
Carbonyl-rich porous organic polymers for cobalt adsorption from water†
Min Chieh
Yang
 a,
Devin S.
Rollins
b,
Dale L.
Huber
c,
Jou-Tsen
Ou
a,
Michael R.
Baptiste
a,
Andrea N.
Zeppuhar
a,
Fu
Chen
a and
Mercedes K.
Taylor
*a
a,
Devin S.
Rollins
b,
Dale L.
Huber
c,
Jou-Tsen
Ou
a,
Michael R.
Baptiste
a,
Andrea N.
Zeppuhar
a,
Fu
Chen
a and
Mercedes K.
Taylor
*a
aDepartment of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA. E-mail: mkt@umd.edu
bDepartment of Chemistry, University of Washington, Seattle, WA 98195, USA
cCenter for Integrated Nanotechnologies, Sandia National Laboratories, Albuquerque, NM 87123, USA
First published on 31st October 2023
Abstract
Transition metals such as cobalt are necessary for various clean-energy technologies, notably electric-vehicle batteries. Global demand for these metals is therefore projected to increase exponentially in coming decades. Metal-ion adsorption from water offers many advantages over mining, as adsorption processes are energy-efficient and compatible with diverse water sources. Porous organic polymers are promising adsorbents: Their covalent nature provides thermal and chemical stability, while their porosity leads to high adsorption capacity. Here, we synthesized a series of amide-linked porous organic polymers denoted TMC-TAPM through the polymerization of a tri-functional acyl chloride monomer with a tetra-functional amine monomer, and we studied the resulting materials for cobalt capture from aqueous solution. By controlling monomer stoichiometry during synthesis, we obtained materials with varying amounts of carbonyl or amino groups. The materials with increasing carbonyl content showed increasing cobalt adsorption capacities, with measured adsorption capacities up to 50 mg Co g−1. Cobalt adsorption capacity was observed to plateau past a certain stoichiometric ratio, indicating an optimal monomer stoichiometry of 1.5-fold excess acyl chlorides relative to amines. The captured cobalt could be desorbed to yield a re-activated adsorbent capable of repeated adsorption cycles, without loss in performance. These results provide design rules for the synthesis of robust, high-capacity transition metal adsorbents.
Introduction
With the transition from fossil fuels to electric energy, demand for certain transition metals used in rechargeable batteries (e.g., cobalt, manganese, nickel) has grown exponentially over the past decade. As this trend continues, cobalt demand is projected to undergo another 20-fold increase over the next 20 years.1,2 To obtain the needed cobalt through mining would lead to irreversible landscape damage and water pollution, along with humanitarian and geopolitical problems.3 Thus, new sources of cobalt and other transition metals are necessary to insure a successful clean-energy transition. In comparison to mining, ion adsorption provides economic advantages such as low maintenance, low capital costs, low energy inputs, and recyclability.4–6 Further, ion capture from water sources offers significant environmental benefits, as the ion-capture process can simultaneously function as a purification step for wastewater.7–10 Transition metals are toxic to animals and plants above certain concentrations, making metal adsorption a doubly-beneficial process as it yields both isolated metal and purified water.11,12 In addition, the release of captured ions from the adsorbent offers a simplified product stream, which alleviates refinement processes in comparison to mining.13Porous organic polymers are a promising class of materials for ion adsorption applications. Their covalent linkages, highly branched structures, and rigid backbones provide permanent porosity and thermal stability suitable for large-scale filtration.14–16 Porous organic polymers have been studied for the adsorption of metal ions such as Cu2+,17 Fe3+,18 Pb2+,19,20 and Hg2+.21 In light of the sustainability problems described above, we focused our materials-design efforts on Co2+ capture. A common strategy to improve the adsorption properties of a porous organic polymer is to install functional groups within the pores,22–24 but functionalization often sacrifices pore space25 and typically entails costly multi-step synthesis.26,27 To circumvent these synthetic challenges, we have shown recently that a simple cobalt-templating strategy can significantly improve the cobalt adsorption capacity of amide-linked porous organic polymers.28 In the present work, we sought to simplify the synthetic route even further: As both amine29–31 and carboxylate32–37 groups are known to chelate metal ions, we hypothesized that an amide-forming polymerization would naturally result in a material with some amino and carboxylate end groups present in the pores. By controlling the amount of unreacted end groups, we aimed to maximize cobalt adsorption capacity without functionalizing monomers, performing a templating step, or post-synthetically modifying the polymer.
Here, a series of porous organic polymers termed TMC-TAPM were synthesized through the reaction of trimesoyl chloride (TMC) with tetrakis(4-aminophenyl)methane (TAPM), as depicted in Fig. 1. Our results show that the polymerization yields products with ranging amounts of unreacted amino or carboxylate end groups, which have significant effects on the cobalt adsorption capacity. This approach provides synthetic chemists with a straightforward way to tune the functionality and ion adsorption properties of a porous organic material.
