
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface chemical mechanisms of trimethyl aluminum in atomic layer deposition of AlN†
Karl
Rönnby
 *,
Henrik
Pedersen
and
Lars
Ojamäe
*,
Henrik
Pedersen
and
Lars
Ojamäe
Department of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden. E-mail: karl.ronnby@tyndall.ie
First published on 26th September 2023
Abstract
Aluminum nitride (AlN) is an important material for semiconductor devices, such as ultraviolet light emitting diodes (LEDs) or high-electron-mobility transistors (HEMTs). One emerging approach to deposit very thin, and highly conformal AlN films is atomic layer deposition (ALD), most commonly by using trimethyl aluminum (TMA) and ammonia. Despite TMA being one of the most common ALD precursors, its surface chemistry in ALD of AlN is poorly understood. By quantum chemical modeling, we have investigated the adsorption and decomposition of TMA on an AlN surface. We have found two competing decomposition pathways, either ligand exchange by protonation of the methyl groups in TMA by a surface amino group, or reductive elimination of the methyl groups as ethane. Reductive elimination is shown to be the dominating reaction step if a TMA molecule adsorbs far from any previously adsorbed molecule. Ligand exchange is preferred if it adsorbs close to an already adsorbed molecule. By comparing the obtained surfaces after decomposition, we find that the saturation coverage is dependent on the preferred pathways with a third of a monolayer saturation for reductive elimination and a full for ligand exchange. The relative low diffusion rate compared to decomposition rate of TMA will cause the molecules to be far apart after adsorption, and we conclude that the dominating decomposition path is reductive elimination.
Introduction
Aluminum nitride (AlN) is a wide band gap semiconductor with a direct band gap of 6.1 eV.1 The large band gap allows AlN to be used in ultraviolet light emitting diodes (LEDs) with wavelengths down to 210 nm.2,3 AlN is also used as a buffer layer in gallium nitride (GaN) based high-electron-mobility transistors (HEMTs), where the high breakdown voltage of AlN allows for high power applications.4 Both applications require AlN to be deposited as a thin and very uniform film. Thin films of AlN can be deposited by chemical vapor deposition (CVD) at temperatures around 1000 °C.5 The need for such high temperature restricts the selection of substrates, and previously deposited films, to more stable materials. It also limits the surface chemical control needed for deposition on topologically complex structures.6Atomic layer deposition (ALD) is a time-resolved form of CVD relying on surface chemical reactions between gaseous precursor molecules and thermodynamically stable monolayers of chemisorbed precursors. ALD is thus an alternative low temperature method that could allow for new types of AlN applications. Some of the earliest reports of ALD of AlN were published in the early 1990s and used trimethyl aluminum (TMA, Al(CH3)3), triethyl aluminum (Al(CH2CH3)3) or aluminum chloride (AlCl3) together with ammonia (NH3) in a thermal process.7–10 Although other aluminum precursors, such as the homoleptic tris-dimethylamido aluminum (TDMAA)11 and tris-diehtylamido aluminum (TDMEA)12 or the heteroleptic AlH2NMe2,13 have been investigated to improve deposition and film properties TMA is still used as the most common precursor. Plasma assisted ALD is often utilized to achieve better film quality where NH3 plasma,14 nitrogen and hydrogen plasma,15 or ammonia and nitrogen plasma have been used.16 However, the films deposited with TMA contain a high amount of carbon impurities and the surface chemical mechanisms are not fully understood, although some investigations on reducing the carbon impurities have been performed.17
The TMA half-cycle in ALD of AlN can be assumed to be similar to the well-studied ALD process for aluminum oxide (Al2O3) using TMA and water, where TMA adsorbs by forming a Lewis adduct with a surface hydroxyl group.18 That mechanism continues by abstraction of hydrogen atoms from the surface hydroxyl groups to the methyl ligands of TMA, which then can be released as methane. The process can continue as long as there are hydrogen atoms left at the surface hydroxyl groups and saturates when they are depleted, or any additional TMA are blocked from adsorption by other surface species. Surface hydroxyl groups are recreated by adsorption of water during the next half-cycle, displacing the remaining methyl groups.
We have in recent works investigated the chemistry of ammonia in ALD of AlN as well as the two other group-13 nitrides gallium nitride (GaN) and indium nitride (InN).19,20 Therein we have investigated different nitrogen containing surface species, the distribution of these at the conditions used for ALD of AlN and investigated an possible adsorption path for ammonia. The results show that the most stable AlN surface after NH3 chemisorption at ALD temperatures is covered by amino groups (–NH2),19 which, like hydroxyl groups, are Lewis bases and can thus form adducts with TMA. The groups also contain acidic protons, which can be transferred to the methyl ligands, facilitating methane release, albeit the acidity of amino groups is weaker than that of hydroxyl groups. Quantum chemical studies of the similar material gallium nitride (GaN) have shown the existence of such adducts with trimethyl gallium (TMG, Ga(CH3)3) as well as a proposed surface protonation of methyl ligands and release of methane.21 Adsorption of trimethyl indium (TMI, In(CH3)3) on indium nitride (InN) has also been shown by computational investigations to lead to such adduct formation and methane release. In addition, reductive elimination of methyl groups as ethane from the TMI adduct is a viable route.22
In this study we have investigated the adsorption of TMA onto AlN by density functional theory (DFT) modeling to elucidate the adsorption mechanism of the aluminum half-cycle of the ALD process. We investigate initial adsorption and diffusion of TMA onto a pristine amino terminated AlN surface. The possible methyl protonation and reductive elimination pathways are compared to find the preferred decomposition route. We also investigate if there is any difference in the adsorption path of a TMA molecule if it adsorbs in close proximity to an already adsorbed and decomposed TMA molecule or not.
Computational methods
Density functional theory (DFT) was used to investigate the adsorption geometries, energies, and mechanisms, using the Vienna Ab initio Simulation Package (VASP) for all quantum-chemical calculations.23–25 The generalized gradient approximation (GGA) functional PBE was used to calculate the exchange–correlation energy and the third version of Grimme's dispersion correction was used to improve description of non-covalent interactions.26,27 Projector augmented wave (PAW) potentials were used with 1 valence electron for hydrogen, 4 for carbon, 5 for nitrogen and 3 for aluminum.28 A kinetic cutoff energy of 700 eV was used for the plane wave basis. Transition states (TS) were found by the nudge elastic band (NEB) method, using climbing image if necessary, the dimer method or constrained optimization.29–31A surface slab was created from an optimized unit cell of wurtzite AlN. The unit cell was extended in the [0001] direction and padded by vacuum, yielding a surface slab of AlN layers with approximately 20 Å of vacuum separation. A surface supercell was created from the unit slab by extending it along [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0] and [1
0] and [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 00], thereby obtaining a (3 × 4) supercell with 12 adsorption sites for TMA. The size of the supercell was selected to allow adsorption of multiple TMA molecules without any self-interactions across the periodic boundary. Each dangling bond on the nitrogen atoms at the bottom of the slab was saturated by hydrogen atoms, 12 in total for the supercell, and the dangling bonds on the top aluminum atoms were saturated by amino groups (NH2). The supercell was optimized after construction with all atoms free to move with the cell parameters kept held constant. For all further optimizations all but the two topmost layers were kept frozen at their initially optimized coordinates. A 2 × 2 × 1 Monkhorst–Pack k-point mesh was used for the surface slabs. The convergence criteria for geometry optimization were that all unconstrained forces were to be less than 0.01 eV Å−1 for minima and 0.05 eV Å−1 for transition states. Energies of gas phase molecules were calculated in a 10 × 10 × 10 Å3 cube with the Γ-point as the only k-point.
00], thereby obtaining a (3 × 4) supercell with 12 adsorption sites for TMA. The size of the supercell was selected to allow adsorption of multiple TMA molecules without any self-interactions across the periodic boundary. Each dangling bond on the nitrogen atoms at the bottom of the slab was saturated by hydrogen atoms, 12 in total for the supercell, and the dangling bonds on the top aluminum atoms were saturated by amino groups (NH2). The supercell was optimized after construction with all atoms free to move with the cell parameters kept held constant. For all further optimizations all but the two topmost layers were kept frozen at their initially optimized coordinates. A 2 × 2 × 1 Monkhorst–Pack k-point mesh was used for the surface slabs. The convergence criteria for geometry optimization were that all unconstrained forces were to be less than 0.01 eV Å−1 for minima and 0.05 eV Å−1 for transition states. Energies of gas phase molecules were calculated in a 10 × 10 × 10 Å3 cube with the Γ-point as the only k-point.
