
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiethoxycarbonyl-BODIPYs as heavy-atom-free photosensitizers for holographic recording in cellulose acetate photopolymer†
Aimee
Sheehan
 a,
Tatsiana
Mikulchyk
b,
Catherine S. P.
De Castro
c,
Safakath
Karuthedath
c,
Wejdan
Althobaiti
c,
Metodej
Dvoracek
a,
Sabad-e-Gul
b,
Hugh J.
Byrne
d,
Frédéric
Laquai
c,
Izabela
Naydenova
be and
Mikhail A.
Filatov
*a
a,
Tatsiana
Mikulchyk
b,
Catherine S. P.
De Castro
c,
Safakath
Karuthedath
c,
Wejdan
Althobaiti
c,
Metodej
Dvoracek
a,
Sabad-e-Gul
b,
Hugh J.
Byrne
d,
Frédéric
Laquai
c,
Izabela
Naydenova
be and
Mikhail A.
Filatov
*a
aSchool of Chemical and Biopharmaceutical Sciences, Technological University Dublin, City Campus, Grangegorman, Dublin 7, Ireland. E-mail: mikhail.filatov@tudublin.ie
bCentre for Industrial & Engineering Optics, Technological University Dublin, D07 ADY7 Dublin, Ireland
cKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Physical Sciences and Engineering Division (PSE), Material Science and Engineering Program (MSE), Thuwal 23955-6900, Kingdom of Saudi Arabia
dFOCAS Research Institute, Technological University Dublin, City Campus, Camden Row, Dublin 8, D08 CKP1 Dublin, Ireland
eSchool of Physics, Clinical & Optometric Sciences, Technological University Dublin, D07 ADY7 Dublin, Ireland
First published on 16th October 2023
Abstract
A series of new heavy-atom-free photosensitizers based on the 2,6-diethoxycarbonyl-BODIPY scaffold was designed and applied for holographic recording in a photopolymerizable material. Photoinduced electron transfer between the BODIPY and meso-aryl subunits, followed by the formation of BODIPY triplet excited states via spin–orbit charge transfer intersystem crossing (SOCT-ISC) was studied by steady-state and ultrafast pump–probe transient optical spectroscopy. Highly efficient photosensitization was observed for dyads bearing pyrene and anthracene substituents, which exhibited singlet oxygen generation quantum yields (ΦΔ) of up to 94%. Charge transfer and SOCT-ISC were observed in non-polar solvent (toluene) due to the increased electron accepting ability of the diethoxycarbonyl-BODIPY. In combination with N-phenylglycine (NPG) as a co-initiator, new BODIPYs initiate a free-radical polymerization of acrylamide monomers under 532 nm irradiation that was used for creation of volume phase transmission gratings in a photopolymerizable material based on cellulose acetate and polyethylene glycol (CA–PEG). As a result of holographic recording, diffractive structures with diffraction efficiency of up to 56% were obtained for CA–PEG layers sensitized with BODIPY-pyrene dyad as compared to a reference heavy-atom-containing diiodo-BODIPY dye (27%). The developed materials showed refractive index modulation of up to 2.3 × 10−3, which demonstrates the potential of diethoxycarbonyl-BODIPYs photosensitizers for holographic recording applications.
Introduction
Photopolymerization is nowadays used in a number of technologies and its application scope is constantly expanding. In particular, UV curing is a very popular technique used for the assembly of a variety of products and materials. However, UV light has low penetration depth, high energy consumption, and potential to damage materials from overexposure.1 Carrying out photopolymerization using safer and lower energy (i.e. visible light) photons has significant advantages as visible light has greater penetration power which is more suitable for highly pigmented polymers.2 Many organic and inorganic compounds are also sensitive to UV irradiation, and thus curing with visible light avoids the occurrence of unwanted side reactions arising from degradation of the materials.3The development of efficient visible light driven photoinitiating systems is the focus of intense research effort. Such systems are usually based on a combination of organic dyes known as photosensitizers (PSs) and various additives (e.g., iodonium salt, amines, silane, etc.) which act as co-initiators.4 A photosensitizer component undergoes intersystem crossing (ISC) and forms long-living triplet excited states which are able to initiate chemical transformation leading to free radical formation and thus cause polymerization. ISC is a spin-forbidden process and so chemical structures of potential PSs must be rationally designed to enhance triplet state yields (ΦT) and lifetimes (τT).5 ISC can be promoted in organic dyes by the introduction of heavy atoms, such as iodine and bromine, or transition metals which are known to enhance spin–orbit coupling.6 However, this approach has significant drawbacks such as higher costs and unwanted side effects, including negative environmental impact, increased toxicity and shortened triplet state lifetimes.7
Spin–orbit charge transfer intersystem crossing (SOCT-ISC) represents an attractive alternative approach for generating long living triplet excited states in heavy-atom-free molecules. This ISC mechanism has attracted much attention in recent years and corresponding dyes found some unique niches in applications such as photodynamic therapy (PDT)8 and triplet–triplet annihilation upconversion (TTA-UC).9 Boron dipyrromethene (BODIPY) dyes10 are so-far the most well-studied class of compounds undergoing SOCT-ISC. Due to their excellent spectroscopic characteristics and convenient synthesis, introducing substituents in almost any position in the core structure is possible thus allowing to tune molecular geometries and excited state energies, and as a result the SOCT-ISC in BODIPYs can be effectively optimized.11
Heavy-atom-free BODIPY photosensitizers were recently demonstrated by us as efficient photoinitiators for free-radical polymerization of acrylate monomers.12 Notably these PSs showed comparable or even higher photopolymerization rates than the reference heavy-atom-containing diiodo-substituted BODIPY which forms triplet excited states due to enhanced spin–orbit coupling. However, developing BODIPY-based photoinitiating systems that efficiently initiate the free radical polymerization for a wide range of monomers under mild irradiation conditions (low light intensity and under air) is still a challenge.13 One of the complications is a strong dependence of triplet state yields on solvent polarity, which is a known characteristic of SOCT-ISC. Recently Uddin et al. demonstrated this by using several BODIPY donor–acceptor dyads as photoinitiators for the polymerization of monomers of varying polarity.14 A general decrease in polymerization rate was observed as monomer polarity increased, however the magnitude of the polarity effect varied between dyes. This was attributed to differences in the CT state stabilization pathways of the BODIPYs. Dyes capable of undergoing efficient SOCT-ISC in low polarity solvents are beneficial, because monomers and formulations used for polymerization are relatively non-polar. The polarity of the matrix also decreases as the polymerization process occurs due to the conversion of more polar carbon–carbon double bonds into less polar single bonds.15
Efficient SOCT-ISC in low and moderate polarity solvents can be achieved by introducing electron-accepting substituents in the dipyrromethene core.11 However, this often leads to a decrease in triplet state yields due to enhancement of ground state recombination rate (CT → S0), due to stabilization of the CT state by electron-withdrawing groups and narrowing CT–S0 energy gap (Fig. 1(a)).16 It is also difficult to design PSs efficiently operating in a broad range of polarities or predict the triplet state yield for different solvents based on the structure. Although recent efforts have shown that it is possible to accurately predict singlet oxygen generation quantum yields (ΦΔ) for BODIPYs by using quantitative structure–property relationship (QSPR) and machine-learning techniques,17 such computational methods are still being optimized.
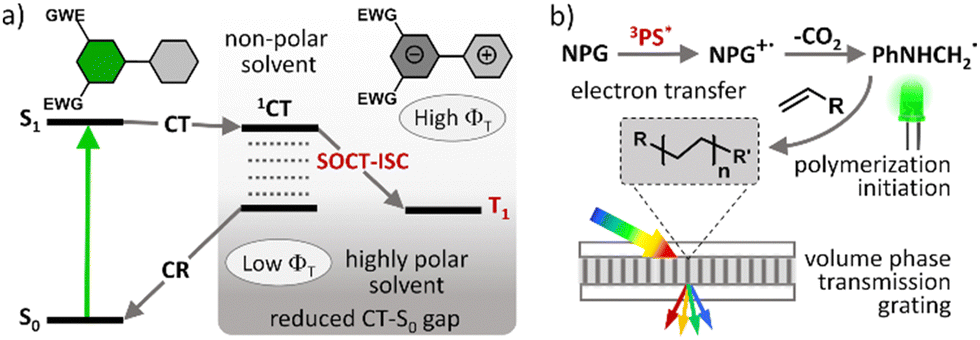 | ||
| Fig. 1 (a) Qualitative Jablonski diagrams depicting photophysical mechanisms of triplet state formation via SOCT-ISC in a donor–acceptor dyad. (b) Schematic representation of volume transmission gratings formation as a result of a spatially controlled photopolymerization. | ||
In this work we investigated new BODIPY dyes containing electron-withdrawing ethoxycarbonyl substituents in positions 2 and 6, which promote electron transfer and allow for efficient SOCT-ISC in both polar and non-polar solvents, such as toluene. Different electron donor substituents, such as anthracenyl, pyrenyl and methoxyphenyl groups, were employed in the meso position to modulate the donor–acceptor capability of the BODIPYs and in turn optimize the formation of long-living triplet excited states via SOCT-ISC. Highly efficient charge transfer and triplet states formation in a non-polar solvent was confirmed by short and long delay transient absorption measurements and singlet oxygen (1O2) quantum yield (ΦΔ) measurements which showed values of up to 94% in toluene. Further, the photosensitizing ability of diethoxycarbonyl-BODIPY dyes was evaluated by employing them as photoinitiators in free-radical polymerization of acrylamide monomers and recording of holographic structures.
