DOI:
10.1039/D3TC02247A
(Paper)
J. Mater. Chem. C, 2023,
11, 12007-12015
Influence of heavy magnesium codoping on emission decay in Ce-doped multicomponent garnet scintillators
Received
27th June 2023
, Accepted 1st August 2023
First published on 1st August 2023
Abstract
This study is aimed at the acceleration of luminescence decay in Ce-doped garnet-type scintillators by heavy aliovalent codoping. Time-resolved photoluminescence spectroscopy over a wide temperature range of LuGAGG:Ce epitaxial layers with up to 6000 ppm of codoping magnesium, and a transient absorption technique probing the population of the emitting Ce centers and energies of optical transitions at Ce3+ ions in the layers, enabled the interpretation of luminescence decay. Heavy Mg-codoping facilitated the luminescence decay acceleration due to the coexistence of two types of emission centers: regular ions of Ce3+; and Ce ions with magnesium in the immediate vicinity, i.e., Ce3+ + Mg2+ centers. In the latter centers, the activation energy for thermal quenching is 0.2 eV, instead of 0.48 eV in regular Ce3+ centers; and a new channel of temperature-independent nonradiative recombination occurs, pushing the low-temperature luminescence decay time from 50 ns – due to radiative recombination at regular Ce3+ centers – down to 10.5 ns. The magnesium content does not change the properties of the emission centers but increases the contribution of the Mg-related centers. Time-resolved cathodoluminescence spectroscopy of the layers evidences a broad applicability of the suggested model for heavy aliovalently codoped Ce-doped garnets beyond the LuGAGG:Ce thin films prospective for X-ray imaging.
1. Introduction
Future applications of scintillation detectors in the two major fields of medical imaging and high energy physics experiments, require substantial improvement in the timing properties of scintillating materials. Ce-doped oxyorthosilicates and garnets are currently among the most promising fast scintillators.1 The Ce-doped LYSO (Lu2(1−x)Y2xSiO5) scintillator is exploited in most modern commercial positron emission tomography (PET) devices and is the scintillator of choice in the barrel timing layer (BTL) for the coming upgrade of Compact Muon Solenoid (CMS) experiments at CERN.2 Garnet-type single crystals, including multicomponent garnets enabling electronic band structure engineering, also serve well as matrixes for Ce-activated scintillators.3–5 In particular, Gd3(Ga,Al)5O12:Ce (GAGG:Ce) will be used in the coming upgrade of the Large Hadron Collider beauty (LHCb) experiments at CERN,6 and is considered for extensive exploitation in other applications.4 The scintillation response time of both types of scintillators might be improved by codoping trivalent Ce3+ ions with divalent ions, usually Ca2+ or Mg2+. Aliovalent codoping has long been exploited to facilitate appropriate crystal growth and to improve optical and luminescence properties of various scintillators.7 A decrease in the emission decay time is observed and interpreted by different emission decay times (43 and 31 ns) for Ce3+ at two lattice sites: one accommodating Ce ions, and one with prevailing Ca2+ dopant occupation, in Ca-codoped lutetium oxyorthosilicate (LSO:Ce).8 X-Ray absorption near edge spectroscopy (XANES) has allowed us to understand the link between the improvement in timing properties of lutetium yttrium oxyorthosilicate (LYSO:Ce) with stabilization of the Ce ions in a Ce4+ oxidation state in the co-doped crystals.9 This stabilization influences the efficiency and increased time of luminescence response, which becomes increasingly important for timing properties when the achieved time resolution is improved down to 100 ps or less. Substantial shortening of the scintillation rise time by aliovalent codoping has been explicitly demonstrated, and is interpreted as due to the Ca-assisted suppression of the influence of traps on the excitation transfer, resulting in the improved coincidence time resolution (CTR),10 an important parameter in the detection of γ-radiation. A beneficial effect of aliovalent codoping for scintillation properties including response time, has also been demonstrated in the Mg-codoped garnet-type single crystal scintillator Lu3Al3O12:Ce, interpreted as again being due to the stabilization of Ce4+.3 The capability of Ce4+ for efficient capturing of electrons to become an excited ion Ce3+* with subsequent photon emission, is beneficial for competing with the capture of electrons in trapping levels routing to the centers of nonradiative recombination. Magnesium-codoping has also been shown to suppress the influence of trapped electrons on the increased time of luminescence response11 and to improve the CTR in GAGG:Ce,Mg.12 The capability of Mg-doping to substantially decrease the decay time of luminescence was studied in GAGG:Ce,Mg single crystal samples with heavy codoping up to Ce and Mg concentrations of approx. 1 at% and 0.5 at%, respectively.13 The luminescence decay time measured after short-pulse X-ray excitation, 511 keV gamma photons and resonant photoexcitation of Ce3+ ions, was found to substantially decrease down to a few nanoseconds, whereas the CTR was constant due to the significant decrease in the scintillation yield, interpreted as due to the formation of closely spaced Ce–Mg pairs accelerating the luminescence decay but also decreasing the emission efficiency.13–15
The current study aims to reveal the properties of recombination centers in aliovalently codoped Ce-activated garnet-type scintillators and the competition of radiative and nonradiative recombination, in order to purposefully exploit the aliovalent codoping to accelerate the response time of Ce-doped scintillators. Epitaxial films of Ce-doped multicomponent lutetium-gadolinium-gallium-aluminum garnets (LuGAGG:Ce,Mg) codoped by magnesium at different levels, are studied in this work. The films were fabricated using liquid phase epitaxy. A higher Ce segregation coefficient in the flux enables congruent incorporation of up to 1 at% of Ce, i.e., by a factor of 8 more than the highest content reported in Czochralski-grown bulk single crystals; where a high concentration of codopant ions without substantial deterioration of the crystalline lattice is also anticipated.16,17 BaO–B2O3–BaF2 flux ensures a higher light yield from the layer, though results in the formation of a high density of macrostructural defects due to the higher viscosity of this flux.16,18,19 In the current study, the possible influence of the structural microdefects is addressed using time-resolved cathodoluminescence spectroscopy. To reveal the dynamics of nonequilibrium carriers, time-resolved photoluminescence spectroscopy was applied and supported by measurements of transient absorption in a pump and probe configuration. Coexistance of two types of centers of radiative recombination were revealed, and the channels of radiative and nonradiative recombination were characterized.
