
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced brightness of ultra-small gold nanoparticles in the second biological window through thiol ligand shell control†
Walaa
Mohammad‡
a,
K. David
Wegner‡
b,
Clothilde
Comby-Zerbino
c,
Vanessa
Trouillet
d,
Marina Paris
Ogayar
e,
Jean-luc
Coll
 a,
Riccardo
Marin
fg,
Daniel Jaque
Garcia
efg,
Ute
Resch-Genger
b,
Rodolphe
Antoine
*c and
Xavier
Le Guével
*a
a,
Riccardo
Marin
fg,
Daniel Jaque
Garcia
efg,
Ute
Resch-Genger
b,
Rodolphe
Antoine
*c and
Xavier
Le Guével
*a
aUniversity of Grenoble Alpes, Institute for Advanced Biosciences, INSERM1209/CNRS-UMR5309, Grenoble, France. E-mail: xavier.le-guevel@univ-grenoble-alpes.fr
bFederal Institute for Materials Research and Testing (BAM), Richard-Willstaetter-Str. 11, 12489 Berlin, Germany
cUniv Lyon, Univ Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622, Villeurbanne, France. E-mail: rodolphe.antoine@univ-lyon1.fr
dInstitute for Applied Materials (IAM) and Karlsruhe Nano Micro Facility (KNMFi), Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen, Germany
eNanomaterials for Bioimaging Group (nanoBIG), Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS) Hospital Ramón y Cajal, Ctra. De Colmenar Viejo km 9.100, Madrid 28034, Spain
fNanomaterials for BioImaging Group (nanoBIG), Departamento de Física de Materiales, Universidad Autónoma de Madrid, C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain
gInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 6th October 2023
Abstract
Gold-based nanoparticles below 2 nm in size are promising as luminescent probes for in vivo bioimaging, owing to their brightness and rapid renal clearance. However, their use as contrast agents in the near-infrared II (NIR-II, 1000–1700 nm) range remains challenging due to their low photoluminescence (PL) quantum yield. To address this, PL enhancement can be achieved by either rigidifying the ligand-shell structure or increasing the size of the ligand shell. In this study, we synthesized ultra-small gold nanoparticles stabilized by co-ligands, namely monothiol and short dithiol molecules. By precisely controlling the amount of reducing agent used during particle preparation, we successfully modulated the physicochemical properties of the co-ligand shell, including its size, composition, and structure. Consequently, we achieved a remarkable 60-fold increase in the absorption cross-section at 990 nm while maintaining the small size of the 1.5-nm metal core. The analytical and optical characterization of our thiol-capped gold nanoparticles indicates that the ligand shell size is governed by the quantity of the reducing agent, which, in turn, impacts the balance between radiative and non-radiative processes, thereby influencing the PL quantum yield.
Introduction
In vivo optical imaging has progressed in the past decade to non-invasively detect early-stage diseases and monitor physiological changes in real time in different organs like brain or heart.1,2 A successful strategy is to perform optical imaging in the second infrared region between 1000 and 1700 nm, which is referred to as NIR-II (or shortwave infrared, SWIR).3 This optical window provides a high spatial resolution – down to a few millimeters – due to reduced scattering and autofluorescence of biological tissue compared to the visible (400–700 nm) and the first near infrared region (NIR-I) between 700 and 900 nm. Following the seminal work of the research groups of Dai and Bawendi,4 a myriad of new NIR-II emitting reporters and probes made from different classes of emitters have been designed. These include organic polymethine, xanthene, and bodipy dyes as well as AIEgens,5,6 carbon based nanomaterials like carbon nanotubes and inorganic nanoparticles like semiconductor quantum dots, lanthanide nanoparticles, and ultra-small gold nanoparticles (us-AuNPs).7–9 NIR-II probes have been meanwhile employed for the diagnosis of several pathologies.4,8,10,11 In oncology especially they have been used for imaging vascular and lymphatic systems and to support tumor resection by optical guided surgery.12–15The requirements for NIR-II probes for biomedical applications are the following: excitability in the infrared to enable high penetration depth in living tissue, a high brightness (i.e., the product of the photoluminescence quantum yield (PLQY) and the molar absorption coefficient at the excitation wavelength), and a high photostability.10,11 For in vivo applications, these emitters should be biocompatible and water-dispersible, with a high colloidal stability at physiological conditions of pH and ionic strength. Moreover, it should be possible to functionalize them with molecules for targeting and/or delivery purposes.
Compared to other inorganic NIR-II probes, luminescent us-AuNPs, also often referred to as gold nanoclusters, have several advantages for photoluminescence (PL) bioimaging, like tunable PL in the 1000–1300 nm range,16–19 PL lifetimes in the microsecond range,16 and PLQY of 0.1 to 6.7% in aqueous solution.14,18,20 Usually, such us-AuNPs are composed of a metal core of a few tens to hundreds of atoms protected/covered by (bio)organic ligands.21–23 Therefore, the pharmacokinetic of these probes can be tailored by the chosen surface ligands.24 Thanks to their ultra-small size, which is usually less than 2 nm and way below the kidney filtration cut-off,25 these us-AuNPs exhibit a high renal clearance. This prevents long-term accumulation in the reticuloendothelial system (RES) leading to severe toxicity.26
Different strategies to tune the emission properties of NIR-II emissive us-AuNPs have been explored. A strong emission in the NIR-II18 can be achieved by controlling the size and the physicochemical properties of the ligand shell, as demonstrated with multishell zwitterionic us-AuNPs.27 Alternatively, us-AuNPs can be encapsulated in polymers or rigid, inorganic nanostructures.28,29 The observed enhanced PL is ascribed to the rigidification of the structure of the us-AuNPs or to an increase in ligand shell size. The former reduces the energy loss caused by intramolecular rotations and vibrations, thus increasing the PL intensity of the nanoclusters and therefore their PLQY. Possible PL quenching by water molecules penetrating into the ligand shell can also be significantly reduced with the multi-shell ligand and encapsulation approaches.30 Surface shell protection can also be obtained with bulky counter-ions or protein templating of us-AuNPs.31,32 In the latter case, a change in the gold–thiolate interface could lead to both a PL enhancement and a red-shift in PL.32 Control of the spatial organization of the organic shell protecting the metal core could also contribute to enhance the NIR absorption and shift the PL emission to longer wavelength.18,33 Engineering the thiol ligand shell by collocation of different charged ligands leading to surface charge anisotropy can boost the NIR absorption of AuNPs.34 A key parameter can be here the tailoring of the thickness of the thiol ligand shell during particle synthesis selecting the nature and strength of the reducing agent that is involved in the formation of the gold thiolate complexes, the nucleation of the gold core, and the formation of disulfide bonds between ligands.
