DOI:
10.1039/D3TC02432F
(Paper)
J. Mater. Chem. C, 2023,
11, 15160-15168
Quinodimethane embedded expanded helicenes and their open-shell diradical dications/dianions†
Received
11th July 2023
, Accepted 19th September 2023
First published on 10th October 2023
Abstract
Helicenes and expanded helical π-conjugated molecules consisting of aromatic benzene rings or heterocycles have been widely studied, but the incorporation of a quinoidal conjugated unit in such π-systems, which is expected to provide intriguing electronic properties, remains limited. Herein, we report the synthesis and redox properties of two quinodimethane-embedded expanded [11]- and [13]helicenes with respectively eleven and thirteen fused rings. Their helical-shaped geometry structures were unambiguously elucidated by X-ray crystallographic analysis. Both molecules exhibit a small energy gap and amphoteric redox behavior with multiple redox waves. Moreover, a fast racemization process for cyclopenta-fused expanded [11]helicene was observed above 173 K by variable temperature nuclear magnetic resonance (NMR), with a small inversion barrier, while cyclopenta-fused expanded [13]helicene possesses a moderate racemization barrier (18.09 ± 1.72 kcal mol−1 at coalescence temperature Tc = 360 K), as demonstrated by dynamic NMR spectroscopy. On the other hand, both compounds can be oxidized or reduced by NOSbF6 or sodium anthracenide to the respective radical cations, radical anions, dications and dianions. Compared with closed-shell neutral compounds, their dications and dianions show significant open-shell singlet diradical character with a small singlet–triplet energy gap. This work provides some insights into the design and synthesis of novel helical π-systems with tunable properties.
Introduction
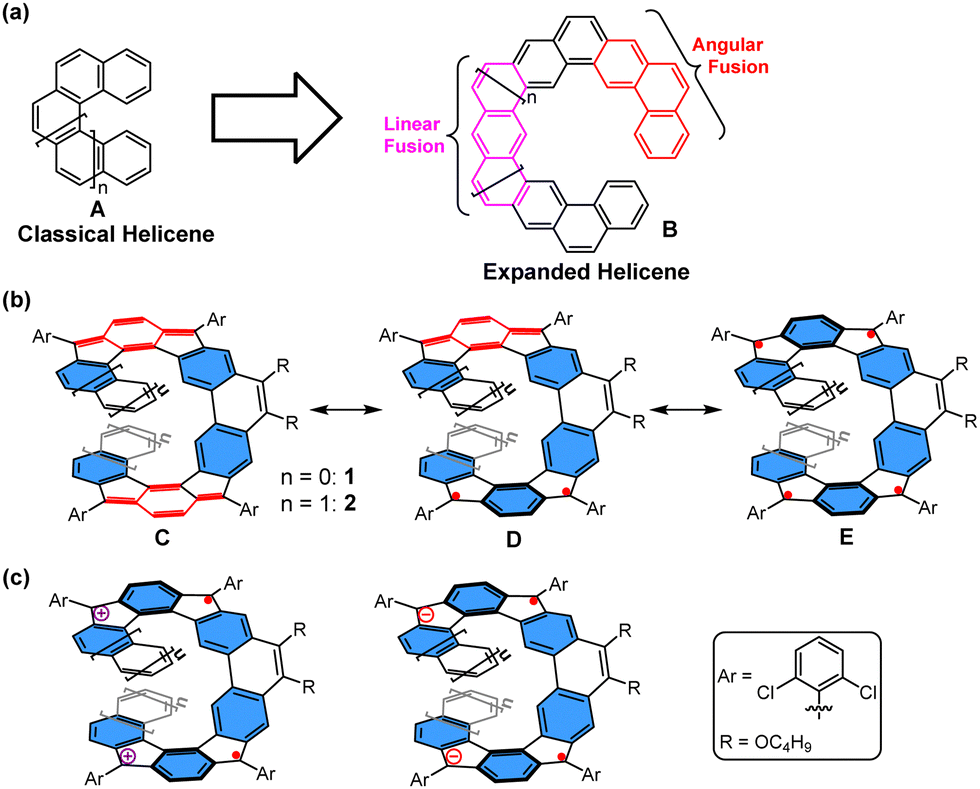
Polycyclic aromatic hydrocarbons (PAHs), which are atomically precise graphene segments, have evolved as important benchmarks in the field of graphene chemistry. Here, their photophysical properties can be fine-tuned by precise control over their size, shape, and peripheral structure.1 Besides planar PAHs, which have made great progress in various practical applications, the design, synthesis, and study of nonplanar PAHs is also of great interest.2 Alteration of π-conjugated systems towards the third dimension is enabled either by the institution of helicity3 or by the introduction of nonhexagonal rings.4–7 Therefore, deviation from planarity will offer novel ways and means to incorporate new physical properties to their flat counterparts.
Helicenes (A, Fig. 1(a)), which are nonplanar PAHs with a screw-shaped helical π-skeleton formed by ortho-fused aromatic rings, have long attracted considerable attention due to their unusual distorted structure, inherent chirality and dynamic behavior.8 These characteristics from helical π-systems also render them promising candidates for diverse applications, including organic electronics,9 supramolecular chemistry,10 and asymmetric catalysis.11 With the increasing number of ortho-fused rings, a multilayer π-electron starts to form, and the configurational stabilities of helicenes increase in most cases. So far, various impressive π-extended helicenes with single helicoid geometries or multihelicity have been reported with intriguing optical and electronic properties.12 On the other hand, increasing the size of a helicene by alternation of linear and angular ring fusion (B, Fig. 1(a)) might result in novel electronic, photophysical, and chiroptical properties.13 In addition to the expanded helical PAHs with solely aromatic benzene rings, the incorporation of a quinoidal conjugated unit in such π-systems is also expected to provide intriguing properties. In this context, herein, we report two new expanded helicenes, 1 and 2, in which two para-quinodimethane (p-QDM) units are fused onto the central phenanthrene moiety and the terminal two benzene or naphthalene rings (Fig. 1(b)). Accordingly, the molecule has eleven and thirteen consecutively fused rings, respectively. 1/2 can be drawn in different resonance forms, for example, an open-shell biradical/tetraradical form with 5/6 aromatic sextet rings (the hexagons shaded in blue) (form D/E, Fig. 1(b)), and a closed-shell form in which the spins are coupled through the 1,4-benzoquinodimethane units with four aromatic sextet rings (form C, Fig. 1(b)). Moreover, oxidation or reduction of 1 and 2 would lead to a dramatic change of the optical properties and aromaticity; in particular, their dications/dianions may display open-shell diradical character (Fig. 1(c)). Thus, the electronic structure, aromaticity, and radical character of both the neutral compounds and their charged species are of great interest. In this article, we report the detailed synthesis and physical characterization of two p-QDM embedded expanded helicenes 1 and 2 and their charged species. We also disclose the dynamic behaviors of both 1 and 2, theoretical studies regarding their isomerization processes, and their physical properties.
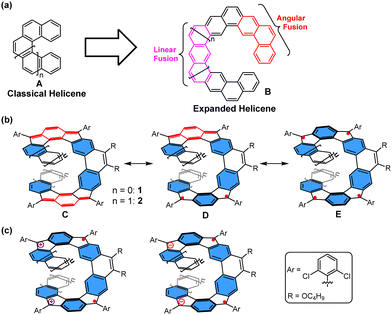 |
| | Fig. 1 (a) Representative structures of classical helicene and expanded helicene. Representative resonance forms of (b) the helical compounds 1 and 2, and (c) their charged species. The rings shaded in blue denote aromatic sextets. | |
Results and discussion
Synthesis
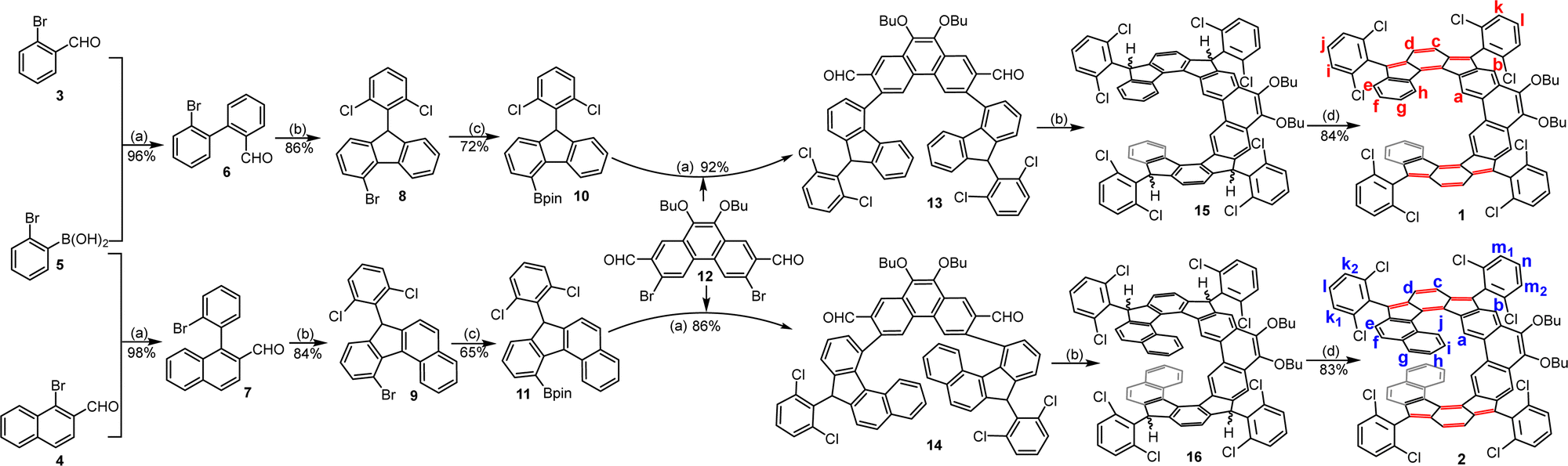
The helical molecules 1 and 2 were synthesized according to Scheme 1. First, Suzuki coupling between 2-bromophenylboronic acid 5 and 3 or 4 gave the aldehyde intermediates 6 and 7, respectively. Treatment of 6/7 with 2,6-dichlorophenylmagnesium chloride reagent afforded the corresponding alcohols, which were subjected to BF3·OEt2-mediated Friedel–Crafts alkylation reaction to generate the precursors 8/9. Pd-catalyzed borylation of 8/9 produced the key intermediates 10 and 11 in 72% and 65% yield, respectively. Next, the key intermediates dialdehydes 13 and 14 were synthesized by Suzuki coupling reaction between 1214 and 10 or 11, respectively. Treatment of 13 and 14 with 2,6-dichlorophenylmagnesium chloride at room temperature gave the respective diols, which then underwent BF3·Et2O mediated intramolecular Friedel–Crafts alkylation to afford the tetrahydro products 15 and 16. Finally, a one-pot deprotonation of 15 and 16 with tBuOK in THF followed by oxidation with p-chloranil furnished the desired helicenes 1 and 2 in 84% and 83% yield (over three steps from 13/14), respectively, which are stable and can be purified by normal silica gel column chromatography. Notably, the bulky 2,6-dichlorophenyl groups are used to block the most reactive sites at the cyclopenta (CP) rings to obtain stable materials, and the long butoxy chains are attached to the 9,10-positions of the phenanthrene units to ensure sufficient solubility.