The photopolymer used in this work is based on a cellulose acetate–polyethylene glycol (CA–PEG, Fig. 2) matrix, containing acrylamide as a monomer and N,N-methylenebisacrylamide as a cross-linker, which provides increased stability of the hologram in water.18 Diethoxycarbonyl-BODIPYs were employed as photosensitizers in a two-component photoinitiating system containing N-phenylglycine (NPG)19 as a co-initiator. NPG was chosen as a co-initiator as its efficiency for the free-radical polymerization has been previously demonstrated in a photosensitive resin for 3D printing20 and in holographic recording materials.21,22 NPG was shown to react with triplet excited states of photosensitizers, undergoing electron transfer and forming corresponding radical cation (NPG˙+)23 which further undergoes deprotonation/decarboxylation to give anilinomethyl radical (PhNHCH2˙) that initiates polymerization of methacrylate and acrylamide monomers (Fig. 1(b)).24 Cellulose acetate was selected as a binder for the following key reasons: (1) the layers produced in this binder are not sensitive to humidity and the optical devices recorded in the layers have unchanged diffraction efficiency after being immersed in water;18 (2) the obtained solid layers are of high optical quality and show very low scattering;18 (3) it is possible to control the porosity of the binder by varying the PEG concentration, which is needed for the development of holographic sensors.18
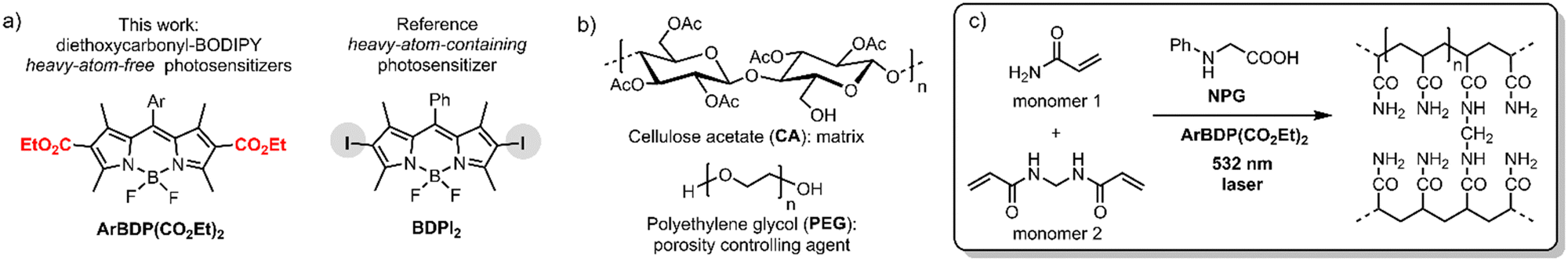 | ||
| Fig. 2 Chemical components of the studied photopolymer: structures of (a) dyes used as photoinitiators, (b) polymeric matrix – cellulose acetate (CA) and polyethylene glycol (PEG). (c) Photopolymerization of acrylamide (monomer 1) and N,N-methylenebisacrylamide (monomer 2, crosslinker) initiated by NPG-BODIPY system under 532 nm excitation. | ||
Holographic recording in photopolymers uses the wave interference of light to create the refractive index modulation and, as a result, form a diffraction grating.25 In this work, transmission gratings with the spatial frequency of 800 lines per mm were recorded in the photopolymer layers sensitized with BODIPYs. Performance of these new photosensitizers was studied through real-time recording of diffraction efficiency growth, which was compared to the reference heavy-atom-containing dye, meso-phenyl-2,6-diiodoBODIPY (BDPI2). The obtained results prove that ethoxycarbonyl BODIPYs are efficient photosensitizers for polymerization of acrylate monomers in the CA–PEG matrix. The resulting photopolymers displayed higher exposure sensitivity achieving diffraction efficiency of up to 56%, compared to 27% for BDPI2. These results expand the arsenal of sustainable, environment-friendly, stable and highly efficient photopolymerizable materials for holographic recording under green light excitation.
Results and discussion
Synthesis of diethoxycarbonyl-BODIPY dyads
To systematically investigate the optical characteristics of diethoxycarbonyl-BODIPY and optimize the triplets formation via SOCT-ISC, a series of compounds containing different aromatic groups were synthesized. Corresponding precursor, 3-ethoxycarbonyl-2,4-dimethylpyrrole (P3) was prepared using a two-step procedure: commercially available diethyl-2,4-dimethylpyrrole-3,5-dicarboxylate (P1) was hydrolysed into a corresponding carboxylic acid (P2) (Scheme 1(a)), followed by a decarboxylation at 210 °C to produce pyrrole P3 in high overall yield.26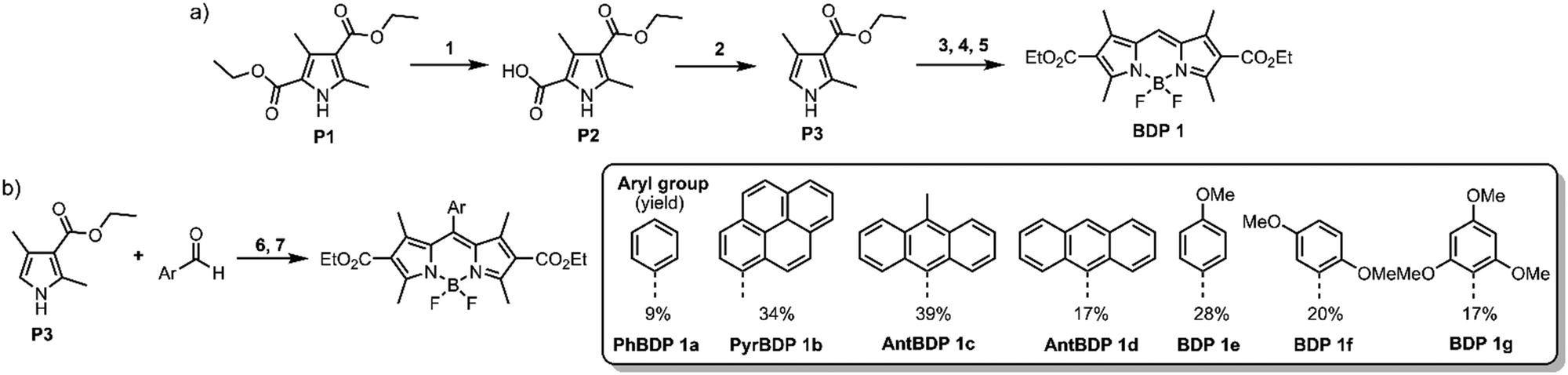 | ||
| Scheme 1 (a) Synthesis of pyrrole precursor and meso-unsubstituted diester BODIPY. Reagents and conditions: (1) KOH, ethanol–H2O, reflux (80%); (2) N2, 1 h, 210 °C (79%); (3) dimethoxymethane, p-toluenesulfonic acid, N2, acetic acid, 18 h, r.t.; (4) DDQ, DCM, 20 min, r.t.; (5) BF3·Et2O, DIPEA, N2, DCM (anh.), 2 h, r.t. (29% over steps 3–5). (b) Synthesis of meso-aryl diethoxycarbonyl-BODIPYs. Reagents and conditions: (6) (i) trifluoroacetic acid, N2, DCM (anh.), 12 h, r.t.; (ii) DDQ, DCM, 10 min, r.t.; (7) BF3·Et2O, DIPEA, N2, DCM (anh.), 2 h, r.t. (9–39% over steps 6–7). | ||
meso-Unsubstituted 2,6-diethoxycarbonyl-BODIPY (BDP 1) was one of the first BODIPYs described by Treibs and Kreuzer in 1968,27 and since then has been used as a precursor for syntheses of ring-fused dyes.28 It was prepared in a three-step reaction (Scheme 1(a)): pyrrole P3 was reacted with dimethoxymethane to give corresponding dipyrromethane which was further oxidised with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), followed by the insertion of boron, using boron trifluoride diethyl etherate (BF3·Et2O) in the presence of N,N-diisopropylethylamine (DIPEA) to give BDP 1 in 29% overall yield.
Steady-state absorption and photoluminescence emission properties of the dyads
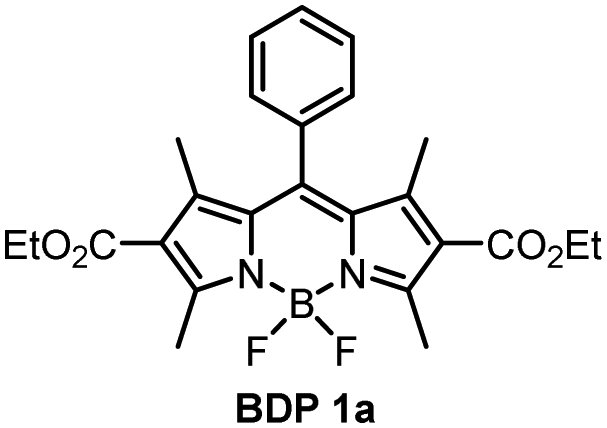
The absorption and fluorescence emission parameters of diethoxycarbonyl BODIPYs in a range of solvents are given in Table 1. Reference compounds BDP 1 and BDP 1a which lack electron donor groups and thus do not undergo intramolecular charge transfer showed optical properties typical for alkyl-substituted BODIPYs, i.e. narrow absorption and emission bands, high fluorescence quantum yields (Φem) both in polar and non-polar solvents. For meso-phenyl BDP 1a (Fig. 3(a)), position of the absorption and emission bands are nearly identical to the parent 1,3,5,7-tetramethyl BODIPY (λabs = 498 nm, λem = 508 nm in methanol),29 indicating that introduction of electron-deficient ethoxycarbonyl in the 2,6 positions has no major effect on optical characteristics. This is in line with the work of Gabe et al. describing the compound BPD 1a and its analogue, meso-phenyl-2,6-diethyl-1,3,5,7-tetramethyl BODIPY.30 While the absorption and emission maxima of the BODIPY are not affected by ethoxycarbonyl groups, the addition of electron-rich ethyl groups to the 2,6-positions causes a strong bathochromic shift (λabs = 520 nm, λem = 534 nm, Fig. 3(a)) due to increasing HOMO energy level of the BODIPY fluorophore.31| Compound | Solventa | λ abs (nm) | λ em (nm) | Φ em | Compound | Solvent | λ abs (nm) | λ em (nm) | Φ em |
|---|---|---|---|---|---|---|---|---|---|