Adsorption energies were calculated as the energy of the surface with adsorbate minus the energy of the surface before adsorption and of the adsorbate in gas phase, eqn (1).
| ΔadsE = Esurf+ads − (Esurf + Egas) | (1) |
Potential energy surfaces (PES) of adsorption were constructed by adding a TMA molecule to the surface cell where the aluminum atom was placed on a grid with its position fixed in the horizontal directions and optimizing the structure at each position. A smooth PES was then obtained by interpolating the computed energies by a periodic piecewise bicubic polynomial. Bader charges were calculated to estimate the oxidation state of different atoms and to investigate possible oxidations and reductions during precursor decomposition. The change of Bader charge for an atom during a reaction was calculated from the charge for the atom in the product subtracted by the charge of the atom in the reactant, eqn (2).
| Δδ = δproduct − δreactant | (2) |
The effect of spin polarization was investigated for the structures named A0, B0, C1, C2, BC1, and BC2, see the ESI.† As the effect of spin polarization was relatively low, non-spin polarized calculations were used.
Results and discussion
Initial TMA adsorption and decomposition
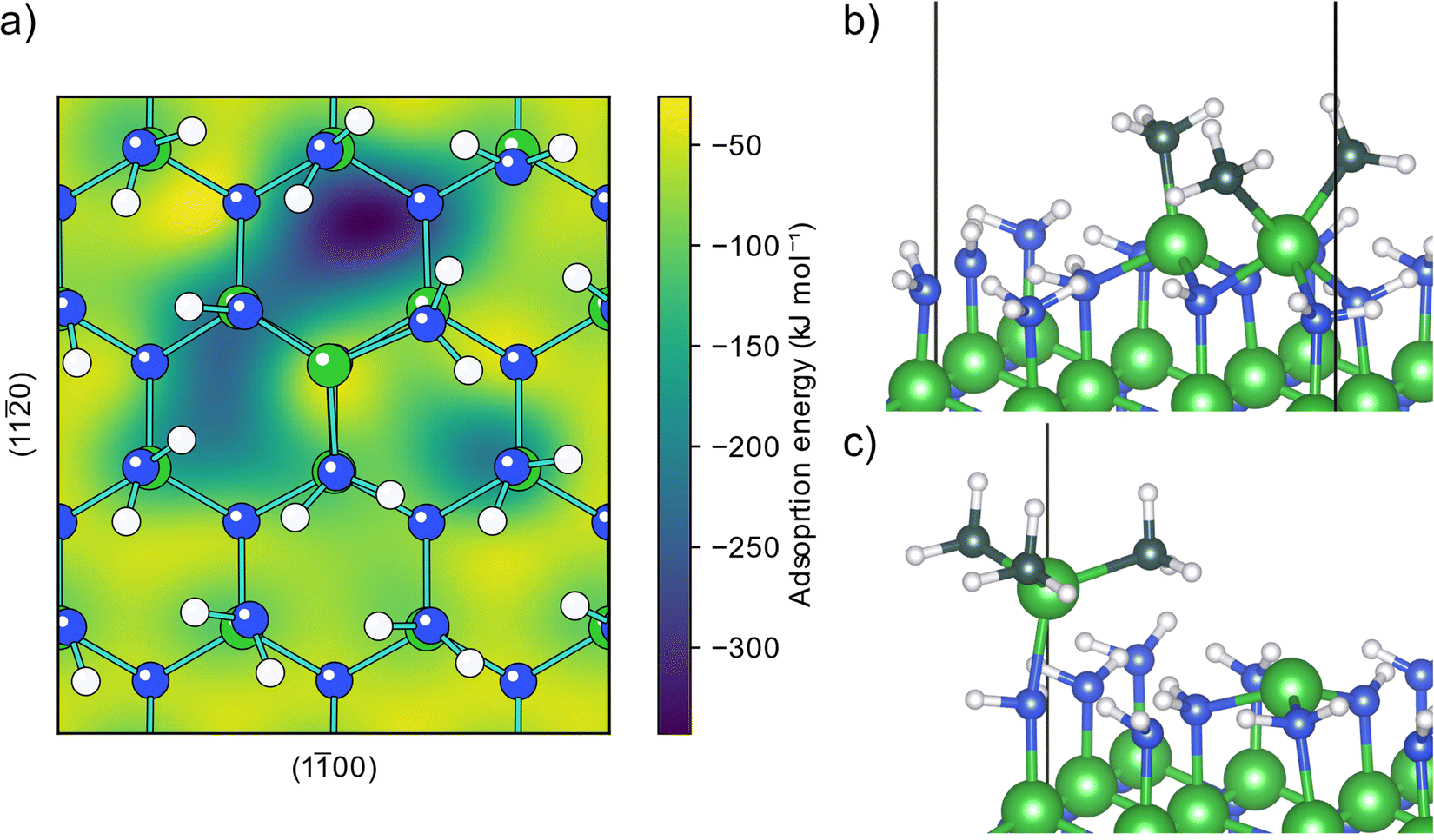
The AlN surface for initial TMA adsorption was modeled as being terminated by amino groups, arranged in a zig-zag pattern forming hydrogen bonds across the surface, (structure A0, Fig. S1 in the ESI†) as this is the surface termination expected after an ideal ammonia ALD half-cycle.19 The PES for a TMA molecule adsorbing onto this surface, Fig. 1a, shows that TMA has a single adsorption configuration, located at the surface amino groups, with a barrierless adsorption process as no bonds are being broken in the adsorption. In this structure, Fig. 1b, the aluminum atom forms a Lewis adduct (B0, Fig. S2, ESI†) with the surface amino group with an adsorption energy of −92 kJ mol−1. The adsorption energy is slightly more negative than the adsorption energy on Al2O3 (−68 kJ mol−1) as reported by Elliott and Greer, although calculated with a different functional,18 indicating that TMA forms a stronger adduct with amino groups compared to hydroxyl groups, in line with the lower electronegativity of nitrogen compared to oxygen.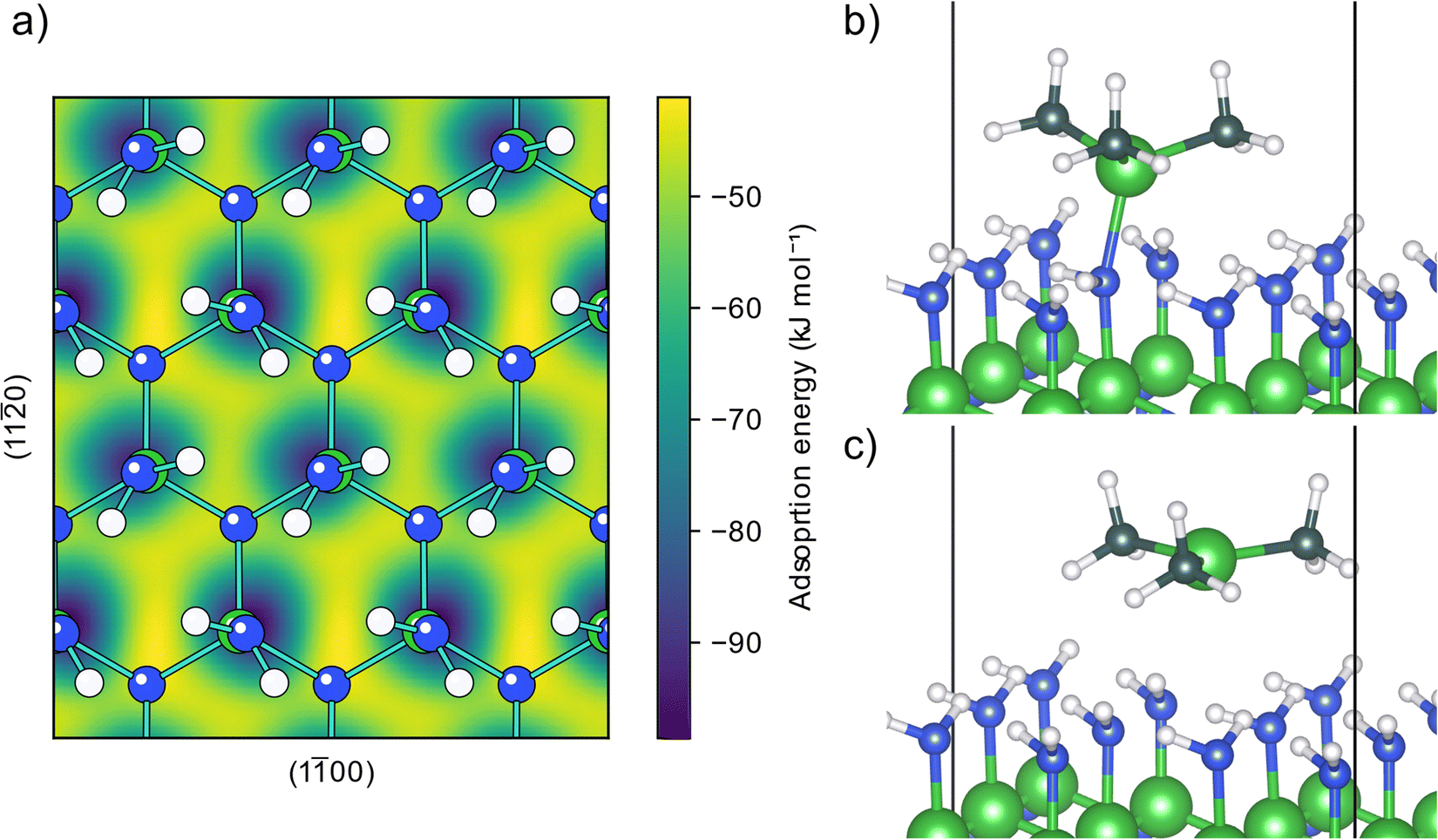 | ||
| Fig. 1 Adsorption of TMA onto a clean amino terminated AlN surface. (a) Potential energy surface for an adsorbing TMA molecule. (b) Structure of TMA adsorbing at an energy minimum forming a Lewis adduct. (c) Transition state structure of TMA diffusing between two Lewis adduct sites. | ||
When forming the adduct the coordination around the aluminum atom changes from its planar geometry in gas phase to a more tetrahedral arrangement. Repulsion between the surface and the methyl groups of TMA forces the nitrogen to adopt a seesaw geometry with an almost linear bend angle (165°) between the surface aluminum and TMA molecule and the bond length (2.14 Å) is slightly longer than the gas phase adduct between TMA and ammonia (2.09 Å).32 Between the different adduct sites of the amino terminated AlN, there are first order saddle points on the PES corresponding to the transition state (TS) of TMA diffusion. When translating between two adduct sites, at the TS TMA almost returns to its planar gas phase geometry, Fig. 1c. The diffusion barrier is 42 kJ mol−1 above the minimum (−50 kJ mol−1 adsorption energy). The negative adsorption energy of the TS indicates that the TMA molecule can stay on the surface while diffusing instead of desorbing and reabsorbed at a different site. The distance between two adjacent amino groups is 3.1 Å. This separation might be too small to fit two undecomposed TMA molecules on adjacent positions (Al–C bond length 1.9 Å), however, one TMA could be able to fit to a site adjacent to a small adsorbent.
