
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganometallic one-pot synthesis of ZnO quantum dots coated by sulfoxides as L-type ligands†
Maria
Jędrzejewska
 ab,
Małgorzata
Wolska-Pietkiewicz
*a,
Zygmunt
Drużyński
a and
Janusz
Lewiński
*ab
ab,
Małgorzata
Wolska-Pietkiewicz
*a,
Zygmunt
Drużyński
a and
Janusz
Lewiński
*ab
aFaculty of Chemistry, Warsaw University of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: malgorzata.pietkiewicz@pw.edu.pl; janusz.lewinski@pw.edu.pl
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 9th October 2023
Abstract
Although an extraordinary amount of research into the chemistry of nanoscale zinc oxide (ZnO) has been conducted over the past three decades, application-driven design and reproducible fabrication of colloidal ZnO quantum dots (QDs) remain a great challenge. The application of low-molecular-weight, non-interfering protecting ligands may be potentially beneficial for the design of quantum-sized ZnO crystals, simultaneously providing colloidal stabilization and long-term functionality. Herein, we pursue the idea of ‘less is more’ and continue our systematic investigations on a non-external-surfactant-assisted organometallic approach for the preparation of colloidal ZnO QDs via direct injection of Et2Zn to ligand-like solvent followed by exposition towards atmospheric air as well as we introduce a novel approach toward ZnO QDs through the transformation of sulfoxide-modulated Et2Zn-based precursors in a THF solution. The application of sulfoxides as L-type protectors contributes to the formation of uniform ZnO QDs with average diameters of 5.4 to 8.2 nm, depending of the character of applied sulfoxide, and the low surface grafting density of the coating ligand without impeding their colloidal as well as solid-state stability. The reported QDs exhibit almost identical absorption parameters and show particularly long, multiexponential PL decays reaching recombination times up to 2.3–2.8 μs. In turn, preliminary control experiments involving photodegradation of methylene blue demonstrate dramatically different photocatalytic performance of QDs derived from the neat ligand-like solvent synthesis and the new approach based on the finetuning of precursors’ reactivity by Lewis-base chemical additives.
Introduction
According to the standard definition, colloidal quantum dots (QDs) are solution-processable semiconductor nanocrystals (NCs) coated with a monolayer of surface ligands. Over three decades ago, Louis E. Brus – one of the pioneers of nanochemistry and colloidal QDs – stated that “an ideal synthesis would prepare pure isolated clusters, monodisperse at the atomic level, with the surface independently derivatized.(…) The enormity of this project is obvious, yet an encouraging start has been made”.1 Until now, the research is constantly moving forward; however, the field of nanocrystal synthesis “is not yet mature” and can evolve in “unexpected directions”.2 The latter statement nicely accentuates the field of colloidal zinc oxide quantum dots (ZnO QDs). Due to its unique physicochemical properties, nanocrystalline ZnO is widely used and highly prospective for various applications spanning from cosmetics, ceramics, sensors to solar cells and advanced electronic devices.3,4 The most commonly used wet-chemical procedure for the preparation of colloidal ZnO QDs is the inorganic sol–gel