| a Concentration of the samples: 5 × 10−6 M. HEX – hexane, TOL – toluene, THF – tetrahydrofuran, DCM – dichloromethane, EtOH – ethanol, ACN – acetonitrile. b Absorption maxima corresponding to BODIPY chromophore. c Fluorescence was recorded by exciting the samples at the vibrational shoulder of the BODIPY absorption. Excitation wavelength: 470 nm. d The fluorescence quantum yields were measured using Rhodamine 6G as a standard (Φem = 0.95 in EtOH).36 e Fluorescence quantum yield value representing the integral emission coming from locally excited (LE) and charge-transfer (CT) excited states. | |||||||||
|
ACN | 496 | 505 | 0.958 |
|
CAN | 497 | 507 | 0.413 |
| EtOH | 497 | 506 | 0.995 | EtOH | 497 | 508 | 0.537 | ||
| THF | 500 | 509 | 0.977 | THF | 499 | 511 | 0.668 | ||
| DCM | 502 | 511 | 0.919 | DCM | 500 | 511 | 0.555 | ||
| TOL | 507 | 517 | 0.966 | TOL | 503 | 516 | 0.646 | ||
| HEX | 504 | 511 | 0.960 | HEX | 501 | 512 | 0.725 | ||
|
CAN | 500 | 517, 704 | 0.002 |
|
ACN | 501 | 513 | 0.040 |
| EtOH | 497 | 510, 673 | 0.015 | EtOH | 501 | 514 | 0.063 | ||
| THF | 503 | 516, 634 | 0.002 | THF | 503 | 516 | 0.074 | ||
| DCM | 504 | 520, 641 | 0.003 | DCM | 504 | 516 | 0.064 | ||
| TOL | 507 | 521 | 0.220 | TOL | 507 | 520 | 0.055 | ||
| HEX | 503 | 514 | 0.773 | HEX | 504 | 515 | 0.225 | ||
|
ACN | 500 | 522 | 0.028 |
|
ACN | 497 | 509 | 0.374 |
| EtOH | 502 | 523 | 0.026 | EtOH | 498 | 510 | 0.545 | ||
| THF | 504 | 525 | 0.041 | THF | 500 | 513 | 0.547 | ||
| DCM | 505 | 519 | 0.008 | DCM | 500 | 513 | 0.630 | ||
| TOL | 508 | 521 | 0.006 | TOL | 503 | 518 | 0.603 | ||
| HEX | 505 | 516 | 0.382 | HEX | 501 | 513 | 0.380 | ||
|
ACN | 499 | 507 | <0.001 |
|
ACN | 501 | 514 | 0.002 |
| EtOH | 500 | 518 | <0.001 | EtOH | 503 | 515 | 0.011 | ||
| THF | 503 | 513 | 0.001 | THF | 505 | 517 | 0.212 | ||
| DCM | 504 | 515 | 0.002 | DCM | 506 | 517 | 0.301 | ||
| TOL | 507 | 522 | 0.158 | TOL | 509 | 521 | 0.941 | ||
| HEX | 504 | 515 | 0.996 | HEX | 506 | 516 | 0.764 | ||
 | ||
| Fig. 3 (a) Normalized absorption of PhBDP 1a, PyrBDP 1b, AntBDP 1c and a reference BODIPY compound in ethanol. (b) Normalized photoluminescence emission spectra of pyrene dyad 1b in a range of solvents (λexc = 470 nm). Concentration of samples: 5 × 10−6 M. TOL – toluene, THF – tetrahydrofuran, EtOH – ethanol. | ||
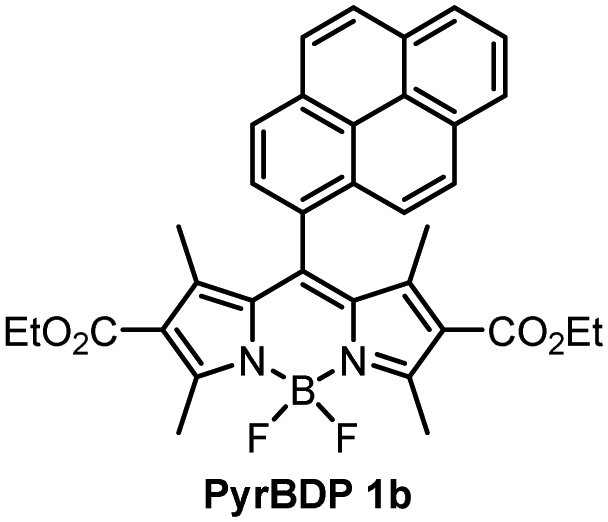
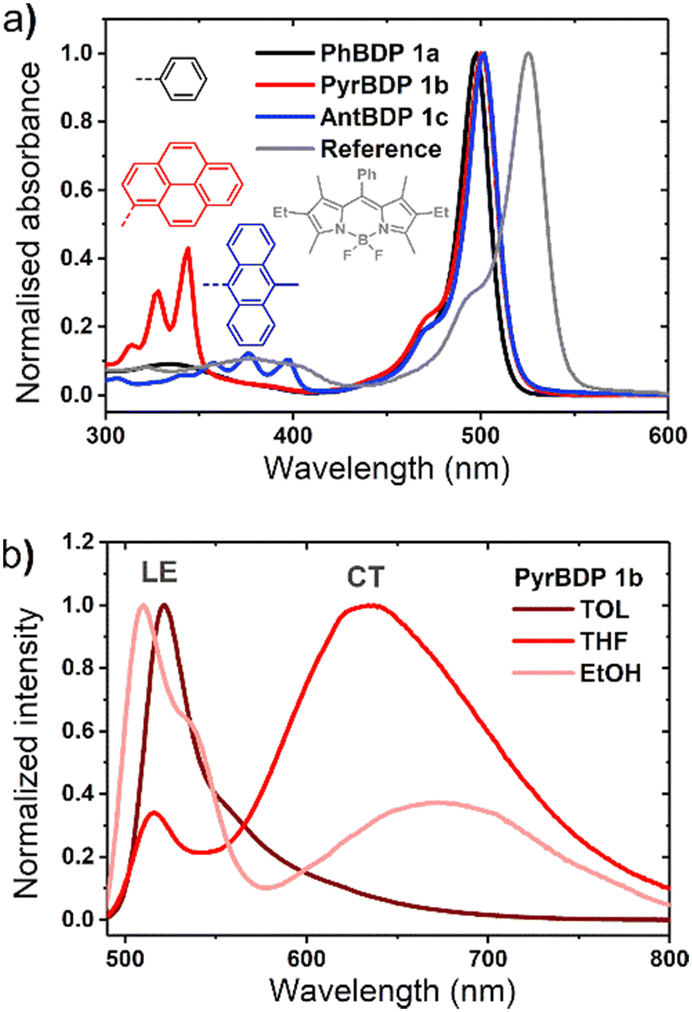
The optical properties of BPD 1a, pyrene and anthracene dyads 1b and 1c, respectively, are presented in Fig. 3 and summarized in Table 1. The absorption spectra show transitions associated with the two subunits, indicating weak coupling of the chromophores in the ground state. Upon changing the solvent polarity, the shape and maxima of the absorption undergo little change (Fig. S10, ESI†), but the emission properties are greatly affected. Fig. 3(b) shows the emission spectra of pyrene dyad 1b in different solvents. In THF the local exciton (LE) peak appears at 516 nm, and is accompanied by broad band centred at 634 nm. This second signal can also be seen in the emission spectrum in EtOH, further red-shifted to 672 nm. These bands are not seen in the emission spectra of PyrBDP 1b in toluene or hexane, indicating that the energy of the corresponding excited state strongly depends on the solvent polarity. The appearance of these broader signals is accompanied by a remarkable drop in emission intensity and quantum yield to 0.002 in THF and 0.015 in EtOH. It should be noted that the observed photoluminescence emission is concentration dependent: bathochromic shift of up to 15 nm was observed both for reference BODIPY and for the studied donor acceptor dyads when increasing the dye concentration (Fig. S11, ESI†); similar behaviour was previously observed for BODIPYs forming J-aggregates.32
In line with previous reports,33 weak emission above 600 nm can be attributed to a charge transfer state between the pyrene and BODIPY subunits. The reported reduction potential EA/A− for the diethoxycarbonyl-BODIPY group is −0.87 eV (MeCN/SCE), which is +0.32 eV more positive than the value reported for the reference 1,3,5,7-tetramethyl BODIPY that has hydrogen atoms at the 2,6-positions.34 This makes diethoxycarbonyl-BODIPY chromophore more susceptible to reduction and facilitates charge transfer. The oxidation potential (ED/D+) of pyrene group in acetonitrile is 1.16 eV,35 making the electron transfer to the BODIPY subunit thermodynamically favourable. In the dyad molecule, pyrene acts as an electron donor and the BODIPY as an acceptor, allowing a dipolar CT state to form. The emission observed for pyrene dyad 1b in THF and ethanol (Fig. 3(b)) can be interpreted as a combination of fluorescence from the LE and CT state. When compared to the previously reported 1,3,5,7-tetramethyl BODIPY-pyrene dyad without ethoxycarbonyl groups, which showed only moderate charge transfer efficiency in polar solvents;33a it can be concluded that the addition of these substituents has promoted intramolecular charge transfer efficiency in the dyad.
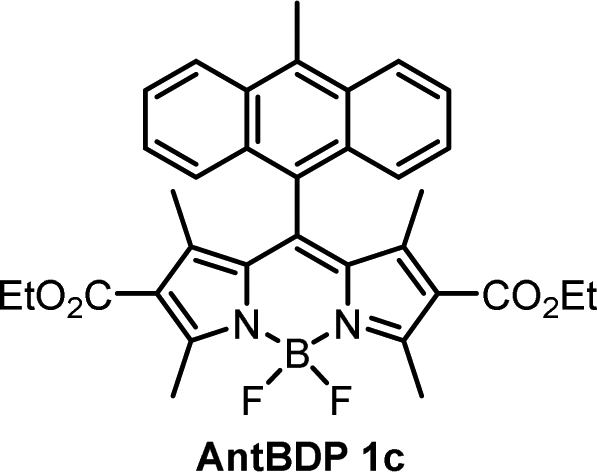
The emission of AntBDP 1c in toluene shows a similar CT fluorescence at 625 nm (Fig. S10, ESI†). The Φem values decreases considerably from 0.225 for nonpolar hexane to 0.04 in ACN. This can be rationalized by taking into account lower oxidation potential of 9-methylanthracene fragment (1.04 eV),16 compared to pyrene (1.16 eV), which increases the driving force of the electron transfer process making it feasible in non-polar toluene. Previously reported BODIPY-anthracene dyads, which lack electron-withdrawing groups in the 2,6 positions, undergo charge transfer only in polar solvents such as ACN and DCM.37
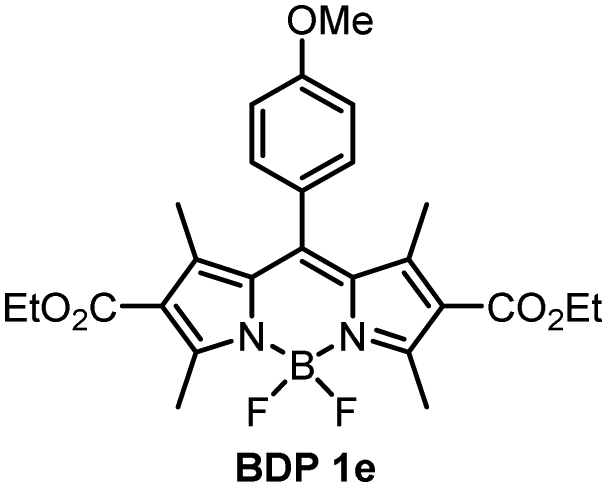
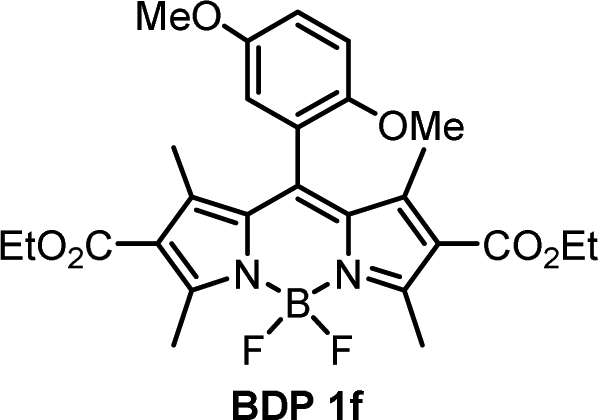
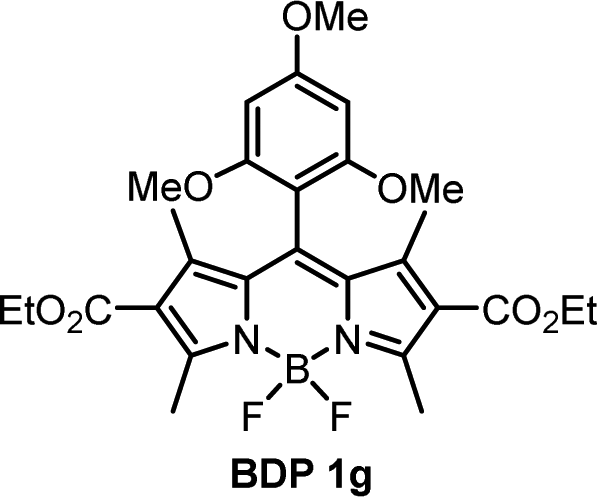
For compounds BDP 1e–g containing different number of methoxy substituents in the meso-phenyl group, the absorption spectra show strong S0 → S1 transitions at around 500 nm, with slight red shifts of 6–9 nm as solvent polarity increases. There is also S0 → S2 transitions at ≈350 nm from the phenyl subunit, indicating that the electronic coupling with the BODIPY core in the ground state is weak. Upon changing the solvent polarity, the shape and maxima of the absorption and emission bands of BDP 1e–g undergo little change. However, changes can be seen in the emission quantum yields (Φem), particularly for BDP 1f where Φem varies from <0.001 in EtOH up to 1 in hexane. As the dyads have the same ethoxycarbonyl-substituted core, the difference in emission yields between BDP 1e, 1f and 1g must be due to the changes in methoxy-substituents in the meso-phenyl group.