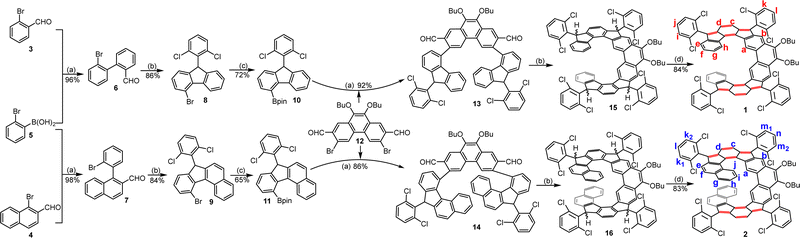 |
| | Scheme 1 Synthesis of expanded helicenes 1 and 2: (a) Pd(PPh3)4, K2CO3, THF/H2O, reflux; (b) (1) 2,6-dichloro-1-bromobenzene, isopropylmagnesium chloride, THF, 0 °C–rt; (2) boron trifluoride etherate, DCM, rt; (c) B(pin)2, Pd(dppf)Cl2, dioxane, 90 °C; (d) (1) potassium tert-butoxide, 18-crown-6, THF, rt; (2) p-chloranil, rt. | |
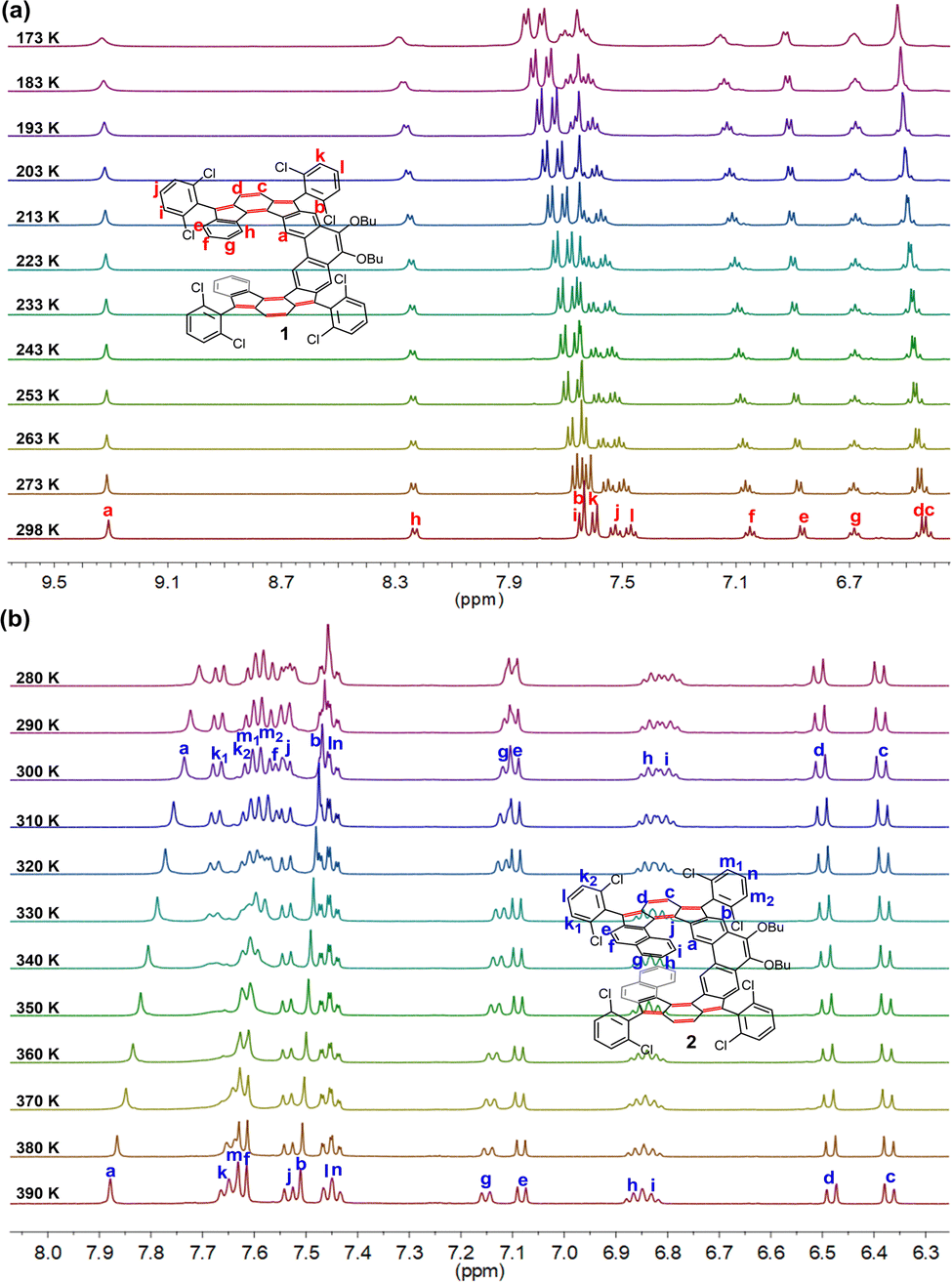
These two compounds are unambiguously characterized by NMR, HRMS, and X-ray crystallography. The 1H NMR analysis showed well-resolved proton signals, which could be clearly assigned with 1H–1H Correlation spectroscopy (COSY) and Rotating-Frame Overhauser spectroscopy (ROESY) measurements (Fig. 2 and Fig. S1–S4, ESI†). As indicated in Fig. 2, the protons c and d in 1 and 2 appeared at relatively higher field (δ = 6.29–6.46 ppm), implicating the shielding effect from the vinylic character along with the help of weak antiaromatic character. In addition, the proton a of 2 that appeared at 7.79 ppm was remarkably shifted upfield by ca. 1.5 ppm compared to the corresponding chemical shift of 1 that appeared at 9.30 ppm, reflecting the shielding effect induced by the spatial overlap of the terminal benzene rings, as revealed by the X-ray structure (vide infra). On the other hand, sharp NMR signals of 1 and 2 at room temperature also indicate their closed-shell natures.