route, involving zinc acetate (and, to a lesser extent, other zinc salts) as the precursor in an alcoholic solution.5–8 This fast, effective, yet uncontrollable process does not meet the expectations in terms of batch-to-batch reproducibility as well as the uniformity of the resulting quantum-sized nanostructures. Moreover, the sol–gel procedure affords ZnO QDs with a high density of core-type and surface-type defects, which manifest themselves via electron paramagnetic resonance (EPR) activity,9,10 the inherently ill-passivated and unstable surface and a heterogenous coating shell composed of the pristine acetate ligand, alcohol molecules and additional stabilizing surfactant molecules (Scheme 1a)7,11–13 that undergoes a time-dependent evolution.14,15 The instability of the inorganic–organic interface might be exceptionally disadvantageous in terms of a wide range of QDs-based applications, and a prolonged (even weeks) post-synthetic treatment is often required to achieve their targeted stable performance and properties.14,16–18 Strikingly, the standard sol–gel procedure does not inherently allow receiving ‘ready-to-use’ and easily redispersible organic ligand-free ZnO QDs.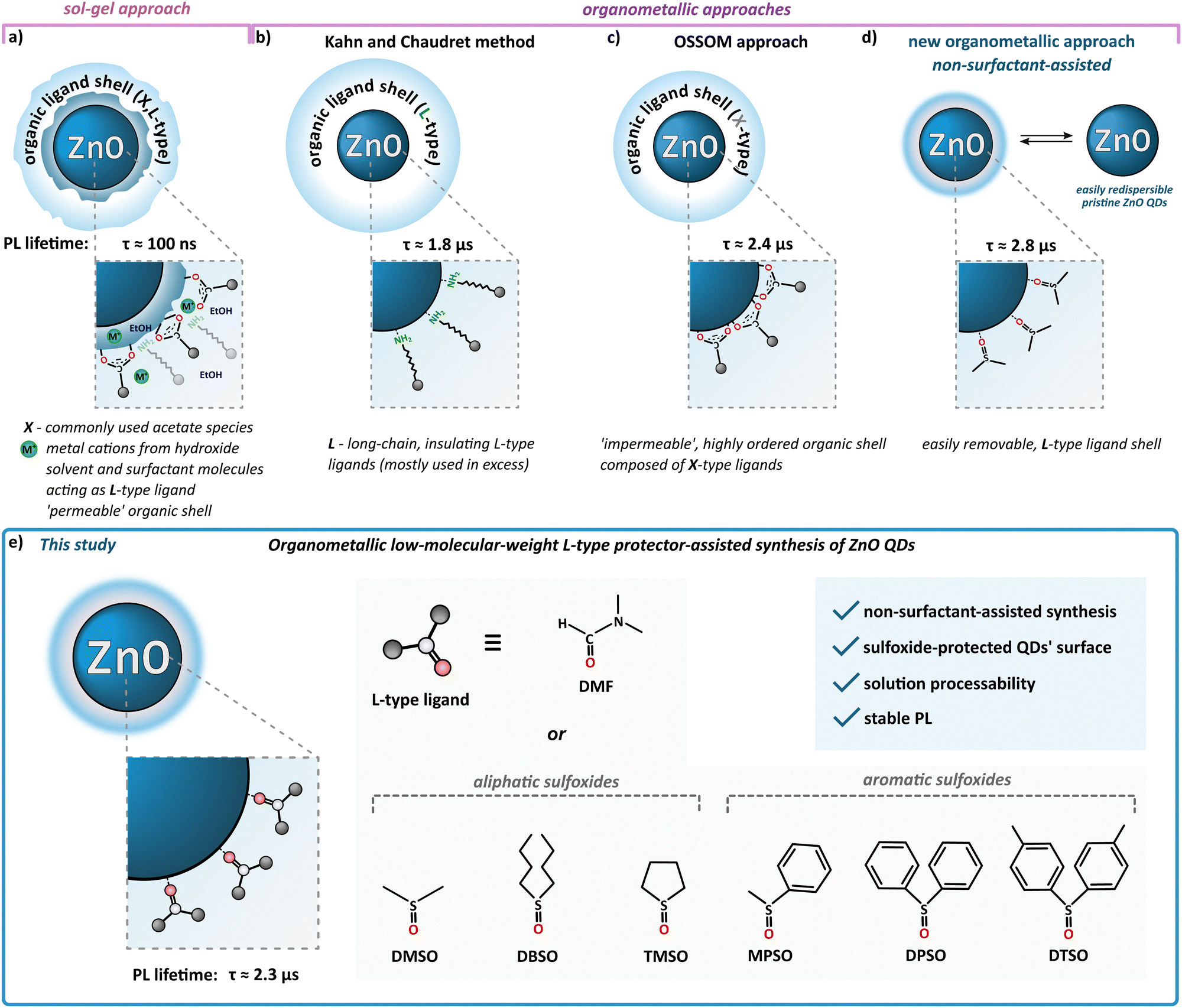 | ||
| Scheme 1 Overview of wet-chemical synthetic routes for the preparation of ZnO QDs; (a) classical sol–gel procedure, and organometallic approaches including (b) hydrolysis of organometallic precursors with L-type ligands developed by Kahn and Chaudret, (c) OSSOM method, (d) one-step non-surfactant assisted method and (e) low-molecular-weight L-type protector-assisted synthesis. | ||
Nowadays, new possibilities have been provided by the development of wet-organometallic approaches, employing homo-19–21 or heteroleptic11,13,22–31 zinc alkyl precursors, affording high-quality monodisperse, quantum-sized ZnO nanocrystals. For instance, Kahn and Chaudret have developed a room-temperature organometallic one-pot preparation of hydrophobic ZnO QDs through the hydrolysis of a dicyclohexyl zinc precursor in the presence of long-chain amines (often applied in great excess) as L-type stabilizing ligands (Scheme 1b).20,21,32–35 Alternatively, a one-pot self-supporting organometallic (OSSOM) procedure involving heteroleptic [RZn–X]-type precursors (where R = Et and X = monoanionic organic ligand, e.g. carboxylate,27 aminoalkoxide29 or phosphinate28), leads to the reproducible formation of essentially bio-safe,30,31 colloidally stable, intrinsic defect-less EPR-silent30 ZnO QDs with ‘impermeable’, well-passivated organic shell composed from the X-type ligands (Scheme 1c) and prone to the effective post-synthetic modification with copper-catalysed ‘click’ chemistry;11,36 note that a mechanochemical variation of the OSSOM approach has also been developed.37,38 Moreover, organometallic methods provide luminescent ZnO QDs with exceptionally long PL lifetimes (up to μs,26,30,31,39 contrary to sol–gel-derived QDs with lifetimes in the range of ns5,40). Nevertheless, there are some potential disadvantages of using X-type protecting ligands. These ligands, covalently bound to the NC surface, can constitute difficult to remove insulating organic shell or restrict access to the surface of the NC which may often disqualify them from the use in many devices requiring, for example, efficient charge transfer41–43 as well as surface-dependent applications