As is seen from Table 1, the fluorescence quantum yields values for BDP 1e in different solvents are very similar to those of meso-phenylBODIPY BDP 1a and, in general, are not affected by solvent polarity. For BDP 1f and 1g, fluorescence is strongly quenched in polar solvents, indicating efficient charge transfer. This difference can also be rationalized taking into account oxidation potentials (Eox) of the meso-methoxybenzene substituents, which are: 1.77 eV for anisole (BDP 1e), 1.55 eV for 1,3-dimethoxybenze (BDP 1f) and 1.54 eV for 1,3,5-trimethoxybenzene (BDP 1g).38 Increasing the number of methoxy-substituents on the phenyl ring leads to lowering the Eox and, consequently, increases the driving force of electron transfer from these subunits to the BODIPY core. This is in line with previous reports on BODIPYs bearing methoxy-, dimethoxy- and trimethoxyphenyl groups in the meso-position.17 In addition, the presence of methoxy groups in the ortho position could potentially hinder the rotation of the meso-aryl donor, making the geometry of the dyad orthogonal and in turn promoting SOCT-ISC.39
To assess the efficiency of triplet state formation in different solvents, the singlet oxygen quantum yields were determined through a chemical-trapping method, employing 1,9-dimethylanthracene (DMA) as a 1O2 sensor. DMA selectively reacts with singlet oxygen giving a corresponding endoperoxide.40 For the measurements, solutions of DMA in air-saturated solvent containing corresponding BODIPY were irradiated with light at 514 nm with a constant 12 mW cm−2 power for periods of 30–120 s, after which the absorption spectra were recorded. The BODIPY absorption shows no change during irradiation, while the DMA absorption decreases linearly over time (Fig. 4, inset). This change is plotted, and the slope is then used to determine ΦΔ as compared to the reference photosensitizer as described in Section S6 of the ESI.†
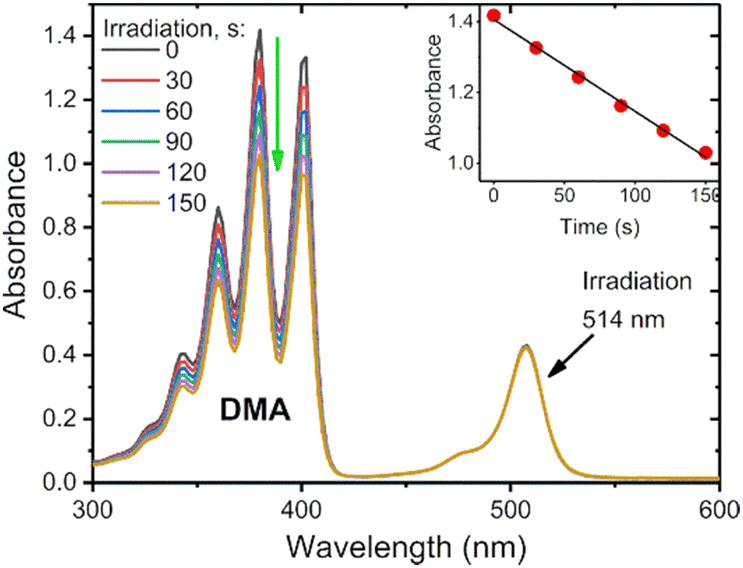 | ||
| Fig. 4 Photosensitized oxidation of 1,9-dimethylanthracene in the presence of AntBDP 1c in air saturated toluene solution irradiated with 514 nm laser (12 mW cm−2). Inset: Change of absorbance at 376 nm with time. | ||
The results from the singlet oxygen quantum yield measurements on the diethoxycarbonyl-BODIPYs can be seen in Table 2 below. The parent compounds BPD 1 and BPD 1a have very low ΦΔ in each solvent, ranging from 0.004–0.027. This correlates with their strong emissive properties discussed previously. Pyrene dyad 1b shows moderate variations of ΦΔ in different solvents, with its highest result of 0.548 obtained in toluene, overall, the second highest yield obtained from these measurements.
| Compound | EtOH | ACN | THF | TOL |
|---|---|---|---|---|
| a Quantum yields were measured using 1,9-dimethylanthracene as a 1O2 trap and 2,6-diiodo-8-phenylBODIPY as a reference photosensitizer (0.85 in toluene).41 | ||||
| BDP 1 | 0.011 | 0.016 | 0.025 | 0.027 |
| BDP 1a | 0.006 | 0.005 | 0.004 | 0.005 |
| PyrBDP 1b | 0.294 | 0.117 | 0.548 | 0.504 |
| AntBDP 1c | 0.211 | 0.075 | 0.511 | 0.941 |
| AntBDP 1d | 0.467 | 0.290 | 0.337 | 0.515 |
| BDP 1e | 0.004 | 0.011 | 0.051 | 0.007 |
| BDP 1f | 0.015 | 0.005 | 0.045 | 0.264 |
| BDP 1g | 0.146 | 0.141 | 0.221 | 0.011 |
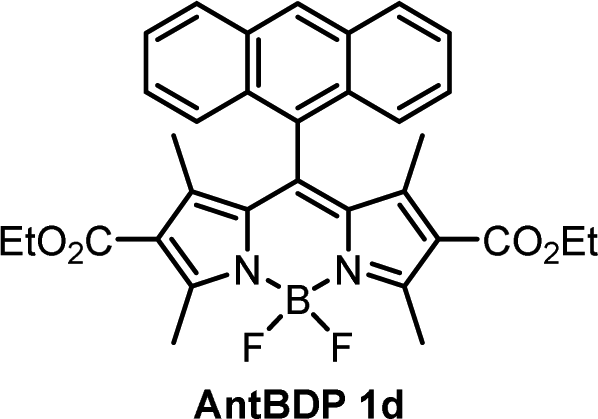
While the ΦΔ varies greatly between solvents for some dyads, unlike Φem, there appears to be no direct correlation between 1O2 quantum yield and solvent polarity. This is evident for AntBDP 1c, which has the highest yield overall of 0.961 in nonpolar toluene, and a much lower yield of 0.075 in more polar acetonitrile. Compound AntBDP 1d, which differs in structure from AntBDP 1c only by the absence of a methyl group in position 9 of the anthracene subunit, does not have the same high yield in toluene. ΦΔ values of AntBDP 1d have less variation than those of AntBDP 1c, and its highest result of 0.515 is also recorded in toluene. Reference BODIPY-pyrene and anthracene dyads which lack ethoxycarbonyl groups in positions 2,6 of the BODIPY scaffold are summarized in Table S1 (ESI†). For 8-pyrenyl-1,3,5,7-tetramethylBODIPY, ΦΔ value of 0.088 was recorded in toluene which is consistent with strong fluorescence (ΦF = 0.926)33a in this solvent and thus can be explained by inefficient charge transfer in non-polar solvent environment.
The methoxybenzene-substituted dyads 1e–g all have relatively low ΦΔ values in all solvents, the highest being recorded for BDP 1f in toluene with a yield of 0.264. BDP 1e is the least efficient 1O2 generator of these three dyads, with a maximum value of 0.051 in THF. BDP 1e also has very low Φem as stated previously, due to inefficient charge transfer.