The removal of the methyl ligands from the adsorbed TMA molecule was investigated by elucidating methyl protonation and reductive elimination pathways. Oxidation of the reduced surface by release of hydrogen gas was also investigated. In the methyl protonation pathway, i.e., a ligand exchange route, one hydrogen is transferred from a surface amino group to a carbon of one of the methyl groups, transforming it to methane, and a covalent bond between the aluminum and the nitrogen atom of the amino group is formed. The dimethyl aluminum (DMA) fragment moves to a bridging site between the deprotonated amino site and the initial adduct amino group during the proton transfer, while the released menthane molecule desorbs spontaneously from the surface (C1), Fig. 2a. The reaction energy of methyl protonation is −96 kJ mol−1, which is slightly smaller than that of the same reaction on Al2O3 (−116 kJ mol−1).18 This indicates that while the adduct binds more strongly to AlN compared to Al2O3, the following elimination of methyl groups and formation of the material is more favorable for the oxide.
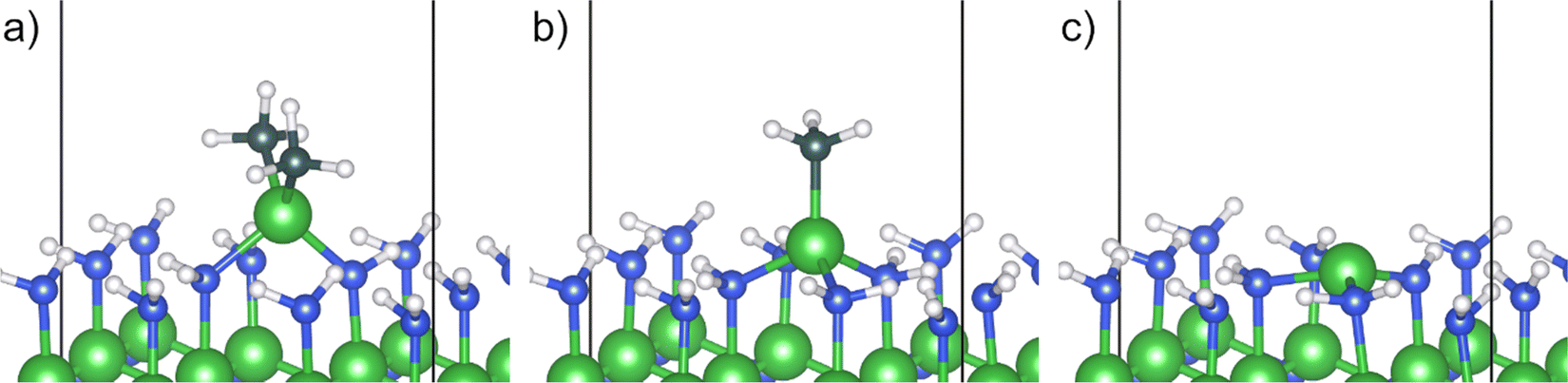 | ||
| Fig. 2 Geometries for (a) DMA fragment after methyl protonation (C1), (b) MMA fragment after reductive elimination (C2) and (c) aluminum adatom after methyl protonation followed by reductive elimination (D1). | ||
Instead of protonation of a second methyl group, the DMA fragment was found to undergo reductive elimination. The two remaining methyl groups are released as ethane, thereby reducing the oxidation state of the surface atoms. The aluminum adatom is positioned at the bulk hcp position, where it binds to the deprotonated amino site and two neighboring amino groups (D1), Fig. 2c. The reductive elimination leads to a significant decrease of energy, −257 kJ mol−1, and has an almost negligible barrier, 14 kJ mol−1, relative to the DMA fragment. The very low barrier would lead to a quick reaction and release of ethane, making it unlikely that other decomposition pathways would be of importance.
Alternatively, the reaction steps can proceed in the opposite order. Ethane can be reductively eliminated from the TMA adduct forming a monomethyl aluminum (MMA) fragment at the surface (C2), Fig. 2b. The MMA fragment translates to the bulk hcp position after the ethane molecule has been released, allowing the aluminum atom to adopt a tetrahedral configuration with the remaining methyl group along the surface normal. In this route, the reductive elimination has a large negative reaction energy of −507 kJ mol−1, indicating a very exothermic reaction. The barrier for the elimination is also quite low at 31 kJ mol−1 leading to a quick reaction. The last methyl group in the MMA fragment can then be protonated from one of the amino groups, releasing it as methane, forming the same aluminum adatom as in the previous pathway (D1). Methyl protonation from an MMA fragment has a positive reaction energy, 153 kJ mol−1, and a barrier of 266 kJ mol−1.
Oxidation of the surface was investigated by the removal of hydrogen atoms from the reduced surfaces at two different steps in the reaction path, either after formation of the aluminum adatom or before methyl elimination from an MMA fragment. In both cases, two surface hydrogen atoms combine to be released as hydrogen gas. The reaction energy is similar in both cases, 389 kJ mol−1 from the aluminum adatom (E3) and 399 kJ mol−1 from the MMA fragment (D3). It is unlikely for these oxidations to proceed as the reaction energy is very high, and TS were not investigated for these steps. After oxidation of the surface with an MMA fragment, methyl protonation can occur, leaving an aluminum adatom (E3), with similar energy as from the reduced surface, i.e., 143 kJ mol−1 compared to 153 kJ mol−1. The energies are very close, implying that the oxidation state of the surface does not affect the methyl elimination significantly.