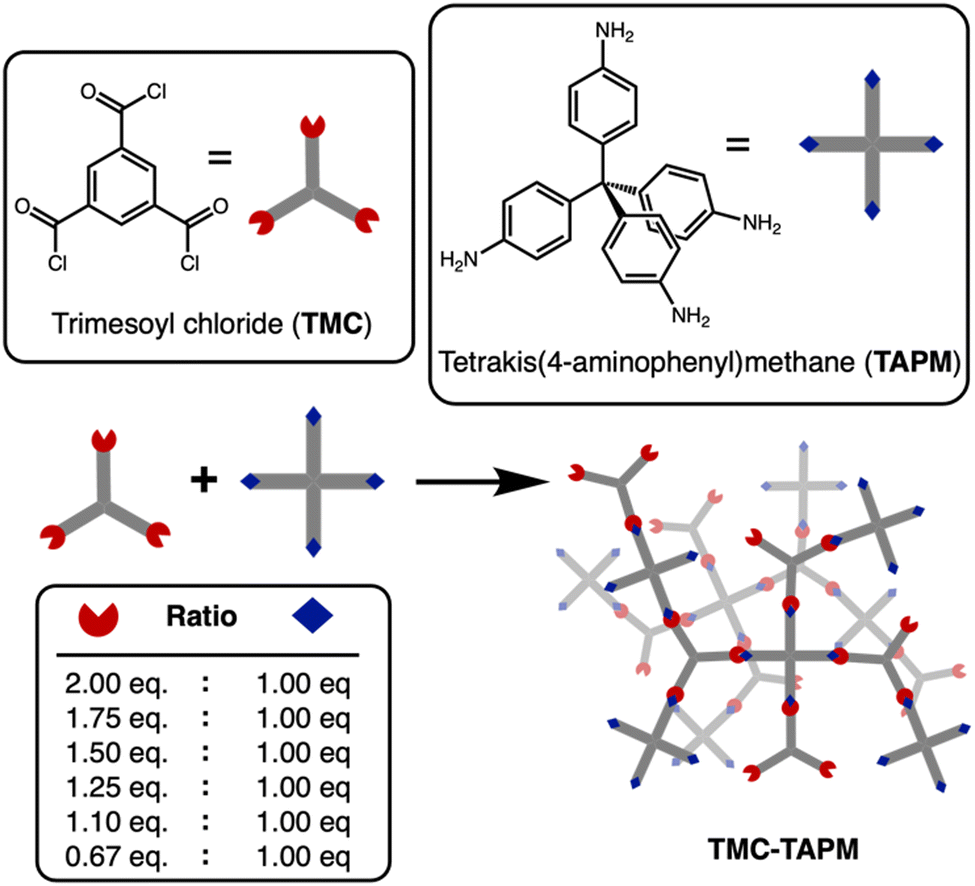 | ||
| Fig. 1 Synthetic route to porous polyamide adsorbents. Trimesoyl chloride (TMC) and tetrakis(4-aminophenyl)methane (TAPM) were combined in varying stoichiometric amounts to control the ratio of acyl chloride groups (red) to amino groups (blue) in the polymerization. | ||
Results and discussion
Amide-linked porous organic polymers were synthesized by combining varying ratios of tetrakis(4-aminophenyl) methane (TAPM) and trimesoyl chloride (TMC) with N,N-diisopropylethylamine in anhydrous tetrahydrofuran under an inert atmosphere. Further synthetic details are provided in the ESI.† The polymerizations were carried out at room temperature to yield insoluble white precipitates. Upon completion, the reactions were quenched with water, which served to hydrolyze any remaining acyl chlorides to the corresponding carboxylates.38 The final products were washed extensively to remove impurities from the pores and were then dried under vacuum to remove residual solvent.A polymerization using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of amine groups to acyl chloride groups should result in an amide-linked polymer with an equal number of amine end groups and carboxylate end groups.39–42 By changing the monomer stoichiometry, we anticipated an uneven end group population between the amines and carboxylates. Our polymer naming scheme refers to the stoichiometric equivalents of acyl-chloride groups in the synthesis, relative to 1 equivalent of amino groups (Table 1). For example, TMC(1.25)-TAPM refers to a polymer synthesized with 1.25 equivalents of acyl-chloride groups and 1 equivalent of amino groups. Note that each TMC monomer contains three acyl chloride groups, while each TAPM monomer contains four amines; our naming scheme refers to ratios of functional groups, not ratios of monomers.