like catalysis. Thus, the challenge of the preparation of colloidally and functionally stable, insulating ligand-free nanomaterials urged us to ‘think outside the box’, and only recently we reported a non-external-surfactant-assisted organometallic procedure via the controlled transformation of commercially available Et2Zn in neat dimethyl sulfoxide (DMSO) upon atmospheric air at ambient temperature.44–47 An ink of these colloidal DMSO-capped ZnO QDs (hereafter denoted as ZnO-DMSO) have been applied for the fabrication of thin film based on organic ligand-less ZnO QDs as an electron transport layer (ETL) in perovskite solar cells (PSCs).46 Remarkably, the introduction of organometallic approach-derived ETL with uniform surface morphology and reduced surface defects led to excellent electron extraction ability. The champion device achieved state-of-the-art performance among reported dopant-free ZnO ETL-based PSCs with a power conversion efficiency of over 20%.46 In turn, our earlier work demonstrated that the controlled hydrolysis of Et2Zn in neat tetrahydrofuran (THF) as a aprotic coordinating solvent afforded a turbid suspension of luminescent ZnO QDs of average size 5.8 ± 1.2 nm.26 Then these as-prepared QDs were successfully applied as nanocrystalline synthons in the subsequent post-synthetic modification of their interfaces with an ethylene glycol oligomer bearing the carboxylate anchoring group. The resulting novel water-soluble ZnO QDs exhibited ultra-long-lived photo-induced charge separation26 and a high propensity for further surface modification and successful application in a hybrid nanosystem for efficient hydrogen evolution48 and the semiconductor-assisted light modulation of supramolecular assemblies.49
Taking into account that a key strategy for tailoring properties of QDs is to modulate surface ligands,50–52 herein we continue the challenge to obtain solution-processable small-molecule ligand-coated QDs and report further systematic investigations of L-type protector-assisted synthesis of ZnO QDs (Scheme 1e). The mentioned successful initial elaboration with DMSO47 prompted us to extend the investigations using other low-molecular organic molecules such as dimethylformamide (DMF), i.e., an aprotic coordinating solvent widely used for the preparation of various hybrid inorganic–organic functional materials, and an array of aliphatic and aromatic sulfoxides (Scheme 1e). More extended investigations concerning the one-pot synthesis in commercially available neat sulfoxides are limited because most of these compounds are solids. Consequently, we use sulfoxides as Lewis base additives to modulate the Et2Zn-based-precursor's reactivity in a THF solution and explore the possibility of achieving a new quality in controlled transformations to sulfoxide-coated ZnO QDs. Surprisingly, the finetuning of precursors’ reactivity by Lewis-base chemical additives has been rarely encountered in preparing semiconductor nanocrystals.53,54 Moreover, while DMSO and DMF have been demonstrated to be very useful solvents in the preparation of various nanoparticles,55–59 including heterogeneously-protected ZnO nanostructures derived from sol–gel procedure60 and other zinc salts-based hydrolytic techniques,56,61 this cannot be said about organosulfoxides.
Experimental
Characterization techniques