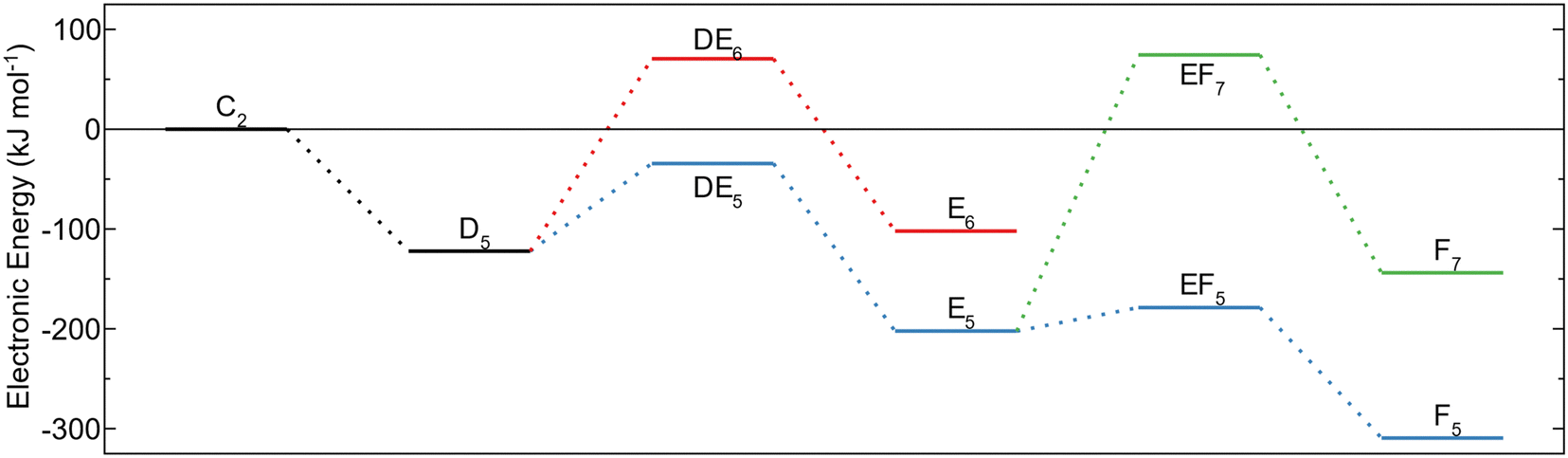
The energy profiles for the reaction paths are shown in Fig. 3. The global energy minimum for the investigated decomposition paths is the MMA fragment (C2) with an energy of −599 kJ mol−1 below the energy of gas phase TMA and the amino terminated AlN surface. Decomposition to form this configuration (BC2) has lower activation energy compared to the surface protonation (BC1) and is thus expected to also be the more dominating decomposition pathway. The high barrier for further methyl protonation makes it unlikely for the MMA fragment to decompose during the time allowed for ALD. The reaction steps are irreversible as they lead to the release of a gas molecule that will be carried away in the gas stream. Thus, even though the reductive elimination to MMA is assumed to be the major pathway for TMA decomposition, a small amount of the TMA–amino adducts could decompose by methyl protonation, followed by a reductive elimination to an aluminum adatom. The energy of this reaction is −446 kJ mol−1, which even though it is higher than the energy for an MMA fragment, is much lower than for TMA in the gas phase.
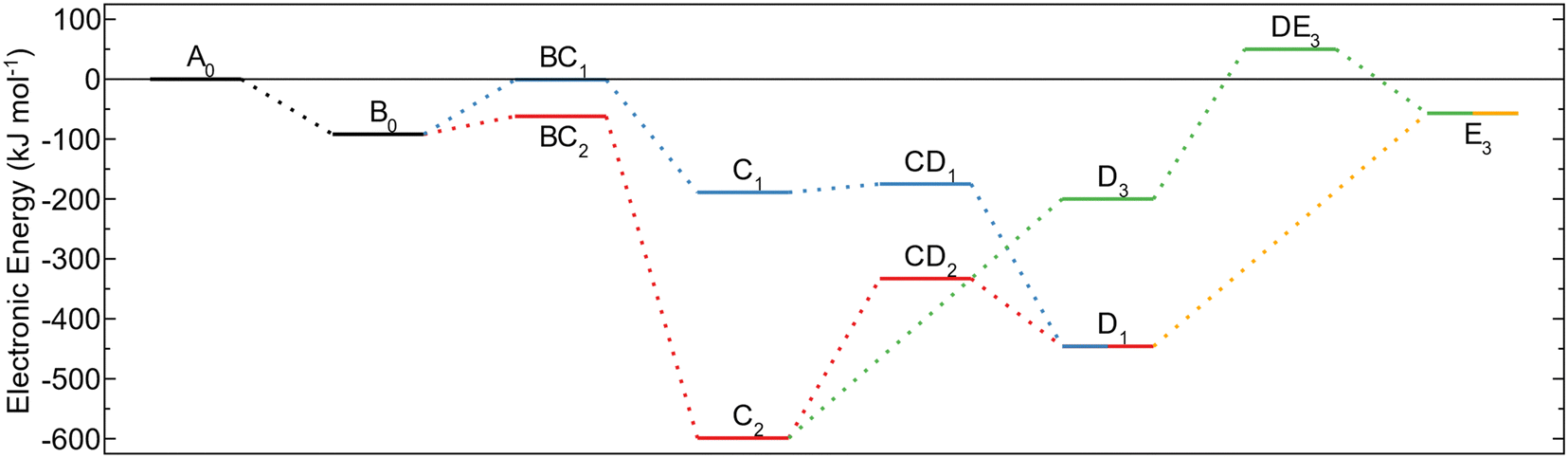 | ||
| Fig. 3 Energy profile for TMA adsorption and decomposition on amino terminated AlN. The black step (A0 to B0) is initial adsorption of TMA. The blue path (B0viaC1 to D1) is methyl protonation followed by reductive elimination and red (B0viaC2 to D1) is reductive elimination followed by methyl protonation. The yellow path (D1 to E3) is surface oxidation, and the green (C2viaD3 to E3) is surface oxidation followed by methyl protonation. | ||
Consecutive TMA adsorption and decomposition
An adsorbed and decomposed TMA molecule will affect how any following TMA molecules can adsorb on the surface. The two surface species described above, an MMA fragment (C2) formed from reductive elimination and an aluminum adatom (D1) formed from a combination of reductive elimination and methyl protonation, were considered as the possible endpoints of the TMA decomposition.The already adsorbed MMA fragment affects the PES of adsorption for any following TMA molecules, Fig. 4a. Close to the fragment, the TMA molecule is blocked from being able to form an adduct with the three closest amino groups and the TMA molecule is only physisorbed, Fig. 4c. The effect of the fragment on the PES is localized to a small area close to it, affecting mainly the nearest amino sites. Further from the fragment, the TMA molecule can form an adduct with the same structure as with the clean surface (D5), Fig. 4b and Fig. S13 (ESI†). The adsorption energy for this adduct is slightly lower, −122 kJ mol−1, implying that the adduct binds more strongly to the reduced surface, possible due to the extra electrons that could be distributed to the aluminum atom.