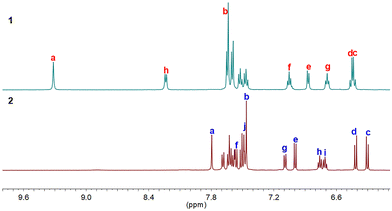 |
| | Fig. 2
1H NMR spectra (aromatic region) of 1 and 2 recorded in THF-d8 (refer to Scheme 1 for labeling). | |
X-ray crystallographic analysis
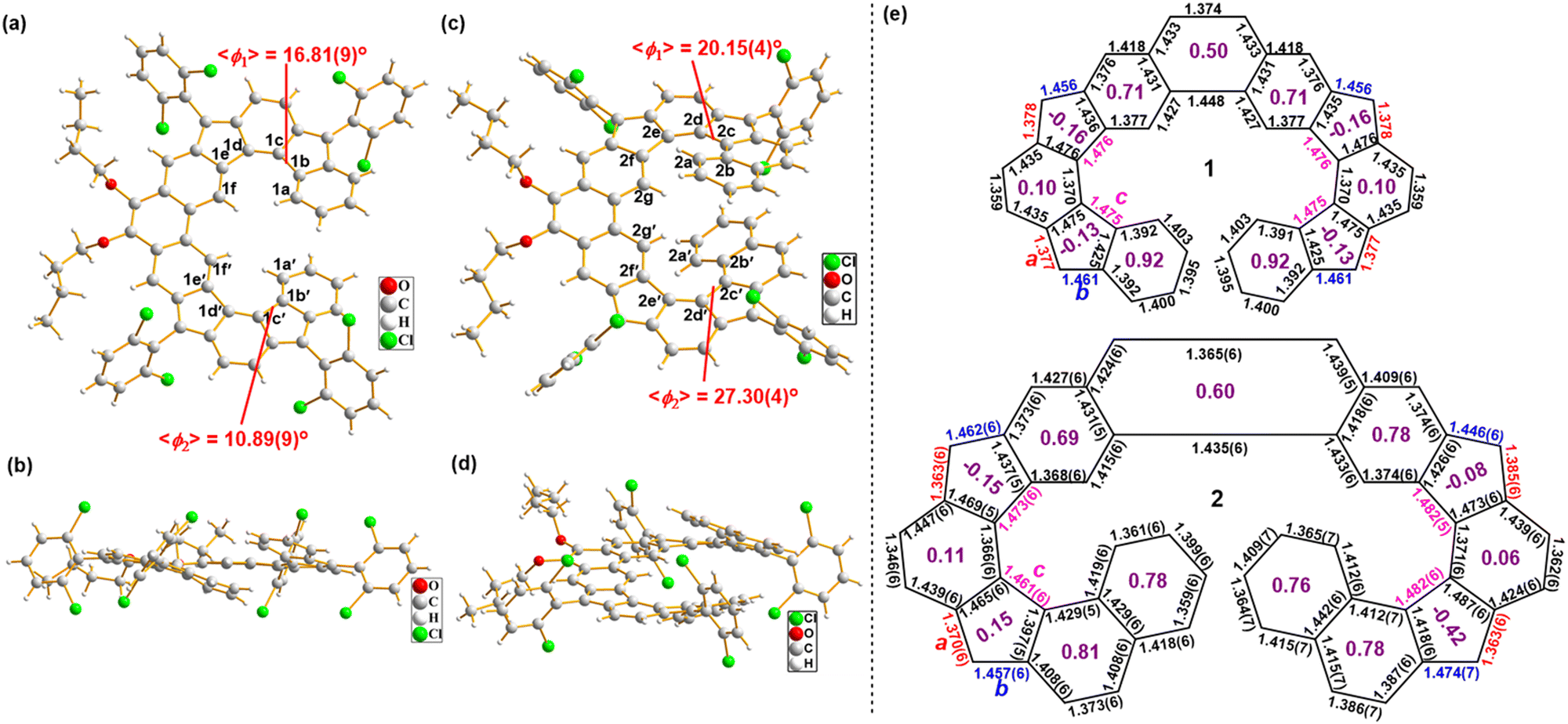
Single crystals of 1/2 suitable for X-ray crystallography were grown by slowly diffusing acetonitrile vapor into the DCM solution of each compound.15 X-ray diffraction data unambiguously discloses their helical structures (Fig. 3). The geometry of 1/2 slightly deviates from the C1 symmetry, which is reflected by the unequal torsion angles of their two cyclopenta-fused helicene substructures. As defined by the selected dihedral angles, the angles of torsion are 16.81° for C1a–C1b–C1c–C1d, 10.89° for C1a′–C1b′–C1c′–C1d′, 20.15° for C2b–C2c–C2d–C2e, and 27.30° for C2b′–C2c′–C2d′–C2e′ (Fig. 3(a) and (c)). In 2, the terminal benzene rings overlap with each other, and the shortest distance between the opposite carbon atoms is about 3.39 Å, well below the sum of van der Waals radius of two carbon atoms (3.4 Å). The more twisted structure of 2 compared to 1 leads to a more significant shielding effect of the relevant protons, as demonstrated by the aforementioned chemical shifts.
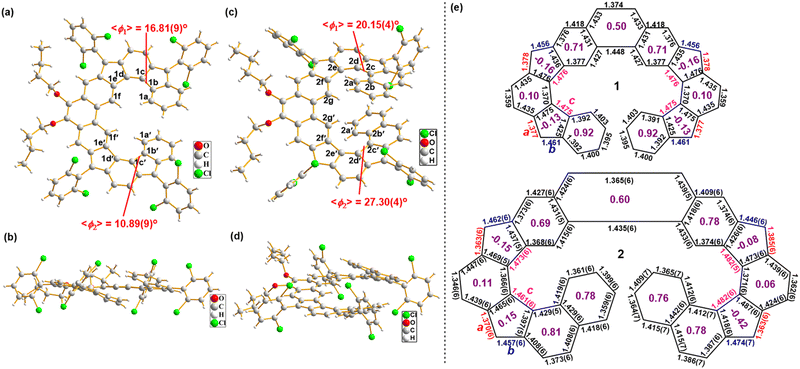 |
| | Fig. 3 Top view (a), (c) and side view (b), (d) of the X-ray crystallographic structures of 1 and 2; (e) selected bond lengths (Å) of the backbone. For 1, the quality of crystallographic data (Rint = 11.98%, R1 = 19.89%, wR2 = 43.06% and max Q peak is 2.9 due to disordered of atoms) is not sufficient for bond length analysis and thus the calculated bond lengths were included. The purple numbers in the individual rings (e) are calculated HOMA values based on the bond lengths. | |
The X-ray diffraction data also revealed the detailed bond parameters of both compounds (Fig. 3(e)). Although the low quality of the crystallographic data of compound 1 does not allow reliable bond length analysis, the backbone of the π-molecule was clearly seen (Fig. S21, ESI†) and thus the calculated bond lengths were included. The bond c (1.461–1.482 Å) in the CP rings is stretched evidently due to steric hindrance. In 1/2, the bond a (1.363–1.385 Å) in the CP rings is obviously shorter than that of a typical C(sp2)–C(sp2) single bond (1.45 Å) and close to that in a typical olefin (1.33–1.35 Å), while the bond b (1.446–1.474 Å) in the CP rings is obviously longer than that of a typical C(sp2)–C(sp2) double bond in benzene (1.39 Å) and close to that of a typical C(sp2)–C(sp2) single bond (1.45 Å) (Fig. 3(e)), all suggesting the formation of a dominate quinoidal structure. Based on bond lengths in the crystal structure, the local aromaticity of individual rings in 1/2 was evaluated using the harmonic oscillator model of aromaticity (HOMA),16 thus implying more aromatic (larger HOMA values) phenanthrene unit and terminal benzene/naphthalene rings versus less aromatic (smaller HOMA values) quinoidal units (Fig. 3(e)). The bond length analysis also suggests that the open-shell radical forms (such as form D/E in Fig. 1(b)) have negligible contribution to the ground-state structure, and indeed, our spin-unrestricted density functional theory (DFT) calculations of both 1 and 2 revealed their closed-shell nature. This is also reasonable considering that conversion from the closed-shell form C to an open-shell diradical form D will only gain one aromatic sextet ring (Fig. 1(b)), which is not sufficient to compensate for the energy required to break a π bond.
Interestingly, both 1 and 2 are chiral molecules, which exist as a pair of enantiomers in the crystals. However, they could not be resolved after some attempts. Moreover, there is no π–π interaction between adjacent molecules in the 3D packing structures of 1 and 2, and each molecule is connected to adjacent molecules through intermolecular [C–Cl⋯π] halogen bonding interactions and [C–Cl⋯H] interactions (Fig. S18, ESI†).
Dynamic process
To study the configurational lability of the expanded helicene frameworks, the kinetics of the isomerization processes were investigated by dynamic variable-temperature (VT) NMR spectroscopy (Fig. 4). The 1H NMR spectrum of 1 in tetrahydrofuran (THF-d8) is well resolved at room temperature and only one set of resonance signals was observed, indicating a fast racemization process. Cooling the solution of 1 in THF-d8 resulted in gradual broadening of the resonances for the protons i and k on the 2,6-dichlorophenyl groups (Fig. 4(a)). However, the coalescent temperature (Tc) was even below 173 K (limit of our measurement), which restricted detailed analysis. Moreover, the 1H NMR spectrum of 2 in 1,1,2,2-tetrachloroethane-d2 (C2D2Cl4) at 390 K is well resolved and can be fully assigned by two-dimensional (2D) COSY and ROESY NMR techniques (Fig. 4(b) and Fig. S5, S6 in ESI†). Only one set of signals for 2 were observed at 390 K due to a rapid interconversion of the two isomers and fast flipping of two helicene units relative to the NMR timescale. As the temperature was lowered, the signals were gradually broadened and became fully coalescent at about 360 K. Upon further cooling, protons k1, k2, m1, and m2, were split into four resonance peaks, indicating that the racemization process of 2 became slow on the NMR time scale. In addition, upon cooling, proton a on the backbone also showed a gradual upfield shift (from δ = 7.88 ppm at 390 K to δ = 7.71 ppm at 280 K), thus indicating that the shielding effect from the terminal benzene rings is enhanced due to the more rigid structure at lower temperature.