In this study, we prepared us-AuNPs stabilized by monothiol and short dithiol molecules that have shown promising optical properties in earlier studies18,35 for in vivo applications.14 Such us-AuNPs are typically obtained by combining metal salt, ligands, and a reducing agent, stirred at room temperature. However, the reducing agent plays a crucial role as it directly reduces Au ions to form the atomically precise metal core in the presence of the ligands. It also controls the growth of the core and the formation of the ligand shell in the resulting us-AuNPs. By adjusting the concentration of the reducing agent, it was possible to fine-tune the physicochemical properties of the co-ligand shell. This resulted in more than a 60-fold enhancement of the absorption cross-section at approximately 990 nm while maintaining a small metal core size below 2 nm. Moreover, we demonstrated the advantages of these us-AuNPs with mixed ligand shells for imaging applications. We successfully detected NIR-II signals from these emitters at a penetration depth of up to 6 mm, exhibiting high sensitivity and a high signal-to-noise ratio even at greater depths in intralipids when excited at 980 nm.
Experimental section
Synthesis
All chemical products were purchased from Sigma-Aldrich (France) and syntheses were conducted using Ultra MiliQ water (16 MΩ).Bi-thiol-protected us-AuNPs, AuMHA/HDT, were synthesized by wet chemistry in alkaline condition. Briefly, 250 μL HAuCl4 solution (20 mM) is added to 2.4 mL of water followed by a slow addition of a mixture of 6-mercaptohexanoic acid (MHA, 1.25 mL, 5 mM) and hexa(ethyleneglycol) dithiol (HDT, 0.75 mL, 5 mM) under mild stirring (350 rpm) keeping a molar ratio Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MHA:HDT = 1:1.4:0.6. 250 μL of NaOH (1 M) was then added dropwise to adjust the pH to 10. Afterwards, the freshly prepared reducing agent, NaBH4 (20 mM), was added dropwise using different amounts (0.5, 2, 4, 6, 10, 12.5, and 16 μmol corresponding to molar ratios Au:NaBH4 of 1:0.1 to 1:3.2) to prepare a series of us-AuNPs that were stirred for 16 hours and 30 minutes at room temperature. The reaction solution was washed several times using an Amicon centrifuge filter (Milipore) 3 kDa to remove unreacted species. Then the pH was adjusted to 7 and the NPs were kept at room temperature in the dark.
:MHA:HDT = 1:1.4:0.6. 250 μL of NaOH (1 M) was then added dropwise to adjust the pH to 10. Afterwards, the freshly prepared reducing agent, NaBH4 (20 mM), was added dropwise using different amounts (0.5, 2, 4, 6, 10, 12.5, and 16 μmol corresponding to molar ratios Au:NaBH4 of 1:0.1 to 1:3.2) to prepare a series of us-AuNPs that were stirred for 16 hours and 30 minutes at room temperature. The reaction solution was washed several times using an Amicon centrifuge filter (Milipore) 3 kDa to remove unreacted species. Then the pH was adjusted to 7 and the NPs were kept at room temperature in the dark.
The monothiol sample AuMHA was prepared using the molar ratio Au:MHA = 1:2 and 2 μmol of NaBH4 as the amount of reducing agent did not influence the optical properties (absorbance, fluorescence, PLQY) of this compound. The reaction solution was stirred for 3 hours, purified and stored using the same protocol described above.
Instrumentations and methods
The sizes of the metal cores were determined by high resolution transmission electron microscopy JEOL2010 (HR-TEM) using a monochromated microscope working at 200 kV. Prior to imaging, the us-AuNPs were dispersed on copper grids covered with a carbon film.X-ray photoelectron spectroscopy (XPS) investigation was performed in a K-Alpha+ spectrometer (ThermoFisher Scientific) using a microfocused, monochromated Al Kα X-ray source (400 μm spot size). The Thermo Avantage software was used for data acquisition and processing. The K-Alpha+ charge compensation system was employed during analysis, using electrons of 8 eV energy, and low-energy argon ions to prevent any localized charge build-up. The spectra were fitted with one or more Voigt profiles (BE uncertainty: ±0.2 eV) and Scofield sensitivity factors were applied for quantification.36 All spectra were referenced to the C 1s peak at 285.0 eV binding energy (C–C, C–H) and controlled by means of the well-known photoelectron peaks of Cu, Ag, and Au, respectively. Measurements were performed on three different locations of dried samples.
Evolution of the us-AuNPs size was determined by positive ion mode nano-electrospray ionization–variable temperature (nanoESI-VT, customized source) on a commercial quadrupole time-of-flight (micro-qTOFII, Bruker-Daltonics, Bremen, Germany, mass resolution ∼10000). The temperature of the solution was set at 17 °C. The samples were prepared by diluting nanoclusters in water, to a final concentration of 0.25–0.5 mg mL−1 in water. External calibration was carried out with a set of synthetic peptides.
The hydrodynamic diameter and the zeta potential of the samples dispersed in phosphate buffer (PBS, pH 7.4) were measured in duplicate on a Zetasizer instrument from Malvern.