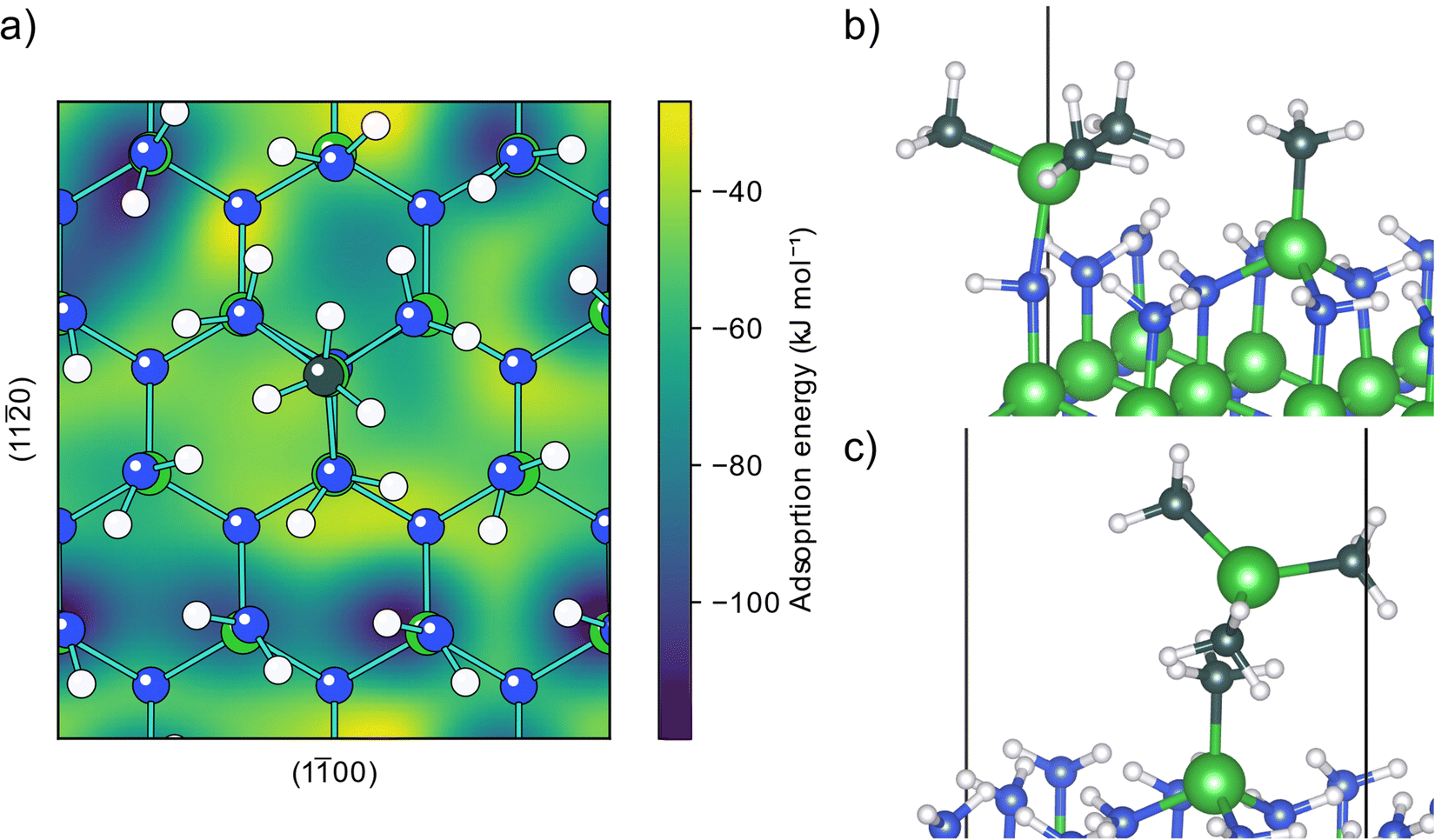 | ||
| Fig. 4 Adsorption of TMA onto AlN with an MMA fragment on the surface. (a) Potential energy surface for an adsorbing TMA molecule. (b) Structure of TMA adsorbing at an energy minimum forming a Lewis adduct. (c) Structure of TMA repelled by being adjacent to the adsorbed MMA fragment. | ||
From the adduct site the second TMA molecule can decompose, similar to the first, by either methyl protonation or reductive elimination. Methyl protonation has a reaction energy of −80 kJ mol−1, a magnitude which is smaller than for the methyl elimination on the initial TMA. The overall energy of TMA adsorption and methyl protonation is, however, more negative for the second TMA due to the more favorable adduct. The formed DMA fragment is positioned at the bulk hcp site instead of bridging two amino groups, with the deprotonated amino group bridging the two aluminum fragments (E5, Fig. S15, ESI†). Reductive elimination can occur from the DMA fragment to form an aluminum adatom (F7, Fig. S21, ESI†). However, due to the surface already being reduced once, this reaction is not energetically favorable, with a reaction energy of 60 kJ mol−1. The difference in reaction energy is significant and shows that the surface becomes much less likely to perform another reductive elimination. A second methyl protonation is a more likely reaction pathway with a reaction energy of −104 kJ mol−1, forming two MMA fragments in neighboring positions on the surface (F5, Fig. S17, ESI†). As with the first TMA, any further methyl protonation is unlikely as the barrier would be too high. Reductive elimination of the second TMA adduct, forming another MMA fragment (E6, Fig. S19, ESI†), is also much higher in both reaction and activation energy than for the first, with the reaction energy being positive (20 kJ mol−1). This again shows that the surface is much less prone to undergo reduction for the second TMA molecule.
An aluminum adatom affects the adsorptions of TMA as well. As with an MMA fragment, the adatom only affects the PES of adsorption, Fig. 5a, close to its absorption site, allowing TMA to adsorb as an adduct at second neighbor amino groups like in the previous cases, Fig. 5c. However, close to the adatom the PES is changed significantly. If the second TMA molecule comes in close enough to the aluminum atom, one of the methyl groups is transferred without any barrier to the adatom, thereby transforming it to an MMA fragment, Fig. 5b. Meanwhile the TMA is changed into a DMA fragment, which can be relocated to a neighboring bulk hcp site. The energy for this adsorption reaction is −356 kJ mol−1, which shows a very favorable reaction, the lack of barrier making it very fast. The produced surface species, a DMA and an MMA fragment in neighboring positions, are the same as those obtained from methyl protonation of a TMA adsorbed close to an MMA fragment (E5) and can thus decompose in the same way.
 | ||
| Fig. 5 Adsorption of TMA onto AlN with an aluminum adatom on the surface. (a) Potential energy surface for an adsorbing TMA molecule. (b) DMA fragment adsorbed after methyl transfer to aluminum adatom. (c) Structure of TMA adsorbing at an energy minimum forming a Lewis adduct. | ||
The reaction energy profile for the following TMA adsorbate, Fig. 6, differs from the profile for the initial TMA molecule, Fig. 3. The main difference is that the energy for reductive elimination is much less favorable which causes these reaction paths to have both a much higher reaction energy and activation energy barrier. The barrier for the reductive elimination step is also higher than the adsorption energy for the TMA adduct causing the adduct to be more likely to desorb rather than decompose reductively. The barrier for the first methyl protonation is larger than for the second, making it the rate determining step for the decomposition.
 | ||
| Fig. 6 Energy profile for TMA adsorption and decomposition on amino terminated AlN with a neighboring MMA fragment. The black step (C2 to D5) is adsorption of TMA. The blue path (D5 to F5) is methyl protonation. The red path (D5 to E6) is reductive elimination of the TMA adduct and the green path (E5 to F7) is reductive elimination of the DMA fragment. | ||
Surface oxidation state