Fourier transformation infrared (FTIR) data for purified ZnO QDs and corresponding pure ligands were collected on Bruker Vertex 800V. Measurements were conducted with ATR attachment. 1H and 13C NMR spectra were acquired on Varian Mercury 400 MHz spectrometer at 303 K. The size and shape of the ZnO QDs were examined using JEOL JEM 2100 Transmission Electron Microscope (Department of Materials and Semiconductor Structures Research, Institute of Electron Technology, Warsaw, Poland) operating at 200 kV with a resolution point of 0.45 nm. QDs’ sample (dispersed in DMF, THF or DMSO) was drop-cast onto carbon-coated copper grids. The size distributions of ZnO QDs were obtained by measuring 100 separated nanocrystals using the ImageJ program. The average diameters and standard deviations were calculated with statistical tools in OriginPro software. Powder X-ray diffraction (PXRD) data were collected on an Empyrean diffractometer (PANalytical). Measurements employed Ni-filtered Cu Kα radiation of a copper sealed tube charged with 40 kV voltage and 40 mA current and Bragg–Brentano geometry with a beam divergence of 1° in the scattering plane. Diffraction patterns were scanned using a step scan mode (step size 0.017°) and measured in the range of 20–80°. The average diameters and standard deviations were calculated with statistical tools in OriginPro software. Solvodynamic diameters of ZnO QDs were determined by Dynamic Light Scattering (DLS) performed on Malvern Zetasizer Nano Z. The ZnO QDs’ solutions were prepared by dispersing 5 mg of sample powder in 5 mL of DMF or DMSO to obtain solutions at a concentration of 1 mg mL−1. Subsequently, as-prepared dispersions were sonicated for 15 minutes. The solutions of ZnO QDs were filtered before the analysis through a 0.45-micron filter to remove dust particles and larger aggregates. The measurements were performed in 3.5 mL quartz cells (Hellma) of path length 10 mm, transparent on all four sides. Thermogravimetric analysis (TGA) was performed using a TA Instruments Q600 under a flow of synthetic air, to a maximum of 650 °C, at a heating rate of 5 °C min−1 (flow rate of 100 mL min−1). Open 5 mm diameter alumina crucibles were used. Optical absorption (UV-Vis) spectra for ZnO QDs colloidal solution in DMSO were collected on the Hitachi U-2910 spectrophotometer. A standard 3.5 mL quartz cell (Hellma) with a 10 mm path length and transparent on all four sides was used. The photoluminescence (PL) measurements were carried out HITACHI Fluorescence Spectrophotometer F-7000. The photoluminescence quantum yields (PLQY) for the solid samples were determined with Quantaurus-QY spectrometer (C11347, Hamamatsu Photonics). The photoluminescence (PL) decays for the solid samples were recorded using Quantaurus-Tau fluorescence lifetime measurement system (C11367, Hamamatsu Photonics) equipped with the LED light source and photon counting measurement system. The samples were excited at 340 nm and PL decays were collected at wavelengths corresponding to emission collected on the instrument. The spectra were collected with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts at the peak. The data were analysed by a least squares reconvolution procedure using the software package provided by Hamamatsu. The goodness of fit (fitting order) was determined by χ2 value and residuals distribution. When lower than 1.3, the χ2 values were taken as appropriate for fitting.