Femto- and nanosecond pump–probe transient absorption spectroscopy
To reveal the photoexcited state dynamics of pyrene and anthracene dyads 1b and 1c dyads, respectively, transient absorption (TA) spectroscopy was employed. Fig. 5 shows the picosecond–nanosecond (ps–ns) and nanosecond–microsecond (ns–μs) TA spectra and kinetics of selected spectral regions shadowed (as insets) of 1c (a and b) and 1b (c and d) in toluene after photoexcitation at 505 nm (ps–ns TA) and 532 nm (ns–μs TA), respectively. The negative ΔT/T signals represent photoinduced absorption (PA), whereas the positive ΔT/T signals represent photo-bleaching (PB). In Fig. 5, we can attribute the band centred at 500–510 nm to the PB of BODIPY (BDP), clearly seen in Fig. 5 panels a, b and d.37 Stimulated emission can be observed in the PB broadening ∼550 nm, for PyrBDP 1b in toluene (see Fig. S15, ESI†). The PA region of AntBPD 1c at ∼420 nm corresponds to the tail of the anthracene exciton-induced absorption (Ant), observable in both toluene (Fig. 5(a) and (b)) and acetonitrile (Fig. S16, ESI†).42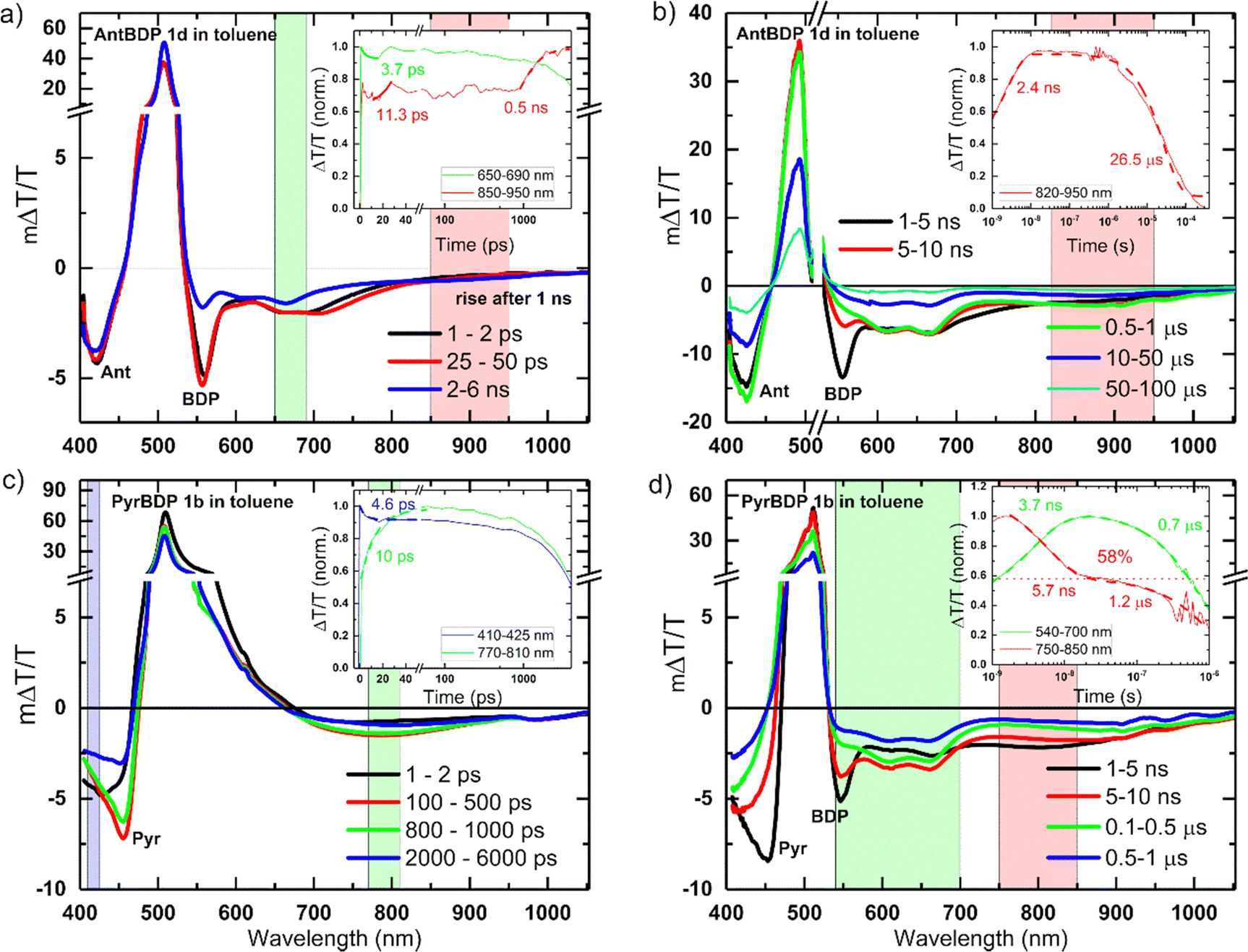 | ||
| Fig. 5 ps–ns (a) and (c) and ns–μs (b) and (d) TA spectra with kinetics (as green, red and blue insets) of the selected spectral region (shadowed in corresponding green, red and blue areas) of pyrene and anthracene dyads PyrBDP 1b and AntBDP 1c in toluene after excitation with 505 nm and 532 nm laser pulses, respectively, under inert atmosphere. Pyr, Ant and BDP stand for pyrene, anthracene and BODIPY, respectively. The spectra are labelled with these where their signatures are clearly seen. When the kinetics were fitted, the fits were shown in insets together with their respective rate constants. | ||
The same can be seen for PyrBDP 1b in acetonitrile with a clear pyrene exciton-induced absorption (Pyr) signature at ∼460 nm readily visible at 1–2 ps (Fig. S16b, ESI†), but visible only at later times (100–500 ps) in toluene (Fig. 5(c)).43
The BODIPY exciton-induced absorption (BDP) can be readily (1–2 ps) seen at around 549–558 nm and 547–556 nm, for AntBDP 1c and PyrBDP 1b, respectively, but only later for dyad PyrBDP 1b in toluene (1–5 ns) due to the overlap with its stimulated emission (Fig. 5(a) and (c)).
The PA regions centred at ∼420 nm and ∼800 nm (clearer in Fig. 5(d) for compound PyrBDP 1b in toluene) showed a spectral evolution with time consistent with the formation of charge-transfer (CT) states, as described in previous work.12 The negligible temporal evolution of the PB band in toluene (except for 1b and 1c at later times) is due to both photoexcited singlet and CT states exhibiting the same photobleaching, and the population of CT states from singlet excited states. The kinetics of the band at 550–570 nm exhibited a clear decay until 10 ps, but no further changes were observed after that up to 500 ps, while the 770–870 nm band exhibited a clear rise due to the generation of CT states (inset Fig. 5(c)).
An exponential fit to the dynamics yielded an inverse rate constant of 4–5 ps for the singlet exciton decay and 10–11 ps for the CT state generation probed at 550–570 nm and 770–870 nm in toluene, respectively (Fig. 5(c)). The ps–ns TA spectra of both dyads 1c and 1b in acetonitrile showed different spectral shapes and evolution (Fig. S16, ESI†) where the PB band in acetonitrile showed an immediate decay, compared to AntBDP 1c in toluene. This is supported by the fast decay of the CT states (band centered at 630 nm). Table S2 (ESI†) summarizes the inverse generation rates of CT states in both toluene and acetonitrile.
The spectral shape and evolution are different for PyrBDP 1b in acetonitrile compared to toluene (Fig. S16, ESI† and Fig. 5, respectively). In acetonitrile, both molecules showed early time (after 10 ps), and faster decay of the CT states compared to the toluene solutions. Dyads 1b and 1c showed a clear difference in the spectra and dynamics of the excited states. In PyrBDP 1b (Fig. 5(d)), the CT state peaked at around 800 nm and decayed within ∼10 ns, causing the emergence of two new long-lived bands at 600 nm and 670 nm. We assigned these bands to the BODIPY triplet state based on previous reports.12 An exponential fit to the kinetics shown in the inset of Fig. 5(d) yielded an inverse rate constant of 5.7 ns for the triplet generation and 0.7–1.2 μs for the triplet decay for PyrBDP 1c much longer than that observed for AntBDP 1b (26.5 μs), in toluene, which are significantly shorter than those obtained for both dyads in acetonitrile (∼65 μs, Fig. S16b and d, ESI†). For AntBDP 1c in toluene, we estimated a triplet yield of 58% (see the horizontal dashed line in the inset Fig. 5(d)), assuming both triplet and singlet states exhibit the same absorption cross-section as reported in previous works9a,12 which agrees with the one measured by relative actinometry (56–73%) using an optically matched (at pump wavelength) Rose Bengal solution in methanol as ref. 44, see Fig. S14 and Table S2 in the ESI.† A similar analysis was done for the remaining materials and solvents combination, see Fig. S16 and Table S2 (ESI†).
Holographic recording of volume transmission gratings
During holographic recording photopolymerizable materials experience spatially varying change of the refractive index due to change in molar refractivity and local density caused by illumination with spatially varying light intensity. Such materials found use in a wide variety of technologies, e.g. data transfer45 and storage,25,46 holographic solar concentrators,47 augmented and virtual reality displays.48 The key component of such materials is a photoinitiating system that triggers polymerization in the illuminated areas leading to the refractive index modulation and, as a result, formation of the diffraction grating. The holographic recording capability (sensitivity and dynamic range) of photopolymerizable materials depends on the photoinitiating system efficiency which is determined by its light absorption properties, initiating radicals quantum yield and radicals reactivity.49,50 Appropriate choice of the photoinitiator component is critical for the performance of photopolymerizable materials in holographic recording applications and the development of such systems is a subject of intense research. Recently, Zheng et al. have demonstrated that the use of two-component photoinitiating system Irgacue® 784 – Rose Bengal improves the stability of the grating in the epoxy resin/acrylate-based photopolymer.51 Rolle et al. have reported a three-component system based on Safranine O dye, an electron acceptor (triazine), and an electron donor (borate salt), which generates up to two initiating radicals from one photon absorption and allows for single pulsed holographic recording in polyurethane/acrylate-based photopolymers.52 Tomita et al. have reported a novel three-component PS consisting of cyanine dye, triazine compound and borate salt for efficient radical generation in photopolymerizable nanoparticle-polymer materials dispersed with hyperbranched polymer.53 These developments highlight the importance of the photoinitiating system choice to achieve the full technological potential of the holographic recording material in a specific application.In this work, holographic recording in cellulose acetate-PEG layers was employed to study the performance of novel diethoxycarbonyl-BODIPY – NPG photoinitiating system for free radical photopolymerization of acrylamide monomers under illumination with 532 nm light. Due to their high triplet state yields, BODIPY-pyrene and BODIPY-anthracene dyads PyrBDP 1b and AntBDP 1d, respectively, were investigated. Although meso-9-methylanthracene dyad AntBDP 1c showed even higher triplet state yield, it can undergo cycloaddition reaction with singlet oxygen similarly to previously reported BODIPY-anthracene dyads.37a Dyad AntBDP 1d was used for photopolymerization instead, due to low reactivity of the anthracene group towards singlet oxygen as compared to 9-methylanthracene.54
Performance of the novel dyes is compared with reference dyes, namely, meso-phenylBODIPY BDP 1a and 2,6-diiodoBODIPY (BDPI2). It is assumed that the binder polymer matrix (CA–PEG) is not reactive and is not involved in the photopolymerization process of acrylamide monomers that are evenly dispersed in the volume. CA–PEG layers containing monomers and photoinitiating system components have been prepared using a modified literature procedure,18 as described in detail in Section S9.1 of the ESI.† The absorption spectra of the resulting materials showed characteristic BODIPY absorption bands at 503–534 nm (Fig. 6(a)). For the recording, layers with 75–105 μm thickness were obtained through a drop-casting method followed by drying under a glass dish to slow solvent evaporation, as described in the ESI.† This was in order to achieve clear, even layers across the substrate as it was observed that allowing rapid drying led to opaque, irregular layers. Dry layers showed low light scattering as seen from a clear image observed through the sample in Fig. 6(b). This allowed for a reduction in losses due to scattering of light during holographic recording and achieving high diffraction efficiency of the grating.
