
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Joint experimental and theoretical study of PbGa2S4 under compression†
Tania
Garcia-Sanchez
 a,
Samuel
Gallego-Parra‡
b,
Akun
Liang§
c,
José Luis
Rodrigo-Ramon
c,
Alfonso
Muñoz
d,
Plácida
Rodriguez-Hernandez
d,
Javier
Gonzalez-Platas
d,
Juán Ángel
Sans
b,
Vanesa Paula
Cuenca-Gotor
b,
Hussien H.
Osman
c,
Catalin
Popescu
e,
Veaceslav
Ursaki
f,
Ion M.
Tiginyanu
f,
Daniel
Errandonea
c and
Francisco Javier
Manjón
b
a,
Samuel
Gallego-Parra‡
b,
Akun
Liang§
c,
José Luis
Rodrigo-Ramon
c,
Alfonso
Muñoz
d,
Plácida
Rodriguez-Hernandez
d,
Javier
Gonzalez-Platas
d,
Juán Ángel
Sans
b,
Vanesa Paula
Cuenca-Gotor
b,
Hussien H.
Osman
c,
Catalin
Popescu
e,
Veaceslav
Ursaki
f,
Ion M.
Tiginyanu
f,
Daniel
Errandonea
c and
Francisco Javier
Manjón
b
aDepartamento de Ingeniería Eléctica, MALTA Consolider Team, Universitat Politècnica de València, Camino de Vera, s/n., Valencia, Spain. E-mail: tagarsan@die.upv.es; Tel: +34 96 387 70 00
bDepartamento de Física, MALTA Consolider Team, Universitat Politècnica de València, Camino de Vera, s/n., Valencia, Spain
cDepartamento de Física Aplicada-ICMUV, MALTA Consolider Team, Universitat de Valencia, Dr Moliner 50, Burjassot, Valencia, 46100, Spain
dDepartamento de Física, Instituto de Materiales y Nanotecnología, MALTA Consolider Team, Universidad de La Laguna, La Laguna, Tenerife 38205, Spain
eALBA-CELLS, MALTA Consolider Team, Cerdanyola del Valles (Barcelona), Cataluña, 08290, Spain
fNational Center for Materials Study and Testing, Technical University of Moldova, Chisinau MD-2004, Republic of Moldova
First published on 28th July 2023
Abstract
The effect of pressure on the structural, vibrational, and optical properties of lead thiogallate, PbGa2S4, crystallizing under room conditions in the orthorhombic EuGa2S4-type structure (space group Fddd), is investigated. The results from X-ray diffraction, Raman scattering, and optical-absorption measurements at a high pressure beyond 20 GPa are reported and compared not only to ab initio calculations, but also to the related compounds α′-Ga2S3, CdGa2S4, and HgGa2S4. Evidence of a partially reversible pressure-induced decomposition of PbGa2S4 into a mixture of Pb6Ga10S21 and Ga2S3 above 15 GPa is reported. Thus, our measurements and calculations show a route for the high-pressure synthesis of Pb6Ga10S21, which is isostructural to the stable Pb6In10S21 compound at room pressure.
1 Introduction
Ternary metal chalcogenides of the AIIBIII2XVI4 family (X = S, Se, Te) can be basically divided into three subfamilies. The first one is constituted by compounds with both A and B cations showing a fourfold coordination. These compounds usually crystallize in the defect chalcopyrite, defect stannite (or defect famatinite), pseudo-cubic, and related structures that are derived from the zinc blende or wurtzite structures. Examples of those compounds are (Zn,Cd,Hg)(Al,Ga)2(S,Se)4 compounds. The second subfamily is constituted by compounds in which there is a mixture of cations with fourfold and sixfold coordination. These compounds crystallize mainly in the spinel (MgAl2O4) or in related structures, such as (Mg,Zn,Cd,Mn)In2(S,Se)4, and show similar structural characteristics to many oxospinels. The third subfamily, and the less studied one, is composed of A cations featuring a coordination much larger than six. Examples of these compounds are those crystallizing in the orthorhombic EuGa2S4-type and related structures, such as (Ca,Sr,Pb,Eu,Sm,Yb)(Al,Ga,In)2 (S,Se,Te)4.1The studies in the last subfamily of EuGa2S4-type compounds have come from the interest in the development first of phosphors,2 and later of mid-infrared (35 μm) solid-state lasers,3–5 due to the large band gap, low-phonon energy, and chemical and thermal stability of these ternary sulphides. In particular, mid-infrared (mid-IR) laser radiation in PbGa2S4 has been consistently reported.6–10 This fact has resulted in recent studies to improve the crystal quality of this mid-IR laser material.11,12
Several works have reported the structural, vibrational, and optical properties of PbGa2S4 under room conditions. From the structural point of view, PbGa2S4 is a layered material that crystallizes in the orthorhombic EuGa2S4-type structure (space group No. 70, D2h24−Fddd).2,13–15 The crystal structure (see Fig. 1 and Fig. S1 and S2 in the ESI†) is built on a framework of GaS4 tetrahedral units and square antiprismatic PbS8 polyhedra. The GaS4 tetrahedra are located in layers stacked along the c-axis, where such layers are constructed from edge-shared Ga2S6 dimers connected to three other dimers via sharing corners. By contrast, the PbS8 polyhedra are linked to two other PbS8 polyhedra by edge-sharing along the c-axis and to four other polyhedra by corner-sharing in the ab-plane. The presence of the lone electron pair of Pb2+ could be related to the crystal structure (S. G. Fddd) of PbGa2S4 in contrast for instance with CdGa2S4 or HgGa2S4 with a defect chalcopyrite structure. However, it must be considered that PbGa2S4 has the orthorhombic EuGa2S4-type structure also common to CaGa2S4 and SrGa2S4. Neither Eu nor Ca or Sr have lone electron pairs, so it is unlikely that the formation of the orthorhombic phase is influenced by the lone electron pair of Pb2+. Instead, the large eight-fold coordination for the A cation in the Fddd phase seems to be related to the large ionic size of Ca(1.12), Eu(1.25), Sr(1.26), and Pb(1.29).16 Curiously, BaGa2S4, with the Ba atom having a much larger ionic size for eight-fold coordination (1.42), does not crystallize in the Fddd structure but in a cubic one described by space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) .2 Note also that the effect of the lone electron pair in Pb when linked to the S atom is very small. In fact, PbS crystallizes in the highly symmetric cubic rock-salt phase while GeS and SnS crystallize in a distorted orthorhombic phase described by space group Pnma.
.2 Note also that the effect of the lone electron pair in Pb when linked to the S atom is very small. In fact, PbS crystallizes in the highly symmetric cubic rock-salt phase while GeS and SnS crystallize in a distorted orthorhombic phase described by space group Pnma.
 | ||
| Fig. 1 Schematic representation of the crystal structure of PbGa2S4. (a) Crystal structure highlighting GaS4 tetrahedral units. (b) Crystal structure highlighting square antiprismatic PbS8 polyhedra. PB, Ga, and S atoms are shown in black, green, and yellow colors. A figure highlighting the layered characteristic of the crystal structure is given in Fig. S2 in the ESI.† | ||
Regarding vibrational properties, phonons from Raman scattering measurements were initially reported by Syrbu et al.17 and infrared (IR) modes were reported from absorption measurements by Neumann et al.18 The first work interpreted the Raman modes in the light of a wrong crystal structure, while the latter work interpreted the IR modes in the light of the good crystal structure of PbGa2S4. Further characterization studies of Raman-active and IR-active modes were also performed, but again were interpreted in the light of the wrong crystal structure.19,20 Fortunately, the Raman spectrum at room temperature was well explained in terms of the right crystal structure by Bletskan et al.15 and more recently by Kamenshchikov et al.21 Regarding the optical properties, the IR spectral region of PbGa2S4 was studied,22 followed by optical-absorption measurements that determined that it is an indirect bandgap semiconductor, whose indirect bandgap energy, Eg, at room temperature was 2.84 eV, and shows a direct bandgap at 2.91 eV.23 Later, Badikov et al. reported the refractive index in the visible region and the lasing by doping with Na and Dy atoms.24 More recently, the reflectance spectrum was analyzed showing excitons with large binding energy and oscillator strength even at room temperature.25,26
The effect of temperature on the properties of PbGa2S4 has been considerably investigated. Regarding structural properties, no phase change has been observed on decreasing the temperature down to 10 K when measuring different optical properties.26 On the other hand, Chilouet et al. found a decomposition of PbGa2S4 above 900 °C13 that was recently confirmed.11 Moreover, a different polymorph of PbGa2S4 (S. G. Pna21) has been recently synthesized at high temperature.27 Regarding vibrational properties, Raman-active phonons were measured at different temperatures between 10 and 300 K.19,28 Finally, regarding optical properties, a decrease of the bandgap was reported on increasing the temperature to between 10 and 300 K.23,29
Unlike the effect of temperature on the properties of PbGa2S4, the effect of pressure on the properties of PbGa2S4, or of any other EuGa2S4-type compounds, has not been investigated yet, to the best of our knowledge. This contrasts with the properties of AIIBIII2XVI4 chalcogenides with spinel, defect chalcopyrite, and their related structures that have been thoroughly studied under compression as recently reviewed.30 In this work, we report a joint experimental and theoretical study of the structural, vibrational, and optical properties of PbGa2S4 under compression of up to 25 GPa. In particular, we report high-pressure (HP) X-ray diffraction (XRD), Raman scattering (RS), and optical-absorption (OA) measurements and compare them to ab initio calculations. We show evidence of a partially reversible pressure-induced decomposition of PbGa2S4 into Pb6Ga10S21 and Ga2S3 above 15 GPa and provide the equation of state of the low-pressure phase and the pressure dependence of Raman-active modes and of the indirect bandgap of PbGa2S4 and Pb6Ga10S21. This study allows a comparison to be made for the first time between the properties of EuGa2S4-type compounds under compression and those of other compounds based on GaS4 polyhedra, such as Ga2S3 and (Zn,Cd,Hg)Ga2(S,Se)4. Hopefully, this study will also help in understanding the properties of PbGa2S4 at room conditions in order to improve its technological applications.