The absorption spectra of diluted us-AuNPs samples were recorded on PerkinElmer Lambda 1050 UV-vis spectrophotometer between 350 and 1300 nm. Steady-state photoluminescence spectra were measured from 700–1600 nm with a calibrated FSP 920 (Edinburgh Instruments, Edinburgh, United Kingdom) spectrofluorometer equipped with a nitrogen-cooled PMT R5509P.
Measurements of the relative PLQYs (Φf,x) were performed using the reference dye IR125 dissolved in dimethylsulfoxide (DMSO). The PLQY of this dye was previously determined absolutely to (Φf,st = 0.23).37 The relative PLQYs were calculated according to the formula of Demas and Crosby,38 see equation below.
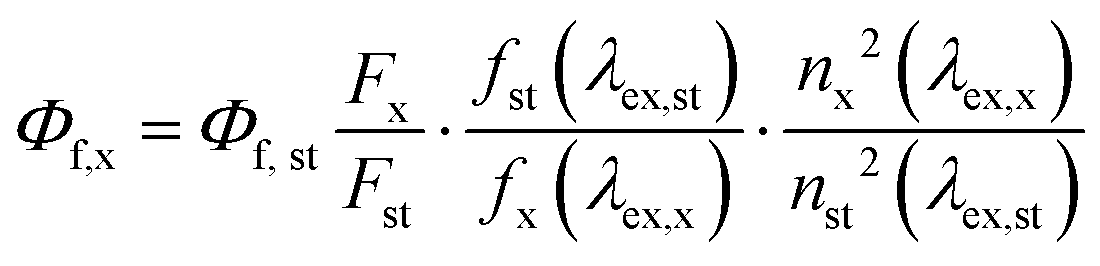
The subscripts x, st, and ex denote sample, standard, and excitation respectively. f(λex) is the absorption factor, F the integrated spectral fluorescence photon flux, and n the refractive index of the solvents used (DMSO for IR125 water for us-AuNPs).
All spectroscopic measurements were done in a 1 cm quartz cuvettes from Hellma GmbH at room temperature using air-saturated solutions.
Lifetime measurements were performed using a thermoelectric cooled NIR Photomultiplier tube (Hamamatsu H10330C-45) connected to a Time-Correlated Single Photon Counting Timeharp 260 system from PicoQuant equipped with two picosecond pulsed diode lasers EPL-640 and EPL-450 (Edinburgh Instruments) with 634.3 nm excitation and 82.4 ps pulse width and 450.8 nm excitation with 79.1 ps pulse width, respectively. An 850 nm long-pass filter (FELH0850 Thorlabs) was used for the 634 nm excitation and a 500 nm long-pass filter (FELH0500 Thorlabs) for the 450 nm excitation. The resulting broadband emission bands were monitored for lifetime analysis using a photomultiplier connected to a time-correlated single photon counting system. All measurements were performed at a fixed temperature of 20 °C using an air-cooled qpod 3e from Quantum Northwest.
NIR-II imaging was performed using a Princeton camera 640ST (900–1700 nm) coupled with a laser excitation source at λ = 980 nm (100 mW cm−2). We used short-pass excitation filter at 1000 nm (Thorlabs) and long pass filters on the NIR-II camera at 1064 nm (Semrock), and at 1250 nm (Thorlabs). A 50 mm lens with 1.4 aperture (Navitar) was used to focus on the samples. Capillaries containing us-AuNPs solution were soaked in intralipid solution (1%vol) at different depths and put in front of the camera before acquisition and analyses were performed using FIJI software.
Results and discussion
1 Physicochemical characterizations
The us-AuNPs were synthesized through a two-step process using wet chemistry. This process involves forming gold complexes with both monothiol molecules MHA and dithiol molecules HDT. Subsequently, the us-AuNPs are formed by utilizing NaBH4 as a reducing agent (Fig. 1). The amount of reducing agent used during the synthesis was varied from 0.5 to 16.0 μmol, corresponding to a molar ratio of gold to reducing agent NaBH4 ranging from 1:0.1 to 1:3.2. The synthesized us-AuNPs were characterized by TEM, and the size distribution of their cores and interparticle distances were evaluated by analyzing at least 200 NPs (Fig. 2 and Fig. S1–S7, ESI†). The analysis revealed a consistent size distribution of the gold cores around 1.5 ± 0.3 nm, independently of the amount of NaBH4 used. This core size was also similar to that of the us-AuNPs prepared using only MHA and 2.0 μmol of the reducing agent (Fig. S8 and Table 1). The TEM images indicated no aggregation of the us-AuNPs, and the interparticle distances ranged between 2.9 and 4.4 nm for all samples.