:1 ratio of amine groups to acyl chloride groups should result in an amide-linked polymer with an equal number of amine end groups and carboxylate end groups.39–42 By changing the monomer stoichiometry, we anticipated an uneven end group population between the amines and carboxylates. Our polymer naming scheme refers to the stoichiometric equivalents of acyl-chloride groups in the synthesis, relative to 1 equivalent of amino groups (Table 1). For example, TMC(1.25)-TAPM refers to a polymer synthesized with 1.25 equivalents of acyl-chloride groups and 1 equivalent of amino groups. Note that each TMC monomer contains three acyl chloride groups, while each TAPM monomer contains four amines; our naming scheme refers to ratios of functional groups, not ratios of monomers.
| Sample | Functional group ratio in synthesis |
|---|---|
| TMC(2)-TAPM | 2 eq. acyl chlorides: 1 eq. amines |
| TMC(1.75)-TAPM | 1.75 eq. acyl chlorides: 1 eq. amines |
| TMC(1.5)-TAPM | 1.5 eq. acyl chlorides: 1 eq. amines |
| TMC(1.25)-TAPM | 1.25 eq. acyl chlorides: 1 eq. amines |
| TMC(1.1)-TAPM | 1.1 eq. acyl chlorides: 1 eq. amines |
| TMC(1)-TAPM | 1 eq. acyl chlorides: 1 eq. amines |
| TMC(0.67)-TAPM | 0.67 eq. acyl chlorides: 1 eq. amines |
We used solid-state nuclear magnetic resonance (ssNMR) spectroscopy to assess polymer composition (Fig. 2a and b).13C cross-polarized magic angle spinning (CP-MAS) solid-state NMR revealed an isolated signal at approximately 65 ppm, corresponding to the tetrahedral carbon in TAPM. Further, we identified the signal at approximately 167 ppm as the carbonyl carbon from TMC. Signals from aromatic carbons on both monomers overlap from 110–150 ppm, with the signal at ∼145 ppm likely corresponding to the aromatic carbon atoms adjacent to nitrogen. Within each spectrum, we compared the intensity of the 65 ppm peak (from TAPM) to the 167 ppm peak (from TMC). The intensity of the TMC peak relative to the TAPM peak increases moving from bottom to top in Fig. 2a; this increase corresponds to increasing acyl chloride content in the reaction. Thus, 13C NMR results support the hypothesis that increased stoichiometric amounts of TMC in a polymerization reaction lead to increased carbonyl content in the product. However, the ratio of TMC peak intensity to TAPM peak intensity appears to plateau at TMC(1.5)-TAPM, indicating a possible upper limit on carbonyl content.
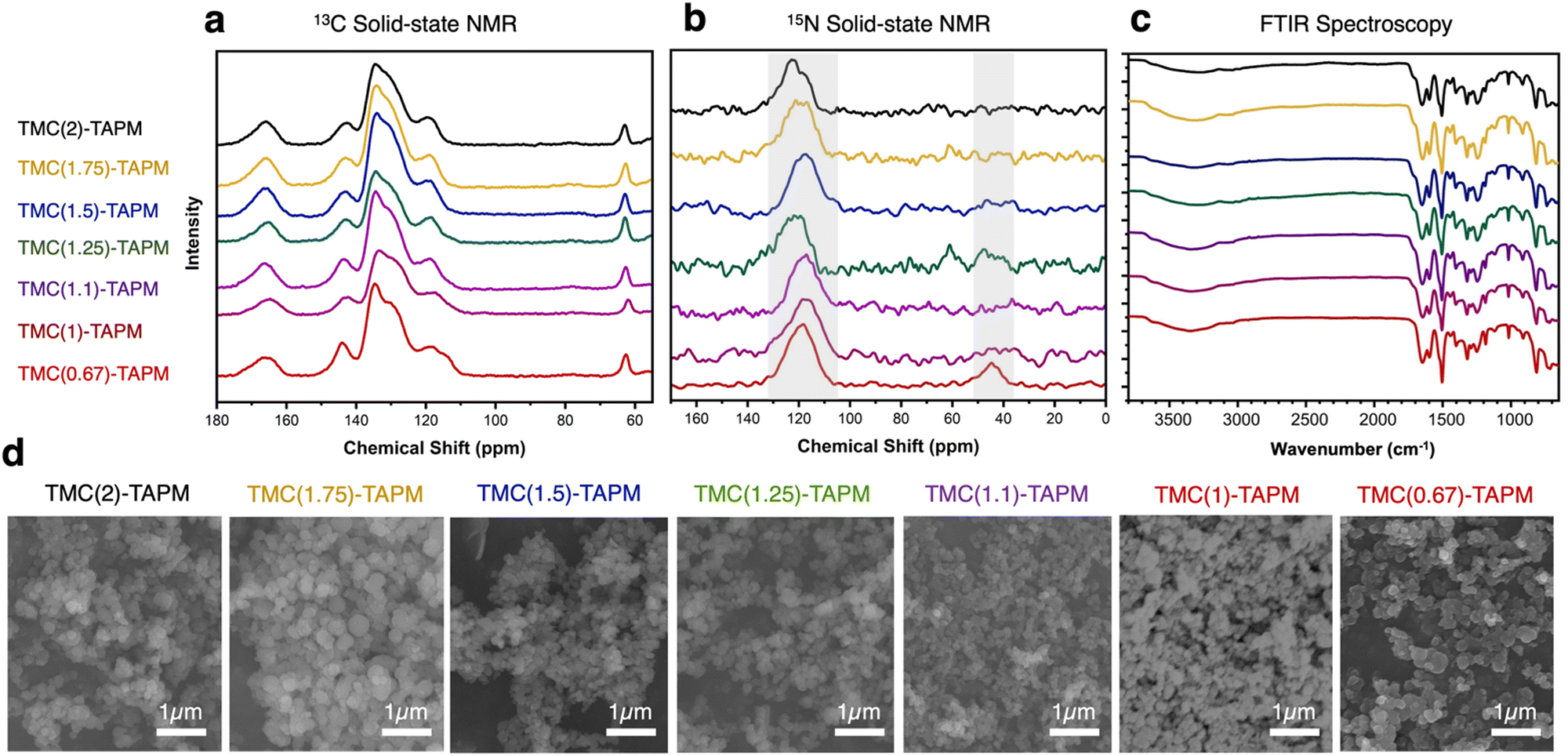 | ||
| Fig. 2 (a) 13C cross-polarized magic angle spinning (CP-MAS) solid-state nuclear magnetic resonance (NMR) spectroscopy, (b) 15N solid-state NMR spectroscopy, and (c) Fourier-transform infrared (FTIR) spectroscopy of the porous organic polymer samples. Spectra are color-coded as follows: TMC(2)-TAPM (black), TMC(1.75)-TAPM (gold), TMC(1.5)-TAPM (blue), TMC(1.25)-TAPM (green), TMC(1.1)-TAPM (purple), TMC(1)-TAPM (maroon), and TMC(0.67)-TAPM (red). (d) Scanning electron microscopy (SEM) images of porous organic polymer samples. Scale bar is 1 μm for all images. Data for TMC(1)-TAPM in parts (c) and (d) was published by us in previous work.28 | ||