2 Experimental and theoretical details
2.1 Experimental details
PbGa2S4 crystals have been grown via the Bridgman–Stockbarger method in quartz ampoules with high purity (99.999%) source components. The temperature of the melt exceeded the PbGa2S4 melting temperature (890 °C) by 50–70 °C. The temperature gradient was 20–30 K cm−1 at a pull down rate of 0.25 mm h−1.31,32 High resistivity p-type crystals with sizes of 2 × 2 × 5 cm3, which are easily cleaved, were grown and the cleaved surfaces were not mechanically processed.25To be sure about the nature of the as-grown sample, structural characterization of the PbGa2S4 sample at room pressure (RP) was performed by single-crystal measurements of an orange plate-shaped crystal with dimensions 0.12 × 0.11 × 0.04 mm3. Data were collected using a SuperNova, Dualflex, EosS2 diffractometer and measured using ω scans with Mo Kα radiation (λ = 0.71073 Å) at a maximum resolution of Θ = 28.278° (0.75 Å). The program CrysAlisPro (Rigaku, V1.171.40.84a, 2020) was used to determine the total number of runs and images for the diffraction pattern as well as to index and refine it. A numerical absorption correction based on Gaussian integration over a multifaceted crystal model was performed using spherical harmonics as implemented in the SCALE3 ABSPACK scaling algorithm.
For HP-RS, HP-XRD, and HP-OA measurements in powder or single crystal samples of PbGa2S4, the samples were placed in a 250 μm-diameter hole in a stainless-steel gasket pre-indented to a thickness of 50 μm inside a membrane-driven diamond-anvil cell (DAC) equipped with diamonds with a culet of 500 μm-diameter. A 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–ethanol (M–E) mixture was used as a pressure-transmitting medium (PTM) in all HP experiments. This medium is quasi-hydrostatic up to 10.5 GPa. Special attention was paid to avoid the sample bridging between the diamond anvils.33 For measuring the pressure, the ruby fluorescence scale was used.34 In addition, Cu grains were loaded close to the sample in the HP-XRD experiments in order to use it as a second pressure scale.35
:1 methanol–ethanol (M–E) mixture was used as a pressure-transmitting medium (PTM) in all HP experiments. This medium is quasi-hydrostatic up to 10.5 GPa. Special attention was paid to avoid the sample bridging between the diamond anvils.33 For measuring the pressure, the ruby fluorescence scale was used.34 In addition, Cu grains were loaded close to the sample in the HP-XRD experiments in order to use it as a second pressure scale.35
Structural characterization at HP was performed by means of angle-dispersive powder HP-XRD measurements at the BL04-MSPD beamline of the ALBA synchrotron, employing a monochromatic X-ray beam with λ = 0.4246 Å focused to 20 × 20 μm2 (full width half-maximum).36 The X-ray beam was focused by Kirkpatrick–Baez mirrors. XRD images were collected using a Rayonix SX165 CCD detector located 240 mm from the sample. The detector parameters were calibrated using LaB6 as the standard. The two dimensional diffraction images were integrated into one-dimensional profiles of intensity versus 2θ using Dioptas.37 XRD profiles were analyzed using GSAS-II by means of Rietveld refinements or by using the Le Bail method.38
Vibrational characterization was carried out by means of polarized and unpolarized RS measurements at RP and HP in order to distinguish between the large number of phonons expected for this compound. All RS measurements were carried out with a Horiba Jobin Yvon LabRAM UV HR microspectrometer equipped with a thermoelectrically cooled multichannel CCD detector that allows a spectral resolution better than 2 cm−1. The Raman signal was excited with a HeNe laser (632.8 nm line) with a power of less than 10 mW and collected in back-scattering geometry using a ULF notch filter that allows signals to be obtained down to 10 cm−1. The frequencies of the Raman-active first-order phonons were obtained after fitting the Raman peaks with Voigt profiles of fixed Gaussian linewidth to the experimental setup resolution (1.6 cm−1).
Optical characterization was performed by means of OA experiments at RP and HP in single crystals using two different home-built optical setups that consist of a tungsten lamp, fused silica lenses, reflecting micro-objectives (15×), and a visible-near-IR spectrometer (Ocean Optics Maya2000 Pro in the first setup and Ocean Optics HR2000+ in the second setup). The experimental transmittance of the sample was obtained with the sample-in, sample-out method,39 and then scaled to the theoretical transmittance value in the spectral range where the sample is completely transparent. Finally, the absorption coefficient α was obtained from the scaled transmittance by taking into account the sample thickness (d ≈ 20 μm) and also the reflectivity obtained from the knowledge of the refractive index (n ≈ 3.0)24 as done in previous works.40–42
2.2 Theoretical details
Ab initio total-energy calculations have been carried out within the density–functional theory (DFT) framework,43 using plane waves and the pseudopotential technique with the Vienna ab initio simulation package (VASP).44 The set of plane waves employed extended up to a kinetic energy cutoff of 400 eV. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof PBE parametrization was used for the description of the exchange and correlation energy.45 Dense special point grids of 4 × 4 × 4 were used to sample the Brillouin zone (BZ) when relaxing the structure at different volumes. Pressure, like other energy derivatives, is obtained simultaneously from the stress tensor.46 Lattice dynamics calculations were performed at the zone center (Γ-point) of the Brillouin zone. The super-cell method with the primitive cell was employed for the calculation of the dynamical matrix at the Γ-point.44 Since GGA–PBE calculations underestimate the bandgap values, we have used the metaGGA47 approach with the modified Becke–Johnson (MBJ) potential,48 which leads to a good agreement with experiments, for electronic band structure calculations.3 Results and discussion
3.1 Structural properties
Single-crystal XRD measurements at RP with R1 = 2.36% confirm that PbGa2S4 crystallizes in the orthorhombic structure (space group Fddd). The unit-cell parameters a = 12.1674(2) Å, b = 20.4180(4) Å, and c = 20.7005(5) Å were refined using 7590 reflections, 31% of the observed reflections. The atomic positions are summarized in Table 1. Further details of the single-crystal XRD analysis are provided in the ESI.† In particular, the final equivalent isotropic displacement parameters, as well as selected bond distances, and angles are provided in Tables S1–S3 in the ESI.† Our structure and values of unit-cell parameters agree with those reported from recent single-crystal XRD measurements.14 As already commented, the structure of PbGa2S4 is composed of a network of GaS4 tetrahedra and PbS8 polyhedra. The large values of Pb–S distances compared to Ga–S distances are related to the soft ionic Pb–S bonds and the stronger covalent Ga–S bonds and are responsible for the easy cleavage of lead thiogallate into layers that run perpendicular to the longest axis (the c-axis in our description).| Atom | Wyckoff position | x | y | z | U eq |
|---|---|---|---|---|---|
| Pb3 | 8a | 0.875 | 0.375 | 0.375 | 0.022(12) |
| Pb2 | 16g | 0.375 | 0.375 | 0.127(2) | 0.022(11) |
| Pb1 | 8b | 0.375 | 0.375 | 0.375 | 0.024(13) |
| Ga1 | 32h | 0.585(4) | 0.304(2) | 0.249(3) | 0.012(12) |
| Ga2 | 32h | 0.626(4) | 0.487(2) | 0.250(3) | 0.012(12) |
| S2 | 32h | 0.501(15) | 0.250(9) | 0.333(5) | 0.012(2) |
| S3 | 32h | 0.748(15) | 0.499(9) | 0.166(5) | 0.012(2) |
| S4 | 32h | 0.765(8) | 0.329(5) | 0.248(10) | 0.011(2) |
| S1 | 32h | 0.503(9) | 0.406(5) | 0.251(11) | 0.013(2) |
Fig. 2 shows the evolution of the powder XRD patterns of PbGa2S4 under compression from 0.3 to 17.6 GPa. As pressure increases, the diffraction peaks move gradually to higher angles due to the reduction of unit-cell parameters. Within this pressure range, all peaks can be indexed with an orthorhombic crystal structure isomorphic to the RP phase. The structure at 0.3 GPa is described by space group Fddd and the unit-cell parameters are a = 12.159(3) Å, b = 20.397(6) Å, c = 20.67(6) Å. The only difference with the structure at RP is the slight reduction of unit-cell parameters. From 0.3 to 17.6 GPa the sample does not present evidence of any structural phase transition. All the XRD patterns can be explained by the low-pressure orthorhombic structure. This conclusion is supported by Rietveld refinements at the lowest and highest pressures. Notice that the accessible 2θ is restricted to 16° (and consequently the number of independent reflections) by the geometrical opening cone of the DAC and the size of the CCD detector. Therefore, the atomic positions were fixed in the refinement at all pressures to the values determined from single-crystal XRD at ambient pressure (see Table 1). In the refinement, we fitted the unit-cell parameters, peak-shape parameters (U, V, and W Caglioti coefficients), the overall displacement factor, and the background (using a 10-term Chebyshev polynomial of the first kind). This approximation usually works quite well for synchrotron HP data.49