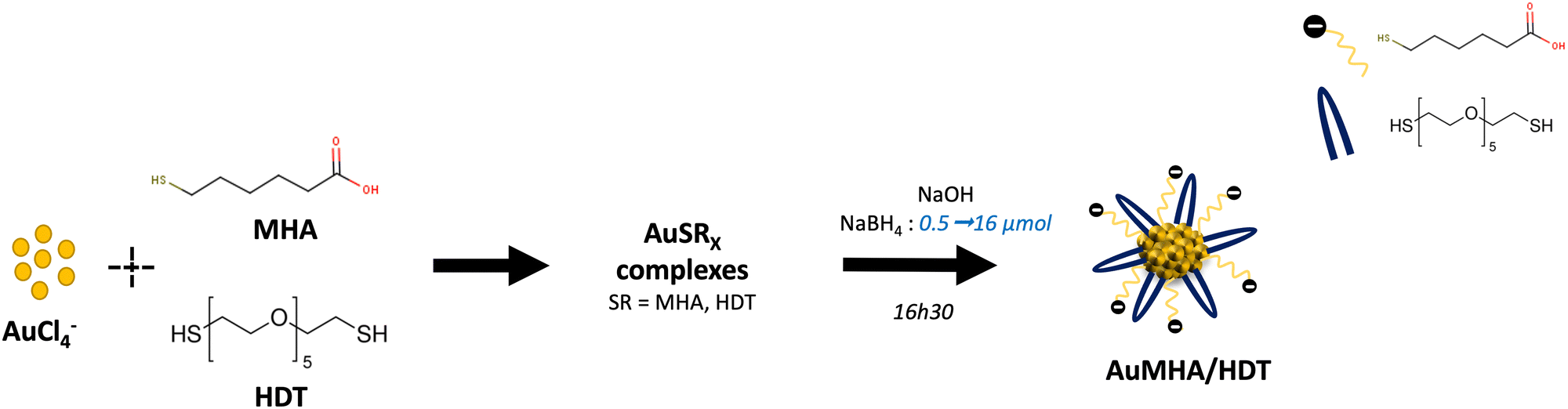 | ||
| Fig. 1 Synthesis of AuMHA/HDT in water using different amounts of NaBH4. | ||
 | ||
| Fig. 2 TEM images and core size analysis of AuMHA/HDT in water prepared with 0.5 (a), 2 (b), 4 (c), 6 (d), 10 (e), 12.5 (f), and 16 (g) μmol of NaBH4. | ||
| AuMHA/HDT NaBH4 (μmol) | Mean zeta potential (mV) | Core seize by TEM (nm) | Molecular weight by ESI-MS (kDa) |
|---|---|---|---|
| 0.5 | −32.9 ± 3.2 | 1.4 ± 0.3 | 8.7 ± 1.0 |
| 2 | −2.73 ± 4.9 | 1.4 ± 0.3 | 10.7 ± 1.4 |
| 4 | −2.8.2 ± 3.1 | 1.5 ± 0.4 | 10.6 ± 2.0 |
| 6 | −23.9 ± 2.1 | 1.4 ± 0.2 | 11.8 ± 3.5 |
| 10 | −25.3 ± 1.8 | 1.2 ± 0.3 | — |
| 12.5 | −31.0 ± 1.7 | 1.6 ± 0.4 | 12.0 ± 5.6 |
| 16 | −18.0 ± 1.5 | 1.5 ± 0.4 | 13.1 ± 5.6 |
Elemental analysis indicated that the gold content in all samples reached 45 wt% of the total weight of the NPs (Fig. 3(a)). This value aligns with many other reports in the literature39,40 and indicates that the overall reaction yield is not affected by the amount of reducing agent used during the synthesis. Only the highest amount of the reducing agent led to a decrease to 26 wt% of Au in the AuMHA/HDT sample. This could be attributed to a lower reaction yield, where unreacted Au was removed during the purification step (Fig. 3(a)).
 | ||
| Fig. 3 (a) Gold concentration in AuMHA/HDT as a function of NaBH4 content during the synthesis determined by elemental analysis. (b) ESI-MS spectra for AuMHA/HDT prepared with different amount of NaBH4 in positive mode except for AuMHA sample in negative mode. (c) Relative HDT concentration on us-AuNPs surface estimated by XPS measurement. | ||
Mass spectrometry using electrospray ionization (ESI†) can provide information about the total molecular weight (MW) of the samples, and we observed an increase in the m/z distributions (Fig. S9a, ESI†), corresponding to a quasi-linear increase in the average MW from 8.7 kDa using 0.5 μmol of NaBH4 to 13.1 kDa for 16.0 μmol of NaBH4 (Fig. S9b, ESI†). Since a consistent size distribution of the gold core around 1.5 ± 0.3 nm was observed regardless of the amount of NaBH4, the mass spectrometry data suggest that the increase in molecular weight for us-AuNPs by increasing the amount of the reducing agent is due to an increase in the number of ligands constituting the organic shell that protects the gold core (Fig. 3(b) and Fig. S9, (ESI†), Table 1).
X-Ray photoelectron spectroscopy (XPS) measurements were performed to obtain information on the oxidation state of gold and the nature of the ligand binding to the gold surface (Fig. 3(c) and Fig. S10, ESI†). Due to the presence of only one doublet Au 4f in the spectrum independently of the presence of the co-ligand HDT or the amount of NaBH4 used during the synthesis, we can affirm that no significant difference in the oxidation of gold could be observed. A binding energy between Au(0) at 84.0 eV and Au(I) at 85.5 eV is commonly reported for us-AuNPs.39 We quantified the amount of HDT by evaluating the intensity of the carbon peak at 286.5 eV, which is attributed to the C–O species present only in the HDT ligand (Fig. 3(c)). This estimation was validated by the significant difference observed when comparing AuMHA/HDT NPs to AuMHA NPs, where the HDT ligand is not present. We observed a linear increase in HDT molecules when increasing NaBH4 to 6.0 μmol, suggesting that NaBH4 promotes the binding of HDT to the gold surface. Above 6.0 μmol, no further correlation was observed and the relative error is large. We hypothesize that this may be due to the thicker organic shell and the difficulty of providing additional HDT to the gold core, possibly due to steric hindrance and the potential rearrangement of the two ligands in the organic shell. Notably, above 6.0 μmol, the increase in average MW is significantly reduced, confirming this saturation effect in the evolution of the ligand shell observed by XPS based on the concentration of C 1s at 286.5 eV (compare Fig. 3(c) and Fig. S9b, ESI†). It was also recently reported that a high amount of the reducing agent can promote the formation of disulfide bonds,41,42 which in our case may lead to the crosslinking of HDT molecules, thereby weakening the thiol interaction with the gold core surface.
The zeta potential of AuMHA/HDT dispersed in water was measured to be approximately −28 ± 3 mV (see Table 1), confirming the negatively charged surface of these samples due to the presence of carboxyl groups from MHA. Only with the highest amount of NaBH4 did we observe a less negative zeta potential (−18 ± 2 mV). This could be attributed to a different distribution of HDT within the ligand shell, which is more neutral than MHA at pH 7.
Therefore, the physicochemical characterizations conducted via elemental analyses and mass spectrometry suggest that increasing the amount of the reducing agent leads to a thicker organic shell that protects the gold core and reduces the surface charge. This thicker organic shell may be accompanied by a restructuring of the co-ligand shell, as indicated by the direct interaction (HDT vs. MHA) with the gold surface.