2. Experimental
2.1 Luminescence kinetics
Epitaxial layers of LuGAGG:Ce,Mg with Ce3+ have the nominal composition, Lu0.8Gd2.2Ga2.5Al2.5O12:Ce,Mg, with a constant Ce content of 1% and varying Mg content from 0 to 6000 ppm, which were grown by isothermal dipping liquid phase epitaxy on the (100)-oriented Czochralski-grown Gd3Ga2.7Al2.3O12 substrate using BaO–B2O3–BaF2 flux at the Technology Laboratory at the Charles University, Prague, Czechia. Both Ce and Mg concentrations are given relative to the number of ions in the dodecahedral sites to mutually compare their concentrations. The composition of all the samples studied were determined using electron probe microanalysis. The Mg content was determined using glow discharge mass spectrometry (GDMS) in one sample and was calculated in other samples from the melt composition and using the segregation coefficient kMg ≈ 0.06. The content of Ba2+ impurities coming from the flux was determined by GDMS, to be less than 20 ppm. Since all the samples were grown under the same conditions and from the same melt, except for the Mg content, the concentrations of Ce, unintentional impurities, or structural defects are virtually the same, enabling a direct comparison of their properties. The thickness of the layers was ∼16 μm. The Mg content in the samples under this study are indicated together with sample labeling in Fig. 1, where the absorption spectra of the samples are presented. Though all the samples nominally contain the same content of Ce ions, the absorption bands due to optical transitions 4f–5d1 peaked at 2.84 eV (438 nm) and 4f–5d2 peaked at 3.65 eV (340 nm), becoming weaker with increasing Mg content. Such a change of absorption spectrum in LYSO:Ce,Mg is attributed to transforming Ce3+ ions into Ce4+ ions by introduction of divalent Mg2+ ions9 and is observed in many codoped garnets including LuAG:Ce,Mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and LuGAGG:Ce,Mg.21 This transformation is accompanied by increasingly broad UV absorption band, probably related to absorption at certain Mg-related centers. Sample S6 with 6000 ppm of magnesium exhibits extremely weak Ce3+-related absorption and emission, probably due to the substantial deterioration of the matrix structure as the Mg content is high, and serves as a reference of the limit of Mg-codoping capability. Sample S1 served as the opposite limiting reference, containing no magnesium. The sharp lines in the absorption spectra are caused by absorption at matrix building Gd ions. Photoluminescence (PL) spectra were analyzed using a Andor Kymera 193i spectrometer and recorded using a Hamamatsu back-thinned charge-coupled device (CCD). The PL decay kinetics were measured with a time resolution of 200 ps ensured using the time correlated single photon counting technique exploited using the Becker & Hickl module SPC-130 coupled with a cooled photomultiplier tube, PMC-150. A Yb:KGW laser (Pharos, Light Conversion) delivering 250 fs pulses at 1030 nm was exploited as an excitation source. To resonantly excite Ce3+ ions to their first excited level 5d1 at 2.84 eV, the laser photon energy was gradually tuned using an optical parametric amplifier (Orpheus, Light Conversion) and stepwise increased using a harmonics generator. The measurements were performed at fixed temperatures in the range 80 to 600 K using a liquid-nitrogen-filled cryosystem from Cryo Industries.
20 and LuGAGG:Ce,Mg.21 This transformation is accompanied by increasingly broad UV absorption band, probably related to absorption at certain Mg-related centers. Sample S6 with 6000 ppm of magnesium exhibits extremely weak Ce3+-related absorption and emission, probably due to the substantial deterioration of the matrix structure as the Mg content is high, and serves as a reference of the limit of Mg-codoping capability. Sample S1 served as the opposite limiting reference, containing no magnesium. The sharp lines in the absorption spectra are caused by absorption at matrix building Gd ions. Photoluminescence (PL) spectra were analyzed using a Andor Kymera 193i spectrometer and recorded using a Hamamatsu back-thinned charge-coupled device (CCD). The PL decay kinetics were measured with a time resolution of 200 ps ensured using the time correlated single photon counting technique exploited using the Becker & Hickl module SPC-130 coupled with a cooled photomultiplier tube, PMC-150. A Yb:KGW laser (Pharos, Light Conversion) delivering 250 fs pulses at 1030 nm was exploited as an excitation source. To resonantly excite Ce3+ ions to their first excited level 5d1 at 2.84 eV, the laser photon energy was gradually tuned using an optical parametric amplifier (Orpheus, Light Conversion) and stepwise increased using a harmonics generator. The measurements were performed at fixed temperatures in the range 80 to 600 K using a liquid-nitrogen-filled cryosystem from Cryo Industries.