 | ||
| Fig. 2 HP-XRD patterns of PbGa2S4 on compression up to 17.6 GPa. In the bottom and top traces, black dots are the experiments, red lines are the results of structural refinements Rietveld, and black lines are the residuals. The black and blue ticks give the positions of calculated reflections for the sample and Cu, respectively. The pressures are indicated in the figure. | ||
Beyond 17.6 GPa, on the upstroke, remarkable changes can be observed in the XRD patterns (Fig. 3) between 17.6 and 25 GPa. We are aware that at these pressures our PTM is not hydrostatic. However, changes start to develop at 17.6 GPa and the quasi-hydrostatic limit of the PTM is 10.5 GPa. This suggests that the observed structural changes are not related to non-hydrostatic effects. Regarding the observed changes, first, we noticed the emergence of extra peaks, denoted by asterisks in the figure, and the weakening of peaks assigned to the RP phase, denoted by the plus symbol in the figure. The patterns measured at 19.7 and 21.4 GPa correspond to the coexistence of the RP and HP phases. At 23.5 and 25 GPa the patterns can be identified with only the HP phase. The DICVOL routine was used to index the Bragg reflections of the HP phase measured at 23.5 GPa and CheckCell was used for space group determination. For these purposes, we used only the peaks below 2θ = 12° to avoid the overlapping of reflections from the HP phase and Cu. It was found that the monoclinic space group C2/m gives the best figure of merit; M(20) = 23.2. A subsequent Le Bail fit, using the structural model obtained from the method described above, provided as unit-cell parameters a = 25.269(9) Å, b = 3.529(2) Å, c = 14.433(5) Å, and β = 96.632°. Fig. 3 shows the good profile match obtained between the Le Bail fit and the measured XRD pattern.
 | ||
| Fig. 3 HP-XRD patterns of PbGa2S4 on compression from 17.6 to 25 GPa. We also include patterns measured upon decompression. These patterns are identified by (r) next to the pressure. We show in blue XRD patterns where the onset of decomposition is detected and in green patterns assigned to Pb6Ga10S21. At 23.6 GPa and 12.1 GPa (r), black dots are the experiments, red lines are the Lebail fits, and black lines are the residuals. The black and blue ticks give the positions of calculated reflections for the sample and Cu, respectively. The red ticks are calculated reflections of Ga2S3. The pressures are indicated in the figure. | ||
Interestingly, the space group and unit-cell parameters obtained for the X-ray diffraction patterns of the HP phase above 21.4 GPa are similar to those of Pb6In10S21.50 Therefore, we performed theoretical calculations on Pb6Ga10S21 at different pressures. For calculations, we have used both the RP phase of Ga2S3, α′-Ga2S3, with a monoclinic Cc structure,51 and the HP phase above 16 GPa, β′-Ga2S3, with a rhombohedral Rm structure.52 Our enthalpy vs. pressure calculations showed that it is thermodynamically favorable for PbGa2S4 to decompose into Pb6Ga10S21 + Ga2S3 above 10 GPa (see Fig. 4) since the enthalpy of the decomposition products becomes smaller than that of the father compound beyond this pressure. In fact, the decomposition process is 6 formula units of PbGa2S4 decompose into Pb6Ga10S21 + Ga2S3. Consequently, we think that both our XRD experiments and calculations support the decomposition hypothesis.
 | ||
| Fig. 4 Calculated enthalpy of the orthorhombic structure of PbGa2S4 (black solid line) and the sum of the enthalpy of decomposition products Pb6Ga10S21 and Ga2S3 (red solid line). | ||
Unfortunately, the XRD peaks of Ga2S3 are much weaker than those of Pb6Ga10S21 (because of the strong X-ray absorption of Pb) and they overlap with those of Pb6Ga10S21 (see Fig. 3). Therefore, the XRD patterns of the HP phase have been explained by a Le Bail fit and not by a Rietveld refinement. In any case, the calculated lattice parameters at 23.5 GPa (a = 25.3061 Å; b = 3.5276 Å; c = 14.4180 Å; and β = 96.7820°) agree with the experimental values we reported in the previous paragraph what gives further support to the decomposition process already commented. For completeness, the theoretical atomic positions of Pb6Ga10S21 at 23.5 GPa are reported in Table 2. The crystal structure of Pb6Ga10S21 is shown in Fig. 5. In contrast with the studied compound, this material is not layered, the crystal structure being formed by zig-zag chains of edge sharing GaS6 octahedra and PbS8 dodecahedra. In summary, we conclude with confidence that PbGa2S4 undergoes a pressure-induced decomposition at HP. This observation is not fully unexpected since it has been recently found that this compound tends to decompose into PbS and Ga2S3 during the growth process of single crystals if temperature is larger than 1250 K.53
| Atom | Wyckoff position | x | y | z |
|---|---|---|---|---|
| S1 | 2a | 0.0000 | 0.0000 | 0.0000 |
| S2 | 4i | 0.9728 | 0.0000 | 0.7662 |
| S3 | 4i | 0.6730 | 0.0000 | 0.7606 |
| S4 | 4i | 0.7647 | 0.0000 | 0.6006 |
| S5 | 4i | 0.5798 | 0.0000 | 0.8778 |
| S6 | 4i | 0.3975 | 0.0000 | 0.9046 |
| S7 | 4i | 0.3796 | 0.0000 | 0.6297 |
| S8 | 4i | 0.5373 | 0.0000 | 0.5955 |
| S9 | 4i | 0.1934 | 0.0000 | 0.9776 |
| S10 | 4i | 0.2790 | 0.0000 | 0.8218 |
| S11 | 4i | 0.1511 | 0.0000 | 0.5670 |
| Ga1 | 4i | 0.3121 | 0.0000 | 0.5110 |
| Ga2 | 4i | 0.2174 | 0.0000 | 0.6926 |
| Ga3 | 4i | 0.7323 | 0.0000 | 0.8966 |
| Ga4 | 4i | 0.4863 | 0.0000 | 0.8711 |
| Ga5 | 4i | 0.0983 | 0.0000 | 0.9850 |
| Pb1 | 4i | 0.0807 | 0.0000 | 0.7200 |
| Pb2 | 4i | 0.9493 | 0.0000 | 0.5615 |
| Pb3 | 4i | 0.8565 | 0.0000 | 0.7601 |
 | ||
| Fig. 5 Schematic representation of the crystal structure of Pb6Ga10S21. GaS6 octahedral units are shown in green and PbS8 bicapped trigonal prismatic polyhedra in black. | ||
Under decompression, Pb6Ga10S21 is still observed at 18.6 GPa; however, the RP phase is recovered at 12.1 GPa and it is also observed at 6.6 GPa. The peaks of the RP phase after decompression are slightly broader that those measured at similar pressures upon compression due to residual stresses in already compressed samples. The reversibility of changes induced by pressure indicates that, in an unexpected way, the pressure-induced decomposition is reversible after pressure release; a fact rather uncommon but that has been previously reported in other compounds.54 In our opinion, the reversibility of the pressure-induced decomposition in PbGa2S4 is favored because 6 formula units of PbGa2S4 (Pb6Ga12S24) decompose into Pb6Ga10S21 + β′-Ga2S3. Therefore, on decompression Pb6Ga10S21 can transform back into 5 PbGa2S4 + PbS, thus resulting a mixture of 5 PbGa2S4 + PbS + γ-Ga2S3 at room conditions from the original 6 formula units of PbGa2S4. Note that the RP phase α′-Ga2S3, with monoclinic Cc structure, transforms into β′-Ga2S3, with rhombohedral Rm structure52 above 16 GPa and on decompression to room-pressure the original structure of Ga2S3 is not recovered and a γ phase is observed instead.52
Let us now analyse the evolution of the structure of the RP phase of PbGa2S4. A good agreement has been found between the experimental and theoretical pressure dependence of the unit-cell parameters and unit-cell volume of PbGa2S4 (see Fig. 6). Curiously, the b- and c-axes tend to become equal at HP, thus indicating and enhancement of the symmetry of the crystal structure upon compression. However, the structure cannot be described as tetragonal above 12 GPa because XRD patterns cannot be indexed by any space group with a fourfold rotation axis. The experimental axial compressibilities have been determined by the following expression  and are κa = 4.33 × 10−3 GPa−1, κb = 6.69 × 10−3 GPa−1, and κc = 8.12 × 10−3 GPa−1. As a result, there is a considerable anisotropic compression of PbGa2S4 since the b- and c-axes are more compressible than the a-axis.