2 Optical characterizations
The variation in the amount of sodium borohydride, which initiates the growth of the us-AuNPs, significantly influences the optical properties of the resulting us-AuNPs. These effects were reproducible across all series of samples (Fig. S11, ESI†). As shown in Fig. 4(a), increasing the amount of NaBH4 from 0.5 to 16.0 μmol led to an increase in the intensity of the absorption band, which was approximately centered at 990 nm. To evaluate the absorption profile, we divided the absorption spectrum into three optical windows: 650–720 nm (I), 720–920 nm (II), and 920–1250 nm (III). The evolution of the area under each spectral window was plotted as a function of the NaBH4 concentration after normalizing the spectra and applying deconvolution (see Fig. 4(b) and Fig. S12, S13, ESI†). The first band (I), centered at 670 nm, was not affected by the amount of NaBH4. Band II exhibited a rapid and strong increase, reaching up to a 4-fold enhancement at low NaBH4 concentrations, followed by a linear decrease. A significant 60-fold enhancement of band III, centered at 990 nm, was observed, reaching its maximum with the use of 12.5 μmol of NaBH4. The molar extinction coefficient determined at 990 nm, using the average molecular weight of each sample from the ESI-MS data, showed a consistent increase from 0.1 × 103 M−1 cm−1 to 1.69 × 104 M−1 cm−1 (Table 2). Changes in the ratio of the ligands MHA:HDT did not affect the impact of NaBH4 content on the optical properties (Fig. S14, ESI†). Additionally, we can exclude the influence of the counterion of the reducing agent by using tetramethylammonium borohydride (Fig. S15, ESI†). Only acidic pH below 4 led to a fall of the band III of these samples (Fig. S16, ESI†). | ||
| Fig. 4 (a) Absorption spectra of AuMHA/HDT prepared using different concentrations of NaBH4. (b) Evolution of the area of the absorption bands in the region 650–720 nm (I), 720–920 nm (II), and 920–1250 nm (III) on normalized and deconvoluted spectra (see ESI†). | ||
| NaBH4 (μmol) | ε 990 (M−1 cm−1 M−1 × 103) | PL QY (%) |
|---|---|---|
| AuMHA (2 μmol) | — | 0.24 |
| 0.5 | 0.15 | 2.00 |
| 2 | 1.47 | 2.82 |
| 4 | 4.05 | 2.56 |
| 6 | 8.70 | 1.52 |
| 10 | 12.41 | 1.04 |
| 12.5 | 15.39 | 0.81 |
| 16 | 16.93 | 0.43 |
TEM data confirmed also that this absorption band (III) was not related to a plasmon band from large AuNPs (size > 5 nm) or anisotropic NPs (Fig. S1–S7, ESI†). Furthermore, interparticle distances were greater than 3 nm, which rules out electron coupling between nanoparticles that could result in NIR absorption.18
Subsequently, a PL study was conducted on AuMHA/HDT dispersed in PBS. All samples exhibited a broad PL emission ranging from 800 to 1400 nm, with three main bands centered at 850 nm, 1050 nm, and 1250 nm. These bands are commonly observed in various types of NIR-II-emitting us-AuNPs.16,35,43 Here, we present the normalized spectra to highlight the influence of excitation wavelengths on the PL emission spectra. The PL emission bands exhibited a dependence on the excitation wavelength in terms of intensity, shape, and spectral position. These effects were also influenced by the amount of NaBH4 used in the synthesis of us-AuNPs. Under short excitation wavelengths (λexc. 450 nm), we observed a shift of the first PL band from 850 to 930 nm and the second band from 1030 nm to 1100 nm with an increase in the NaBH4 amount (see Fig. 5(a)). The third band centered at 1250 nm exhibited a linear increase in PL intensity. A similar trend, though less pronounced, was observed with excitation at 780 nm (Fig. 5(b)). When excited at 980 nm, corresponding to band III in the absorption spectra, we observed a slight shift of the PL band maximum from 1060 nm to 1080 nm, accompanied by a shift of the PL tail towards longer wavelengths, with an increasing NaBH4 amount from 2.0 μmol to 16.0 μmol of NaBH4 (Fig. 5(c)). PL excitation spectra of AuMHA/HDT samples recorded at λem. 925 nm exhibited a single band at 830 nm (Fig. 6(a)), while selecting the PL emission at λem. 1050 nm revealed two bands, one at 780 nm and another at 990 nm (Fig. 6(b)). The band at 780 nm exhibited a consistent decrease with increasing NaBH4 concentration from 4 μmol to 16 μmol. The PLQY of the different AuMHA/HDT samples was determined in water relative to the reference dye IR125 dissolved in DMSO (with a PLQY of 23%). The PLQY values reached a maximum of 2.8% using 2 μmol of NaBH4, which is 10 times higher than that observed for the sample obtained with the monothiol MHA (Table 2).
 | ||
| Fig. 5 (a) PL emission spectra of AuMHA/HDT samples dispersed in PBS buffer (pH 7) at λexc. = 450 nm (a), λexc. = 780 nm (b), and λexc. = 980 nm (c). | ||
 | ||
| Fig. 6 PL excitation spectra of AuMHA/HDT samples dispersed in PBS buffer (pH 7) measured at λem. = 925 nm (a), and λem. = 1050 nm (b). | ||
While changing the pH from 12 to 6 did not result in significant changes in the absorbance spectra (Fig. S16, ESI†), we observed higher PL emission at basic pH (Fig. S17, ESI†). This pH effect has also been reported for various thiol-stabilized gold nanoclusters, attributed to the efficient energy transfer between gold and the thiolate ligand under alkaline conditions.44,45
PL lifetime measurements were performed for three AuMHA/HDT samples prepared with different amounts of NaBH4: 2.0 μmol, 6.0 μmol, and 16.0 μmol using 450 nm and 634 nm as excitation wavelengths and collecting the photons in a broad NIR-II emission window (1050–1700 nm). All decay curves could be fitted with three components τ1 (∼1 ns), τ2 (40 to 110 ns), and τ3 (200 to 750 ns) (Fig. S18, ESI†). Results depicted in Fig. 7 indicated a decrease of the PL lifetime with the increase of NaBH4 concentration at higher excitation wavelengths. This results in the fall of the average PL lifetime ![[small tau, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e5.gif) = 714 ns (2 μmol NaBH4), 501 ns (6 μmol NaBH4), 233 ns (16 μmol NaBH4) using 450.8 nm excitation wavelength and also using 634.3 nm excitation wavelength: = 660 ns (2 μmol NaBH4), 442 ns (6 μmol NaBH4), 216 ns (16 μmol NaBH4).