000 counts at the peak. The data were analysed by a least squares reconvolution procedure using the software package provided by Hamamatsu. The goodness of fit (fitting order) was determined by χ2 value and residuals distribution. When lower than 1.3, the χ2 values were taken as appropriate for fitting.
General remarks
All manipulations involving organozinc compounds were conducted under a dry, oxygen-free, inert gas atmosphere using standard Schlenk techniques (CAUTION!: Volatile dialkylzinc compounds are moisture sensitive and can spontaneously inflame in air and these reagents should be kept under an inert atmosphere and handled with caution). The reagents were purchased from commercial vendors: Et2Zn, 95% (abcr), DMSO and DMF (POCh), dibutyl sulfoxide (DBSO), diphenyl sulfoxide (DPSO) and tetramethylene sulfoxide (TMSO) (TCI), methyl phenyl sulfoxide (MPSO) and di-p-tolyl sulfoxide (DTSO) (Sigma Aldrich), and used as received. THF and hexane were purified with MBraun SPS-800 solvent purification system.Preparation of ZnO QDs in neat DMF
An appropriate amount of Et2Zn (0.44 mL, 2.25 M in hexane, 1 mmol) was injected into DMF (10 mL) with the tip of the needle submerged in the solution. Then, the reaction mixture was stirred vigorously up to 3 days under ambient air conditions at the surrounding temperature (ca. 22–33 °C). After this time, turbid dispersion was obtained. ZnO-DMF: IR (ATR): v = 3426 (w), 2928 (w), 2859 (w), 2361 (w), 2160 (w), 2028 (w), 1979 (vw), 1668 (s), 1505 (w), 1435 (w), 1386 (m), 1255 (w), 1153 (vw), 1093 (w), 1063 (w), 904 (vw), 865 (vw), 844 (vw), 658 (vw), 616 (vw), 424 (vs) cm−1. Elemental analysis found (%): C 2.87 ± 0.09, H 0.89 ± 0.05, N 0.61 ± 0.02, O 21.68 ± 0.09.Preparation of ZnO QDs in neat DBSO
An appropriate amount of Et2Zn (0.22 mL, 2.25 M in hexane, 0.5 mmol) was injected into DBSO (4 mL) with the tip of the needle submerged in the solution. Then, the reaction mixture was stirred vigorously for 24 h under ambient air. After this time, turbid dispersion was obtained. ZnO nanocrystallites were precipitated with acetone and centrifuged (10 min at 12500 rpm). The supernatant was discarded, and the procedure of dispersion in hexane and centrifugation was repeated. ZnO-DBSO: IR (ATR): v = 3438 (vw), 2960 (vw), 2863 (vw), 1586 (w), 1464 (vw), 1420 (vw), 1382 (vw), 1358 (vw), 1246 (vw), 1206 (vw), 1158 (vw), 1041 (vw), 973 (vw), 870 (vw), 782 (vw), 734 (vw), 719 (vw), 661 (vw), 626 (vw), 412 (vs) cm−1. Elemental analysis found (%): C 4.35 ± 0.07, H 1.36 ± 0.08, S 0.15 ± 0.04, O 24.71 ± 0.27.