The oxidation state of the surface was estimated by analysis of the change of Bader charges during the adsorption and decomposition reactions. An increase in Bader charge corresponds to oxidation of the atom while a decrease corresponds to reduction. The changes of Bader charge for atoms at the surface level and below were negligible for all investigated reactions. All transfer of electron charge took place at either the surface amino groups or adatoms. For adsorption of TMA there is no significant change in the oxidation state, Fig. 7a. The nitrogen atom gets a slightly more negative charge which mainly corresponds with an electron withdrawal from the methyl groups. For methyl protonation there is also no large change in the oxidation state of the surface species, Fig. 7b. The deprotonated amino nitrogen gains a partial electron from the loss of the proton, while the hydrogen atoms on the methyl group become more positive as they share their charge with the newly obtained proton.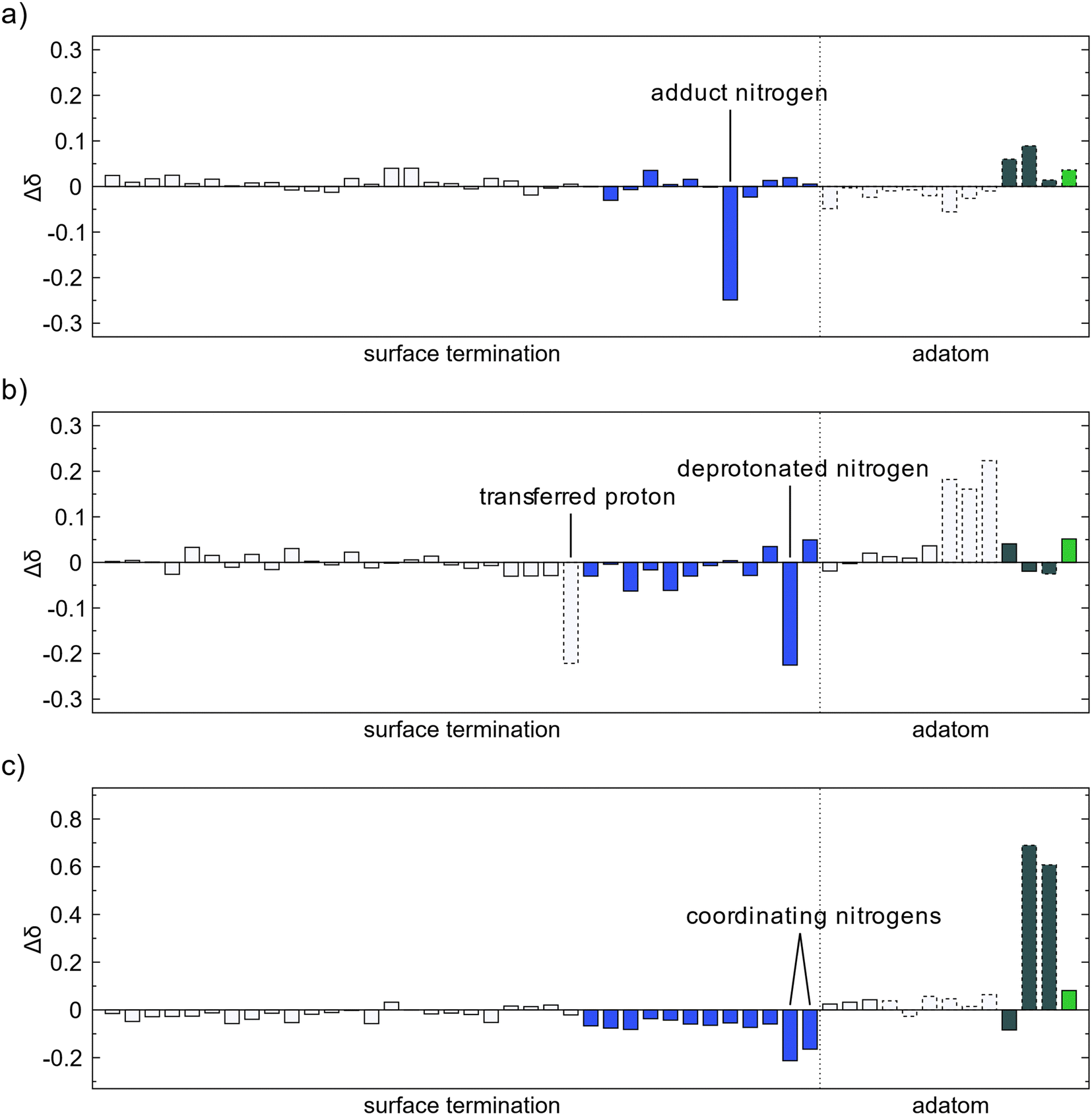 | ||
| Fig. 7 Change of Bader charge after (a) TMA forms a Lewis adduct with an amino group, (b) methyl protonation, and (c) reductive elimination. The colors of the bars correspond to the element, white for hydrogen, blue for nitrogen, dark gray for carbon and green for aluminum. Bars with a dashed outline correspond to atoms adsorbing in (a) or desorbing in (b) and (c). | ||
The ethane elimination changes the oxidation state of the surface to a much higher degree than methyl protonation, Fig. 7c. The two eliminated carbon atoms gain a partial charge of about 0.6–0.7 each, close to the expected oxidation state change as given by the simple Lewis structures, -IV for carbon in methyl to -III for carbon in ethane. The electrons gained by the surface from the elimination is however not localized at any specific atoms, and instead they are distributed over all surface amino groups giving the nitrogen atoms a more negative charge. The two nitrogen atoms gaining slightly more charge than the others are the two that form a new coordination to the formed MMA species.
Bader charges for the adsorption of TMA onto a surface with an MMA fragment were also investigated. The charge differences for adsorption and methyl protonation follow the same pattern as for the initial adsorbed TMA with only small changes in charge and is mostly due to electrons being displaced in the newly formed bonds, Fig. 8a and b. The ethane elimination does show some difference, Fig. 8c. The carbon atoms that are eliminated as ethane are oxidized with a 0.6–0.7 charge each, as before. However, the electrons obtained by the surface are no longer evenly distributed at the amino nitrogen atoms as they already have gained electronic charge in the previous elimination. The electrons are instead more distributed to the hydrogen atoms in the amino groups. This would explain the much lower affinity for the second ethane elimination, as the surface cannot carry the extra electrons as well. Only one TMA molecule per nucleation site is thus expected to be able to decompose by reductive elimination while the following adsorbed TMA would decompose mainly by methyl protonation.
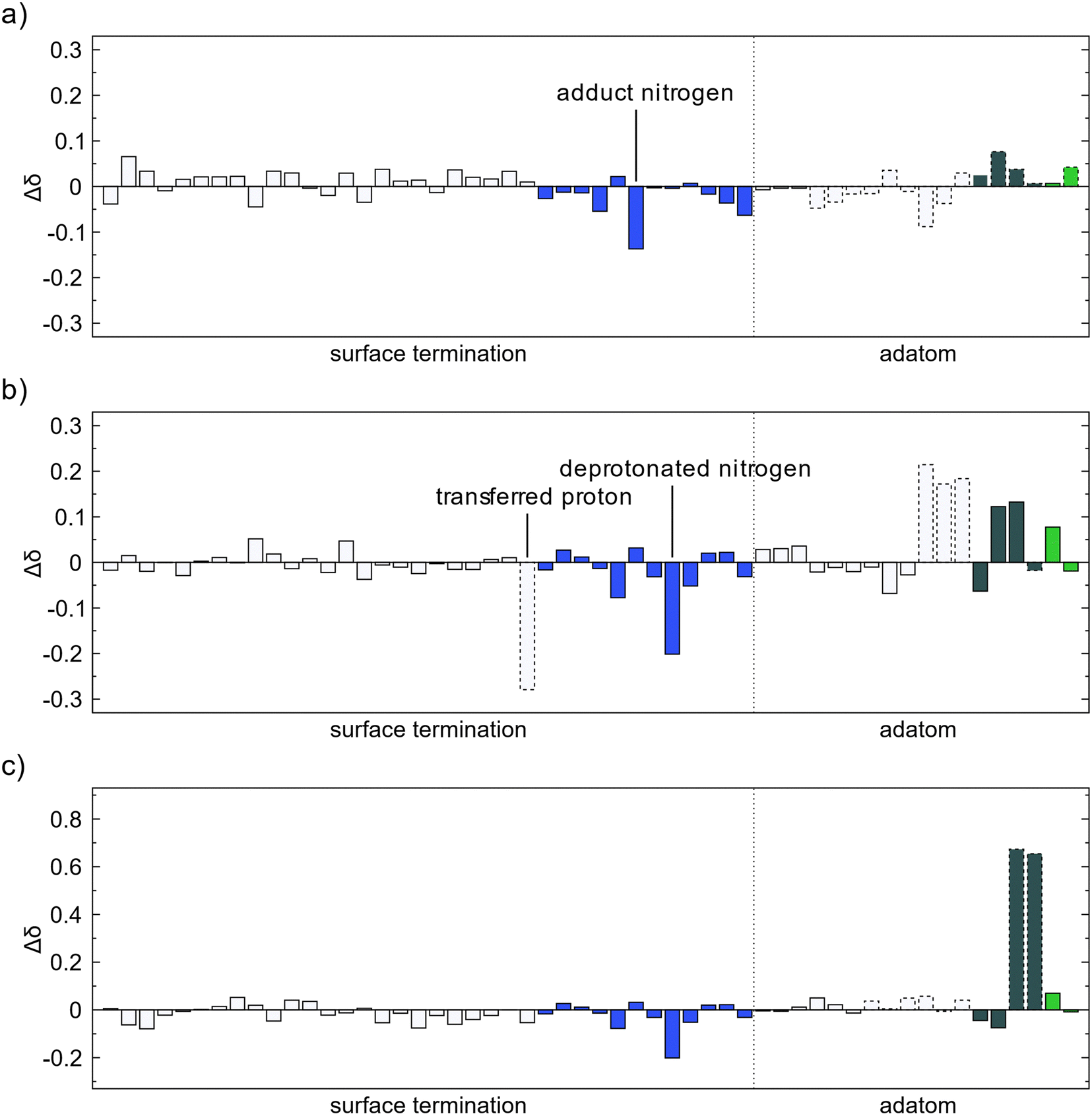 | ||
| Fig. 8 Change of Bader charge on a surface with an MMA fragment after (a) TMA forms a Lewis adduct with an amino group, (b) methyl protonation, and (c) reductive elimination. The colors of the bars correspond to the element, white for hydrogen, blue for nitrogen, dark gray for carbon and green for aluminum. Bars with a dashed outline correspond to atoms adsorbing in (a) or desorbing in (b) and (c). | ||
Surface coverage
Three surface decomposition pathways were found for TMA, reductive elimination (R1), methyl protonation followed by reductive elimination (R2) and methyl protonation twice (R3).| TMA(s) → MMA(s) + C2H6(g) | (R1) |
| TMA(s) + 2H(s) → MMA(s) + 2CH4(g) | (R2) |
| TMA(s) + H(s) → Al(s) + CH4(g) + C2H6(g) | (R3) |
Reactions (R1) and (R3) are available for TMA adducts decomposing if adsorbed far away from an already adsorbed and decomposed MMA fragment. The very low barrier for reductive elimination from the DMA intermediate in reaction (R3) would hinder it from decomposing by another methyl protonation. By comparing the energy barriers for the rate determining step of these reactions, it was determined that around 98% of the TMA adducts would decompose by reaction (R1) compared to reaction (R3). Reaction (R2) is instead the decomposition path for a TMA molecule absorbing close to an already adsorbed and decomposed MMA fragment as no reductive eliminations are possible from this surface. The two dominating decomposition pathways for a TMA adduct are then assumed to be Reactions (R1) and (R2), depending on how close they adsorb, and decompose, to each other.