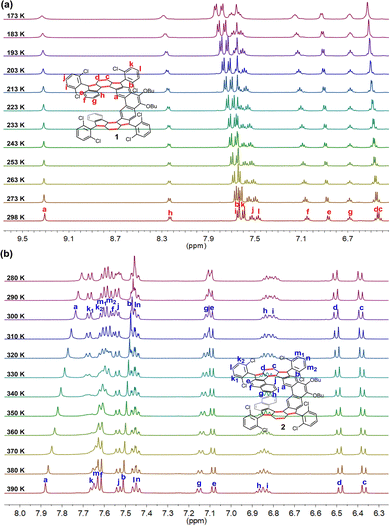 |
| | Fig. 4 VT 1H NMR spectra (aromatic region) of (a) 1 in THF-d8 and (b) 2 in C2D2Cl4. | |
Next, the interconversion rate constant (k [s−1]) was estimated by line-shape analysis17 of the pair of peaks k1 and k2, in the range from 360 to 290 K (see details in the SI). At temperatures above the coalescence temperature (Tc = 360 K), only one set of averaged resonances was observed due to the fast interconversion, and the rate constant at 300 K was estimated to be 0.24 s−1. The exchange rate constants k were then plotted versus the reciprocal number of temperature (1/T) and fitted by the Eyring equation:
where
R is the gas constant,
T is the measured temperature, Δ
H‡ is the activation enthalpy,
kB is the Boltzmann constant,
h is the Planck constant, and Δ
S‡ is the activation entropy. These plots provided the thermodynamic parameters Δ
H‡ = 18.32 ± 0.82 kcal mol
−1 and Δ
S‡ = 0.63 ± 2.49 cal (mol K)
−1 (
Fig. 5). The racemization energy barrier of
2 at the coalescence temperature
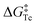
was then estimated as 18.09 ± 1.72 kcal mol
−1, which is much lower than that of the [6]helicene (37.3 kcal mol
−1)
18 and [7]helicene (41.7 kcal mol
−1).
19 The low barrier should be attributed to the fact that the distortion required for racemization is spread over a larger number of bonds and angles (as for [7]heliphene
20).
 |
| | Fig. 5 Kinetic analysis of the exchange rate constants of 2 at different temperatures by the Eyring equation. | |
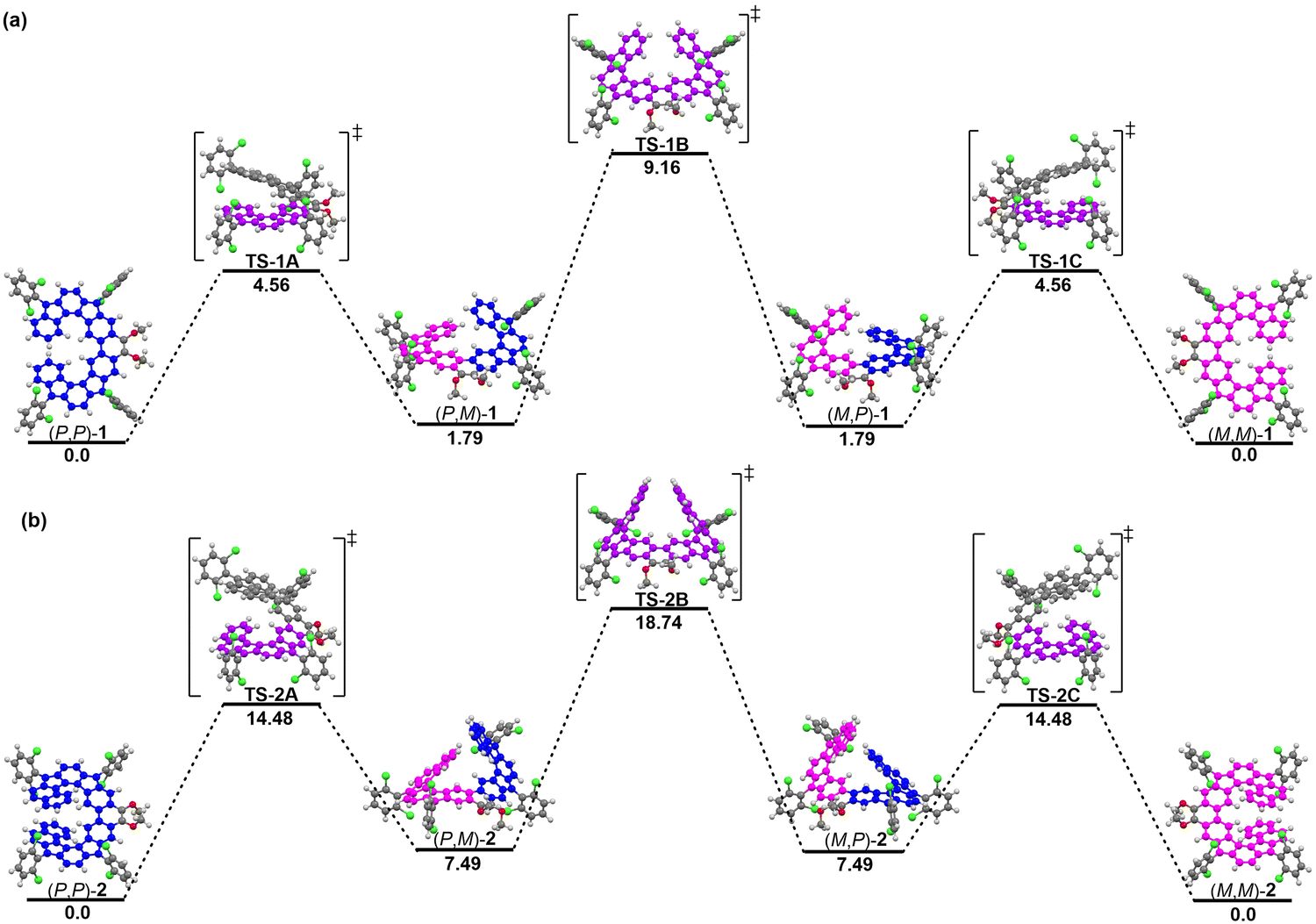
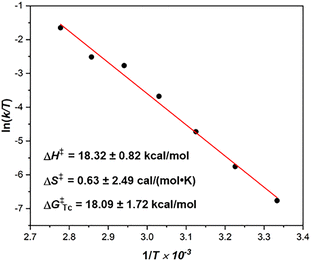
To uncover the thermal stability and dynamic behavior of racemic 1 and 2, DFT calculations were conducted to understand these dynamic processes (Fig. 6). Based on the calculations, (P,P)-1 and (P,P)-2 are thermodynamically more stable than (P,M)-1 and (P,M)-2 by 1.79 kcal mol−1 and 7.49 kcal mol−1, respectively. Furthermore, all possible transition states with face-to-face oriented aromatic rings of the helical substructures associated with diastereomers were fully optimized at the B3LYP/6-31G(d) level of theory. The most plausible isomerization pathway from (P,P)- to (M,M)-forms consists of an inversion of the two helicities labeled TS1A-1C/TS2A-2C with the lowest total activation energy of ΔG‡ = 9.16 kcal mol−1 (from (P,P)-1 to (M,M)-1) and ΔG‡ = 18.74 kcal mol−1 (from (P,P)-2 to (M,M)-2), respectively (Fig. 6). The first step starts from the inversion of lower helicenes TS1A/TS2A to afford (P,M)-1/2 (ΔG‡ = 4.56 kcal mol−1 (from (P,P)-1 to (P,M)-1) and ΔG‡ = 14.48 kcal mol−1 (from (P,P)-2 to (P,M)-2)). The second step includes the further inversion of lower helicenes TS1B/TS2B to furnish (M,P)-1/2 (ΔG‡ = 9.16 kcal mol−1 (from (P,P)-1 to (M,P)-1) and ΔG‡ = 18.74 kcal mol−1 (from (P,P)-2 to (M,P)-2)), which is the rate-determining step of the overall inversion process. The final step consists of an inversion of the upper helicenes TS1C/TS2C to generate (M,M)-1/2. Although both the (P,M)-1 and (P,M)-2 intermediates could not be detected by 1H NMR measurements during the thermal isomerization, they can be elucidated by theoretical calculations. It should also be noted that the racemization of 1 proceeds more easily via intermediates TS1B (ΔG‡ = 9.16 kcal mol−1), indicating that optical resolution of the two enantiomers at room temperature is difficult, which is consistent with our unsuccessful efforts. In addition, the calculated racemization energy (ΔG‡ = 18.74 kcal mol−1) of 2 is comparable to that experimentally obtained (ΔG‡ = 18.09 ± 1.72 kcal mol−1). Chiral separation of the two enantiomers of 2 was achieved on an analytical HPLC with a chiral stationary phase column (column, CHIRALPAK ID; mobile phase, n-hexane/isopropanol = 65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35) (Fig. S7, ESI†). However, the preparatory scale separation of 2 was unsuccessful due to its limited solubility in the mobile phase.