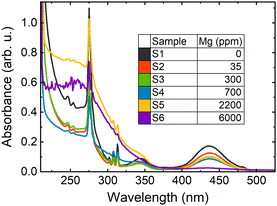 |
| | Fig. 1 Optical absorption spectra of studied (LuGd)3(GaAl)5O12:Ce,Mg layers with different Mg content, with the sample labeling indicated in the inset. | |
The population of nonequilibrium carriers was also studied using the transient absorption (TA) technique in a pump and probe configuration. In the TA experiments, the output beam of the Yb:KGW laser was split into pump and probe beams. The optical parametric amplifier and a harmonics generator were used to tune the pump photon energy for resonant excitation of Ce3+, whereas the probe beam was converted in a sapphire plate to a white light supercontinuum to simultaneously probe the transient absorption in the spectral range from 1.3 to 2.7 eV (950–460 nm). The probe beam was led through an optomechanical delay stage to tunably change the delay between pump and probe pulses from 0 to 10 ns with subpicosecond precision limited just by the laser pulse duration (see ref. 12 for more details). Secondary electron (SE) and cathodoluminescence (CL) imaging was performed at room temperature using a hybrid cathodoluminescence-scanning electron microscope (Chronos, Attolight) operated at an acceleration voltage of 10 kV in a continuous wave or a pulsed mode with a pulse width of 20 ps and a repetition rate of 80.7 MHz. The emitted light was collected by a Cassegrain-type objective and then dispersed using a Horiba iHR spectrometer. The spectra of time-integrated CL intensity were recorded using an Andor Newton UV-VIS CCD camera, whereas the time evolution of spectrally-integrated CL intensity was measured using a Hamamatsu streak camera.
3. Results
3.1 Structure of the layers
As seen in Fig. 2, the films have nearly flat, smooth platforms decorated with square-shaped pits. In all the samples studied, the pits have sides in the order of tens of nanometers, and are surrounded by a round-shaped indentation. The average length of the square sides is different in different areas of the same layer and in different samples, however, they show no evident dependence on the level of Mg-doping. The intensity of the light emitted from the pits is up to two times as large as the intensity from the flat sample surface. Meanwhile, the spectra selectively recorded from the pits and the flat areas are identical (see inset in Fig. 2c) and the corresponding emission intensities decay at the same rate (see Fig. 2c) within the entire spectral range of the emission. These observations are an indication that the enhanced light emission from the pits is caused by increased light extraction.
 |
| | Fig. 2 Sample S6 with Mg content of 6000 ppm: spatial distribution of panchromatic cathodoluminescence intensity presented in color (a) and secondary electron images in greyscale (b) of the same areas at increasing magnification (from left to right); (c) time evolution of normalized spectrally-integrated cathodoluminescence intensity and, in the inset, normalized time-integrated CL spectra collected from flat areas (violet line) and pits (orange dots). | |
The PL spectra of the Mg-free sample (S1) and heavily Mg-codoped sample (S5) are presented at several temperatures in the range 90 to 600 K, in Fig. 3. The spectra are dominated by a broad asymmetric band that is typical of Ce3+ emission due to optical transitions from level 5d1 to two ground levels 2F7/2 and 2F5/2. The origin of the broad long-wavelength band peaking at 1.65 eV has no unambiguous interpretation. This band is substantially more pronounced at 2.84 eV excitation, thus, the corresponding emission center in the layer or substrate is probably excited resonantly. The temperature dependence of the PL intensity spectrally-integrated within the Ce-emission band is presented as an Arrhenius plot in Fig. 5 for samples with different Mg content. The lines in Fig. 5a are the best fits of the experimental points with the Arrhenius expression:
| |  | (1) |
Here,
VlowT is the low-temperature PL intensity,
A is a fitting constant,
T is the temperature, and
Ea is the activation energy.