and are κa = 4.33 × 10−3 GPa−1, κb = 6.69 × 10−3 GPa−1, and κc = 8.12 × 10−3 GPa−1. As a result, there is a considerable anisotropic compression of PbGa2S4 since the b- and c-axes are more compressible than the a-axis.
 | ||
| Fig. 6 Pressure dependence of the lattice parameters and volume in PbGa2S4. Symbols represent the experiments, lines represent the calculations and dashed lines represent the equations of states determined from experiments. The same color is used for the same parameter in experimental and theoretical results. Green symbols represent the experiment at ambient pressure. | ||
From the experimental and theoretical data given in Fig. 6, the pressure–volume (P–V) equation of state (EoS) for the orthorhombic phase has been obtained with a third-order Birch–Murnaghan EoS (BM3-EOS),57 using the software EosFit.58 The zero-pressure unit-cell, V0, bulk modulus, B0, and its pressure derivative,  are summarized in Table 3. It can be observed that the experimental and theoretical results are in relatively good agreement. For comparison purposes, Table 3 also shows the corresponding experimental parameters obtained in other AGa2S4 compounds. Since both parameters are correlated59 and they may depend on experimental conditions,60 the pressure-transmitting medium (PTM) used in each experiment is also reported in Table 3. It can be observed that for PbGa2S4 the experimental B0 (47.3(1) GPa) obtained with the BM3-EOS is similar to that obtained for HgGa2S4, but the bulk modulus is 40% smaller than in CdGa2S4. The reason for the larger bulk modulus of CdGa2S4 could be the poorer hydrostatic conditions of the experiments performed in this compound, as discussed by Errandonea et al.61 This hypothesis is consistent with the fact that DFT calculations give for CdGa2S4 a bulk modulus of 40.8–46.0 GPa.62,63 In any case, a slightly smaller value of B0 is expected for EuGa2S4-type compounds than for defect chalcopyrite AGa2S4 (A = Cd, Hg) compounds since a larger compressibility is expected for eightfold-coordinated Pb atoms in the orthorhombic Fddd structure than for fourfold-coordinated Cd and Hg atoms in the defect chalcopyrite structure described by the space group I
are summarized in Table 3. It can be observed that the experimental and theoretical results are in relatively good agreement. For comparison purposes, Table 3 also shows the corresponding experimental parameters obtained in other AGa2S4 compounds. Since both parameters are correlated59 and they may depend on experimental conditions,60 the pressure-transmitting medium (PTM) used in each experiment is also reported in Table 3. It can be observed that for PbGa2S4 the experimental B0 (47.3(1) GPa) obtained with the BM3-EOS is similar to that obtained for HgGa2S4, but the bulk modulus is 40% smaller than in CdGa2S4. The reason for the larger bulk modulus of CdGa2S4 could be the poorer hydrostatic conditions of the experiments performed in this compound, as discussed by Errandonea et al.61 This hypothesis is consistent with the fact that DFT calculations give for CdGa2S4 a bulk modulus of 40.8–46.0 GPa.62,63 In any case, a slightly smaller value of B0 is expected for EuGa2S4-type compounds than for defect chalcopyrite AGa2S4 (A = Cd, Hg) compounds since a larger compressibility is expected for eightfold-coordinated Pb atoms in the orthorhombic Fddd structure than for fourfold-coordinated Cd and Hg atoms in the defect chalcopyrite structure described by the space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) .
.
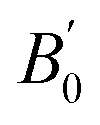 dimensionless) for several AGa2S4 compounds. The pressure-transmitting medium used in experiments is indicated
dimensionless) for several AGa2S4 compounds. The pressure-transmitting medium used in experiments is indicated

Curiously, the experimental and theoretical bulk moduli of PbGa2S4 are also similar to those of the RP phase of Ga2S3 (α′-Ga2S3, experimental B0 = 47 GPa and theoretical B0 = 38 GPa). In fact, also the axial compressibilities of monoclinic α′-Ga2S3 are also similar to those of PbGa2S4.51 Noteworthy, α′-Ga2S3 is composed of GaS4 tetrahedra with 1/3 empty cation sites and it was shown that the two GaS4 tetrahedra in α′-Ga2S3 feature a much higher polyhedral bulk modulus (99.1 and 123.0 GPa) than the compound bulk modulus. On the basis of this data, we expect a similar polyhedral bulk modulus for the GaS4 tetrahedra in PbGa2S4 and other AGa2S4 compounds. Consequently, our results suggest that the bulk modulus of PbGa2S4 is mainly determined by the compressibility of the PbS8 polyhedra and that, surprisingly, they compress at a similar rate than cation vacant sites in Ga2S3, what is in agreement with the weak Pb–S bonds and easy cleavage of layers in PbGa2S4.
3.2 Vibrational properties
The orthorhombic Fddd structure of PbGa2S4 at RP contains 32 formula units in the unit cell (224 atoms) and its primitive unit cell contains 8 formula units (56 atoms). Therefore, according to group theory, PbGa2S4 should have 168 vibrational modes at the Γ point with the mechanical representation:64| Γ = 19Ag + 19Au + 21B1g + 21B1u + 22B2g + 22B2u + 22B3g + 22B3u |
Due to the large number of Raman-active modes allowed in PbGa2S4, polarised and unpolarised RS measurements were performed in order to better distinguish their symmetry. From the polarized measurements at RP, we found the 6 different types of symmetry, Ag (xx), Ag (zz), Ag (yy), B1g (xz), B2g (xz) and B3g (xz) (see Fig. S3 in the ESI†), that are in good agreement with those already published by Kamenshchikov et al.21 A comparison of the frequencies of the different modes observed at room conditions in this work and by Kamenshchikov et al. is reported in Table S4 in the ESI.†
The Raman spectrum of PbGa2S4 can be divided into two regions: the low- (high-) frequency region below (above) 200 cm−1. Our calculations confirm that modes of the high-frequency region are related to the internal Ga–S stretching modes of the GaS4 tetrahedra, while modes of the low-frequency region are related to Ga–S bending modes of the GaS4 tetrahedra (between 100 and 200 cm−1) and to modes related to Pb vibrations as well as to rigid translations and rotations of GaS4 tetrahedra (below 100 cm−1). In general, the modes related to Pb vibrations show a smaller intensity and a narrower linewidth than those corresponding to internal (Ga–S bending and stretching) modes of the GaS4 tetrahedra. This classification of vibrational modes in PbGa2S4 according to internal and external modes of GaS4 tetrahedra agrees with the one performed in α′-Ga2S3, where a separation between low- and high-frequency regions is observed below and above 200 cm−1.51 In particular, the study of the vibrational properties of α′-Ga2S3 showed that all internal modes of GaS4 tetrahedra are above 200 cm−1 and that all external modes of GaS4 tetrahedra are between 70 and 200 cm−1. Similarly, the vibrational modes in CdGa2S4 (HgGa2S4) extend from 80 (60) to 400 cm−1.62,65 Therefore, we can conclude that since Pb has a similar mass as Hg, the existence of vibrational modes below 60 cm−1 in PbGa2S4 at RP must be exclusively ascribed to the vibrations of Pb atoms. This conclusion agrees with the comments of Kamenshchikov et al. that attributed the modes related to Pb–S bonds to those observed between 20 and 55 cm−1 (note that these authors did not report modes below 20 cm−1).21
Regarding the pressure dependence of the Raman-active modes in PbGa2S4, Fig. 7 shows a selection of unpolarized HP-RS spectra of PbGa2S4 under compression up to 22 GPa and decompression down to RP. Since samples are exfoliated perpendicular to the c-axis, our Raman measurements obtained in back-scattering geometry inside the DAC correspond to a mixture of modes with Ag, B2g and B3g symmetry. This information has been obtained from our polarized RS measurements at room conditions (see Fig. S4 in the ESI†) and also from HP-RS measurements in which only modes of Ag symmetry or a mixture of modes of B2g and B3g symmetry have been distinguished (see Fig. S3 in the ESI†).