Because peak integrations from 13C solid-state NMR data can provide only approximate values, C/H/N combustion analysis was performed for each polymer sample (see Table S2, ESI†). The results of combustion analysis show that the C:N ratio increases as TMC content in the synthesis increases, but this trend tapers off after TMC(1.5)-TAPM. These results are consistent with the 13C NMR spectra, indicating that the ratio of carbonyl groups (from TMC) to amino groups (from TAPM) is maximized in TMC(1.5)-TAPM. Thus, by varying the ratios of monomers used in synthesis, we are indeed able to vary the chemical composition of the products.
We were able to use the natural abundance of 15N to obtain 15N solid-state NMR spectra for the porous organic polymers (without synthesizing isotopically-enriched samples). All polymer samples show peaks corresponding to amide nitrogen atoms at approximately 120 ppm, confirming the formation of amide-linked materials (Fig. 2b).43 However, only TMC(0.67)-TAPM has an unambiguous second signal at approximately 45 ppm, indicating an unreacted amino group.44 These spectra indicate a somewhat surprising finding: Even a minor excess of acyl chloride groups is sufficient to react with all available amines present in the polymerization. We recognize that there may be some unreacted amines present in all samples that are not visible in the 15N solid-state NMR spectra. However, the data indicate a high degree of amide formation, and thus a high degree of connectivity, within the amide-linked polymers.
Fourier-transform infrared spectroscopy (FTIR) also provides evidence of amide formation in the crosslinked polymers; the peaks at approximately 1660 cm−1 are consistent with amide carbonyl stretches.45 The FTIR spectra show minimal differences between the various polymers, suggesting the pore environment of the porous organic polymers is chemically similar. Likewise, scanning electron microscopy (SEM) underscored the structural similarities of all polymer samples (Fig. 2d). SEM images show that the porous organic polymers are composed of nanoscale spheres that are agglomerated to form a rough surface. These results indicate that the morphology of the materials is independent of their chemical composition and functional group ratios.
We used N2 adsorption isotherms at 77 K to calculate the BET surfaces areas of the porous organic polymers,46 which vary from 44 m2 g−1 to 102 m2 g−1. The N2 adsorption results show that all the polymer samples are permanently porous and have BET surface areas within the same order of magnitude (discussed further below). The N2 adsorption isotherms were also used to determine the pore size distribution for each polymer (Fig. S8–S14, ESI†), showing a broad range of nanoscale pores for all polymer samples.
Considered in total, the NMR, C/H/N analysis, FTIR, SEM, and BET data indicate that the porous organic polymers studied here possess similar morphologies, porosities, and chemical backbones. Consequently, we hypothesized that differences in ion adsorption among the polymers would likely be a result of the different composition of unreacted end groups (carboxylate vs. amine). To test this hypothesis, we performed adsorption experiments in which polymer samples were submerged in aqueous solutions of known cobalt concentration. The samples were shaken for 24 hours, and the polymer was subsequently removed from solution by filtration. We quantified the amount of cobalt remaining in solution by microwave plasma atomic emission spectrometry (MP-AES) or inductively coupled optical emission spectrometry (ICP-OES), which allowed us to calculate the amount of cobalt adsorbed by the polymers.