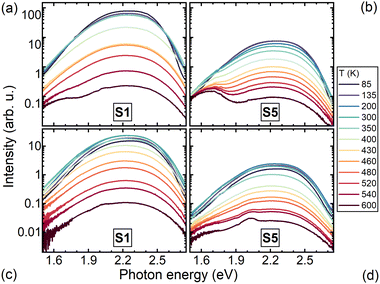 |
| | Fig. 3 Photoluminescence spectra of Mg-free (S1) and heavily Mg-codoped (S5) LuGAGG:Ce samples at excitation to the first (4f–5d1), (a) and (b), and the second (4f–5d2), (c) and (d), excited states of Ce3+ ion. | |
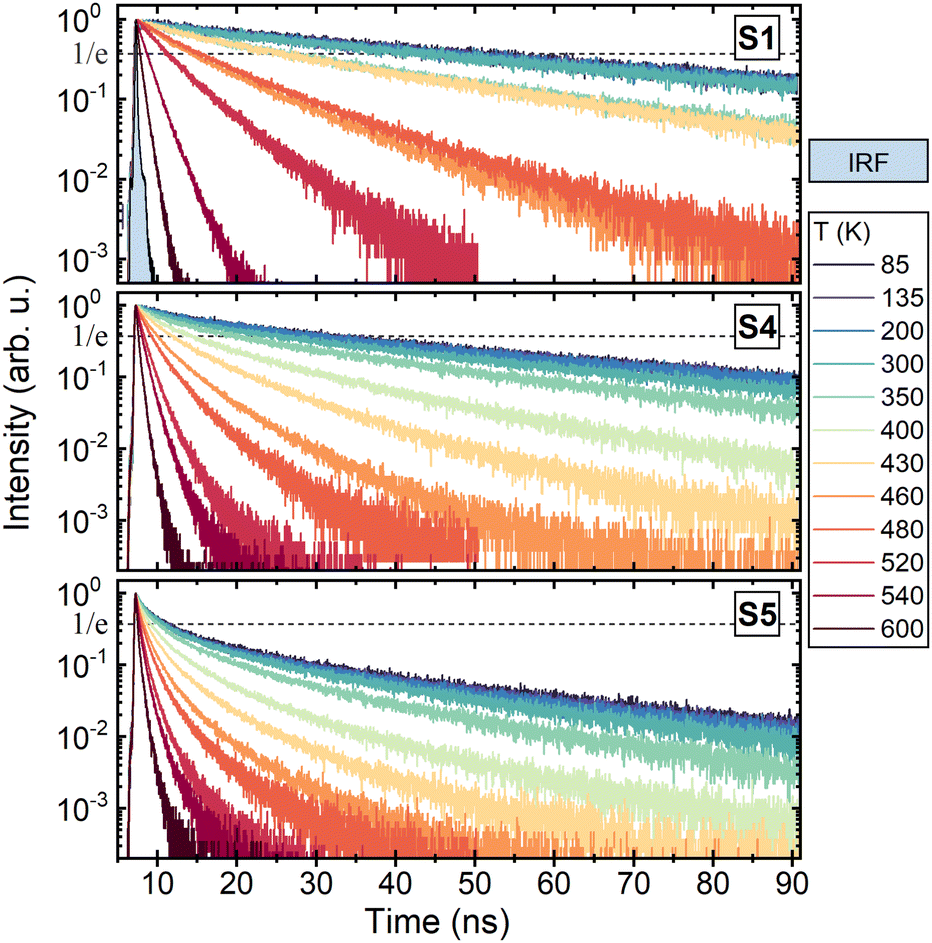
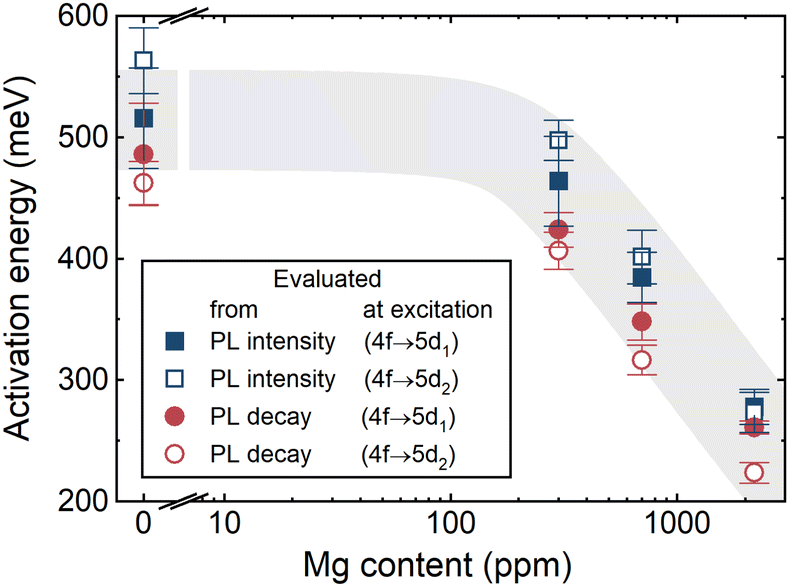
The temperature dependence of PL kinetics is shown for Mg-free (S1), moderately (S4) and heavily Mg codoped (S5) samples in Fig. 4. In the Mg-free sample, the decay is close to single-exponential decay at all temperatures in the range 90–600 K. Magnesium codoping results in an increasing contribution of a fast component. The temperature dependence of the time period of the decay to the level of 1/e is presented as an Arrhenius plot in Fig. 5b for samples with different Mg content. The lines in Fig. 5b are the best fits of the experimental points with the Arrhenius eqn (1). The activation energies extracted from the Arrhenius plots for the temperature dependences of PL intensity and decay time are presented in Fig. 5(a) and (b), respectively. The values of Ea evaluated using both measurements, coincide within the experimental error, and evidence a substantial decrease of Ea with increasing Mg content. The activation energies as a function of Mg content are plotted in Fig. 6. Spreading slightly outside the determination error, the activation energies for each sample are similar when extracted from the temperature dependences of PL intensity or decay time and at the excitation to the first or second excited level of Ce3+. The value of Ea decreases slightly as Mg content increases to hundreds of ppm and drops sharply with a further increase in Mg content.