 | ||
| Fig. 7 Normalized Raman spectra of PbGa2S4 at high pressure up to 22 GPa. (a) Upstroke, (b) downstroke. Crosses indicate the main peaks of the Fddd phase that disappear with increasing pressure. Stars indicate new peaks of the high-pressure phase. | ||
Raman spectra of Fig. 7(a) and Fig. S4 (ESI†) show that some of the Raman modes of the RP phase (marked with crosses in Fig. 7) disappear above 16.6 GPa together with the appearance of some extra peaks (marked with asterisks). The changes observed in HP-RS measurements above 16 GPa agree with those from HP-XRD measurements above 17 GPa, thus suggesting the occurrence of a phase transition above 16 GPa. On downstroke, Fig. 7(b) shows similar Raman spectra to those of the RP phase, but with peaks of smaller intensity and larger linewidth, appear below 7.4 GPa, thus supporting the hypothesis that the sample partially reverses to the original RP phase on downstroke from 25 GPa.
From the analysis of the unpolarized HP-RS spectra of PbGa2S4, 21 of the 84 modes of the compound were distinguished and followed under pressure. The symmetry of those modes was tentatively attributed thanks to the reasonable comparison of the pressure dependence of the experimental and theoretical frequencies (also reported in Fig. 8). The experimentally observed Raman modes and their tentatively assigned theoretical symmetries with their zero-pressure frequencies and pressure coefficients are summarized in Table 4. For the sake of completeness, the zero-pressure frequencies and pressure coefficients of all the theoretically predicted Raman-active modes are summarized in Table S5 in the ESI.†
 | ||
| Fig. 8 Pressure dependence of the experimental (symbols) and theoretical (lines) Raman frequencies of PbGa2S4 during compression. We use black color for Ag modes, blue color for B2g modes, orange color for B3g modes, and green color for the mode of the HP phase. | ||
| Mode | PbGa2S4 th. | PbGa2S4 exp. | ||||
|---|---|---|---|---|---|---|
| ω 0 | a 1 | a 2 | ω 0 | a 1 | a 2 | |
| B12g | 17.7(2) | 3.81(0) | −0.1(0) | 20.3(1) | 4.1(0) | 0.0(0) |
| B13g | 33.8(1) | 0.7(0) | −0.1(0) | 34.7(1) | 0.2(0) | 0.0(0) |
| B23g | 39.2(0) | 1.6(0) | −0.1(0) | 40.8(0) | 1.5(0) | −0.1(0) |
| A1g | 44.0(1) | 2.4(0) | −0.1(0) | 45.2(0) | 2.7(0) | −0.1(0) |
| A2g | 84.3(0) | 0.1(0) | 0.0(0) | 86.6(0) | 0.2(0) | 0.0(0) |
| A3g | 88.7(1) | 1.1(0) | 0.0(0) | 104.7(1) | −1.4(0) | 0.0(0) |
| B73g | 108.3(0) | −0.4(0) | 0.0(0) | 109.9(2) | −2.3(0) | −0.1(0) |
| B62g | 110.4(0) | −0.7(0) | 0.0(0) | 112.3(0) | −0.4(0) | 0.0(0) |
| B83g | 112.7(0) | 0.0(0) | 0.0(0) | 125.6(4) | −2.4(1) | 0.1(0) |
| A5g | 145.4(1) | 0.8(0) | 0.0(0) | 150.5 (0) | 1.6(0) | −0.2(0) |
| B103g | 153.5(2) | 3.7(0) | −0.1(0) | 152.6(1) | 3.9(0) | −0.1(0) |
| A8g | 160.2(1) | 3.2(2) | 0.0(0) | 160.9(1) | 4.4(0) | −0.1(0) |
| B123g | 162.8(1) | 4.7(0) | 0.0(0) | 158.1(2) | 5.6(1) | −0.1(0) |
| B143g | 173.9(0) | 5.8(0) | 0.0(0) | 174.9(0) | 5.1(0) | −0.1(0) |
| A10g | 182.3(0) | 5.4(0) | 0.0(0) | 181.8(1) | 6.6(0) | −0.2(0) |
| A14g | 283.3(0) | 5.1(0) | −0.1(0) | 289.9(4) | 5.0(1) | −0.1(0) |
| B183g | 292.7(0) | 3.1(0) | 0.0(0) | 295.5(1) | 4.2(0) | −0.1(0) |
| A15g | 320.0(1) | 2.4(0) | 0.0(0) | 299.6(1) | 5.3(0) | −0.1(0) |
| B193g | 334.4(0) | 3.0(0) | 0.0(0) | 325.6(1) | 4.1(0) | −0.1(0) |
| B192g | 339.1(0) | 3.2(0) | 0.0(0) | 352.4(0) | 5.2(0) | −0.1(0) |
| A18g | 379.2(0) | 5.6(0) | −0.1(0) | 394.7(0) | 7.0(0) | −0.2(0) |
| A19g | 380.0(0) | 6.1(0) | −0.1(0) | 400.8(0) | 7.8(0) | −0.2(0) |
Regarding the pressure dependence of the Raman-active modes, the modes above 250 cm−1 show pressure coefficients similar to those of CdGa2S4, HgGa2S4, and α′-Ga2S3 above 200 cm−1.51,62,65 In fact, the modes with the largest pressure coefficient are those close to 180 cm−1 in PbGa2S4, and at slightly larger frequencies between 200 and 250 cm−1 in the other three compounds. Similarly, modes below 150 cm−1 in PbGa2S4 show very small or even negative pressure coefficients in the same way as modes below 180 cm−1 in the other three compounds. Therefore, there is a clear correspondence between the vibrational modes in PbGa2S4 and in other thiogallates.
Above 16 GPa, new Raman modes were observed together with modes that seem to come from the original RP phase. The pressure dependence of the frequencies of all modes measured above 16 GPa are shown in Fig. 8 and also in more detail in Fig. S5 in the ESI.† Assuming that most of the Raman-active modes observed above 16 GPa correspond to monoclinic Pb6Ga10S21, we have calculated the vibrational modes of this compound at different pressure between 16 and 23 GPa. According to group theory,64 Pb6Ga10S21 should have 111 vibrational modes at Γ with the mechanical representation:
| Γ = 36Ag + 19Au + 18Bg + 38Bu |
The theoretical frequencies and pressure coefficients of Pb6Ga10S21 at 16 GPa are summarized in Table S6 and plotted in Fig. S5 (ESI†) for their comparison with experimental modes. As can be observed, there is no match between the experimental and theoretical modes since theoretical modes of Pb6Ga10S21 between 16 and 23 GPa are distributed in a smaller frequency range (between 40 and 410 cm−1) as compared to experimental modes (between 20 and 470 cm−1). The reason for the smaller high-frequency modes in Pb6Ga10S21 compared to PbGa2S4 is because Ga attains a sixfold coordination in the former compound while it has fourfold coordination in the latter. The larger coordination of Ga in Pb6Ga10S21 leads to larger Ga–S bond distances and smaller stretching frequencies. Note that a similar decrease of stretching frequencies occurs upon the transformation of α′-Ga2S3 to β′-Ga2S3.51,52 Therefore, we think that modes above 16 GPa are likely due to a mixture of modes of the original PbGa2S4 compound and of the new Pb6Ga10S21 compound.