We first selected three porous organic polymers for comparison across a range of cobalt concentrations (Fig. 3a). The resulting adsorption isotherms show a clear trend: As the carbonyl content of the polymer grows from TMC(0.67)-TAPM to TMC(1.5)-TAPM, the cobalt adsorption capacity increases by nearly an order of magnitude. The isotherms rise quickly at low cobalt concentrations (below approximately 30 ppm) and subsequently plateau, approaching saturation capacity by 400 ppm. This preliminary data strongly indicates that excess carbonyl content, resulting in a greater population of carboxylate end groups, is more important than excess amine content in maximizing cobalt adsorption capacity.
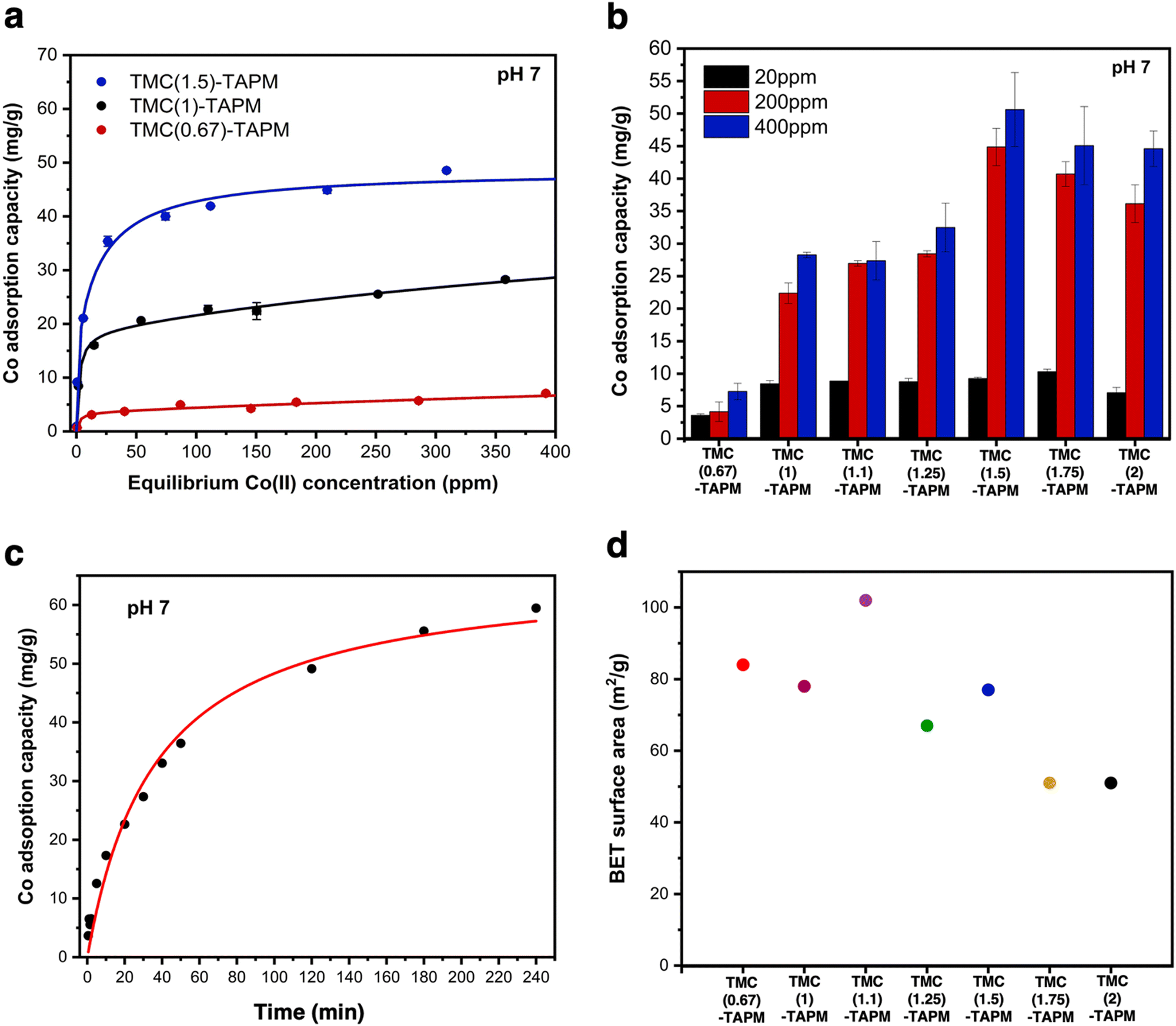 | ||
| Fig. 3 (a) Cobalt adsorption isotherms for TMC(0.67)-TAPM (red), TMC(1)-TAPM (black), and TMC(1.5)-TAPM (blue). (b) Cobalt adsorption experiments at 20 ppm (black), 200 ppm (red), and 400 ppm (blue) for all porous organic polymers. (c) Time-dependent cobalt adsorption capacities for TMC(1.5)-TAPM, with an initial cobalt concentration of 200 ppm. (d) BET surface area measurements for porous polyamides. Data for TMC(1)-TAPM in parts (a), (b), and (d) was published by us in previous work.28 | ||
To further investigate this trend, we tested all porous organic polymers at three representative cobalt concentrations (20 ppm, 200 ppm, and 400 ppm). In agreement with the previous results, polymer samples with excess carbonyl groups show higher cobalt uptake at all three concentrations (Fig. 3b). The measured cobalt adsorption capacity of the best performer, TMC(1.5)-TAPM, represents a relatively high value compared to other cobalt adsorbents in the literature.47–50 However, the improvements in cobalt uptake reach a plateau after the acyl chloride excess surpasses 1.5 equivalents; as the acyl chloride excess is pushed to 1.75 or 2 equivalents, cobalt capacity begins to decrease. Thus, these results provide a clear recipe for the optimal monomer ratio: 1.5 equivalents acyl chloride relative to 1 equivalent amine.