 |
| | Fig. 4 Normalized photoluminescence decay kinetics at different temperatures in Mg-free (S1), moderately (S4) and heavily Mg codoped (S5) LuGAGG:Ce at excitation to the first (4f–5d1) excited state of Ce3+. Blue-filled black curve presents the instrumental function. | |
 |
| | Fig. 5 Arrhenius plots of photoluminescence intensity (a) and decay time of fast component (b), in LuGAGG:Ce,Mg with different Mg content at excitation to the first (5d1) excited state of Ce3+. The Mg content and activation energies extracted by fitting the curves using eqn (1) are indicated. | |
 |
| | Fig. 6 Dependence of activation energies for photoluminescence intensity quenching and shortening of decay time of fast component at excitation to the first (5d1) and the second (5d2) excited states of Ce3+ ion on Mg content in LuGAGG:Ce,Mg. The grey field indicates the observed tendency. | |
3.2 Study of optical transitions in Ce3+ activator ions
The dynamics of the emitting Ce3+ 5d1 level population was investigated at resonant excitation conditions using the transient absorption technique. The transient absorption after short-pulse (250 fs) resonant excitation of Ce3+ ions as a function of the probe photon energy and the delay between pump and probe pulses at room temperature are presented for Mg-free (S1) and Mg-codoped at 2200 ppm (S5) LuGAGG:Ce epitaxial layers in Fig. 7a and b, respectively. A transient absorption band peaked at 1.6 eV for all the samples and no significant shift of the band after incorporation of Mg was observed (see Fig. 7c). The position of the TA band peak corresponds to the energy distance between the lower 5d1 and triplet t2g level observed in other scintillators based on Ce-doped garnets.22 It is worth noting that the spectral position of the optical transition between doublet 5d1 and 5d2 at Ce3+ is also not influenced by Mg-codoping. The TA decay becomes faster with increasing Mg content and proceeds at the same rate as the PL decay, as illustrated in Fig. 7d.
 |
| | Fig. 7 Data carpets presenting transient absorption (color scale) after short-pulse resonant excitation of Ce3+ ions as a function of probe photon energy and the delay between pump and probe pulses in (a) Mg-free (S1) and (b) Mg-codoped (S5) LuGAGG:Ce, epitaxial layers. Transient absorption spectra of samples with different Mg content (c), and decay kinetics (d) of PL (points) and TA (lines) in the layers. | |
4. Discussion
The substantial acceleration of luminescence decay by aliovalent codoping of Ce-doped scintillators with a garnet-type matrix, has already been observed in many scintillators containing Ce ions in garnet-type matrixes and is interpreted as due to the formation of Ce3+ emitting centers modified by close proximity Mg ions.13–15 To test this assumption, we attempted to interpret our experimental results using a model consisting of two types of emitting center: the conventional Ce3+ emitting center and a Ce3+ emitting ion with Mg ion in close proximity (Ce3+ + Mg2+ center). Due to their ionic radius, Mg2+ are preferentially located in octahedral sites, but part of the Mg ions can also be accommodated in dodecahedral sites.23 Each Ce3+ ion is stabilized in a dodecahedral site and is surrounded by 10 possible positions for Mg2+ separated by distances of 3.05/3.41/3.74 Å (in the third coordination sphere). These distances are close enough to consider just one type of Ce–Mg center.
The PL intensity decay in a Mg-free sample can be fairly well fitted using a single exponent (see Fig. 8). The decay times 1 extracted this way at different temperatures are well described using the Arrhenius formula in eqn (1), with activation energy of 0.48 eV and radiative decay time of 50 ns. To describe the PL kinetics at different temperatures in LuGAGG:Ce codoped by magnesium, we introduced a second emission center Ce3+ + Mg2+, and expressed the time dependence of the PL intensity I(t) as
| |  | (2) |
with factor
w1 as the weight coefficient for the contribution of the first channel (Ce
3+ center). The emission decay times
τ1 and
τ2 at different temperatures were extracted in samples with a Mg content of 300 and 700 ppm by fitting the measured PL kinetics using expression
(2). They were used for fitting the Arrhenius-type expression
(1), as presented in
Fig. 9. Note that the temperature-independent time in the fit for
τ2 is equal to 10.6 ns,
i.e., is substantially shorter than that for
τ1. The low-temperature time of 50 ns for Ce
3+ centers is due to 5d
1-4f radiative recombination. Meanwhile, for Ce
3+ + Mg
2+ centers, the decay rate is faster than the rate of radiative recombination, even at temperatures low enough for thermally activated quenching to be neglected. Thus, a new recombination route occurs in Ce
3+ + Mg
2+ centers. The rate of this decay is temperature independent. Thus, the emission decay times can be expressed as
| |  | (3) |
and
| |  | (4) |
here,
τq1,
τq2 and
τTINR are the decay time constants due to thermal quenching at Ce
3+ and Ce
3+ + Mg
2+ centers and temperature-independent nonradiative recombination, respectively.
 |
| | Fig. 8 Temperature dependence of emission decay time τ1 in Mg-free LuGAGG:Ce (points) and the best fit using eqn (1) (line). In the inset, time evolution of normalized photoluminescence intensity measured at different temperatures (colors as indicated) and the best fit using single-exponential decay (black lines). | |
 |
| | Fig. 9 Temperature dependence of emission decay times τ1 and τ2 (points) extracted in samples with Mg content of 300 and 700 ppm from measured PL kinetics using expression (2). The lines present the Arrhenius plots with corresponding parameters, as shown in the insets. | |
The analysis shows that the activation energies for thermal quenching are different for the two recombination centers: 0.48 eV for Ce3+ centers and 0.20 eV for Ce3+ + Mg2+ centers, whereas the effective activation energy extracted from PL intensity decay decreases with increasing Mg content due to the increasing contribution of Ce3+ + Mg2+ centers. The weight coefficient of the Ce3+ component, w1, of the luminescence decay rate as described by eqn (2), is presented as a function of magnesium content in Fig. 10, where the parameters extracted for centers Ce3+ and Ce3+ + Mg2+ are also provided.
