang, H. Tan, W. Zhu and S. Su, Org. Electron., 2017, 50, 317–324 CrossRef CAS.
- W. Xiong, F. Meng, C. You, P. Wang, J. Yu, X. Wu, Y. Pei, W. Zhu, Y. Wang and S. Su, J. Mater. Chem. C, 2019, 7, 630–638 RSC.
- P. Mandapati, J. D. Braun, I. B. Lozada, J. A. G. Williams and D. E. Herbert, Inorg. Chem., 2020, 59, 12504–12517 CrossRef CAS PubMed.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- S. F. Wang, Y. Yuan, Y. Wei, W. Chan, L. Fu, B. Su, I. Chen, K. Chou, P. Chen, H. Hsu, C. Ko, W. Hung, C. Lee, P. Chou and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002173 CrossRef CAS.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- D. L. Rochester, S. Develay, S. Záliš and J. A. G. Williams, Dalton Trans., 2009, 1728 RSC.
- J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575–588 CAS.
- D. Kim and J. L. Brédas, J. Am. Chem. Soc., 2009, 131, 11371–11380 CrossRef CAS PubMed.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- K. Mori, T. P. M. Goumans, E. van Lenthe and F. Wang, Phys. Chem. Chem. Phys., 2014, 16, 14523–14530 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- S. F. Wang, L.-W. Fu, Y.-C. Wei, S.-H. Liu, J.-A. Lin, G.-H. Lee, P.-T. Chou, J.-Z. Huang, C.-I. Wu, Y. Yuan, C.-S. Lee and Y. Chi, Inorg. Chem., 2019, 58, 13892–13901 CrossRef CAS PubMed.
- J. Kang, X. Zhang, H. Zhou, X. Gai, T. Jia, L. Xu, J. Zhang, Y. Li and J. Ni, Inorg. Chem., 2016, 55, 10208–10217 CrossRef CAS PubMed.
- R. J. Salthouse, P. Pander, D. S. Yufit, F. B. Dias and J. A. G. Williams, Chem. Sci., 2022, 13, 13600–13610 RSC.
- B. Ma, P. I. Djurovich, S. Garon, B. Alleyne and M. E. Thompson, Adv. Funct. Mater., 2006, 16, 2438–2446 CrossRef CAS.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D’Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171–1178 RSC.
- B. Ma, P. I. Djurovich and M. E. Thompson, Coord. Chem. Rev., 2005, 249, 1501–1510 CrossRef CAS.
- B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28–29 CrossRef CAS PubMed.
- S. E. Brown-Xu, M. S. J. Kelley, K. A. Fransted, A. Chakraborty, G. C. Schatz, F. N. Castellano and L. X. Chen, J. Phys. Chem. A, 2016, 120, 543–550 CrossRef CAS PubMed.
- Z. Wen, Y. Xu, X. Song, J. Miao, Y. Zhang, K. Li and C. Yang, Adv. Opt. Mater., 2013, 11, 2300201 CrossRef.
- C. Sukpattanacharoen, P. Kumar, Y. Chi, N. Kungwan and D. Escudero, Inorg. Chem., 2020, 59, 18253–18263 CrossRef CAS PubMed.
- W. Chen, C. Sukpattanacharoen, W. Chan, C. Huang, H. Hsu, D. Shen, W. Hung, N. Kungwan, D. Escudero, C. Lee and Y. Chi, Adv. Funct. Mater., 2020, 30, 2002494 CrossRef CAS.
- C.-H. Tseng, M. A. Fox, J.-L. Liao, C.-H. Ku, Z.-T. Sie, C.-H. Chang, J.-Y. Wang, Z.-N. Chen, G.-H. Lee and Y. Chi, J. Mater. Chem. C, 2017, 5, 1420–1435 RSC.
- P. Pander, A. Sil, R. J. Salthouse, C. W. Harris, M. T. Walden, D. S. Yufit, J. A. G. Williams and F. B. Dias, J. Mater. Chem. C, 2022, 10, 15084–15095 RSC.