The surface concentration of hydrogen atoms at a pristine amino terminated surface is two atoms per amino group, corresponding to a surface concentration of 39.6 μmol m−2 assuming a full amino monolayer. All surface hydrogen atoms must be removed during film growth to render stochiometric AlN without contamination of trapped hydrogen. If adsorbed TMA is decomposed by ethene elimination reaction (R1), no hydrogen atoms are removed from the surface and the surface concentration of hydrogen remains unchanged. In contrast, two surface hydrogen atoms are removed and transferred to the methyl groups when an adsorbed TMA molecule decomposes twice by methyl protonation reaction (R2). From these two pathways for TMA adsorption, the concentration of hydrogen (cH) on the surface can be expressed in terms of the concentration of TMA decomposing by reductive elimination (c1), the concentration of TMA decomposing by methyl protonation (c2) and the initial concentration of amino groups (cam), eqn (3).
| cH = 2cam − 0c1 − 2c2 | (3) |
Each surface amino group can form up to three bonds with other surface species. At the initial amino covered surface two of these bonds are occupied by hydrogen atoms while the third can be formed by forming the Lewis adduct with TMA by donating electron density from the lone pair on the nitrogen atom. After decomposition of TMA by either reaction (R1) or (R2), the formed MMA fragment will have one bond to each of its three neighboring amino groups, with a total of three bonds per MMA fragment. The number of surface sites occupied by MMA fragments is thus 3(c1 + c2). The remaining surface hydrogen atoms will occupy one surface site each, i.e., CH. For a TMA molecule to be able to form the initial adduct there must be an amino group on the surface with a lone electron pair. The concentration of TMA adsorption sites can thus be expressed by eqn (4).
| csite = 3cam − 3(c1 + c2) − cH = cam − 3c1 − c2 | (4) |
The right hand of eqn (4) is obtained by inserting eqn (3). From the PES of adsorption, it can be seen that TMA molecules can adsorb close to each other, not sterically blocking any adsorption sites. Thus, at full saturation the number of sites is zero, i.e., csite = 0, yielding eqn (5).
| cam = 3c1 + c2 | (5) |
The coverage at saturation can then be expressed as the total concentration of MMA, c1 + c2, divided by initial coverage of adsorption sites, equal to the concentration of amino groups, eqn (6)
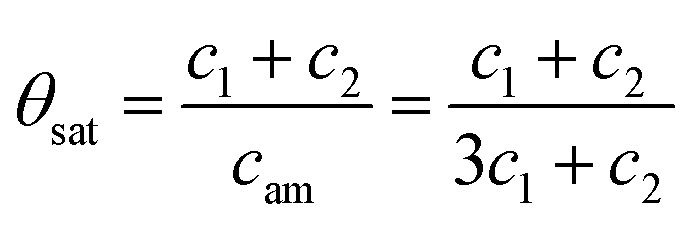 | (6) |
Assuming that very few TMA molecules can decompose by reductive elimination, and most decompose by methyl protonation at already formed nucleation points (c1 ≪ c2) the coverage approaches 1. If instead TMA forms many initial islands that decomposes mainly by reductive elimination (c1 ≫ c2) the coverage instead approaches 1/3. Growth per cycle (GPC) can be predicted from the saturation coverage by the thickness of a monolayer (ML). For AlN growth in the [0001]-direction this corresponds to 2.499 Å per ML, since the c-axis of the AlN crystal is 4.98 Å and there are two monolayers per AlN unit cell in this direction.33 The predicted GPC for different ratios of the decomposition paths is given in Fig. 9.
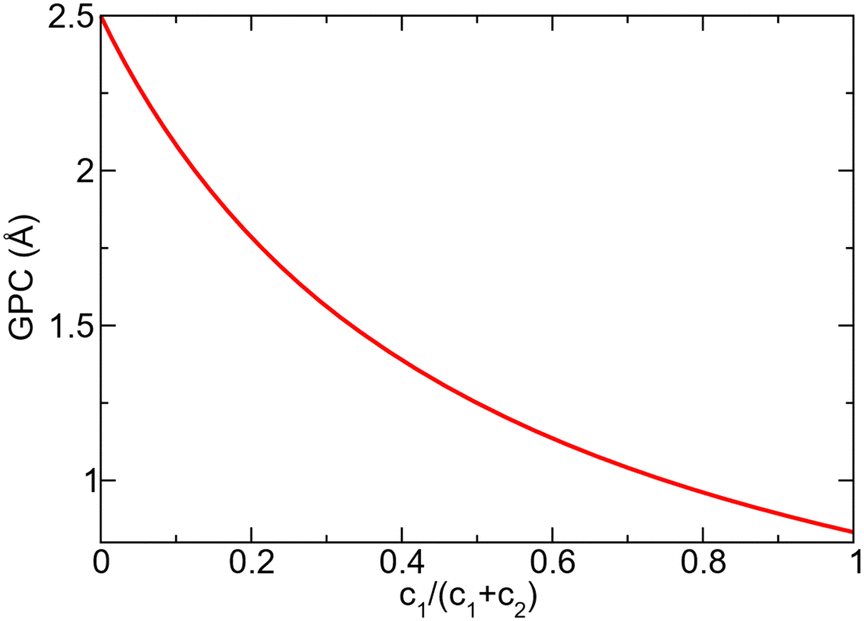 | ||
| Fig. 9 Variation of predicted growth per cycle (GPC) with different ratios of MMA produced by the different decomposition pathways. | ||
An incoming TMA molecule could adsorb directly or diffuse until it reaches an already adsorbed molecule. As the barriers for diffusion and decomposition are similar, the two processes are expected to have similar rate and a TMA molecule is thus expected to only diffuse a few adsorption sites before decomposing and becoming more strongly bonded to the site. This would in turn lead to a large amount of nucleation sites and formation of many small islands during the initial time of each TMA half cycle. The TMA decomposition should then be dominated by reductive elimination, and low saturation coverage with a large amount of hydrogen still at the surface is to be expected. These hydrogen atoms must then be removed either by the following ammonia adsorption or in the following TMA half-cycles to produce contamination free films. The experimental growth rate has been reported as around 1 Å/cycle.34,35 This growth rate corresponds to low saturation coverage and the assumption of TMA molecules at the surface mainly decomposing by reductive elimination.
Conclusions
From DFT calculations we suggest that TMA easily adsorbs as a Lewis adduct on the amino covered surface that is left after an ammonia half-cycle and can then decompose in two different ways. For a TMA adsorbing far away from any previously adsorbed TMA, the main decomposition pathway is a reductive elimination of ethane while a TMA adsorbing close to previously decomposed TMA will mainly decompose by methyl protonation, i.e., ligand exchange where methyl groups leave as methane. The final decomposition product in both pathways is an MMA fragment with one methyl group remaining on each aluminum, but the amount of surface hydrogen left after decomposition is different. The reductive elimination leave hydrogen on the surface that at a high coverage would block additional TMA from adsorbing and contributing to the film growth. The maximum surface coverage at saturation could be expressed by the ratio of the TMA decomposing by each pathway. If the dominating decomposition pathway is methyl protonation the surface can be fully saturated as the removed hydrogen allows for additional adsorption sites for TMA adducts. In the other case, if the dominating adsorption pathway is reductive elimination only a third of all adsorption sites are available. By comparing experimental growth per cycle with the predicted coverage the ratio between the two decomposition paths can be estimated.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was funded by the Swedish foundation for Strategic Research through the project “Time-resolved low temperature CVD for III-nitrides” (SSF-RMA 15-0018). L.O. acknowledges financial support from the Swedish Research Council (VR). High-performance computing resources made available through the National Supercomputer Centre at Linköping University (NSC) and the National Academic Infrastructure for Supercomputing in Sweden (NAISS).References
- J. Li, K. B. Nam, M. L. Nakarmi, J. Y. Lin, H. X. Jiang, P. Carrier and S. H. Wei, Band Structure and Fundamental Optical Transitions in Wurtzite AlN, Appl. Phys. Lett., 2003, 83(25), 5163–5165, DOI:10.1063/1.1633965.