:35) (Fig. S7, ESI†). However, the preparatory scale separation of 2 was unsuccessful due to its limited solubility in the mobile phase.
 |
| | Fig. 6 Energy diagram for the racemization of (a) 1 and (b) 2. The relative Gibbs free energy (unit: kcal mol−1) was calculated at the B3LYP/6-311G(d) level. | |
Optical and electrochemical properties
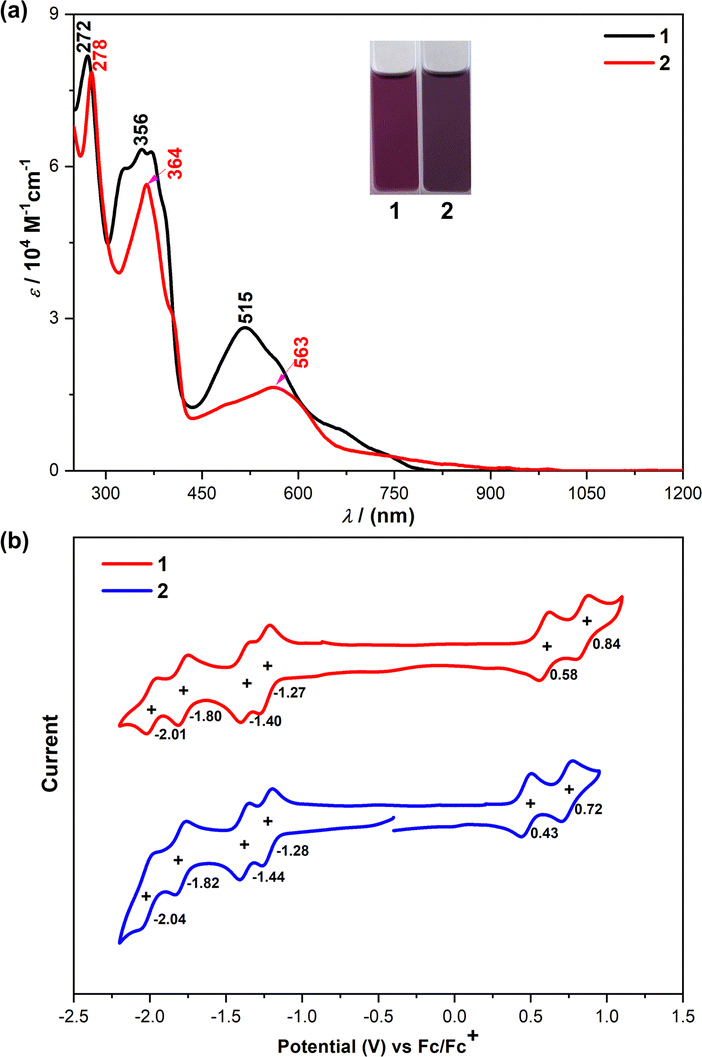
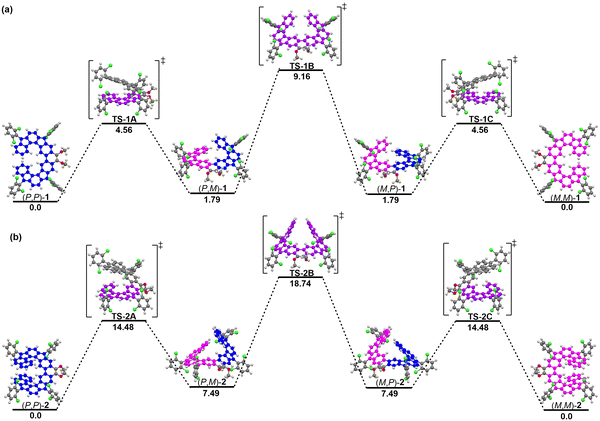
To evaluate the optical properties of both 1 and 2, UV/vis absorption spectra in dichloromethane were measured (Fig. 7(a)). The absorption spectrum of 1 showed a long-wavelength absorption band centered at 515 nm, together with a long tail extended to 800 nm. After extension of the π-systems by benzoannulation, the maximum absorption peak of 2 is red-shifted to 563 nm with the tail extended to 1050 nm. The observed absorption bands were in good agreement with the simulation by time-dependent DFT (TD DFT) calculations (see ESI†). The optical energy gaps (Eoptg) of 1 and 2 were estimated to be 1.51 eV and 1.23 eV, respectively, from the lowest energy absorption onset.
 |
| | Fig. 7 (a) UV-vis-NIR spectra of 1 and 2 in DCM solutions. (b) Cyclic voltammograms of 1 and 2 in DCM solutions. Inset are photos of the solutions. | |
Subsequently, we carried out cyclic voltammetry (CV) measurements to experimentally investigate the electrochemical properties of 1 and 2 (Fig. 7(b)). Both compounds exhibited two reversible oxidation waves with half-wave potential (Eox1/2) at 0.58 and 0.84 V for 1, and 0.43 and 0.72 V for 2 (vs. Fc/Fc+, Fc = ferrocene), and four reversible reduction waves with half-wave potential (Ered1/2) at −1.27, −1.40, −1.80 and −2.01 V for 1, and −1.28, −1.44, −1.82 and −2.04 V for 2. The HOMO/LUMO energy levels of 1 and 2 were determined to be −5.30/−5.17 and −3.63/−3.64 eV, respectively, from the onset of the first oxidation/reduction wave, and the electrochemical energy gaps (EECg) are estimated to be 1.67 and 1.53 eV, respectively, in agreement with the trend of the optical energy gaps. These results revealed that the LUMO level remains unchanged upon the π-extension from 1 to 2, while the HOMO level changes. As such, DFT calculations were performed at the B3LYP/6-31G(d,p) level of theory to elucidate the significant difference in the physical properties observed in 1 and 2 (Fig. S11, ESI†). The LUMO coefficients of both molecules are mainly localized on the quinodimethane units, while their HOMO coefficients are delocalized along their molecular backbones (Fig. S11, ESI†), so 2 has an enhanced HOMO energy level due to the π-extension, leading to the bathochromic shift of its absorption spectrum compared to that of 1.
Chemical oxidation and reduction
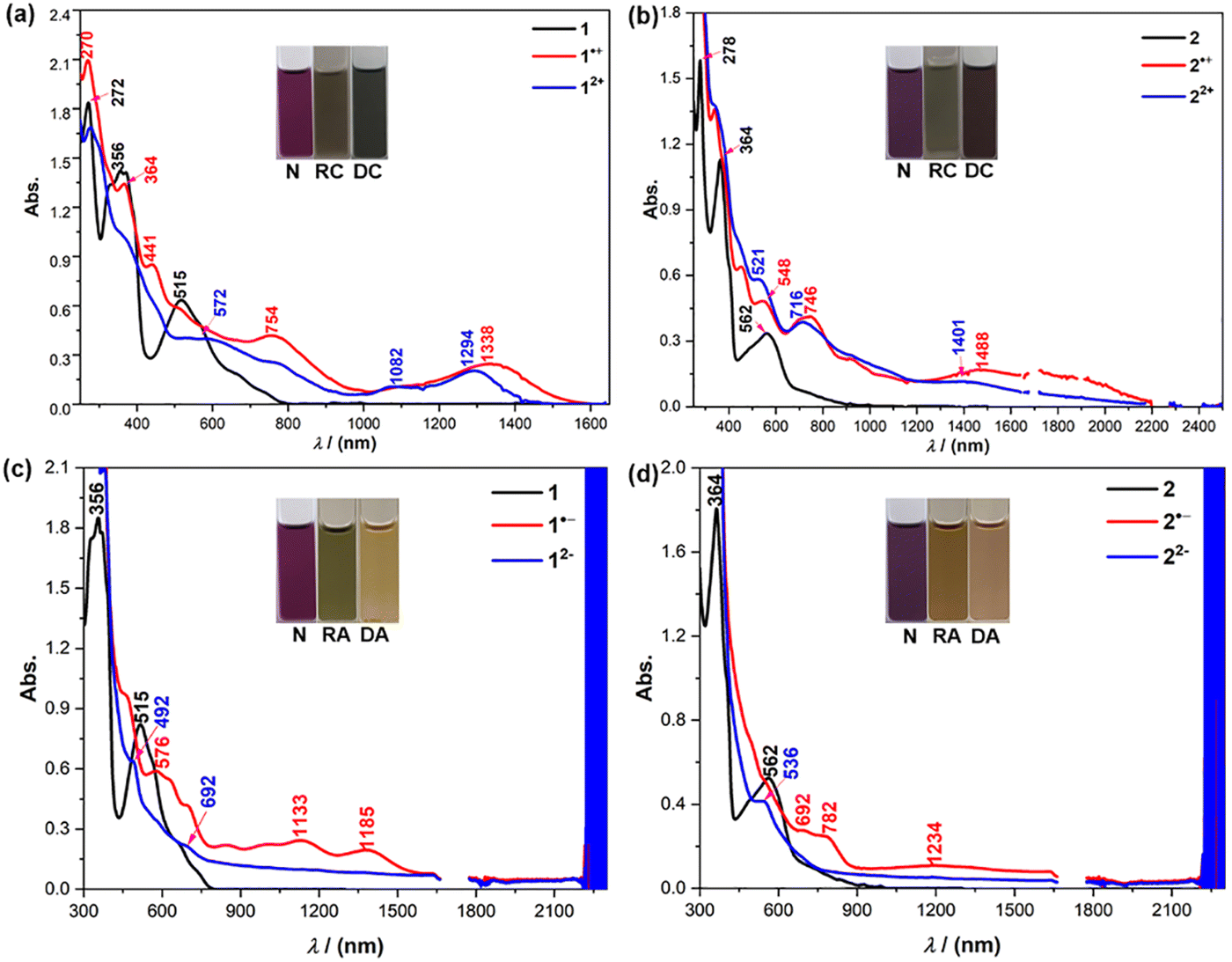
The oxidative and reductive titration experiments were carried out to study the redox behaviors of 1 and 2. Stepwise oxidation of 1/2 can be achieved by titration with NO·SbF6 in anhydrous DCM as monitored by UV-vis-NIR absorption spectroscopy (Fig. 8(a) and (b)). The radical cations display long wavelength absorption with maximum at 1338 nm for 1˙+, and 1488 nm for 2˙+. The dications exhibit blue-shifted absorption compared to the respective radical cations, with a moderate absorption band at 1294 nm for 12+ and 1401 nm for 22+ in the near-infrared (NIR) region. The addition of an excess amount of the oxidant did not generate the trications or tetracations. Reductive titration of 1/2 with freshly prepared sodium anthracenide in dry THF (Fig. 8(c) and (d)) gave their respective radical anions and dianions. Both the radical anions and dianions display long wavelength absorption (Fig. 8(c) and (d)). However, the addition of an excess amount of the reductant did not generate the trianions and tetraanions due to the high reduction potentials (Fig. 7(b)). This trend is in agreement with the time-dependent DFT calculations (see ESI†).