 |
| | Fig. 10 Dependence of the weight coefficient of the Ce3+ component of the luminescence decay rate, as defined in eqn (2), on magnesium content. The table presents parameters extracted for centers Ce3+ and Ce3+ + Mg2+: activation energies Ea1 and Ea2, frequency factors c1 and c2, and nonradiative decay time τTINR for three samples with different Mg content. The point colors correspond to the colors highlighting different samples in the table. | |
The key processes governing the dynamics of nonequilibrium carriers and, consequently, the luminescence decay are schematically depicted in Fig. 11. To estimate the importance of the Mg-enhanced nonradiative recombination at the radiative recombination centers Ce3+ + Mg2+, we compared the emission intensities at selective photoexcitation of the emitting Ce3+ centers and at excitation by high energy electrons. In the latter case, the generated nonequilibrium electron–hole pairs are distributed across the entire matrix, some them might be lost due to nonradiative recombination before they reach the radiative centers, whereas at the resonant excitation of Ce3+, the stage of excitation transfer to the emission center does not exist. The dependences presented in Fig. 12 show a substantial decrease for Mg content above a few hundred ppm. The dependence for PL intensity measured at nominally the same excitation power density for all samples is affected by decreasing absorption at the excitation photon energy corresponding to the 4f–5d1 transition due to valency transformation from Ce3+ to Ce4+, whereas in CL both ions might emit photons, either after capturing an electron by Ce4+ or after excitation transfer to Ce3+.24 The red circles in Fig. 12 present the dependence corrected for emission intensity per absorbed photon using the measured absorbance values (see Fig. 1). After the correction, the decrease in luminescence intensity with increasing Mg content coincides in heavily codoped samples within the experimental error. This result shows that the excitation losses due to nonradiative recombination during the excitation transfer to Ce-related emission centers, which might be expected in CL but not in PL at selective excitation of Ce ions, is small or unaffected by Mg codoping. The nonradiative recombination of the nonequilibrium electron–hole pairs at the emission centers is dominant.
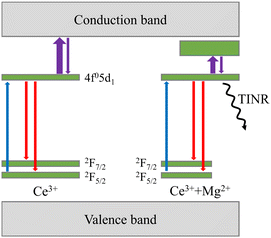 |
| | Fig. 11 Energy scheme of Ce3+ and Ce3+ + Mg2+ centers in LuGAGG:Ce,Mg (not to scale) with excitation (blue arrow), thermal activation (violet), radiative recombination (red), and temperature-independent nonradiative recombination (TINR), indicated. | |
 |
| | Fig. 12 Normalized luminescence intensity in LuGAGG:Ce,Mg samples with different Mg content under electron beam excitation (blue squares) and resonant photoexcitation to the emitting 5d1 level of Ce3+ without (open triangles) and with (red circles) correction taking into account the change in Ce3+/Ce4+ ratio at increasing Mg content. The grey field indicates the observed tendency. | |
Our study shows that the codoping-related recombination centers force nonradiative losses due to thermal quenching over the decreased potential energy barrier, and have a temperature-insensitive nonradiative recombination channel. Consequently, the emission decay acceleration by Mg codoping can be achieved but with a trade-off in efficiency. For certain applications, the efficiency decrease with increasing codoping level might be tolerated to a certain extent, due to a comparatively high light yield of the scintillation crystals of Ce-doped garnets in general (up to 58000 ph MeV−1 in GAGG:Ce25) and of LuGAGG:Ce in particular (up to 30900 ph MeV−1).26
5. Conclusions
The influence of magnesium codoping on the luminescence properties of Ce3+ ions was studied in garnet scintillator (LuGd)3(GaAl)5O12:Ce,Mg thin films with different Mg content. Time-resolved cathodoluminescence spectroscopy evidenced that the square-shaped pits of size ∼100 nm inherent to films grown by liquid phase epitaxy, serve to provide better light extraction from the film.
Our results obtained using time-resolved PL spectroscopy and transient absorption technique probing of the population of the emitting centers at temperatures in the range 80 to 600 K, show that the luminescence behavior in Mg-codoped LuGAGG:Ce might be due to the coexistence of two types of emission centers: regular ions of Ce3+ and Ce ions with magnesium in close vicinity, i.e., Ce3+ + Mg2+ centers. Magnesium has no significant effect on the energy position of the emitting 5d1 level and, consequently, does not affect the emission spectrum even up to Mg contents of 6000 ppm. Instead, the Mg2+ ions modify the nonradiative recombination at Ce3+ by introducing a barrier-free channel of nonradiative recombination and a level with an activation energy of 200 meV serving for thermal quenching. Regular Ce3+ centers are thermally quenched over a barrier of 480 meV, which is equal to the energy gap between the 5d1 level and the bottom of the conduction band. Thus, the quenching level in Ce3+ + Mg2+ centers is well below the bottom of the conduction band. The activation energy and the low-temperature emission decay rate (a sum of the rates of radiative recombination and temperature-independent nonradiative recombination) do not depend on Mg content. The introduction of more magnesium, results in an increasing share of Ce3+ ions affected by magnesium transforming the ions into modified centers, Ce3+ + Mg2+. The emission at these centers decays faster than that in regular Ce3+ centers due to the stronger influence of nonradiative recombination because of a smaller energy barrier for thermal quenching and the existence of a barrier-free channel for nonradiative recombination.
Comparison of luminescence intensity dependence on the level of Mg codoping with electron beam and selective photoexcitation to the emitting level 5d1 of the activator ion Ce3+, shows that the excitation losses due to nonradiative recombination of electron–hole pairs already populating the emitting centers, especially the Ce3+ + Mg2+ center, dominates over the excitation losses during the excitation transfer to the centers.