3.3 Optical properties
HP-OA measurements showing OA spectra with unpolarized light perpendicular to the c-axis of the sample are plotted in Fig. 9 at selected pressures up to 23.9 GPa. At RP, an abrupt absorption edge near 2.9 eV is observed. The OA spectra and the value of the bandgap energy were obtained from the original transmittance spectra in the same way as that reported in ref. 23. We found that the fundamental absorption edge energy (2.85 eV) extrapolated from the (α × hν)1/2vs. hν curves to the abscissa (Tauc plot) at RP corresponds to the indirect bandgap energy. Both the energy and character of the fundamental absorption edge we have obtained agree with the data previously reported (2.84 eV) by Neumann et al. at RP.23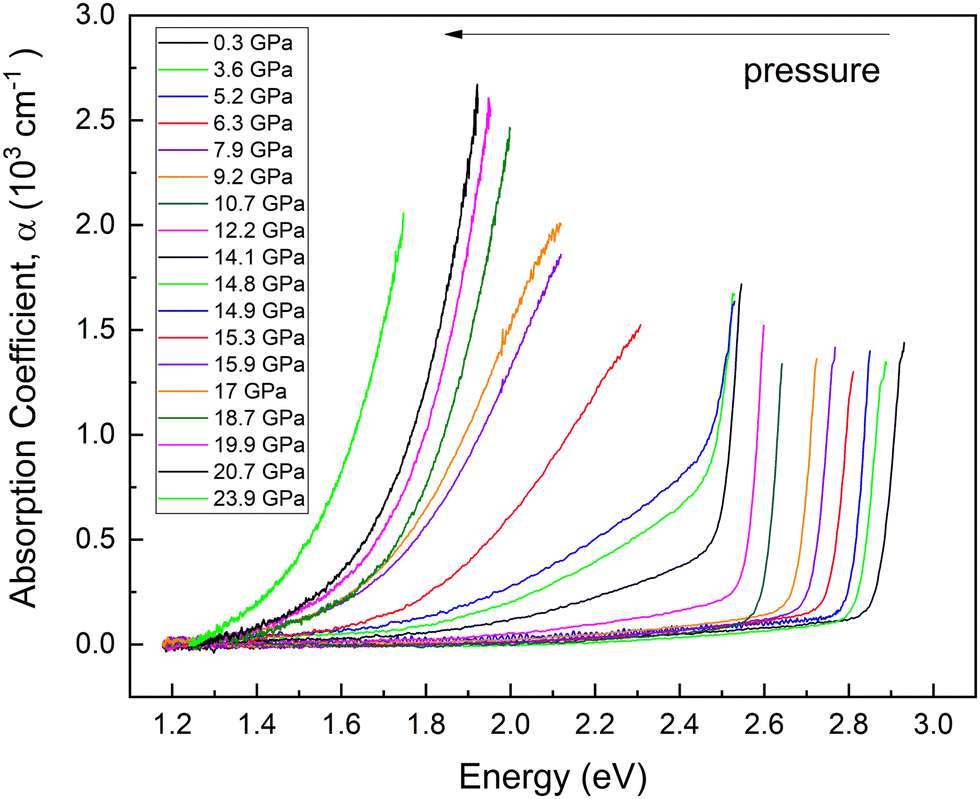 | ||
| Fig. 9 Optical absorption spectra measured at different pressures. Each pressure is shown in a different color. Pressures are indicated in the inset. | ||
As pressure increases the bandgap red shifts and develops a low-energy tail above 14 GPa. In addition, we observed an abrupt shift of the OA edge towards low energies when pressure is increased from 14.9 to 15.3 GPa. The changes in this pressure range are consistent with changes observed in HP-XRD and HP-RS measurements at similar pressures and can be attributed to the already commented pressure-induced decomposition of PbGa2S4. In fact, we consider that the appearance of the low-energy tail in the OA spectra above 14 GPa is likely due to the creation of defects in the crystal that favour the transmission of diffuse light and/or to the coexistence of the RP phase of PbGa2S4 and the HP phase corresponding to Pb6Ga10S21.
In Fig. 10 we report the pressure dependence of the bandgap energy obtained from two HP-OA measurements by fitting the high-energy part of the OA spectra with the Tauc plot.66 In both experiments, we observed a non-linear pressure dependence which is followed by a collapse of the indirect bandgap of nearly 0.55 eV at the pressure where PbGa2S4 undergoes the pressure-driven decomposition. In the pressure range of stability of PbGa2S4, we found that the bandgap first slightly opens and then closes under compression. This behavior agrees with the theoretical pressure dependence of the optical bandgap from our DFT calculations. In this context, our metaGGA calculations predict indirect and direct bandgap energies of 2.89 and 3.00 eV at RP for PbGa2S4; i.e. the calculated bandgap is in good agreement with experiments. In contrast, our PBEsol calculations underestimated both bandgaps by ca. 0.32 eV. Calculations also support that the optical bandgap of PbGa2S4 first increases and then decreases with increasing pressure following a qualitatively similar dependence than observed in experiments.
 | ||
| Fig. 10 Pressure dependence of the indirect band-gap energy in both compounds as measured from two experiments (shown with black and red dots). The solid line represents the results of calculations. At 15 GPa there is an abrupt decrease of the bandgap of 0.56 eV associated with the observed pressure-induced decomposition of PbGa2S4. | ||
Fig. 11 shows the electronic band structure and electronic density of states in PbGa2S4 at 0 and 15.3 GPa. At 0 GPa, both the valence and conduction band are not very dispersive. The bottom of the conduction band is at the C0/A0 point of the Brillouin zone and the top of the valence band is between the Γ and Σ0 points of the Brillouin zone. Therefore, our calculations confirm that PbGa2S4 is an indirect-gap semiconductor. This is not surprising because this compound crystallizes in a space group which is centrosymmetric. In centrosymmetric crystals, no p–d mixing takes place at the Γ point, but it does in less symmetrical points leading to upwards (downwards) dispersion in the valence (conduction) band when moving away from the Γ point. At 15.3 GPa, the situation is similar to that of 0 GPa (the points where there is the minimum of the conduction band and the maximum of the valence band are the same), but there is a much higher dispersion of the electronic bands.
 | ||
| Fig. 11 (a) Band-structure at 0 GPa. (b) Band-structure at 15.3 GPa. (c) Total and partial electronic density of states at 0 GPa (d) Total and partial electronic density of states at 15.3 GPa. In Fig. S6 in the ESI,† we zoom on the partial density of states for density values inferior to 10 states per eV. | ||
Regarding the electronic density of states at both 0 and 15.3 GPa, we can observe that the top of the valence band is basically contributed by S 3p states, which are slightly hybridized with Pb 6s states. On the other hand, the bottom of the conduction band is dominated by Pb 6p orbitals. This is the main difference between PbGa2S4 on one hand and CdGa2S4 and HgGa2S4 on the other hand. In the latter compounds, the states near the Fermi level in both the valence and conduction bands are dominated by Ga and S orbitals with no contribution from the divalent cations. Therefore, our calculations show that the interpretation of the assumed similarity of the electronic band structure of Cd and Pb thiogallates made by Neumann et al. is not correct.
The difference in orbital contribution to states near the Fermi level between the three thiogallates makes the bandgap of PbGa2S4 much smaller than that of the other two thiogallates.67 In CdGa2S4 and HgGa2S4, the increase of the crystal field under compression cause the bandgap to open,68,69 and the same occurs in α′-Ga2S3.51 In PbGa2S4, the contribution of Pb 6s states to the top of the valence band is enhanced when pressure increases. The same happens to the contribution of Ga 3s states to the bottom of the conduction band. Since the increase of the energy of the top of the valence band with pressure in PbGa2S4 is larger than the increase of the energy of the bottom of the conduction band due to the larger compressibility of the Pb–S bonds in PbS8 units than of the Ga–S bonds in GaS4 units, after an initial opening, both the indirect and direct bandgaps close as indicated in Fig. 11 and in agreement with our HP-OA measurements once the underestimation of the calculated indirect bandgap is corrected. In summary, the decrease of the bandgap of PbGa2S4 under compression is due to two effects: the delocalized character of Pb 6s states and the higher compressibility of dodecahedral units, PbS8, compared to tetrahedral units, GaS4. The same behavior has been previously observed in other lead compounds, such as PbWO4, PbMoO4, and PbCrO4.42,70–72
In Fig. 10 we have also shown that the indirect optical bandgap of Pb6Ga10S21 also decreases under compression. This behavior is also consistent with the theoretical pressure dependence of the indirect optical bandgap of this compound from our metaGGA calculations as plotted in Fig. 10. Our calculations show that Pb6Ga10S21 is also a semiconductor with an indirect bandgap of 0.74 eV followed by a direct bandgap at 0.95 eV at 18.2 GPa. This means that our calculations underestimate the value of the indirect bandgap by 0.8 eV. The mechanism exposed in the previous paragraph to explain the decrease of the bandgaps of PbGa2S4 with pressure is also valid to explain the decrease of the bandgaps of Pb6Ga10S21, as evidenced by the similarity between the theoretical electronic band structure and electronic density of states of Pb6Ga10S21 at 15.3 GPa (shown in Fig. S7 in the ESI†) and those already described for PbGa2S4.