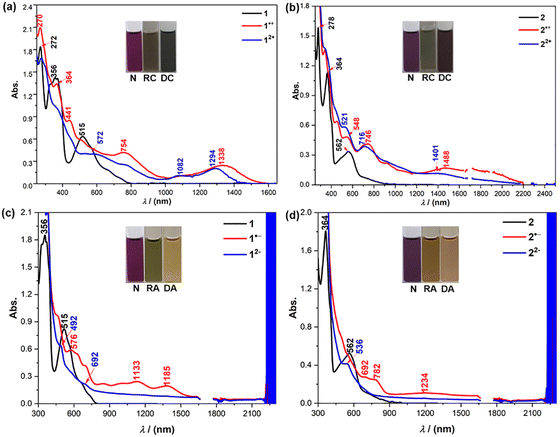 |
| | Fig. 8 (a), (b) UV-vis-NIR absorption spectra of 1/2, the radical cation (RC) and dication (DC) species in DCM. (c), (d) UV-vis-NIR absorption spectra of 1/2, the radical anion (RA) and, dianion (DA) species in THF; the background absorbance at ca. 2250 nm may arise from the overtone of CH vibration of the solvent. Inset are photos of the solutions. | |
The dications and dianions of 1 and 2 did not show obvious NMR signals for the protons in their backbones at room temperature and even after cooling to 178 K. In fact, the diradical character (y0) of 12+, 22+, 12− and 22− was calculated to be 70.1%, 75.1%, 84.7% and 80.2%, respectively (Fig. S12 and S13, ESI†), explaining the significant NMR broadening.21 This is further validated by VT ESR measurements of 12+, 22+, 12− and 22− in frozen solution, which showed a featureless broad signal (ge = 2.0025–2.0029), and the intensity decreased as the temperature was lowered (Fig. S8a, c, e and g, ESI†), indicating that they have a singlet diradical ground state, and can be regarded as diradical dications/dianions. Fitting of the data by the Bleaney–Bowers equation22 gave a singlet–triplet energy gap (ΔES–T) of −1.36 kcal mol−1 for 12+, −1.04 kcal mol−1 for 22+, −0.43 kcal mol−1 for 12− and −0.66 kcal mol−1 for 22− (Fig. S8b, d, f and h, ESI†).
Computational studies
To further study the electronic structures and aromaticity of 1/2 and their charged species, nucleus-independent chemical shift (NICS)23 and anisotropy of the induced current density (ACID)24 calculations were conducted at the B3LYP/6-31G(d,p) level of theory by using the Gaussian 09 program package. In neutral compounds 1 and 2, the central phenanthrene unit (rings A and B) and terminal benzene (rings F)/naphthalene rings (rings F and G) have negative NICS(1)zz values, indicating their aromatic character. Meanwhile, as-indacene moieties (rings C, D and E) possess positive NICS(1)zz values, indicating their anti-aromatic character (Fig. 9(a) and (c)). This is further validated by ACID calculations, which show clockwise diatropic ring current flow along the central phenanthrene ring and the terminal benzene/naphthalene units while counter-clockwise paratropic ring current for the three rings in the as-indacene unit (Fig. 9(b) and (d)). Accordingly, the protons c and d appear at high field (Fig. 2). In the dications of 1 and 2, rings C, D and E still maintain the anti-aromatic nature of the as-indacene units, which display local paratropic ring current and large positive NICS(1)zz value (Fig. 9(e)–(h)). The two benzene rings nearby as-indacene units (rings B and F) and the central benzene ring (ring A) are nearly non-aromatic. The ring G is still aromatic with the reduced NICC(1)zz value. Furthermore, NICS and ACID calculations on the dianions 12−/22− show that only the terminal benzene/naphthalene rings and central benzenoid rings are aromatic, the four CP rings are highly antiaromatic, while the other rings are nearly non-aromatic (Fig. 9(i)–(l)). The calculated electrostatic potential maps (Fig. S14 and S15, ESI†) and Mulliken charge distribution (Fig. S16 and S17, ESI†) suggest that the charges in 12+/22+ and 12−/22− are well delocalized along the backbone to minimize Coulomb repulsion.
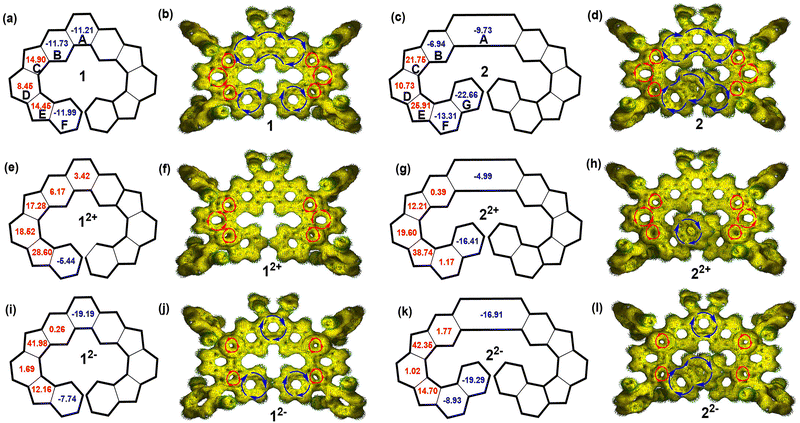 |
| | Fig. 9 (a) Calculated NICS(1)zz values (the numbers in the rings) of 1 (a), 12+ (e), 12− (i), 2 (c), 22+ (g), and 22− (k). Calculated ACID plots of 1 (b), 2 (d), 12+ (f), 22+ (h), 12− (j), and 22− (l). The red and blue arrows indicate the counterclockwise (paratropic) and clockwise (diatropic) current flow, respectively. | |
Conclusions
In summary, we have described the synthesis and characterization of two expanded quinodimethane embedded helicenes 1 and 2 with consecutively eleven and thirteen fused rings. Single-crystal X-ray analysis clearly reveals their helical geometry structures and significant overlaps at the helical end in molecule 2 due to the extended π-conjugation. It was elucidated in detailed experimental and computational studies that both 1 and 2 have a closed-shell ground state. The VT NMR and theoretical studies revealed that the isomerization process of 1 is fast on the NMR time scale, while compound 2 possesses a moderate racemization barrier. The dications and dianions of 1 and 2 show significant open-shell diradical character with small singlet–triplet energy gaps. In addition, these compounds also displayed size- and structure-dependent optical and electrochemical properties. Therefore, our studies shed some light on the design and synthesis of new helical π-systems with tunable properties.
Author contributions
C. Chi supervised the project. Q. Jiang synthesized and characterized the compounds. Y. Han performed crystallographic analysis. Y. Zou performed the ESR measurements. All authors discussed the results and contributed to the manuscript writing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Singapore MOE Tier 1 grant (A-8000992-00-00) and Tier 2 grant (MOE-MOET2EP10120-0006). We thank Dr Tan Geok Kheng and Voon Kunn Ng from the Department of Chemistry, National University of Singapore for her help with the X-ray crystallographic analysis.
Notes and references
-
(a) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC;
(b) Q. Ye and C. Chi, Chem. Mater., 2014, 26, 4046 CrossRef CAS;
(c) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed;
(d) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed;
(e) A. N. Lakshminarayana, A. Ong and C. Chi, J. Mater. Chem. C, 2018, 6, 3551 RSC;
(f) X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203 CrossRef CAS;
(g) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed;
(h) J. Luo, B. Zhao, J. Shao, K. A. Lim, H. S. O. Chan and C. Chi, J. Mater. Chem., 2009, 19, 8327 RSC;
(i) X. Shi and C. Chi, Chem. Rec., 2016, 16, 1690 CrossRef CAS PubMed;
(j) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS PubMed;
(k) W. Zeng, M. Ishida, S. Lee, Y. M. Sung, Z. Zeng, Y. Ni, C. Chi, D. Kim and J. Wu, Chem. – Eur. J., 2013, 19, 16814 CrossRef CAS PubMed;
(l) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857 RSC;
(m) Q. Jiang, H. Wei, X. Hou and C. Chi, Angew. Chem., Int. Ed., 2023, 62, e202306938 CrossRef CAS PubMed;
(n) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed;
(o) J.-J. Shen, Y. Han, S. Dong, H. Phan, T. S. Herng, T. Xu, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2021, 60, 4464 CrossRef CAS PubMed;
(p) W. Zeng and J. Wu, Chem, 2021, 7, 358 CrossRef CAS.