- Y. Taniyasu, M. Kasu and T. Makimoto, An Aluminium Nitride Light-Emitting Diode with a Wavelength of 210 Nanometres, Nature, 2006, 441(7091), 325–328, DOI:10.1038/nature04760.
- A. Khan, K. Balakrishnan and T. Katona, Ultraviolet Light-Emitting Diodes Based on Group Three Nitrides, Nat. Photonics, 2008, 2(2), 77–84, DOI:10.1038/nphoton.2007.293.
- S. Ozaki, J. Yaita, A. Yamada, Y. Kumazaki, Y. Minoura, T. Ohki, N. Okamoto, N. Nakamura and J. Kotani, First Demonstration of X-Band AlGaN/GaN High Electron Mobility Transistors Using Free-Standing AlN Substrate over 15 W Mm−1 Output Power Density, Appl. Phys. Express, 2021, 14(4), 041004 CrossRef CAS.
- Z. Chen, S. Newman, D. Brown, R. Chung, S. Keller, U. K. Mishra, S. P. Denbaars and S. Nakamura, High Quality AlN Grown on SiC by Metal Organic Chemical Vapor Deposition, Appl. Phys. Lett., 2008, 93(19), 191906, DOI:10.1063/1.2988323.
- J. R. Abelson and G. S. Girolami, New Strategies for Conformal, Superconformal, and Ultrasmooth Films by Low Temperature Chemical Vapor Deposition, J. Vac. Sci. Technol., A, 2020, 38(3), 030802, DOI:10.1116/6.0000035.
- T. M. Mayer, T. A. Michalske and J. W. Rogers, Mechanism of Nucleation and Atomic Layer Growth of AIN on Si, Chem. Mater., 1991, 3(4), 641–646, DOI:10.1021/cm00016a016.
- M. E. Bartram, T. A. Michalske, J. W. Rogers and R. T. Paine, Nucleation and Growth of AlN: Self-Limiting Reactions and the Regeneration of Active Sites Using Sequential Exposures of Trimethylaluminum and Ammonia on Silica at 600 K, Chem. Mater., 1993, 5(10), 1424–1430 CrossRef CAS.
- M. Asif Khan, J. N. Kuznia, R. A. Skogman, D. T. Olson, M. Mac Millan and W. J. Choyke, Low Pressure Metalorganic Chemical Vapor Deposition of AIN over Sapphire Substrates, Appl. Phys. Lett., 1992, 61(21), 2539–2541, DOI:10.1063/1.108144.
- K.-E. Elers, M. Ritala, M. Leskelä and L.-S. Johansson, Atomic Layer Epitaxy Growth of AIN Thin Films, J. Phys. IV, 1995, 05(C5), C5–1021, DOI:10.1051/JPHYSCOL:19955120.
- A. I. Abdulagatov, R. R. Amashaev, K. N. Ashurbekova, K. N. Ashurbekova, M. K. Rabadanov and I. M. Abdulagatov, Atomic Layer Deposition of Aluminum Nitride and Oxynitride on Silicon Using Tris(Dimethylamido)Aluminum, Ammonia, and Water, Russ. J. Gen. Chem., 2018, 88(8), 1699–1706, DOI:10.1134/S1070363218080236/METRICS.
- A. I. Abdulagatov, S. M. Ramazanov, R. S. Dallaev, E. K. Murliev, D. K. Palchaev, M. K. Rabadanov and I. M. Abdulagatov, Atomic Layer Deposition of Aluminum Nitride Using Tris(Diethylamido)Aluminum and Hydrazine or Ammonia, Russ. Microelectron, 2018, 47(2), 118–130, DOI:10.1134/S1063739718020026/METRICS.
- S. C. Buttera, P. Rouf, P. Deminskyi, N. J. O’Brien, H. Pedersen and S. T. Barry, Resolving Impurities in Atomic Layer Deposited Aluminum Nitride through Low Cost, High Efficiency Precursor Design, Inorg. Chem., 2021, 60(15), 11025–11031 CrossRef CAS PubMed.
- C. Detavernier, D. Deduytsche, J. Musschoot and J. Dendooven, Thermal Versus Plasma-Enhanced ALD: Growth Kinetics and Conformality, ECS Trans., 2008, 16(4), 239, DOI:10.1149/ma2008-02/24/1913.
- K. H. Kim, N. W. Kwak and S. H. Lee, Fabrication and Properties of AlN Film on GaN Substrate by Using Remote Plasma Atomic Layer Deposition Method, Electron. Mater. Lett., 2009, 5(2), 83–86, DOI:10.3365/eml.2009.06.083.
- M. Bosund, T. Sajavaara, M. Laitinen, T. Huhtio, M. Putkonen, V. M. Airaksinen and H. Lipsanen, Properties of AlN Grown by Plasma Enhanced Atomic Layer Deposition, Appl. Surf. Sci., 2011, 257(17), 7827–7830, DOI:10.1016/J.APSUSC.2011.04.037.
- P. Rouf, P. Sukkaew, L. Ojamäe and H. Pedersen, Reduction of Carbon Impurities in Aluminum Nitride from Time-Resolved Chemical Vapor Deposition Using Trimethylaluminum, J. Phys. Chem. C, 2020, 124(26), 14176–14181, DOI:10.1021/acs.jpcc.0c01724.
- S. D. Elliott and J. C. Greer, Simulating the Atomic Layer Deposition of Alumina from First Principles, J. Mater. Chem., 2004, 14(21), 3246–3250, 10.1039/b405776g.
- K. Rönnby, H. Pedersen and L. Ojamäe, Surface Structures from NH3 Chemisorption in CVD and ALD of AlN, GaN, and InN Films, J. Phys. Chem. C, 2022, 126(13), 5885–5895, DOI:10.1021/acs.jpcc.2c00510.
- K. Rönnby, H. Pedersen and L. Ojamäe, On the Limitations of Thermal Atomic Layer Deposition of InN Using Ammonia, J. Vac. Sci. Technol., A, 2023, 41(2), 020401, DOI:10.1116/6.0002355.
- Q. An, A. Jaramillo-Botero, W. G. Liu and W. A. Goddard, Reaction Pathways of GaN (0001) Growth from Trimethylgallium and Ammonia versus Triethylgallium and Hydrazine Using First Principle Calculations, J. Phys. Chem. C, 2015, 119(8), 4095–4103, DOI:10.1021/jp5116405.
- G. B. Damas, K. Rönnby, H. Pedersen and L. Ojamäe, Understanding Indium Nitride Thin Film Growth under ALD Conditions by Atomic Scale Modelling: From the Bulk to the In-Rich Layer, Appl. Surf. Sci., 2022, 592, 153290, DOI:10.1016/j.apsusc.2022.153290.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47(1), 558–561, DOI:10.1103/PhysRevB.47.558.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- G. Henkelman and H. Jónsson, Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points, J. Chem. Phys., 2000, 113(22), 9978, DOI:10.1063/1.1323224.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths, J. Chem. Phys., 2000, 113(22), 9901–9904, DOI:10.1063/1.1329672.
- G. Henkelman and H. Jónsson, A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives, J. Chem. Phys., 1999, 111(15), 7010, DOI:10.1063/1.480097.
- K. Rönnby, S. C. Buttera, P. Rouf, S. T. Barry, L. Ojamäe and H. Pedersen, Methylamines as Nitrogen Precursors in Chemical Vapor Deposition of Gallium Nitride, J. Phys. Chem. C, 2019, 123(11), 6701–6710, DOI:10.1021/acs.jpcc.9b00482.
- Y. N. Xu and W. Y. Ching, Electronic, Optical, and Structural Properties of Some Wurtzite Crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(7), 4335–4351, DOI:10.1103/PhysRevB.48.4335.
- S. Banerjee, A. A. I. Aarnink, R. van de Kruijs, A. Y. Kovalgin and J. Schmitz, PEALD AlN: Controlling Growth and Film Crystallinity, Phys. Status Solidi, 2015, 12(7), 1036–1042, DOI:10.1002/PSSC.201510039.
- A. P. Perros, H. Hakola, T. Sajavaara, T. Huhtio and H. Lipsanen, Influence of Plasma Chemistry on Impurity Incorporation in AlN Prepared by Plasma Enhanced Atomic Layer Deposition, J. Phys. D. Appl. Phys., 2013, 46(50), 505502, DOI:10.1088/0022-3727/46/50/505502.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02328a |
| This journal is © The Royal Society of Chemistry 2023 |