4 Conclusions
We have reported the effect of pressure on the structural, vibrational, and optical properties of lead thiogallate by means of powder HP-XRD, HP-RS, and HP-OA measurements beyond 20 GPa. Those measurements have been complemented with ab initio calculations at HP. First of all, we have checked that PbGa2S4 crystallizes at room conditions in the EuGa2S4-type orthorhombic (space group Fddd) structure by means of single-crystal XRD measurements. Then, we have shown by means of powder HP-XRD measurements that PbGa2S4 is an anisotropic material, as expected from its layered-like structure. Its axial and bulk moduli are of the same order as those of monoclinic α′-Ga2S3 and tetragonal CdGa2S4 and HgGa2S4; i.e. semiconductors with similar GaS4 tetrahedra.After checking the complex vibrational pattern of PbGa2S4 by means of polarized RS measurements at room conditions, we have shown that the vibrational modes of PbGa2S4 show phonon spectra that show a considerable similarity to those of α′-Ga2S3, CdGa2S4, and HgGa2S4. In fact, we have shown that the pressure dependence of the Raman-active modes in PbGa2S4 is similar to those of the mentioned semiconductors and have made a tentative assignment of the symmetry of the Raman-active modes experimentally observed.
Finally, we have measured the pressure dependence of the optical bandgap of PbGa2S4 by means of HP-OA measurements. We have confirmed that PbGa2S4 is an indirect bandgap semiconductor, whose bandgap decreases as pressure increases, unlike what happens in α′-Ga2S3, CdGa2S4, and HgGa2S4. The different behavior is explained by the contribution of the 6s lone electron pair of Pb to the topmost valence band and the strong decrease of the Pb–S bond distance upon compression that leads to a strong increase of the energy of the topmost valence band under pressure.
To finish, we want to stress that all our measurements have shown evidence of a partially reversible pressure-induced decomposition of PbGa2S4 into a mixture of Pb6Ga10S21 and β′-Ga2S3 above 16 GPa. This decomposition is supported by enthalpy vs. pressure calculations of the three compounds and makes sense because both compounds show sixfold-coordinated Ga atoms in comparison with the fourfold-coordinated Ga atoms in PbGa2S4. The structure of the new compound Pb6Ga10S21 at HP, which is isostructural to already known Pb6In10S21 at RP, is reported at 23.5 GPa since it seems not to be stable at RP. Moreover, we have determined its Raman-active phonons and optical bandgap above 16 GPa. In summary, this work shows the first HP study of a compound with EuGa2S4-type (orthorhombic Fddd) structure and the route for the synthesis of Pb6Ga10S21. Therefore, this work will be of interest for the study of the EuGa2S4-type subfamily of AIIBIII2XVI4 compounds, in which the effect of pressure is far from being understood.
Author contributions
Tania Garcia-Sanchez: investigation, formal analysis, discussion, writing, review and editing Samuel Gallego-Parra: investigation, formal analysis Akun Liang: investigation, formal analysis José Luis Rodrigo-Ramon: investigation, formal analysis Alfonso Muñoz: investigation, formal analysis Plácida Rodriguez-Hernandez: investigation, formal analysis Javier Gonzalez-Platas: investigation, formal analysis Juan Ángel Sans: investigation Vanesa Paula Cuenca-Gotor: investigation Hussien H. Osman: investigation Catalin Popescu: investigation Veaceslav Ursaki: investigation Ion M. Tiginyanu: investigation Daniel Errandonea: formal analysis, validation, funding acquisition, writing, review, and editing Francisco Javier Manjón: conceptualization, investigation, formal analysis, validation, writing, review, and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study forms part of the Advanced Materials programme and was supported by MCIN with funding from European Union Next Generation EU (PRTR-C17.I1) and by Generalitat Valenciana through projects MFA/2022/024 (ARCANGEL), PROMETEO CIPROM/2021/075 (GREENMAT) and MFA/2022/007 (MATGREEN) and from the Spanish Ministerio de Ciencia e Innovación and Agencia Estatal de Investigación (MCIN/AEI/10.13039/501100011033) under grant No. PID2019-106383GB-41/42/43, PID2021-125927NB-C21, and RED2022-134388-T (MALTA-Consolider Team network). T.G.-S. thanks Universitat Politecnica de Valencia for the support through the program “Ayudas para la recualificacion del profesorado universitario”, financial support provided by Ministerio de Universidades, funding from the European Union-Next generation EU. T. G.-S. and V. P. C.-G. thanks Primeros proyectos de investigación 2022 (PAID-06-22), en el marco de ayudas del Vicerrectorado de Investigación de la Universitat Politècnica de València. The authors also thank the ALBA synchrotron light source for providing beamtime under proposal number 2021085226. H. H. O. and S. G. P. acknowledge PRACE for awarding access to the Fenix Infrastructure resources at CINECA, which are partially funded from the European Union's Horizon 2020 research and innovation programme through the ICEI project under the grant agreement No. 800858. We also acknowledge the computer resources of the Centro de Supercomputación de Castilla y León (SCAYLE)”.Notes and references
- A. N. Georgobiani, B. G. Tagiev, O. B. Tagiev, R. B. Djabbarov, N. N. Musaeva and U. F. Kasumov, Jpn. J. Appl. Phys., 2000, 39, 434 CrossRef CAS.
- T. Peters and J. Baglio, J. Electrochem. Soc., 1972, 119, 230 CrossRef CAS.
- A. A. Kaminskii, Laser Photonics Rev., 2007, 1, 93–177 CrossRef CAS.
- V. Shiryaev and M. Churbanov, J. Non-Cryst. Solids, 2017, 475, 1–9 CrossRef CAS.
- S. B. Mirov, I. S. Moskalev, S. Vasilyev, V. Smolski, V. V. Fedorov, D. Martyshkin, J. Peppers, M. Mirov, A. Dergachev and V. Gapontsev, IEEE J. Sel. Top. Quantum Electron., 2018, 24, 1–29 Search PubMed.
- T. Basiev, M. Doroshenko, V. Osiko and D. Badikov, Adv. photonics, 2005, 75 CAS.
- M. E. Doroshenko, T. T. Basiev, V. V. Osiko, V. V. Badikov, D. V. Badikov, H. Jelnková, P. Koranda and J. Šulc, Opt. Lett., 2009, 34, 590–592 CrossRef CAS PubMed.
- J. Šulc, H. Jelnková, M. E. Doroshenko, T. T. Basiev, V. V. Osiko, V. V. Badikov and D. V. Badikov, Opt. Lett., 2010, 35, 3051–3053 CrossRef PubMed.
- H. Jelnková, M. Doroshenko, M. Jelnek, J. Šulc, T. Basiev, V. Osiko, V. Badikov and D. Badikov, Laser Phys. Lett., 2011, 8, 349 CrossRef.
- H. Jelnková, M. E. Doroshenko, M. Jelnek, J. Šulc, V. V. Osiko, V. V. Badikov and D. V. Badikov, Opt. Lett., 2013, 38, 3040–3043 CrossRef PubMed.
- X. Yu, C. Huang, Y. Ni, Z. Wang and H. Wu, CrystEngComm, 2022, 24, 5149–5155 RSC.
- C. Huang, Y. Ni, H. Wu, Z. Wang, P. Jiang and W. Han, Cryst. Growth Des., 2020, 20, 845–850 CrossRef CAS.
- A. Chilouet, A. Mazurier and M. Guittard, Mater. Res. Bull., 1979, 14, 1119–1124 CrossRef CAS.
- K. Wu, S. Pan, H. Wu and Z. Yang, J. Mol. Struct., 2015, 1082, 174–179 CrossRef CAS.
- D. Bletskan, Y. V. Voroshilov, L. Durdinets and V. Kabacij, Sci. Her. Uzhhorod Univ. Ser. Phys., 1999, 4, 168–176 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- N. Syrbu, V. Lvin, B. Zadnipui, M. Golovei and F. T. Poluprov, Phys. Tech. Semicond, 1991, 25, 1721–1730 CAS.
- H. Neumann, H. Sobotta, N. Syrbu and V. Golovei, Cryst. Res. Technol., 1994, 29, 289–296 CrossRef CAS.
- N. Syrbu and V. Cebotari, J. Phys.: Condens. Matter, 1998, 10, 3467 CrossRef CAS.
- V. Zalamai, N. Syrbu, N. Bejan and I. Hirjeu, Mater. Sci., 2016, 8, 114 Search PubMed.
- V. Kamenshchikov, V. Stefanovich, Z. Gadmashi, V. Side and L. Suslikov, Phys. Solid State, 2007, 49, 351–355 CrossRef CAS.
- V. Golovey, L. Ivanchenko, A. Knyazev, V. Obolonchik and E. Troyan, Ukr. fiz. ž., 1981, 26, 1037–1039 Search PubMed.
- H. Neumann, W. Hörig, G. Nooke and N. Syrbu, Solid State Commun., 1988, 65, 155–157 CrossRef CAS.