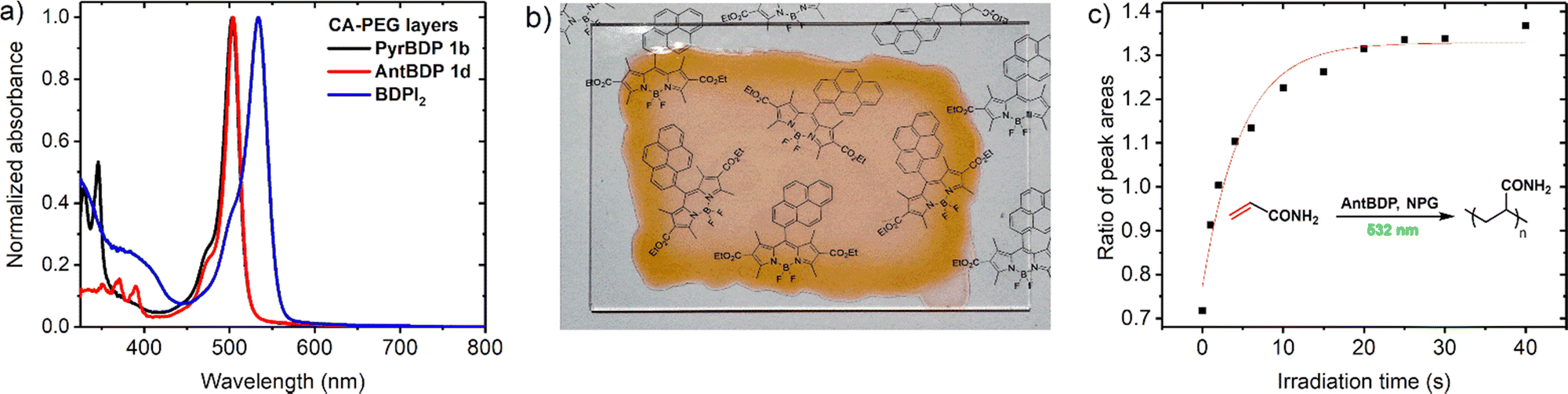 | ||
Fig. 6 (a) UV-Vis absorption spectra of layers containing PyrBDP 1b, AntBDP 1d, and BDPI2 as photosensitizers; (b) photograph of a sample of CA–PEG layers containing PyrBDP 1b on a glass substrate; (c) graph of ratio between Raman scattering peaks for monomers C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds at ≈1600 and ≈1623 cm−1vs. irradiation time for CA–PEG layer containing AntBDP 1d. The sample was exposed to 532 nm light source (10 mW cm−2). The red solid line is a mono-exponential growth fitting curve. C bonds at ≈1600 and ≈1623 cm−1vs. irradiation time for CA–PEG layer containing AntBDP 1d. The sample was exposed to 532 nm light source (10 mW cm−2). The red solid line is a mono-exponential growth fitting curve. | ||
The polymerization of acrylamide and N,N′-methylenebisacrylamide in the CA–PEG materials under 532 nm excitation was confirmed by using Raman microspectroscopy, which has been previously used to study photopolymerization of these monomers.55 The reaction progress was monitored by changes in the characteristic vibrational peaks at 1600 cm−1 and 1623 cm−1, which correspond to the CC double bonds of acrylamide and methylenebisacrylamide, respectively. The spectrometer is equipped with both 785 nm and 532 nm lasers as options for Raman source (Fig. S18, ESI†), and therefore the 532 nm laser was used as a polymerization source and 785 nm as a Raman probe beam. During consumption of the monomers, the CC double bond present in the monomer structure is converted into a C–C single bond leading to a decrease of the intensity of the associated Raman peak. Relative polymerization rates were compared by monitoring the decrease in the intensity of these two characteristic acrylamide Raman peaks as a function of 532 nm irradiation time. The peak area ratio was used in order to negate the effect of fluctuations in intensity due to background, scatter, and laser focus.55b The peaks at 1600 cm−1 and 1623 cm−1 were each fitted using a Gaussian–Lorentzian function, and the ratio of the peak areas was plotted versus time (Fig. 6(c)). The experimental setup and results are shown in more detail in Section S9.3 of the ESI† (Fig. S18 and S19). The results demonstrate that photopolymerization in these materials can be driven under 532 nm irradiation, and that both pyrene and anthracene dyads exhibited comparable photopolymerization rates relative to BDPI2 (Fig. S20, ESI†).
Fig. 7(a) represents the real-time diffraction efficiency growth curves of the transmission gratings with the spatial frequency of 800 lines per mm recorded in the layers containing pyrene and anthracene dyads, and reference photosensitizer BDPI2 as described in Section S10 of the ESI.† Thickness of the gratings has been estimated from the Bragg selectivity curves (Fig. 7(b) and (c)). Video in the ESI† demonstrates the performance of the transmission gratings recorded in CA–PEG layer sensitized with pyrene dyads under illumination with ambient light. The gratings selectively diffract light with different wavelengths of the spectrum at the specific angles (the Bragg angle).
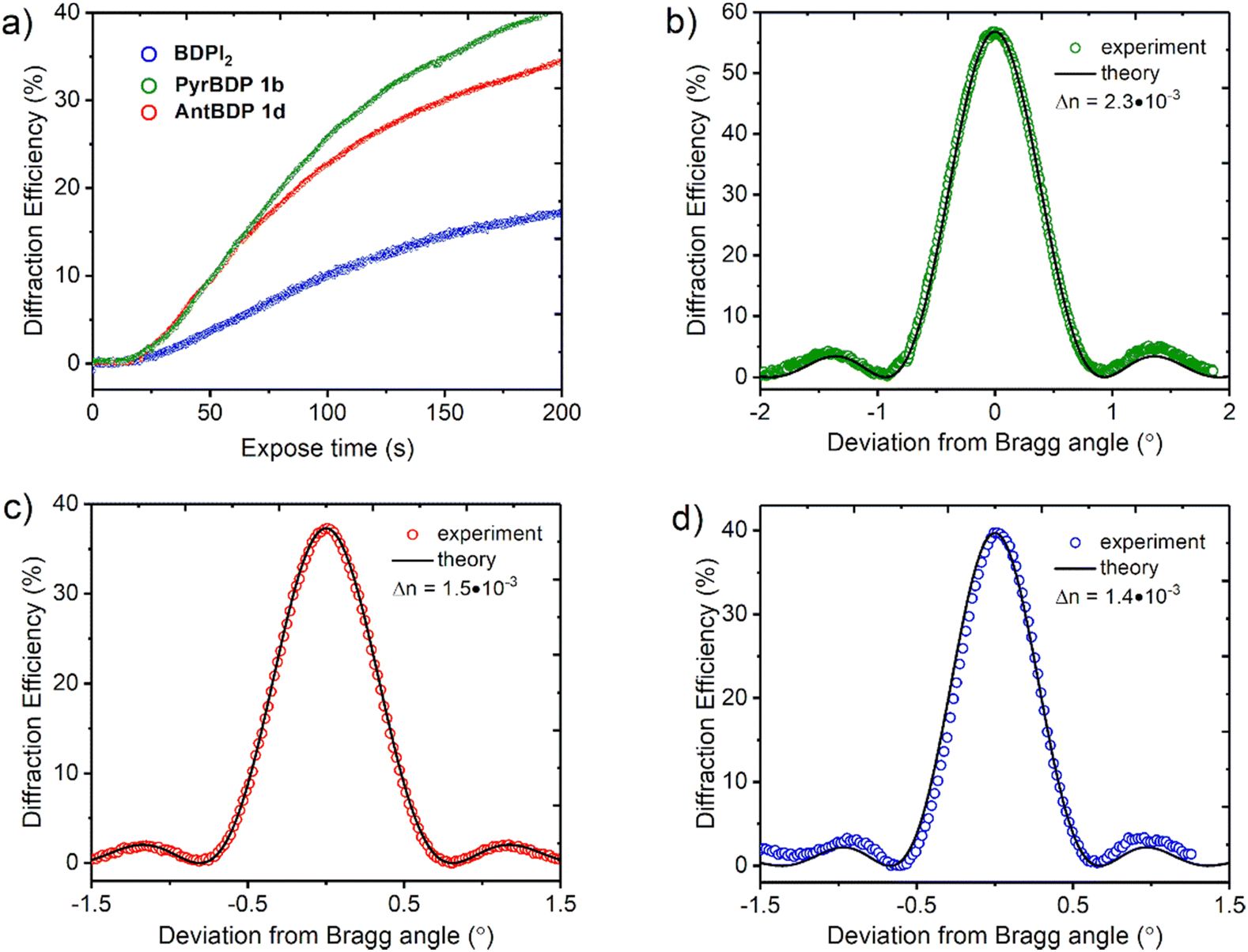 | ||
| Fig. 7 (a) Real time diffraction efficiency evolution for transmission gratings recorded on CA–PEG layers sensitized with dyads PyrBDP 1b (74 μm), AntBDP 1d (87 μm) and BDPI2 (104 μm). The total recording intensity is 20 mW cm−2; (b)–(d) typical Bragg selectivity curves of transmission gratings recorded on CA–PEG layers sensitized with PyrBDP 1b, AntBDP 1d and BDPI2, respectively. Experimental data are fitted by means of coupled-wave theory (eqn (S5)–(S9), ESI†). | ||
We attempted to perform holographic patterning of CA–PEG layer containing BDP 1a and NPG. After exposure to the total recording intensity of 74 mW cm−2 for 100 s, no diffracted intensity was detected, and no diffracted beam was observed. This indicates that BDP 1a – NPG system has too low free radical generation capability to cause polymerization of acrylamide monomers. This can be explained by inefficient ISC in BDP 1a which is manifested by low singlet oxygen generation quantum yields measured for this compound (Table 2).
The photosensitivity of the holographic recording material is directly linked to the polymerization rate, which depends on the ability of the photoinitiator to generate free radicals which lead to the formation of polymer chains and, as a result, the diffraction efficiency increase. The photosensitivity has been estimated from the linear part of growth curves (ESI,† Section S10.3). As seen from Fig. 7(a), the linear part of the curves for PyrBDP 1b and AntBDP 1d have identical slopes, which denotes equal exposure sensitivity of the two compositions, and it is found to be 4 × 10−4 mJ−1 cm2. The slope of the BDPI2 curve is lower and corresponds to the exposure sensitivity of 2 × 10−4 mJ−1 cm2. The lower photosensitivity of layers containing BDPI2 can be explained by lower efficiency of the photoinitiating system containing BDPI2 and NPG to generate free radicals that initiate the formation of polymer chains. Thus, donor–acceptor dyads PyrBDP 1b and AntBDP 1d have demonstrated a higher capability to initiate free radical polymerization in CA–PEG photopolymer than the reference heavy-atom containing photosensitizer.
Fig. 7(b)–(d) represents typical Bragg selectivity curves of the transmission gratings recorded on layers containing PyrBDP 1b, AntBDP 1d and BDPI2 using an exposure energy of 4 J cm−2. Shelf-life study has showed unchanged diffraction efficiency for all samples for 4 months (see Section S10.4 of the ESI† for details). Further shelf-life study is in progress. Refractive index modulation has been estimated by eqn (S5) (ESI†) and it has been found to be 2.3 × 10−3, 1.5 × 10−3 and 1.4 × 10−3 for PyrBDP 1b (74 μm thick layer), AntBDP 1d (87 μm thick layer) and BDPI2 (104 μm thick layer), respectively. The obtained refractive index modulation is comparable to what was achieved in CA–PEG sensitized with Erythrosine B and NPG as reported in our previous work,18 and in polyvinyl alcohol-based photopolymers sensitized with methylene blue58 and Erythrosine B21 (Table 3).
| Refractive index modulation (× 10−3) | ||||
|---|---|---|---|---|
| Thickness (μm) | PVA/AA/MB58 | PVA/NIPA/ErB21 | Cellulose/AA/ErB18 | This work |
| PVA – polyvinyl alcohol; AA – acrylamide; MB – methylene blue; NIPA – N-isopropyl acrylamide; ErB – erythrosine B. N/A – not described. | ||||
| 70 | 1.4 | 2.4 | 2.3 | 2.3 (PyrBDP 1b) |
| 85 | N/A | 2.1 | N/A | 1.5 (AntBPD 1d) |
| 100 | N/A | 1.8 | 1.1 | 1.4 (BDPI2) |
The grating formation mechanism and creation of the refractive index modulation during holographic recording is discussed in the following section.