For crosslinked networks, an increase in the number of unreacted carbonyl end-groups means that carboxylate functional groups are present within the pore environment, and likewise for unreacted amino groups. We hypothesize that the improvements in cobalt capacity with increasing carbonyl content can be explained by the fact that the anionic character of the carboxylate group leads to a stronger interaction with Co2+ ions than does a neutral amino group. (Cobalt adsorption experiments were performed at pH 7; the aniline-like amino groups will not be protonated, and carboxylic acid groups will be deprotonated.) Further, the C/H/N analyses for TMC(1.75)-TAPM or TMC(2)-TAPM show that when an overwhelming excess of the acyl chloride monomer is present in the polymerization reaction, this does not translate to a corresponding excess of carbonyl groups relative to amino groups in the resulting polymers. This upper bound on the carbonyl content of the polymers is reflected in the cobalt adsorption trends in Fig. 3b.
A real-world cobalt extraction process will require the desorption of absorbed cobalt ions from the porous organic polymer, which not only liberates the desired cobalt but regenerates the adsorbent for subsequent reuse. We performed a sequence of preliminary regeneration experiments using TMC(1.75)-TAPM. First, a typical adsorption experiment was performed: The porous organic polymer was incubated with 400ppm aqueous cobalt solution, the polymer was removed from solution via filtration, and the change in cobalt concentration of the water sample was measured by MP-AES. The porous organic polymer was then washed with 1 M HCl (aq.) to remove the bound cobalt ions. This process was repeated twice, for a total of three adsorption–desorption cycles (Fig. S15, ESI†). Over the course of three cycles, cobalt uptake remained constant without loss in performance. We used 1H NMR experiments in 1 M DCl in D2O to confirm that the amide linkages in the polymer were stable under the acidic regeneration conditions, as detailed in the ESI.†
To investigate the kinetics of cobalt adsorption in the highest-capacity material, TMC(1.5)-TAPM, we removed aliquots of the supernatant from a 200-ppm adsorption experiment at set time intervals and quantified the cobalt concentration of the aliquots by MP-AES. The results provide insight into the time-dependent adsorption capacity of the polymer, shown in Fig. 3c. The data was fit to a linearized pseudo-second-order rate model (eqn (1)),41 in which Qt is the cobalt adsorption capacity at a given time point (in units of mg g−1), Qe is the cobalt adsorption capacity at equilibrium (in units of mg g−1), and k2 is the rate constant (in units of g (mg × min)−1).
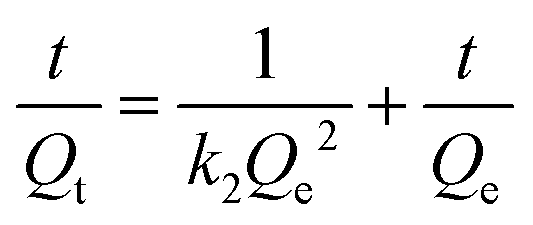 | (1) |
This fit predicts a relatively modest rate constant of 4.17 × 10−4 g (mg × min)−1 for TMC(1.5)-TAPM. As has been shown for ion adsorption in other porous polymers, increased surface area can improve the rate of adsorption.51 To further improve the ion adsorption kinetics of TMC-TAPM polymers in future work, increases in surface area may be achieved through particle size control techniques such as ball milling.
The BET surface areas for the series of polyamides are plotted in Fig. 3d, revealing a rough downward trend with increasing equivalents of acyl chloride. Notably, the trend in BET surface area does not mirror the trend observed for cobalt adsorption shown in Fig. 3b. This observation supports the role of chemical functionality (i.e., carboxylate content), rather than BET surface area, in determining the cobalt adsorption capacity of the adsorbents in this study. We also observed a correlation between BET surface area and particle size, as imaged by SEM (Fig. 2d). In particular, TMC(1.1)-TAPM stands out as having notably small particles and the highest BET surface area, while TMC(1.75)-TAPM stands out as having notably large particles and low surface area. These trends further support particle size control as a possible future strategy to maximize cobalt capacity: While maintaining optimal carboxylate content, BET surface area may be improved by tuning particle size, leading to an even greater cobalt adsorption capacity.