= 714 ns (2 μmol NaBH4), 501 ns (6 μmol NaBH4), 233 ns (16 μmol NaBH4) using 450.8 nm excitation wavelength and also using 634.3 nm excitation wavelength: = 660 ns (2 μmol NaBH4), 442 ns (6 μmol NaBH4), 216 ns (16 μmol NaBH4).
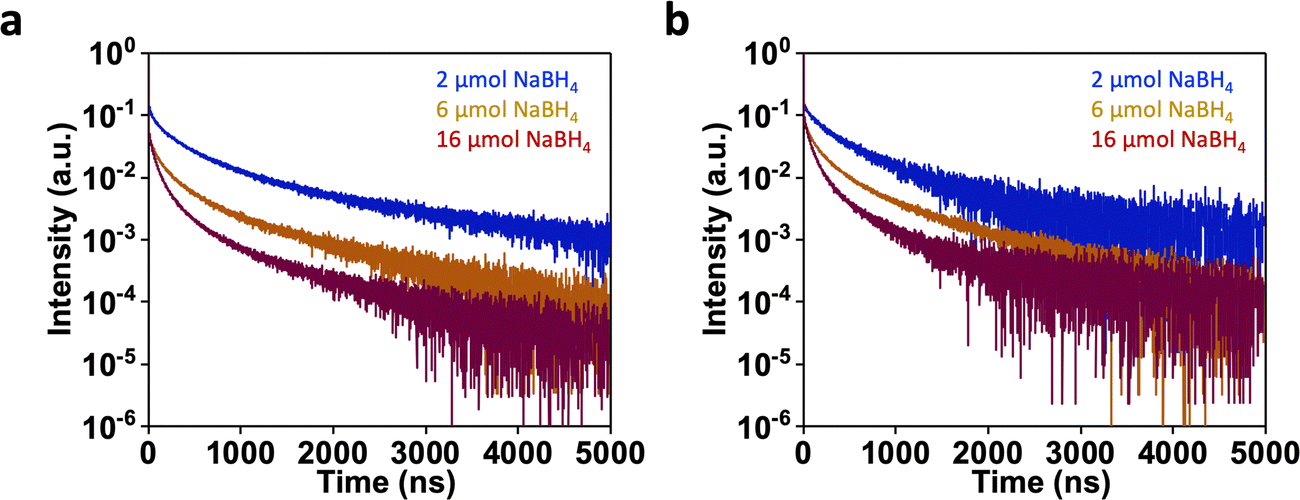 | ||
| Fig. 7 (a) PL decay curves at 20 °C for AuMHA/HDT samples dispersed in PBS (pH 7.4) using 450.8 nm excitation (a) and 634.3 nm excitation (b). | ||
At this stage, we can only speculate about how a thicker organic shell could enhance NIR absorption. One possible explanation is an increase in the rigidity of the ligand shell, as previous studies have shown that this can significantly alter optical properties.32,34,39,44 Another hypothesis is that the change in the surface shell could induce variations in the charge states of the gold core. In fact, Zhu et al. discovered that the absorption spectra in the NIR region are altered when the charge state of Au25 changes from −1 to 0, likely due to a slight distortion of the cluster structures.46
The PL properties of the us-AuNPs exhibit a similar trend to that observed in other luminescent ultra-small NPs.47 This trend involves a shift in PL towards longer wavelengths through the rigidification of the surface, which limits the access of water molecules to the gold surface and reduces non-radiative processes.35 These findings are supported by the decrease in PL lifetime, indicating more efficient energy transfer to water molecule. Furthermore, the PL emission of these emitters is dependent on the excitation wavelength, confirming the occurrence of multiple energy transfers within nanoparticles of potentially different sizes. This is illustrated by the attenuation of a specific excitation band at 830 nm. We observed PL excitation band at 780 nm and 980 nm, which clearly indicate optical transitions at these wavelengths and contribute to the PL of these emitters. We also confirm the absorption at 980–995 nm corresponds to an excited state with PL emission exhibiting a small Stokes shift of approximately 60 nm (emission centered at 1050 nm). This suggests that these transition energies likely occur within the core and involve aurophilic interactions. Conversely, shorter excitations at 450 nm involve both energy transfer from the core and from the surface, as reported in the literature.48
3 NIR-II imaging in intralipids
To demonstrate the potential of these new NIR-II emitters for bioimaging, we filled capillaries with different samples of AuMHA/HDT submerged in an intralipid solution (1%)49 at various depths and measured the PL intensity that mimics living tissue using a 980 nm excitation. Two sub-NIR-II windows were selected using a long-pass filter at 1064 nm and 1250 nm to evaluate the sensitivity of detection, as it is known to be optimal at longer wavelengths.49 Images (Fig. 8) were obtained, and the PL signal for all samples could be detected down to a depth of 6 mm in intralipids. The sensitivity of detection was better when selecting PL emission above 1250 nm (Fig. 9), where the PL signal was still 10 times higher than the background for AuMHA/HDT samples prepared with a high NaBH4 content. These samples exhibited the highest absorption at 980 nm, which was significantly above the detection limit (threshold SNR = 3). Despite a shorter PL emission range using 980 nm instead of 780 nm excitation wavelength, the strong absorption at 980 nm still provided a high number of photons, with a reasonable PLQY ranging from 0.41 to 2.85%. This enables a sensitive detection of PL in the capillaries down to 6 mm in intralipids. | ||
| Fig. 8 NIR-II imaging of capillaries filled with AuMHA/HDT samples at different depth in intralipid solution (1%) using long pass filter at 1064 nm and 1250 nm. | ||
 | ||
| Fig. 9 Sensitivity of detection of AuMHA/HDT samples in capillaries by NIR-II imaging at different depth in intralipid solution (1%). Signal-to-noise ratio (SNR) threshold for limit of detection is fixed at 3. | ||
Conclusion and outlook
In this work, we demonstrated the ability to design ultra-small gold nanoparticles (us-AuNPs) with a size smaller than 2 nm, which exhibit efficient absorption in the infrared region around 1000 nm. Physicochemical characterizations, including elemental analyses and mass spectrometry, were performed. These characterizations suggest that an increase in the amount of the reducing agent leads to the formation of a thicker organic shell, which serves to protect the gold core while maintaining a relatively constant diameter and reducing the negative charge on the surface. By conducting comprehensive characterizations of these us-AuNPs, we can better understand the structural factors that contribute to their optical properties. It is possible that the arrangement of the organic shell induces distortions and/or changes in the charge state of the gold core, which may be responsible for the significant increase in NIR absorption.Importantly, these us-AuNPs are dispersible in water and exhibit efficient photoluminescence emission in the NIR-II window, specifically between 1000 and 1400 nm. When excited at 980 nm, PL signals from these particles can be detected through a 6 mm thick intralipid solution that mimics biological tissue. This discovery opens up new avenues for utilizing such ultra-small probes in bioimaging applications. However, further studies are required to fully understand the nature of this near-infrared absorption and its correlation with the ligand shell.