Grating formation mechanism
In photopolymerizable materials, grating formation mechanism depends on the material composition and the permeability of the matrix which influence the diffusion of mobile species and the resultant refractive index modulation.56 In volume gratings, the refractive index modulation (Δn) is created by the impact of three mechanisms such as change in density, molar refraction and monomer concentration gradient driven diffusion as described in details further.57 Firstly, in bright fringe areas (illuminated areas) the density increases due to polymerization, and it has the minimum value in dark fringe areas (not illuminated areas). This leads to different refractive indexes in bright and dark areas, nbrightdensity and ndarkdensity, respectively, according to the distribution of the light intensity (Fig. 8(a)–(c)). Secondly, the different degree of polymerization in bright and dark areas causes variation in polarizability due different degree of monomers’ CC bonds conversion which is shifted with respect to the modulation due to density variation by 180°. This provides variation in molar refraction and, as a result, in the refractive index too, such as Δnrefraction = nbrightrefraction − ndarkrefraction (Fig. 8(c)). Thirdly, the concentration gradient that occurs during recording leads to the diffusion of monomers. If the diffusion is high then most of the monomers manage to reach bright areas, and the density variation is the main contributor to the refractive index modulation. In case of low diffusion, only a small fraction of monomer molecules reaches the bright area, and structural changes in the monomer molecules play a major role in the formation of refractive index modulation.
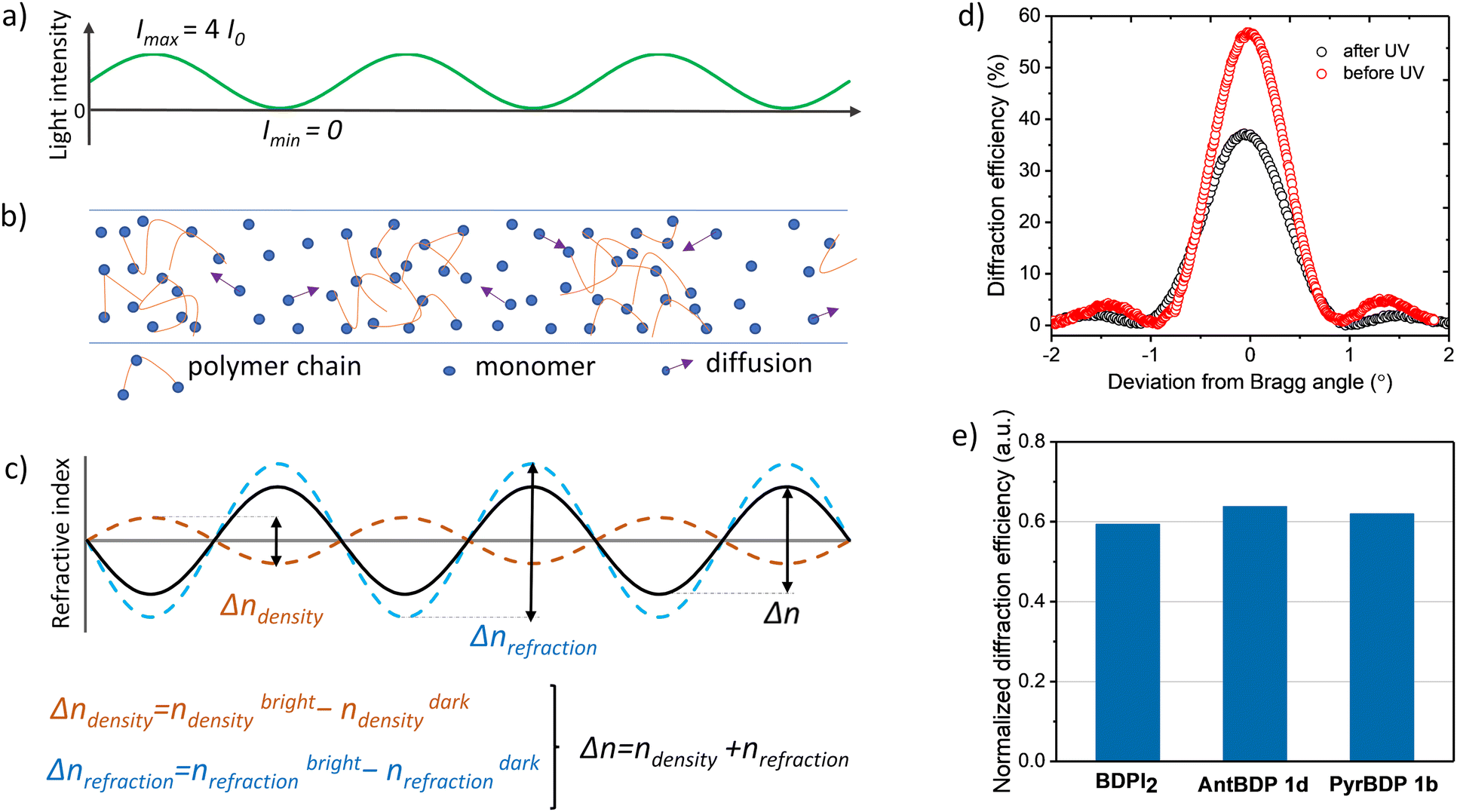 | ||
| Fig. 8 (a) Spatial variation of the recording light intensity created by two recording beams (with I0 intensity) during holographic exposure using the set-up shown in Fig. S21 (ESI†); (b) distribution of monomers/polymer chains in the volume of the photopolymerizable material after holographic exposure. Arrows show the diffusion of monomers due to the concentration gradient created by holographic exposure; (c) mechanism of obtaining refractive index modulation in the photopolymerizable material with low diffusion (monomer molecules diffuse slowly and only a small fraction of them reach the bright fringe area); (d) typical Bragg selectivity curves of PyrBDP 1b sample measured before and after exposure to uniform UV irradiation; (e) normalised diffraction efficiency shows the change in the diffraction efficiency of transmission gratings recorded in cellulose-PEG layers sensitized with BDPI2, PyrBDP 1b and AntBDP 1d after UV exposure. | ||
Here, we present preliminary results on the grating formation mechanism in CA–PEG sensitized with new BODIPYs and reference BDPI2. Our approach is based on the evaluation of the main contributor to the grating formation by applying UV-post exposure with uniform intensity as was previously demonstrated for the acrylamide-based photopolymer containing Erythrosine B and NPG as photoinitiators.58 UV post-exposure (after holographic recording) induces changes in the refractive index modulation due to further bond conversion evenly through the thickness. Changes in the refractive index modulation can be estimated by evaluating the changes in the diffraction efficiency as these parameters are correlated according to the coupled wave theory as described in Section S10.2 of the (ESI†).59
Gratings recorded on 80 ± 5 μm thick layers (PyrBDP 1b, AntBDP 1d and BDPI2) using the exposure energy of 4 J cm−2 were exposed to UVA light with a Dymax UV-curing System (ECE Series). During UV exposure, an energy of 26.3 J cm−2 was used and the diffraction efficiency was measured before and after UV exposure. The normalized diffraction efficiency was calculated as their ratio. Typical Bragg selectivity curves before and after UV-exposure are presented in Fig. 8(d). As seen from Fig. 8(e), the diffraction efficiency of PyrBDP 1b, AntBDP 1d and BDPI2 decreases to 38, 36 and 40.6%, respectively, after UV exposure. The observed decrease of the refractive index modulation indicates that the bond conversion is the main contributor to the grating formation which assumes that the matrix has low permeability and that the mobile species’ diffusion is restricted.58 Further investigation of the photopolymerization kinetics, relationship between the rate of polymerization and the concentrations of monomer, polymer, dye, and initiator is required in order to develop the model of the grating formation mechanism in CA–PEG sensitized with the studied BODIPY dyes. The model will allow to predict the material response at different special frequencies and achieve its full potential. This will be the focus of our future work.
Conclusion
In this work, we designed and synthesized a series of novel heavy-atom-free SOCT-ISC photosensitizers based on 2,6-diethoxycarbonyl-BODIPY scaffold as an electron acceptor. Due to increased electron acceptor ability of this BODIPY scaffold, dyads with pyrene and anthracene groups as electron donors undergo efficient charge transfer and triplets’ formation in non-polar solvent, such as toluene, which is desired for application of these dyes as photoinitiators for polymerization. Transient absorption spectroscopy data confirmed that intramolecular charge separation takes place on a picosecond timescale, followed by recombination of CT states into triplet excited states of BODIPY via SOCT-ISC. For the lead BODIPY-anthracene compound 1c, excellent photosensitization ability was observed as is evidenced by singlet oxygen generation quantum yields of up to 94%.In combination with NPG as a co-initiator, diethoxycarbonyl-BODIPY have showed the ability to initiate free-radical polymerization of acrylamide monomers under 532 nm irradiation, allowing for the formation of volume transmission gratings in a cellulose acetate-based photopolymer. The novel heavy-atom-free photosensitizers provide higher exposure sensitivity than a reference heavy-atom-containing dye, BDPI2. High diffraction efficiency (up to 56%) was obtained for CA–PEG layers sensitized with pyrene and anthracene dyads, proving that they can efficiently operate as a photosensitizers and initiate recording of diffractive structures. The developed photopolymerizable material showed high refractive index modulation (up to 2.3 × 10−3), low scattering, and good mechanical and chemical stability. In addition, unchanged diffraction efficiency was observed for 4 months after the recording. These results provide further perspective for the utilisation of heavy-atom-free BODIPYs as components of photoinitiating systems and expand the arsenal of environment-friendly and sustainable photopolymerizable materials for holography.
Author contributions
A. S. and M. F. synthesized new dyes, studied steady-state optical properties and singlet oxygen generation, prepared CA–PEG layers and performed photopolymerization tests using Raman spectroscopy. T. M. and I. N. performed holographic recording experiments and interpreted the results. C. D. C., S. K., W. A. and F. L. performed spectroscopic measurements and interpreted the results. M. D. assisted in synthesis and characterization of new compounds. S. G. and I. N. developed the procedure for CA–PEG layers preparation and assisted in synthesis of materials. H. J. B. assisted with interpretation of Raman spectroscopy data. A. S., T. M., C. D. C., and M. F. wrote the manuscript. M. F. conceived the idea of this study and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. F. and A. S. acknowledge TU Dublin Research Scholarship Programme. This work was supported by a grant from Science Foundation Ireland (SFI award 21/FFP-A/9214). This publication is based on work supported by the KAUST Office of Sponsored Research (OSR) under award no. OSR-CRG2018-3746 and OSR-CRG2019-4025. The authors thank Dr Luke O'Neill for the help with Raman microspectroscopy experiments.References
- J. Shao, Y. Huang and Q. Fan, Polym. Chem., 2014, 5, 4195–4210 RSC.
- (a) P. Chittavanich, K. Miller and M. D. Soucek, Prog. Org. Coatings, 2012, 73, 392–400 CrossRef CAS; (b) N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef CAS PubMed.
- P. Lederhose, Z. Chen, R. Müller, J. P. Blinco, S. Wu and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 12195–12199 CrossRef CAS PubMed.