Finally, we performed multicomponent adsorption experiments to determine the affinity of TMC(1.5)-TAPM for other transition metals. A solution containing 40 ppm Mn(II), 40 ppm Ni(II), 40 ppm Co(II), and 40 ppm Zn(II) was prepared and used for a series of adsorption experiments (Fig. S17, ESI†). The results show that TMC(1.5)-TAPM has comparable adsorption capacity for all four transition metal ions tested, with a slight selectivity for Zn(II) (which may arise from the increased charge density of Zn(II) and its lack of a preference for a given coordination geometry, as a d10 ion).52 Thus, the trends described herein for cobalt adsorption capacity in carbonyl-rich polymers may be more broadly applicable to the capture of other critical metals.
Conclusions
In summary, we have synthesized a library of amide-linked porous organic polymers with varying monomer ratios. Through a series of materials characterization techniques, we have shown that the chemical composition and pore functionality of the polymers can be tuned by simply varying the monomer ratios, while porosity, morphology, and nanostructure remain relatively constant. The differences between amine-rich and carbonyl-rich materials are apparent in the cobalt adsorption capacities of the polymers: Comparing the extremes within this study, cobalt uptake improves 10-fold by increasing carbonyl content. Our results further show that carbonyl content and cobalt uptake reach a plateau past a given monomer ratio. The reported polymers are stable to acidic regeneration conditions, allowing for successful adsorption–desorption cycles, and are capable of adsorbing other transition metal ions such as Mn(II) and Ni(II). These insights can guide synthetic chemists in preparing high-capacity ion adsorbents for applications in water purification and transition metal capture.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the University of Maryland College Park for funding. We thank the National Science Foundation (NSF-1726058) for funding a solid-state NMR spectrometer. We acknowledge the support of the Maryland NanoCenter and its AIMLab. We gratefully acknowledge the support of the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the US DOE Office of Science. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the US DOE's National Nuclear Security Administration (contract no. DE-NA-0003525). The views expressed in the article do not necessarily represent the views of the US DOE or the US government.References
- Executive summary – The Role of Critical Minerals in Clean Energy Transitions – Analysis – IEA, https://www.iea.org/reports/the-role-of-critical-minerals-in-clean-energy-transitions/executive-summary, (accessed 20 January 2023).
- Lithium-ion batteries need to be greener and more ethical, Nature, 2021, 595, 7, DOI:10.1038/d41586-021-01735-z.
- The Dark Side of Congo's Cobalt Rush | The New Yorker, https://www.newyorker.com/magazine/2021/05/31/the-dark-side-of-congos-cobalt-rush, (accessed 20 January 2023).
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- Powering the Blue Economy: Exploring Opportunities for Marine Renewable Energy in Maritime Markets | Tethys, https://tethys.pnnl.gov/publications/powering-blue-economy-exploring-opportunities-marine-renewable-energy-maritime-markets, (accessed 20 January 2023).
- W.-W. Li, H.-Q. Yu and B. E. Rittmann, Nature, 2015, 528, 29–31 CrossRef CAS PubMed.
- P. Gikas and A. N. Angelakis, Desalination, 2009, 248, 1049–1064 CrossRef CAS.
- M. Qadir, B. R. Sharma, A. Bruggeman, R. Choukr-Allah and F. Karajeh, Agric. Water Manage., 2007, 87, 2–22 CrossRef.
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS PubMed.
- E. Ahmadi, B. McLellan, B. Mohammadi-Ivatloo and T. Tezuka, Sustainability, 2020, 12, 5233 CrossRef.
- P. C. Nagajyoti, K. D. Lee and T. V. M. Sreekanth, Environ. Chem. Lett., 2010, 8, 199–216 CrossRef CAS.
- L. Leyssens, B. Vinck, C. Van Der Straeten, F. Wuyts and L. Maes, Toxicology, 2017, 387, 43–56 CrossRef CAS PubMed.
- S. H. Farjana, N. Huda and M. A. P. Mahmud, J. Sustainability Min., 2019, 18, 150–161 CrossRef.
- N. Chaoui, M. Trunk, R. Dawson, J. Schmidt and A. Thomas, Chem. Soc. Rev., 2017, 46, 3302–3321 RSC.
- S. Qiu and T. Ben, Porous Polymers, 2015 Search PubMed.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- S. Lee, G. Barin, C. M. Ackerman, A. Muchenditsi, J. Xu, J. A. Reimer, S. Lutsenko, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 7603–7609 CrossRef CAS PubMed.
- S. Lee, A. Uliana, M. K. Taylor, K. Chakarawet, S. R. S. Bandaru, S. Gul, J. Xu, C. M. Ackerman, R. Chatterjee, H. Furukawa, J. A. Reimer, J. Yano, A. Gadgil, G. J. Long, F. Grandjean, J. R. Long and C. J. Chang, Chem. Sci., 2019, 10, 6651–6660 RSC.
- M. X. Tan, Y. N. Sum, J. Y. Ying and Y. Zhang, Energy Environ. Sci., 2013, 6, 3254–3259 RSC.
- Y. Yang, Z. Yan, L. Wang, Q. Meng, Y. Yuan and G. Zhu, J. Mater. Chem. A, 2018, 6, 5202–5207 RSC.