-
(a) M. Rickhaus, M. Mayor and M. Juríĉek, Chem. Soc. Rev., 2016, 45, 1542 RSC;
(b) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267 CrossRef CAS PubMed;
(c) Y. Han, S. Dong, J. Shao, W. Fan and C. Chi, Angew. Chem., Int. Ed., 2021, 60, 2658 CrossRef CAS PubMed;
(d) K. Y. Cheung, K. Watanabe, Y. Segawa and K. Itami, Nat. Chem., 2021, 13, 255 CrossRef CAS PubMed;
(e) K. Y. Cheung, S. Gui, C. Deng, H. Liang, Z. Xia, Z. Liu, L. Chi and Q. Miao, Chem, 2019, 5, 838 CrossRef CAS.
-
(a) M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86 CrossRef CAS PubMed;
(b) K. Baumgärtner, A. L. M. Chincha, A. Dreuw, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15594 CrossRef PubMed;
(c) R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315 CrossRef CAS.
- Bharat, R. Bhola, T. Bally, A. Valente, M. K. Cyrański, Ł. Dobrzycki, S. M. Spain, P. Rempała, M. R. Chin and B. T. King, Angew. Chem., Int. Ed., 2010, 49, 399 CrossRef CAS PubMed.
-
(a) Y. Zou, W. Zeng, T. Y. Gopalakrishna, Y. Han, Q. Jiang and J. Wu, J. Am. Chem. Soc., 2019, 141, 7266 CrossRef CAS PubMed;
(b) Q. Wang, T. Y. Gopalakrishna, H. Phan, T. S. Herng, S. Dong, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2017, 56, 11415 CrossRef CAS PubMed;
(c) K. Shoyama and F. Würthner, J. Am. Chem. Soc., 2019, 141, 13008 CrossRef CAS PubMed;
(d) M.-K. Chen, H.-J. Hsin, T.-C. Wu, B.-Y. Kang, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Chem. – Eur. J., 2014, 20, 598 CrossRef CAS PubMed;
(e) T.-C. Wu, M.-K. Chen, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 1289 CrossRef CAS PubMed;
(f) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868 CrossRef CAS PubMed;
(g) Q. Wang, P. Hu, T. Tanaka, T. Y. Gopalakrishna, T. S. Herng, H. Phan, W. Zeng, J. Ding, A. Osuka, C. Chi, J. Siegel and J. Wu, Chem. Sci., 2018, 9, 5100 RSC;
(h) G. Liu, L. Gao, Y. Han, Y. Xiao, B. Du, J. Gong, J. Hu, F. Zhang, H. Meng, X. Li, X. Shi, Z. Sun, J. Wang, G. Dai, C. Chi and Q. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301348 CrossRef CAS PubMed.
-
(a) T. Kirschbaum, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2020, 59, 270 CrossRef CAS PubMed;
(b) J. M. Farrell, V. Grande, D. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 16504 CrossRef CAS PubMed;
(c) S. H. Pun, Y. Wang, M. Chu, C. K. Chan, Y. Li, Z. Liu and Q. Miao, J. Am. Chem. Soc., 2019, 141, 9680 CrossRef CAS PubMed;
(d) S. H. Pun, C. K. Chan, J. Luo, Z. Liu and Q. Miao, Angew. Chem., Int. Ed., 2018, 57, 1581 CrossRef CAS PubMed;
(e) X. Gu, H. Li, B. Shan, Z. Liu and Q. Miao, Org. Lett., 2017, 19, 2246 CrossRef CAS PubMed;
(f) Y. Han, Z. Xue, G. Li, Y. Gu, Y. Ni, S. Dong and C. Chi, Angew. Chem., Int. Ed., 2020, 59, 9026 CrossRef CAS PubMed.
-
(a) H. Chen and Q. Miao, ChemPlusChem, 2019, 84, 627 CrossRef CAS PubMed;
(b) K. Y. Cheung, C. K. Chan, Z. Liu and Q. Miao, Angew. Chem., Int. Ed., 2017, 56, 9003 CrossRef CAS PubMed;
(c) K. Y. Cheung, S. Yang and Q. Miao, Org. Chem. Front., 2017, 4, 699 RSC;
(d) S. Nobusue, K. Fujita and Y. Tobe, Org. Lett., 2017, 19, 3227 CrossRef CAS PubMed;
(e) J.-X. Chen, J.-W. Han and H. N. C. Wong, Org. Lett., 2015, 17, 4296 CrossRef CAS PubMed;
(f) R. W. Miller, A. K. Duncan, S. T. Schneebeli, D. L. Gray and A. C. Whalley, Chem. – Eur. J., 2014, 20, 3705 CrossRef CAS PubMed;
(g) C.-N. Feng, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 7791 CrossRef CAS PubMed;
(h) Y. Sakamoto and T. Suzuki, J. Am. Chem. Soc., 2013, 135, 14074 CrossRef CAS PubMed.
-
(a) M. Gingras, Chem. Soc. Rev., 2013, 42, 968 RSC;
(b) M. Gingras, Chem. Soc. Rev., 2013, 42, 1051 RSC;
(c) M. Gingras, G. Felix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007 RSC;
(d) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS PubMed.
-
(a) J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 45 CrossRef CAS;
(b) Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. Correa da Costa, G. J. Hedley, D.-M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-dela-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, ACS Nano, 2017, 11, 8329 CrossRef CAS PubMed;
(c) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743 CrossRef CAS PubMed;
(d) J. Vacek, J. V. Chocholousova, I. G. Stara, I. Stary and Y. Dubi, Nanoscale, 2015, 7, 8793 RSC.
-
(a)
C.-F. Chen and Y. Shen, Helicene Chemistry, Springer, Berlin, Heidelberg, 2017, pp. 201–220 CrossRef;
(b) M. A. Shcherbina, X. Zeng, T. Tadjiev, G. Ungar, S. H. Eichhorn, K. E. S. Phillips and T. J. Katz, Angew. Chem., Int. Ed., 2009, 48, 7837 CrossRef CAS PubMed;
(c) C. Nuckolls, T. J. Katz and L. Castellanos, J. Am. Chem. Soc., 1996, 118, 3767 CrossRef CAS.
-
(a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680 RSC;
(b) M. J. Narcis and N. Takenaka, Eur. J. Org. Chem., 2014, 21 CrossRef CAS;
(c) P. Aillard, A. Voituriez, D. Dova, S. Cauteruccio, E. Licandro and A. Marinetti, Angew. Chem., Int. Ed., 2014, 53, 861 CrossRef PubMed;
(d) J. Misek, F. Teply, I. G. Stara, M. Tichy, D. Saman, I. Cisarova, P. Vojtisek and I. Stary, Angew. Chem., Int. Ed., 2008, 47, 3188 CrossRef CAS PubMed;
(e) M. R. Crittall, H. S. Rzepa and D. R. Carbery, Org. Lett., 2011, 13, 1250 CrossRef CAS PubMed;
(f) T. Lu, R. Zhu, Y. An and S. E. Wheeler, J. Am. Chem. Soc., 2012, 134, 3095 CrossRef CAS PubMed;
(g) N. Takenaka, R. S. Sarangthem and B. Captain, Angew. Chem., Int. Ed., 2008, 47, 9708 CrossRef CAS PubMed;
(h) J. Chen, B. Captain and N. Takenaka, Org. Lett., 2011, 13, 1654 CrossRef CAS PubMed;
(i) M. Hasan and V. Borovkov, Symmetry, 2018, 10, 10/1 CrossRef CAS.