- V. Badikov, D. Badikov, M. Doroshenko, V. Panyutin, V. Chizhikov and G. Shevyrdyaeva, Opt. Mater., 2008, 31, 184–188 CrossRef CAS.
- N. Syrbu, V. Parvan and V. Ursaki, Opt. Mater., 2012, 34, 691–695 CrossRef CAS.
- I. Stamov, N. Syrbu, V. Ursaki, V. Parvan and V. Zalamai, Opt. Commun., 2012, 285, 5198–5204 CrossRef CAS.
- W.-F. Chen, B.-W. Liu, X.-M. Jiang and G.-C. Guo, J. Alloys Compd., 2022, 905, 164090 CrossRef CAS.
- N. Syrbu and V. Zalamai.
- N. Musaeva, R. Dzhabbarov, U. Kasumov and K. B. Ganbarova, J. Opt. Technol., 2003, 70, 676–679 CrossRef CAS.
- F. J. Manjon, I. Tiginyanu and V. Ursaki, Pressure-induced phase transitions in AB2X4 chalcogenide compounds, Springer, 2014 Search PubMed.
- L. Isaenko, A. Yelisseyev, A. Tkachuk and S. Ivanova, NATO Science for Peace and Security Series B: Physics and Biophysics, 2008, 3–63 Search PubMed.
- Y. V. Orlovskii, T. T. Basiev, K. K. Pukhov, M. E. Doroshenko, V. V. Badikov, D. V. Badikov, O. K. Alimov, M. V. Polyachenkova, L. N. Dmitruk and V. V. Osiko, et al. , Opt. Mater., 2007, 29, 1115–1128 CrossRef CAS.
- D. Errandonea, Phys. Status Solidi B, 2017, 254, 1700016 CrossRef.
- H. Mao, J.-A. Xu and P. Bell, J. Geophys. Res. Solid Earth, 1986, 91, 4673–4676 CrossRef CAS.
- A. Dewaele, P. Loubeyre and M. Mezouar, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 094112 CrossRef.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS.
- C. Prescher and V. B. Prakapenka, High Press. Res., 2015, 35, 223–230 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- R. Letoullec, J. Pinceaux and P. Loubeyre, High Press. Res., 1988, 1, 77–90 CrossRef.
- O. Gomis, R. Vilaplana, F. J. Manjón, J. Ruiz-Fuertes, E. Pérez-González, J. López-Solano, E. Bandiello, D. Errandonea, A. Segura and P. Rodrguez-Hernández, et al. , Phys. Status Solidi B, 2015, 252, 2043–2051 CrossRef CAS.
- D. Errandonea, C. Popescu, A. B. Garg, P. Botella, D. Martinez-García, J. Pellicer-Porres, P. Rodríguez-Hernández, A. Muñoz, V. Cuenca-Gotor and J. A. Sans, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 035204 CrossRef.
- D. Errandonea, D. Martinez-Garcia, R. Lacomba-Perales, J. Ruiz-Fuertes and A. Segura, Appl. Phys. Lett., 2006, 89, 091913 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. Mujica, A. Rubio, A. Munoz and R. Needs, Rev. Mod. Phys., 2003, 75, 863 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2006, 124, 221101 CrossRef PubMed.
- D. Errandonea, R. S. Kumar, O. Gomis, F. J. Manjón, V. V. Ursaki and I. M. Tiginyanu, J. Appl. Phys., 2013, 114, 233507 CrossRef.
- V. Krämer and K. Berroth, Mater. Res. Bull., 1980, 15, 299–308 CrossRef.
- S. Gallego-Parra, R. Vilaplana, O. Gomis, E. L. da Silva, A. Otero-de-la Roza, P. Rodrguez-Hernández, A. Muñoz, J. González, J. Sans and V. P. Cuenca-Gotor, et al. , Phys. Chem. Chem. Phys., 2021, 23, 6841–6862 RSC.
- S. Gallego-Parra, R. Vilaplana, O. Gomis, P. Rodríguez-Hernández, A. Munoz, J. A. González, J. A. Sans, C. Popescu and F. J. Manjón, Chem. Mater., 2022, 34, 6068–6086 CrossRef CAS.
- Q. Jiang, R. Li, F. Wang, X. Shi, F. Chen, Y. Huang, B. Wang, W. Zhang, X. Wu and F. Wei, et al. , Adv. Mater., 2022, 34, 2107062 CrossRef CAS PubMed.
- M. Dunuwille, M. Kim and C.-S. Yoo, J. Chem. Phys., 2016, 145, 084701 CrossRef PubMed.
- D. Errandonea, R. S. Kumar, F. J. Manjón, V. Ursaki and I. Tiginyanu, J. Appl. Phys., 2008, 104, 063524 CrossRef.
- O. Gomis, D. Santamaria-Perez, R. Vilaplana, R. Luna, J. Sans, F. J. Manjón, D. Errandonea, E. Perez-Gonzalez, P. Rodrguez-Hernández and A. Muñoz, et al. , J. Alloys Compd., 2014, 583, 70–78 CrossRef CAS.
- F. Birch, Phys. Rev., 1947, 71, 809 CrossRef CAS.
- R. J. Angel, M. Alvaro and J. Gonzalez-Platas, Z. Kristallogr. - Cryst. Mater., 2014, 229, 405–419 CrossRef CAS.
- R. Angel, J. Mosenfelder and C. Shaw, Phys. Earth Planet. Inter., 2001, 124, 71–79 CrossRef CAS.
- O. Gomis, R. Vilaplana, F. J. Manjón, D. Santamaria-Perez, D. Errandonea, E. Perez-Gonzalez, J. Lopez-Solano, P. Rodriguez-Hernandez, A. Munoz and I. M. Tiginyanu, et al. , Mater. Res. Bull., 2013, 48, 2128–2133 CrossRef CAS.
- D. Errandonea, A. Muñoz and J. Gonzalez-Platas, J. Appl. Phys., 2014, 115, 216101 CrossRef.
- S. Gallego-Parra, O. Gomis, R. Vilaplana, H. M. Ortiz, E. Perez-Gonzalez, R. Luna, P. Rodrguez-Hernández, A. Muñoz, V. Ursaki and I. Tiginyanu, et al. , J. Appl. Phys., 2019, 125, 115901 CrossRef.
- M. Fuentes-Cabrera, J. Phys.: Condens. Matter, 2001, 13, 10117 CrossRef CAS.
- E. Kroumova, M. Aroyo, J. Perez-Mato, A. Kirov, C. Capillas, S. Ivantchev and H. Wondratschek, Phase Transitions, 2003, 76, 155–170 CrossRef CAS.
- R. Vilaplana, M. Robledillo, O. Gomis, J. Sans, F. J. Manjón, E. Perez-Gonzalez, P. Rodrguez-Hernández, A. Muñoz, I. Tiginyanu and V. Ursaki, J. Appl. Phys., 2013, 113, 093512 CrossRef.
- A. B. Garg, D. Vie, P. Rodriguez-Hernandez, A. Muñoz, A. Segura and D. Errandonea, J. Phys. Chem. Lett., 2023, 14, 1762–1768 CrossRef CAS PubMed.
- A. Liang, F. Rodríguez, P. Rodríguez-Hernandez, A. Muñoz, R. Turnbull and D. Errandonea, Phys. Rev. B: Condens. Matter Mater. Phys., 2022, 105, 115204 CrossRef CAS.
- A. Liang, L. Shi, S. Gallego-Parra, O. Gomis, D. Errandonea, I. Tiginyanu, V. Ursaki and F. Manjón, J. Alloys Compd., 2021, 886, 161226 CrossRef CAS.
- F. J. Manjón, O. Gomis, P. Rodrguez-Hernández, E. Perez-Gonzalez, A. Muñoz, D. Errandonea, J. Ruiz-Fuertes, A. Segura, M. Fuentes-Cabrera and I. Tiginyanu, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 195201 CrossRef.
- D. Errandonea, E. Bandiello, A. Segura, J. Hamlin, M. Maple, P. Rodriguez-Hernandez and A. Muñoz, J. Alloys Compd., 2014, 587, 14–20 CrossRef CAS.
- V. Monteseguro, J. Ruiz-Fuertes, J. Contreras-Garca, P. Rodrguez-Hernández, A. Muñoz and D. Errandonea, Appl. Phys. Lett., 2019, 115, 012102 CrossRef.
- E. Bandiello, D. Errandonea, D. Martinez-Garcia, D. Santamaria-Perez and F. J. Manjón, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 024108 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02288a |
| ‡ Present address: European Synchrotron Radiation Facility, 71 Avenue des Martyrs, 38000 Grenoble, France. |
| § Present address: Centre for Science at Extreme Conditions and School of Physics and Astronomy, University of Edinburgh, Edinburgh EH9 3FD, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2023 |