- B. Li, Y. Zhang, D. Ma, Z. Shi and S. Ma, Nat. Commun., 2014, 5, 5537 CrossRef CAS PubMed.
- Y. He, Q. Liu, J. Hu, C. Zhao, C. Peng, Q. Yang, H. Wang and H. Liu, Sep. Purif. Technol., 2017, 180, 142–148 CrossRef CAS.
- A. Sen, S. Sharma, S. Dutta, M. M. Shirolkar, G. K. Dam, S. Let and S. K. Ghosh, ACS Appl. Mater. Interfaces, 2021, 13, 34188–34196 CrossRef CAS PubMed.
- A. K. Sekizkardes, P. Wang, J. Hoffman, S. Budhathoki and D. Hopkinson, Mater. Adv., 2022, 3, 6668–6686 RSC.
- S. Canossa and S. Wuttke, Adv. Funct. Mater., 2020, 30, 2003875 CrossRef CAS.
- N. Enjamuri, S. Sarkar, B. M. Reddy and J. Mondal, Chem. Rec., 2019, 19, 1782–1792 CrossRef CAS PubMed.
- A. Modak, P. Bhanja, M. Selvaraj and A. Bhaumik, Environ. Sci.: Nano, 2020, 7, 2887–2923 RSC.
- D. S. Rollins, C. P. Easterling, A. N. Zeppuhar, J. A. Krawchuck, T. A. Dreier, J. Watt, D. L. Huber and M. K. Taylor, Nanoscale, 2022, 14, 299–304 RSC.
- F. S. Awad, A. M. Bakry, A. A. Ibrahim, A. Lin and M. S. El-Shall, Ind. Eng. Chem. Res., 2021, 60, 12675–12688 CrossRef CAS.
- F. Ahmadijokani, S. Tajahmadi, A. Bahi, H. Molavi, M. Rezakazemi, F. Ko, T. M. Aminabhavi and M. Arjmand, Chemosphere, 2021, 264, 128466 CrossRef CAS PubMed.
- R. R. Navarro, K. Sumi and M. Matsumura, Water Sci. Technol., 1998, 38, 195–201 CrossRef CAS.
- B. N. Bhadra, I. Ahmed, H. J. Lee and S. H. Jhung, Coord. Chem. Rev., 2022, 450, 214237 CrossRef CAS.
- G. E. Decker, G. R. Lorzing, M. M. Deegan and E. D. Bloch, J. Mater. Chem. A, 2020, 8, 4217–4229 RSC.
- A. Mangalum, C. D. McMillen and A. G. Tennyson, Inorg. Chim. Acta, 2015, 426, 29–38 CrossRef CAS.
- B.-H. Ye, M.-L. Tong and X.-M. Chen, Coord. Chem. Rev., 2005, 249, 545–565 CrossRef CAS.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- E. Cevik and A. Bozkurt, J. Energy Chem., 2021, 55, 145–153 CrossRef CAS.
- A. Queen, Can. J. Chem., 1967, 45, 1619–1629 CrossRef CAS.
- V. Gabara, Ullmann's Encyclopedia of Industrial Chemistry, M. Bohnet, John Wiley & Sons, 2016, pp. 1–22 Search PubMed.
- L. Cosimbescu, D. Malhotra, M. R. Pallaka and M. S. Swita, ACS Omega, 2022, 7, 32026–32037 CrossRef CAS PubMed.
- D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899–7925 RSC.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, John Wiley & Sons, 2000 Search PubMed.
- G. R. Hatfield, Y. Guo, W. E. Killinger, R. A. Andrejak and P. M. Roubicek, Macromolecules, 1993, 26, 6350–6353 CrossRef CAS.
- A. Dokalik, H. Kalchhauser, W. Mikenda and G. Schweng, Magn. Reson. Chem., 1999, 37, 895–902 CrossRef CAS.
- V. F. Mandyuk and N. P. Lushina, J. Appl. Spectrosc., 1968, 9, 1121–1123 CrossRef.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X.-H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. M. D’Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- W. Luo, Z. Bai and Y. Zhu, RSC Adv., 2018, 8, 13370–13387 RSC.
- M. Abbas, S. Kaddour and M. Trari, J. Ind. Eng. Chem., 2014, 20, 745–751 CrossRef CAS.
- M. He, Y. Zhu, Y. Yang, B. Han and Y. Zhang, Appl. Clay Sci., 2011, 54, 292–296 CrossRef CAS.
- E. Demirbaş, Adsorpt. Sci. Technol., 2003, 21, 951–963 CrossRef.
- J. Kamcev, M. K. Taylor, D.-M. Shin, N. N. Jarenwattananon, K. A. Colwell and J. R. Long, Adv. Mater., 2019, 31, 180802748 CrossRef PubMed.
- S. J. Archibald, Comprehensive Coordination Chemistry II, ed. D. E. Fenton, Elsevier, 2nd edn, 2003, vol. 6, pp. 1147–1251 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc03320a |
| This journal is © The Royal Society of Chemistry 2023 |