The conclusions of this study based on the properties of (LuGd)3(GaAl)5O12:Ce,Mg thin films with different Mg content are not related to a small thickness of the films studied, therefore should also be valid for bulk crystals and other scintillators based on Ce–Mg codoped garnet-type matrixes. On the other hand, the Mg-codoped LuGAGG:Ce thin epitaxial layers are of interest for applications as scintillating films in X-ray screens,27 thus, the recombination mechanisms revealed and the extracted parameters of the emitting centers, can be exploited for the optimization of the scintillation screens.
Author contributions
S. Nargelas: investigation, formal analysis, visualization, writing – review & editing; A. Solovjovas: investigation, formal analysis; Y. Talochka: methodology, software, formal analysis, writing – original draft; Ž. Podlipskas: investigation, formal analysis, visualization; M. Kucera: sample preparation, funding acquisition, resources, writing – review & editing; Z. Lucenicova: investigation, formal analysis; G. Tamulaitis: funding acquisition, supervision, writing – original draft, writing – review & editing. All authors have read and agreed on the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research has been carried out in the framework of Crystal Clear Collaboration at CERN and received funding via the HORIZON 2020 RIA project AIDAinnova. The work at Charles University was supported by the Czech Science Foundation via project no. 21-17731S.
Notes and references
- M. Nikl and A. Yoshikawa, Adv. Opt. Mater., 2015, 3, 463–481 CrossRef CAS.
-
R. Abbott, A. Abreu, F. Addesa, M. Alhusseini, T. Anderson, Y. Andreev, A. Apresyan, R. Arcidiacono, M. Arenton, E. Auffray, D. Bastos, L. Bauerdick, R. Bellan, M. Bellato, A. Benaglia, M. Benettoni, R. Bertoni, M. Besancon, S. Bharthuar, A. Bornheim, E. Brücken, J. Butler, C. Campagnari, M. Campana, R. Carlin, P. Carniti, N. Cartiglia, M. Casarsa, O. Cerri, P. Checchia, H. Chen, S. Chidzik, F. Chlebana, F. Cossutti, M. Costa, B. Cox, I. Dafinei, F. De Guio, P. Debbins, D. Del Re, A. Dermenev, E. Di Marco, K. Dilsiz, K. Di Petrillo, G. Dissertori, S. Dogra, U. Dosselli, I. Dutta, F. Caleb, C. Fernandez Madrazo, M. Fernandez, M. Ferrero, Z. Flowers, W. Funk, M. Gallinaro, S. Ganjour, M. Gardner, F. Geurts, A. Ghezzi, S. Gninenko, F. Golf, J. Gonzalez, C. Gotti, L. Gray, F. Guilloux, S. Gundacker, E. Hazen, S. Hedia, A. Heering, R. Heller, T. Isidori, R. Isocrate, R. Jaramillo, M. Joyce, K. Kaadze, A. Karneyeu, H. Kim, J. King, G. Kopp, M. Korjik, O. Koseyan, A. Kozyrev, N. Kratochwil, M. Lazarovits, A. Ledovskoy, H. Lee, J. Lee, A. Li, S. Li, W. Li, T. Liu, N. Lu, M. Lucchini, W. Lustermann, C. Madrid, M. Malberti, I. Mandjavize, J. Mao, Y. Maravin, D. Marlow, B. Marsh, P. Martinez Del Arbol, B. Marzocchi, R. Mazza, C. McMahon, V. Mechinsky, P. Meridiani, A. Mestvirishvili, N. Minafra, A. Mohammadi, F. Monti, C. Moon, R. Mulargia, M. Murray, Y. Musienko, J. Nachtman, S. Nargelas, L. Narvaez, O. Neogi, C. Neu, T. Niknejad, M. Obertino, H. Ogul, G. Oh, I. Ojalvo, Y. Onel, G. Organtini, T. Orimoto, J. Ott, I. Ovtin, M. Paganoni, F. Pandolfi, R. Paramatti, A. Peck, C. Perez, G. Pessina, C. Pena, S. Pigazzini, O. Radchenko, N. Redaelli, D. Rigoni, E. Robutti, C. Rogan, R. Rossin, C. Rovelli, C. Royon, M. Sahin, W. Sands, F. Santanastasio, U. Sarica, I. Schmidt, R. Schmitz, J. Sheplock, J. Silva, F. Siviero, L. Soffi, V. Sola, G. Sorrentino, M. Spiropulu, D. Spitzbart, A. Stahl Leiton, A. Staiano, D. Stuart, I. Suarez, T. Tabarelli De Fatis, G. Tamulaitis, Y. Tang, B. Tannenwald, R. Taylor, E. Tiras, M. Titov, S. Tkaczyk, D. Tlisov, I. Tlisova, M. Tornago, M. Tosi, R. Tramontano, J. Trevor, C. Tully, B. Ujvari, J. Varela, S. Ventura, I. Vila, T. Wamorkar, C. Wang, X. Wang, M. Wayne, J. Wetzel, S. White, D. Winn, S. Wu, S. Xie, Z. Ye, G. Yu, G. Zhang, L. Zhang, Y. Zhang, Z. Zhang and R. Zhu, J. Inst., 2021, 16, P07023 Search PubMed.
- M. Nikl, K. Kamada, V. Babin, J. Pejchal, K. Pilarova, E. Mihokova, A. Beitlerova, K. Bartosiewicz, S. Kurosawa and A. Yoshikawa, Cryst. Growth Des., 2014, 14, 4827–4833 CrossRef CAS.