Author contribution section
WM conducted the synthesis and the imaging experiments. WM, KDW, CZZ, VT and MPO performed spectroscopic characterizations and analyses. XLG and RA participate to the conceptualization of this work. All the authors discussed the results and wrote the article.Conflicts of interest
All authors declare no conflict of interests.Acknowledgements
XLG would like to thank “ligue contre le cancer” (R22004CC), ANR SIREN (ANR-20-CE92-0039-01) for their financial support. This work was financed by the Spanish Ministerio de Ciencia e Innovación under project NANONERV PID2019-106211RB-I00. R. M. is grateful to the Spanish Ministerio de Ciencia e Innovación for support to research through a Ramón y Cajal Fellowship (RYC2021-032913-I). WM and XLG would like to thank GDR Or-Nano for their financial support. URG acknowledges financial support by the DFG-ANR grant SIREN (DFG RE 1203/38-1).References
- O. T. Bruns, T. S. Bischof, D. K. Harris, D. Franke, Y. Shi, L. Riedemann, A. Bartelt, F. B. Jaworski, J. A. Carr, C. J. Rowlands, M. W. B. Wilson, O. Chen, H. Wei, G. W. Hwang, D. M. Montana, I. Coropceanu, O. B. Achorn, J. Kloepper, J. Heeren, P. T. C. So, D. Fukumura, K. F. Jensen, R. K. Jain and M. G. Bawendi, Nat. Biomed. Eng., 2017, 1, 0056 CrossRef CAS PubMed.
- Y. Cai, Z. Wei, C. Song, C. Tang, W. Han and X. Dong, Chem. Soc. Rev., 2019, 48, 22–37 RSC.
- H. Wan, J. Yue, S. Zhu, T. Uno, X. Zhang, Q. Yang, K. Yu, G. Hong, J. Wang, L. Li, Z. Ma, H. Gao, Y. Zhong, J. Su, A. L. Antaris, Y. Xia, J. Luo, Y. Liang and H. Dai, Nat. Commun., 2018, 9, 1171 CrossRef PubMed.
- H. Du, H. Wan and H. Dai, in Near Infrared-Emitting Nanoparticles for Biomedical Applications, ed. A. Benayas, E. Hemmer, G. Hong and D. Jaque, Springer International Publishing, Cham, 2020, pp. 83–101 DOI:10.1007/978-3-030-32036-2_5.
- Q. Yang, H. Ma, Y. Liang and H. Dai, Acc. Mater. Res., 2021, 2, 170–183 CrossRef CAS.
- W. Xu, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 7476–7487 CrossRef CAS PubMed.
- J. Cao, B. Zhu, K. Zheng, S. He, L. Meng, J. Song and H. Yang, Front. Bioeng. Biotechnol., 2020, 7, 1–21 CAS.
- Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16294–16308 CrossRef CAS PubMed.
- J. Huang and K. Pu, Chem. Sci., 2021, 12, 3379–3392 RSC.
- B. Li, M. Zhao, J. Lin, P. Huang and X. Chen, Chem. Soc. Rev., 2022, 51, 7692–7714 RSC.
- H. Dai, X. Wang, J. Shao, W. Wang, X. Mou and X. Dong, Small, 2021, 17, 2102646 CrossRef CAS PubMed.
- S. Wang, J. Liu, C. C. Goh, L. G. Ng and B. Liu, Adv. Mater., 2019, 31, 1904447 CrossRef CAS PubMed.
- Z. Cai, L. Zhu, M. Wang, A. W. Roe, W. Xi and J. Qian, Theranostics, 2020, 10, 4265–4276 CrossRef CAS PubMed.
- Z. Yu, B. Musnier, K. D. Wegner, M. Henry, B. Chovelon, A. Desroches-Castan, A. Fertin, U. Resch-Genger, S. Bailly, J.-L. Coll, Y. Usson, V. Josserand and X. Le Guével, ACS Nano, 2020, 14, 4973–4981 CrossRef CAS PubMed.
- K. Zhang, F.-R. Chen, L. Wang and J. Hu, Small, 2023, 19, 2206044 CrossRef CAS PubMed.
- L. Haye, P. I. Diriwari, A. Alhalabi, T. Gallavardin, A. Combes, A. S. Klymchenko, N. Hildebrandt, X. Le Guével and A. Reisch, Adv. Opt. Mater., 2022, 2201474 Search PubMed.