- A. Balcerak and J. Kabatc, Polym. Chem., 2022, 13, 1787–1812 RSC.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- J. Zhao, W. Wu, J. Suna and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- (a) S. Callaghan, M. A. Filatov, H. Savoie, R. W. Boyle and M. O. Senge, Photochem. Photobiol. Sci., 2019, 18, 495–504 CrossRef CAS PubMed; (b) J. Deckers, T. Cardeynaels, H. Penxten, A. Ethirajan, M. Ameloot, M. Kruk, B. Champagne and W. Maes, Chem. – Eur. J., 2020, 26, 15212–15225 CrossRef CAS PubMed; (c) J. Jiménez, R. Prieto-Montero, B. L. Maroto, F. Moreno, M. J. Ortiz, A. Oliden-Sánchez, I. López-Arbeloa, V. Martínez-Martínez and S. de la Moya, Chem. – Eur. J., 2020, 26, 601–605 CrossRef PubMed; (d) V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed; (e) R. Prieto-Montero, A. D. Andres, A. Prieto-Castañeda, A. Tabero, A. Longarte, A. R. Agarrabeitia, A. Villanueva, M. J. Ortiz, R. Montero, D. Casanova and V. Martínez-Martínez, J. Mater. Chem. B, 2023, 11, 169–179 RSC.
- (a) N. Kiseleva, M. A. Filatov, M. Oldenburg, D. Busko, M. Jakoby, I. A. Howard, B. S. Richards, M. O. Senge, S. M. Borisov and A. Turshatov, Chem. Commun., 2018, 54, 1607–1610 RSC; (b) Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Phys. Chem. C, 2019, 123, 22793–22811 CrossRef CAS; (c) Y. Hu, Y. Hou, Z. Wang, Y. Li and J. Zhao, J. Chem. Phys., 2020, 153, 224304 CrossRef CAS PubMed; (d) N. Kiseleva, D. Busko, B. S. Richards, M. A. Filatov and A. Turshatov, J. Phys. Chem. Lett., 2020, 11, 6560–6566 CrossRef CAS PubMed; (e) R. A. Arellano-Reyes, A. Prabhakaran, R. C. E. Sia, J. Guthmuller, K. K. Jha, T. Yang, B. Dietzek-Ivanšić, V. McKee and T. E. Keyes, Chem. – Eur. J., 2023, 29, e202300239 CrossRef CAS PubMed.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- (a) M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC; (b) E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- T. Mikulchyk, S. Karuthedath, C. S. P. De Castro, A. A. Buglak, A. Sheehan, A. Wieder, F. Laquai, I. Naydenova and M. A. Filatov, J. Mater. Chem. C, 2022, 10, 11588–11597 RSC.
- F. Dumur, Eur. Polym. J., 2023, 196, 112241 CrossRef CAS.
- A. Uddin, S. R. Allen, A. K. Rylski, C. J. O’Dea, J. T. Ly, T. A. Grusenmeyer, S. T. Roberts and Z. A. Page, Angew. Chem., Int. Ed., 2023, 62, e202219140 CrossRef CAS PubMed.
- M. Topa, F. Petko, M. Galek, K. Machowski, M. Pilch, P. Szymaszek and J. Ortyl, Polymers, 2019, 11, 1756 CrossRef CAS PubMed.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2018, 5, 1 Search PubMed.
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem. – Eur. J., 2021, 27, 9934–9947 CrossRef CAS PubMed.
- S.-e.- Gul, J. Cassidy and I. Naydenova, Photonics, 2021, 8, 329 CrossRef CAS.
- J. Zhang, J. Lalevée, X. Mou, F. Morlet-Savary, B. Graff and P. Xiao, Macromolecules, 2018, 51, 3767 CrossRef CAS.
- A. Al Mousawi, P. Garra, M. Schmitt, J. Toufaily, T. Hamieh, B. Graff, J. P. Fouassier, F. Dumur and J. Lalevée, Macromolecules, 2018, 51, 4633 CrossRef CAS.
- T. Mikulchyk, S. Martin and I. Naydenova, Appl. Opt., 2017, 56, 6348 CrossRef CAS PubMed.
- Q. Gong, S. Wang, M. Huang and F. Gan, Mater. Lett., 2005, 59, 2969 CrossRef CAS.
- G. Qiaoxia, H. Mingju and G. Fuxi, Dyes Pigm., 2006, 69, 204 CrossRef CAS.
- S. Ikeda, S. Murata, K. Ishii and H. Hamaguchi, Chem. Lett., 1999, 1009 CrossRef CAS.
- (a) R. Malallah, H. Li, D. Kelly, J. Healy and J. Sheridan, Polymers, 2017, 9, 337 CrossRef PubMed; (b) F.-K. Bruder, R. Hagen, T. Rölle, M.-S. Weiser and T. Faecke, Angew. Chem., Int. Ed., 2011, 50, 4552 CrossRef CAS PubMed.
- G. A. Cordell, J. Org. Chem., 1975, 40, 3161–3169 CrossRef CAS.
- A. Treibs and F.-H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS.
- (a) H. Falk and G. Schoppel, Monatsh. Chem., 1990, 121, 67–76 CrossRef CAS; (b) N. O. Didukh, V. P. Yakubovskyi, Y. V. Zatsikha, V. N. Nemykin and Y. P. Kovtun, Dyes Pigm., 2018, 149, 774–782 CrossRef CAS; (c) N. O. Didukh, V. P. Yakubovskyi, Y. V. Zatsikha, G. T. Rohde, V. N. Nemykin and Y. P. Kovtun, J. Org. Chem., 2019, 84, 2133–2147 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- Y. Gabe, Y. Urano, K. Kikuchi, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2004, 126, 3357–3367 CrossRef CAS PubMed.
- J.-J. Shie, Y.-C. Liu, Y.-M. Lee, C. Lim, J.-M. Fang and C.-H. Wong, J. Am. Chem. Soc., 2014, 136, 9953–9961 CrossRef CAS PubMed.
- A. B. Descalzo, P. Ashokkumar, Z. Shen and K. Rurack, ChemPhotoChem, 2020, 4, 120–131 CrossRef CAS.
- (a) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606–615 CrossRef CAS; (b) N. Kiseleva, M. A. Filatov, J. C. Fischer, M. Kaiser, M. Jakoby, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, Phys. Chem. Chem. Phys., 2022, 24, 3568 RSC.
- D. P. Kennedy, C. M. Kormos and S. C. Burdette, J. Am. Chem. Soc., 2009, 131, 8578–8586 CrossRef CAS PubMed.
- E. S. Pysh and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 2124–2130 CrossRef CAS.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- (a) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282 CrossRef CAS PubMed; (b) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016–8031 RSC.
- P. Luo, E. C. Feinberg, G. Guirado, S. Farid and J. P. Dinnocenzo, J. Org. Chem., 2014, 79, 9297–9304 CrossRef CAS PubMed.
- (a) Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194 CrossRef CAS PubMed; (b) Z. E. X. Dance, Q. Mi, D. W. McCamant, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2006, 110, 25163 CrossRef CAS PubMed.
- E. Gross, B. Ehrenberg and F. M. Johnson, Photochem. Photobiol., 1993, 57, 808 CrossRef CAS PubMed.
- Y. Zhao, R. Duan, J. Zhao and C. Li, Chem. Commun., 2018, 54, 12329–12332 RSC.
- B. Lang, S. Mosquera-Vázquez, D. Lovy, P. Sherin, V. Markovic and E. Vauthey, Rev. Sci. Instrum., 2013, 84, 73107 CrossRef PubMed.
- P. Foggi, L. Pettini, I. Santa, R. Righini and S. Califano, J. Phys. Chem., 1995, 99, 7439–7445 CrossRef CAS.
- L. Ludvíková, P. Friš, D. Heger, P. Šebej, J. Wirza and P. Klán, Phys. Chem. Chem. Phys., 2016, 18, 16266–16273 RSC.
- S. Vyas, P.-H. Wang and Y. Luo, Opt. Express, 2017, 25, 23726 CrossRef CAS PubMed.
- P. Hu, J. Li, J. Jin, X. Lin and X. Tan, ACS Appl. Mater. Interfaces, 2022, 14, 21544–21554 CrossRef CAS PubMed.
- N. Vorzobova and P. Sokolov, Polymers, 2019, 11, 2020 CrossRef CAS PubMed.
- (a) Z. Shen, Y. Weng, Y. Zhang, C. Wang, A. Liu and X. Li, Polymers, 2021, 13, 936 CrossRef CAS PubMed; (b) J. Xiong, K. Yin, K. Li and S.-T. Wu, Adv. Photonics Res., 2021, 2, 2000049 CrossRef; (c) X. Duan, J. Liu, X. Shi, Z. Zhang and J. Xiao, Opt. Express, 2020, 28, 31316 CrossRef PubMed; (d) M. V. Shishova, S. B. Odinokov, A. Y. Zherdev and D. S. Lushnikov, Appl. Opt., 2021, 60, A140–A144 CrossRef CAS PubMed; (e) C. Jang, C. Lee, J. Jeong, G. Li, S. Lee, J. Yeom, K. Hong and B. Lee, Appl. Opt., 2016, 55, A71–A85 CrossRef CAS PubMed; (f) J. Xiong, K. Yin, K. Li and S.-T. Wu, Adv. Photonics Res., 2021, 2, 2000049 CrossRef.
- M. R. Gleeson, J. V. Kelly, D. Sabol, C. E. Close, S. Liu and J. T. Sheridan, J. Appl. Phys., 2007, 102, 023108 CrossRef.
- A. Ibrahim, X. Allonas, C. Ley, K. Kawamura, H. Berneth, F.-K. Bruder, T. Fäcke, R. Hagen, D. Hönel, T. Rölle, G. Walze and M. S. Weiser, Chem. – Eur. J., 2014, 20, 15102–15107 CrossRef CAS PubMed.
- Y. Liu, T. Shen, B. Kang, S. Li, Z. Zheng, H. Lv and J. Zheng, Opt. Laser Technol., 2022, 156, 108580 CrossRef CAS.
- H. Berneth, F. K. Bruder, T. Fäcke, S. Hansen, K. Kawamura, L. Pitzer, S. Kern, B. Wewer and T. A. Rölle, Polymers, 2021, 13, 3517 CrossRef CAS PubMed.
- A. Narita, J. Oshima, Y. Iso, S. Hasegawa and Y. Tomita, Opt. Mater. Express, 2021, 11, 614–628 CrossRef.
- J. M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668 CrossRef CAS PubMed.
- (a) R. Jallapuram, I. Naydenova, H. J. Byrne, S. Martin, R. Howard and V. Toal, Appl. Opt., 2008, 47, 206 CrossRef CAS PubMed; (b) D. Cody, E. Mihaylova, L. O’Neill and I. Naydenova, Opt. Mater., 2015, 48, 12–17 CrossRef CAS.
- S. Blaya, L. Carretero, R. F. Madrigal, M. Ulibarrena, P. Acebal and A. Fimia, Appl. Phys. B, 2003, 77, 639–662 CrossRef CAS.
- E. S. Gyulnazarov, T. N. Smirnova and E. A. Tikhonov, Opt. Spectrosc., 1989, 67, 99 Search PubMed.
- T. Mikulchyk, S. Martin and I. Naydenova, Opt. Mater., 2014, 37, 810–815 CrossRef CAS.
- H. Kogelnik, Bell Syst. Tech. J., 1969, 48, 2909–2947 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02640j |
| This journal is © The Royal Society of Chemistry 2023 |