- C. Dujardin, E. Auffray, E. Bourret-Courchesne, P. Dorenbos, P. Lecoq, M. Nikl, A. N. Vasil'ev, A. Yoshikawa and R.-Y. Zhu, IEEE Trans. Nucl. Sci., 2018, 65, 1977–1997 CAS.
- C. Kim, W. Lee, A. Melis, A. Elmughrabi, K. Lee, C. Park and J.-Y. Yeom, Crystals, 2021, 11, 669 CrossRef CAS.
- M. Pizzichemi, J. Inst., 2020, 15, C05062–C05062 Search PubMed.
- M. Nikl, Meas. Sci. Technol., 2006, 17, R37–R54 CrossRef CAS.
- M. A. Spurrier, P. Szupryczynski, K. Yang, A. A. Carey and C. L. Melcher, IEEE Trans. Nucl. Sci., 2008, 55, 1178–1182 CAS.
- S. Blahuta, A. Bessiere, B. Viana, P. Dorenbos and V. Ouspenski, IEEE Trans. Nucl. Sci., 2013, 60, 3134–3141 CAS.
- G. Tamulaitis, E. Auffray, A. Gola, M. Korzhik, A. Mazzi, V. Mechinski, S. Nargelas, Y. Talochka, A. Vaitkevičius and A. Vasil’ev, J. Phys. Chem. Solids, 2020, 139, 109356 CrossRef CAS.
- G. Tamulaitis, A. Vaitkevičius, S. Nargelas, R. Augulis, V. Gulbinas, P. Bohacek, M. Nikl, A. Borisevich, A. Fedorov, M. Korjik and E. Auffray, Nucl. Instrum. Methods Phys. Res., Sect. A, 2017, 870, 25–29 CrossRef CAS.
- G. Tamulaitis, A. Vasil’ev, M. Korzhik, A. Mazzi, A. Gola, S. Nargelas, A. Vaitkevicius, A. Fedorov and D. Kozlov, IEEE Trans. Nucl. Sci., 2019, 66, 1879–1888 CAS.
-
L. Martinazzoli, S. Nargelas, P. Boháček, R. Calá, M. Dušek, J. Rohlíček, G. Tamulaitis, E. Auffray and M. Nikl, Mater. Adv., 2022, 3, 6842–6852 Search PubMed.
-
V. Babin, P. Boháček, K. Jurek, M. Kučera, M. Nikl and S. Zazubovich, Optical Materials, 2018, 83, 290–299 Search PubMed.
- V. Babin, P. Herman, M. Kucera, M. Nikl and S. Zazubovich, J. Lumin., 2019, 215, 116608 CrossRef CAS.
- M. Kučera, K. Nitsch, M. Nikl and M. Hanuš, Radiat. Meas., 2010, 45, 449–452 CrossRef.
- P. Průša, M. Kučera, V. Babin, P. Brůža, D. Pánek, A. Beitlerová, J. A. Mareš, M. Hanuš, Z. Lučeničová and M. Nikl, Adv. Opt. Mater., 2017, 5, 1600875 CrossRef.
- M. Kučera, K. Nitsch, M. Nikl, M. Hanuš and S. Daniš, J. Cryst. Growth, 2010, 312, 1538–1545 CrossRef.
- M. Kucera, K. Nitsch, M. Kubova, N. Solovieva, M. Nikl and J. A. Mares, IEEE Trans. Nucl. Sci., 2008, 55, 1201–1205 CAS.
- P. Prusa, M. Kučera, V. Babin, P. Bruza, T. Parkman, D. Panek, A. Beitlerova, J. A. Mares, M. Hanus, Z. Lucenicova, M. Pokorny and M. Nikl, Cryst. Growth Des., 2018, 18, 4998–5007 CrossRef CAS.
- O. Lalinsky, P. Schauer and M. Kucera, Phys. Status Solidi A, 2019, 216, 1801016 CrossRef.
- S. Nargelas, Y. Talochka, A. Vaitkevičius, G. Dosovitskiy, O. Buzanov, A. Vasil’ev, T. Malinauskas, M. Korzhik and G. Tamulaitis, J. Lumin., 2022, 242, 118590 CrossRef CAS.
- D. Mateika, E. Völkel and J. Haisma, J. Cryst. Growth, 1990, 102, 994–1013 CrossRef CAS.
- S. R. Rotman, H. L. Tuller and C. Warde, J. Appl. Phys., 1992, 71, 1209–1214 CrossRef CAS.
- K. Kamada, S. Kurosawa, P. Prusa, M. Nikl, V. V. Kochurikhin, T. Endo, K. Tsutumi, H. Sato, Y. Yokota, K. Sugiyama and A. Yoshikawa, Opt. Mater., 2014, 36, 1942–1945 CrossRef CAS.
- M. Korzhik, V. Retivov, A. Bondarau, G. Dosovitskiy, V. Dubov, I. Kamenskikh, P. Karpuk, D. Kuznetsova, V. Smyslova, V. Mechinsky, V. Pustovarov, D. Tavrunov, E. Tishchenko and A. Vasilev, Crystals, 2022, 12, 1196 CrossRef CAS.
- A. Koch, C. Raven, P. Spanne and A. Snigirev, J. Opt. Soc. Am. A, 1998, 15, 1940 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Arnoldas
Solovjovas
a,
Yauheni
Talochka
*a,
Arnoldas
Solovjovas
a,
Yauheni
Talochka