- Y. Kong, D. Santos-Carballal, D. Martin, N. N. Sergeeva, W. Wang, G. Liu, B. Johnson, B. Bhayana, Z. Lin, Y. Wang, X. Le Guével, N. H. de Leeuw, D. Zhou and M. X. Wu, Mater. Today, 2021, 51, 96–107 CrossRef CAS.
- B. Musnier, K. D. Wegner, C. Comby-Zerbino, V. Trouillet, M. Jourdan, I. Hausler, R. Antoine, J. L. Coll, U. Resch-Genger and X. Le Guevel, Nanoscale, 2019, 11, 12092–12096 RSC.
- Y. Chen, D. Montana, H. Wei, J. Cordero, M. Schneider, X. Le Guevel, O. Chen, O. Bruns and M. Bawendi, Nano Lett., 2017, 17, 6330–6334 CrossRef CAS PubMed.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan, L. Li, X. Mu, J. Wang, W. Mi, J. Luo, J. Xie and X.-D. Zhang, Adv. Mater., 2019, 31, 1901015 CrossRef CAS PubMed.
- V. Bonačić-Koutecký, X. Le Guével and R. Antoine, ChemBioChem, 2022, e202200524 Search PubMed.
- T. Chen, H. Lin, Y. Cao, Q. Yao and J. Xie, Adv. Mater., 2022, 34, 2103918 CrossRef CAS PubMed.
- J. Xu, J. Li, W. Zhong, M. Wen, G. Sukhorukov and L. Shang, Chin. Chem. Lett., 2021, 32, 2390–2394 CrossRef CAS.
- C. Colombe, X. Le Guevel, A. Martin-Serrano, M. Henry, E. Porret, C. Comby-Zerbino, R. Antoine, I. Atallah, B. Busser, J. L. Coll, C. A. Righini and L. Sancey, Nanomedicine, 2019, 20, 102011 CrossRef CAS PubMed.
- W. Poon, Y.-N. Zhang, B. Ouyang, B. R. Kingston, J. L. Y. Wu, S. Wilhelm and W. C. W. Chan, ACS Nano, 2019, 13, 5785–5798 CrossRef CAS PubMed.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS PubMed.
- D. Shen, M. Henry, V. Trouillet, C. Comby-Zerbino, F. Bertorelle, L. Sancey, R. Antoine, J. L. Coll, V. Josserand and X. Le, Guével, APL Mater., 2017, 5, 053404 CrossRef.
- X. Le Guével, B. Hötzer, G. Jung and M. Schneider, J. Mater. Chem., 2011, 21, 2974–2981 RSC.
- F. Cao, E. Ju, C. Liu, W. Li, Y. Zhang, K. Dong, Z. Liu, J. Ren and X. Qu, Nanoscale, 2017, 9, 4128–4134 RSC.
- M. Perić, Ž. Sanader Maršić, I. Russier-Antoine, H. Fakhouri, F. Bertorelle, P.-F. Brevet, X. le Guével, R. Antoine and V. Bonačić-Koutecký, Phys. Chem. Chem. Phys., 2019, 21, 23916–23921 RSC.
- F. Bertorelle, C. Moulin, A. Soleilhac, C. Comby-Zerbino, P. Dugourd, I. Russier-Antoine, P.-F. Brevet and R. Antoine, ChemPhysChem, 2018, 19, 165–168 CrossRef CAS PubMed.
- F. Bertorelle, K. D. Wegner, M. Perić Bakulić, H. Fakhouri, C. Comby-Zerbino, A. Sagar, P. Bernadó, U. Resch-Genger, V. Bonačić-Koutecký, X. Le Guével and R. Antoine, Chem. – Eur. J., 2022, 28, e202200570 CrossRef CAS PubMed.
- Y. Zhong, J. Zhang, T. Li, W. Xu, Q. Yao, M. Lu, X. Bai, Z. Wu, J. Xie and Y. Zhang, Nat. Commun., 2023, 14, 658 CrossRef CAS PubMed.
- X. Yuan, N. Goswami, W. Chen, Q. Yao and J. Xie, Chem. Commun., 2016, 52, 5234–5237 RSC.
- X. Le Guével, K. D. Wegner, C. Würth, V. A. Baulin, B. Musnier, V. Josserand, U. Resch-Genger and J.-L. Coll, Chem. Commun., 2022, 58, 2967–2970 RSC.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- X. Le Guével, O. Tagit, C. E. Rodríguez, V. Trouillet, M. Pernia Leal and N. Hildebrandt, Nanoscale, 2014, 6, 8091–8099 RSC.
- Z. Liu, Z. Wu, Q. Yao, Y. Cao, O. J. H. Chai and J. Xie, Nano Today, 2021, 36, 101053 CrossRef CAS.
- S. Tamang, G. Beaune, I. Texier and P. Reiss, ACS Nano, 2011, 5, 9392–9402 CrossRef CAS PubMed.
- M. La Rosa, T. Avellini, C. Lincheneau, S. Silvi, I. A. Wright, E. C. Constable and A. Credi, Eur. J. Inorg. Chem., 2017, 5143–5151 CrossRef CAS.
- Q. Li, C. J. Zeman Iv, Z. Ma, G. C. Schatz and X. W. Gu, Small, 2021, 17, 2007992 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- K. He, Y. Tan, Z. Zhao, H. Chen and J. Liu, Small, 2021, 17, 2102481 CrossRef CAS PubMed.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS.
- M. Guo, G. Zhang, R. Zhao, H. Ma, Y. Yan, S. Yang, J. Meng, Y. Huang, X.-D. Zhang, H. Wang and R. Zhang, ACS Appl. Nano Mater., 2023, 6, 15945–15958 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- B. Musnier, M. Henry, J. Vollaire, J.-L. Coll, Y. Usson, V. Josserand and X. Le Guével, J. Biophotonics, 2020, e202000345 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc03021k |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2023 |