- A. Iwakiri, Y. Konno and K. Shinozaki, J. Lumin., 2019, 207, 482–490 CrossRef CAS.
- S. Develay and J. A. G. Williams, Dalton Trans., 2008, 4562 RSC.
- R. E. Cook, B. T. Phelan, R. J. Kamire, M. B. Majewski, R. M. Young and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 1607–1615 CrossRef CAS PubMed.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- G. R. Freeman and J. A. G. Williams, Top. Organomet. Chem., 2013, 40, 89–129 CrossRef CAS PubMed.
- M. Cocchi, J. Kalinowski, V. Fattori, J. A. G. Williams and L. Murphy, Appl. Phys. Lett., 2009, 94, 073309 CrossRef.
- S. J. Farley, D. L. Rochester, A. L. Thompson, J. A. K. Howard and J. A. G. Williams, Inorg. Chem., 2005, 44, 9690–9703 CrossRef CAS PubMed.
- Y. J. Cho, S. Y. Kim, H. J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 5486–5494 RSC.
- K. Li, G. S. Ming Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC.
- W. Lu, M. C. W. Chan, N. Zhu, C.-M. Che, C. Li and Z. Hui, J. Am. Chem. Soc., 2004, 126, 7639–7651 CrossRef CAS PubMed.
- S. Chakraborty, A. Aliprandi and L. De Cola, Chem. – Eur. J., 2020, 26, 11007–11012 CrossRef CAS PubMed.
- Z. Guo, S. M. Yiu and M. C. W. Chan, Chem. – Eur. J., 2013, 19, 8937–8947 CrossRef CAS PubMed.
- M. Chen, Y. J. Bae, C. M. Mauck, A. Mandal, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 9184–9192 CrossRef CAS PubMed.
- H. Liu, L. Shen, Z. Cao and X. Li, Phys. Chem. Chem. Phys., 2014, 16, 16399–16406 RSC.
- H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed.
- E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2011, 21, 15501–15510 RSC.
- Z. Chen, A. K.-W. Chan, V. C.-H. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 11204–11211 CrossRef CAS PubMed.
- S. Y. L. Leung, A. Y. Y. Tam, C. H. Tao, H. S. Chow and V. W. W. Yam, J. Am. Chem. Soc., 2012, 134, 1047–1056 CrossRef CAS PubMed.
- W. A. Tarran, G. R. Freeman, L. Murphy, A. M. Benham, R. Kataky and J. A. G. Williams, Inorg. Chem., 2014, 53, 5738–5749 CrossRef CAS PubMed.
- E. Rossi, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, A. Valore, L. Falciola, P. Brulatti, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2012, 22, 10650–10655 RSC.
- Z. Wang, E. Turner, V. Mahoney, S. Madakuni, T. Groy and J. Li, Inorg. Chem., 2010, 49, 11276–11286 CrossRef CAS PubMed.
- R. Muñoz-Rodríguez, E. Buñuel, J. A. G. Williams and D. J. Cárdenas, Chem. Commun., 2012, 48, 5980–5982 RSC.
- M. Cocchi, D. Virgili, V. Fattori, J. A. G. Williams and J. Kalinowski, Appl. Phys. Lett., 2007, 90, 023506 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- J. D. Villada, R. F. D’Vries, M. Macías, F. Zuluaga and M. N. Chaur, New J. Chem., 2018, 42, 18050–18058 RSC.
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold and A. P. Monkman, Nat. Commun., 2017, 8, 14987 CrossRef CAS PubMed.
- G. Zhang, H. Zhang, Y. Gao, R. Tao, L. Xin, J. Yi, F. Li, W. Liu and J. Qiao, Organometallics, 2014, 33, 61–68 CrossRef CAS.
- M. T. Walden, P. Pander, D. S. Yufit, F. B. Dias and J. A. G. Williams, J. Mater. Chem. C, 2019, 7, 6592–6606 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2063389, 2063391 and 2063392. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc03432a |
| This journal is © The Royal Society of Chemistry 2023 |