-
(a) Y. Zhu and J. Wang, Acc. Chem. Res., 2023, 56, 363 CrossRef CAS PubMed;
(b) Y.-Y. Ju, L. Chai, K. Li, J.-F. Xing, X.-H. Ma, Z.-L. Qiu, X.-J. Zhao, J. Zhu and Y.-Z. Tan, J. Am. Chem. Soc., 2023, 145, 2815 CrossRef CAS PubMed;
(c) J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A.-H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958 CrossRef CAS PubMed;
(d) Y. Chen, C. Lin, Z. Luo, Z. Yin, H. Shi, Y. Zhu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 7796 CrossRef CAS PubMed;
(e) G. Liu, Y. Liu, C. Zhao, Y. Li, Z. Wang and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202214769 Search PubMed;
(f) Q. Jiang, Y. Han, Y. Zou, H. Phan, L. Yuan, T. S. Herng, J. Ding and C. Chi, Chem. – Eur. J., 2020, 26, 15613 CrossRef CAS PubMed;
(g) G. Li, T. Matsuno, Y. Han, S. Wu, Y. Zou, Q. Jiang, H. Isobe and J. Wu, Angew. Chem., Int. Ed., 2021, 60, 10326 CrossRef CAS PubMed;
(h) S. K. Pedersen, K. Eriksen and M. Pittelkow, Angew. Chem., Int. Ed., 2019, 58, 18419 CrossRef CAS PubMed;
(i) Y. Nakakuki, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 15461 CrossRef CAS PubMed;
(j) Y. Zhu, Z. Xia, Z. Cai, Z. Yuan, N. Jiang, T. Li, Y. Wang, X. Guo, Z. Li, S. Ma, D. Zhong, Y. Li and J. Wang, J. Am. Chem. Soc., 2018, 140, 4222 CrossRef CAS PubMed;
(k) K. Kato, Y. Segawa, L. T. Scott and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 1337 CrossRef CAS PubMed;
(l) Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Mullen and A. Narita, Angew. Chem., Int. Ed., 2017, 56, 3374 CrossRef CAS PubMed;
(m) V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, J. Am. Chem. Soc., 2017, 139, 18508 CrossRef CAS PubMed;
(n) S. Míguez-Lago, I. F. A. Mariz, M. A. Medel, J. M. Cuerva, E. Maçôas, C. M. Cruz and A. G. Campaña, Chem. Sci., 2022, 13, 10267 RSC;
(o) N. J. Schuster, D. W. Paley, S. Jockusch, F. Ng, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2016, 55, 13519 CrossRef CAS PubMed;
(p) H. Huang, Y.-C. Hsieh, P.-L. Lee, C.-C. Lin, Y.-S. Ho, W.-K. Shao, C.-T. Hsieh, M.-J. Cheng and Y.-T. Wu, J. Am. Chem. Soc., 2023, 145(18), 10304 CrossRef CAS PubMed;
(q) Y. Chen, C. Lin, Z. Luo, Z. Yin, H. Shi, Y. Zhu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 7796 CrossRef CAS PubMed;
(r) Y.-J. Shen, N.-T. Yao, Y. Yang, X.-L. Chen and H.-Y. Gong, Angew. Chem., Int. Ed., 2023, 62, e202300840 CrossRef CAS PubMed;
(s) S. H. Pun, K. M. Cheung, D. Yang, H. Chen, Y. Wang, S. V. Kershaw and Q. Miao, Angew. Chem., Int. Ed., 2022, 61, e202113203 CrossRef CAS PubMed.
-
(a) G. R. Kiel, S. C. Patel, P. W. Smith, D. S. Levine and T. D. Tilley, J. Am. Chem. Soc., 2017, 139, 18456 CrossRef CAS PubMed;
(b) G.-F. Huo, T. M. Fukunaga, X. Hou, Y. Han, W. Fan, S. Wu, H. Isobe and J. Wu, Angew. Chem., Int. Ed., 2023, 62, e202218090 CrossRef CAS PubMed;
(c) G. R. Kiel, H. M. Bergman, A. E. Samkian, N. J. Schuster, R. C. Handford, A. J. Rothenberger, R. Gomez-Bombarelli, C. Nuckolls and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 23421 CrossRef CAS PubMed;
(d) I. G. Stará and I. Starý, Acc. Chem. Res., 2020, 53, 144 CrossRef PubMed;
(e) Hn. M. Bergman, D. D. Beattie, G. R. Kiel, R. C. Handford, Y. Liu and T. D. Tilley, Chem. Sci., 2022, 13, 5568 RSC;
(f) M. Toya, T. Omine, F. Ishiwari, A. Saeki, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 11553 CrossRef CAS PubMed;
(g) Y. Nakakuki, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2018, 140, 15461 CrossRef CAS PubMed.
- X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052 CrossRef CAS PubMed.
- CCDC 2289867 (1) and 1984797 (2)†.
-
(a) J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839 CrossRef;
(b) T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70 CrossRef CAS;
(c) T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385 CrossRef CAS PubMed.
- F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 258 CrossRef CAS.
- R. H. Janke, G. Haufe, E.-U. Würthwein and J. H. Borkent, J. Am. Chem. Soc., 1996, 118, 6031 CrossRef CAS.
- R. H. Martin and M. J. Marchant, Tetrahedron, 1974, 30, 347 CrossRef CAS.
- S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhardt and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3223 CrossRef CAS PubMed.
-
(a) Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC;
(b) T. Kubo, Chem. Rec., 2015, 15, 218 CrossRef CAS PubMed;
(c) A. Konishi and T. Kubo, Top. Curr. Chem., 2017, 375, 83 CrossRef PubMed;
(d) Q. Jiang, T. Tao, H. Phan, Y. Han, T. Y. Gopalakrishna, T. S. Herng, G. Li, L. Yuan, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2018, 57, 16737 CrossRef CAS PubMed;
(e) X. Shi, E. Quintero, S. Lee, L. Jing, T. S. Herng, B. Zheng, K.-W. Huang, J. T. López Navarrete, J. Ding, D. Kim, J. Casado and C. Chi, Chem. Sci., 2016, 7, 3036 RSC;
(f) Y. Tobe, Top. Curr. Chem., 2018, 376, 107 Search PubMed;
(g) S. Dong, T. S. Herng, T. Y. Gopalakrishna, H. Phan, Z. L. Lim, P. Hu, R. D. Webster, J. Ding and C. Chi, Angew. Chem., Int. Ed., 2016, 55, 9316 CrossRef CAS PubMed;
(h) S. Dong, T. Y. Gopalakrishna, Y. Han, H. Phan, T. Tao, Y. Ni, G. Liu and C. Chi, J. Am. Chem. Soc., 2019, 141, 62 CrossRef CAS PubMed;
(i) H. Hayashi, J. E. Barker, A. C. Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S.-I. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444 CrossRef CAS PubMed;
(j) T. Xu, Y. Han, Z. Shen, X. Hou, Q. Jiang, W. Zeng, P. W. Ng and C. Chi, J. Am. Chem. Soc., 2021, 143, 20562 CrossRef CAS PubMed;
(k) A. Ong, T. Tao, Q. Jiang, Y. Han, Y. Ou, K.-W. Huang and C. Chi, Angew. Chem., Int. Ed., 2022, 61, e202209286 CrossRef CAS PubMed;
(l) Z. Li, X. Hou, Y. Han, W. Fan, Y. Ni, Q. Zhou, J. Zhu, S. Wu, K.-W. Huang and J. Wu, Angew. Chem., Int. Ed., 2022, 61, e202210697 CrossRef CAS PubMed;
(m) J. E. Barker, T. W. Price, L. J. Karas, R. Kishi, S. N. MacMillan, L. N. Zakharov, C. J. Gómez-García, J. I. Wu, M. Nakano and M. M. Haley, Angew. Chem., Int. Ed., 2021, 60, 22385 CrossRef CAS PubMed;
(n) T. Xu, X. Hou, Y. Han, H. Wei, Z. Li and C. Chi, Angew. Chem., Int. Ed., 2023, 62, e202304937 CrossRef CAS PubMed;
(o) W. Kueh, X. Shi, T. W. Phua, H. Kueh, Y. C. Liau and C. Chi, Org. Lett., 2022, 24, 5935 CrossRef CAS PubMed;
(p) B. Huang, H. Kang, C.-W. Zhang, X.-L. Zhao, X. Shi and H.-B. Yang, Commun. Chem., 2022, 5, 127 CrossRef CAS PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic studies, computational analysis and crystal structures. CCDC 2289867 (1) and 1984797 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc02432f |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*

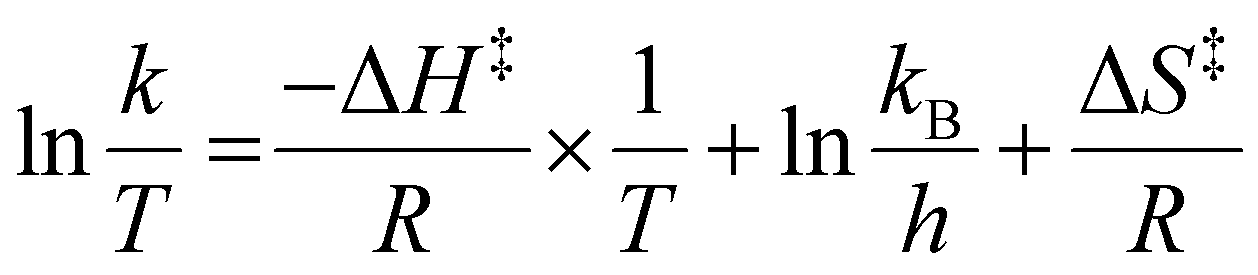
 was then estimated as 18.09 ± 1.72 kcal mol−1, which is much lower than that of the [6]helicene (37.3 kcal mol−1)18 and [7]helicene (41.7 kcal mol−1).19 The low barrier should be attributed to the fact that the distortion required for racemization is spread over a larger number of bonds and angles (as for [7]heliphene20).
was then estimated as 18.09 ± 1.72 kcal mol−1, which is much lower than that of the [6]helicene (37.3 kcal mol−1)18 and [7]helicene (41.7 kcal mol−1).19 The low barrier should be attributed to the fact that the distortion required for racemization is spread over a larger number of bonds and angles (as for [7]heliphene20).