Stoichiometric reactions of Et2Zn with DMF and selected sulfoxides
To a THF solution of selected low-molecular-weight donor ligand (1 mmol), an appropriate amount of Et2Zn (0.46 mL of a 2.19 M solution in hexane, 1 mmol) was added dropwise at −78 °C under nitrogen atmosphere. Then the reaction mixture was allowed to warm gradually to room temperature and the volatiles were removed under reduced pressure. The reaction afforded oily colourless products, which were characterized by NMR and FTIR spectroscopy. The corresponding spectra are shown in the ESI.†The general synthetic protocol for ZnO-L QDs
The preparation of sulfoxide-protected ZnO QDs involves a two-step procedure performed in a one-pot manner. Et2Zn (0.5 mL of 2 M solution in hexane, 1 mmol) was added dropwise to a solution of the corresponding sulfoxide (1 mmol) in THF (10 mL) at −78 °C. The solution was allowed to warm gradually to room temperature, and then the solution was exposed to ambient air and moisture to initiate slow (5–8 days) transformation to ZnO QDs. The resulting ZnO QDs were precipitated from THF as a white solid intended for further purification.Isolation and purification of ZnO-L QDs
The resulting crude suspensions of sulfoxide-protected ZnO QDs were centrifuged for 10 min at 12500 rpm. The supernatant was discarded and the precipitate was dispersed in 5 mL of hexane and centrifuged again for 10 min at 12500 rpm. Again, the supernatant was discarded and the procedure of dispersion in hexane and centrifugation was repeated. Finally, the supernatant was discarded and the resulting precipitate was dried under vacuum. ZnO QDs were collected as the final white solid.
Photocatalytic degradation of methylene blue (MB) by ZnO QDs
ZnO QDs (2 mg) were dispersed in 9.5 mL of deionised water and then 500 μL of a MB solution (50 mg L−1) was added (final QDs’ concentration of 200 μg mL−1). The suspension was stirred in the dark for approx. 5 min at room temperature (approx. 22 °C) to obtain a good dispersion. The concentration of MB was monitored by collecting 2 mL aliquots of the suspension into a quartz cuvette and measuring at 10-minute intervals. After 30 minutes, the suspension was irradiated with UV light source (6 W, 365 nm) and measurements were continued with 10-minute intervals for 90 minutes (total measurement time: 120 minutes). MB photodegradation was monitored by measuring the absorbance at the characteristic band at 664 nm, using a UV-vis spectrophotometer (Hitachi U-2910) over the wavelength range 280–800 nm. The percentage of MB degradation was calculated as follows: %MBdegradation = [(A0 = At)/A0] × 100%, where A0 is the initial absorbance, At is the absorbance after irradiation at various time intervals. The photodegradation of MB was fitted as a pseudo-first-order kinetics reaction (ln(C0/C) = kt), where C0 is the initial MB concentration, C is MB concentration after irradiation at various time intervals, and k is the reaction rate constant.Results and discussion
For systematic investigations of L-type protector-assisted synthesis of ZnO QDs, we selected liquid organic molecules like DMF, an aprotic coordinating solvent widely used for the preparation of various hybrid inorganic–organic functional materials,53,62 and DBSO as a commercially available di-n-alkyl sulfoxide. In this case, DMF or DBSO can serve as both the solvent and a stabilizing ligand, and the one-pot synthesis of QDs can be performed via the direct injection of Et2Zn into a ligand-like solvent under ambient air conditions (where both H2O and O2 act as oxygen sources). In turn, in separate experiments, we employ DMSO, DBSO and an array of other solid aliphatic and aromatic sulfoxides (see Scheme 1e) as Lewis bases additives in order to potentially modulate the Et2Zn-based-precursor's reactivity in a THF solution under exposition towards the air to induce slow transformation to ZnO QDs. Below, we present various one-pot organometallic approaches, i.e., synthesis in neat ligand-like solvents and L-type protector-assisted organometallic synthesis, allowing comparative investigation on the preparation and characterization of ZnO QDs. In addition, preliminary control experiments of photocatalytic activity have been carried out to establish the synthesis-property-relationship of the selected ZnO QDs.One-pot synthesis of ZnO QDs in neat ligand-like solvents
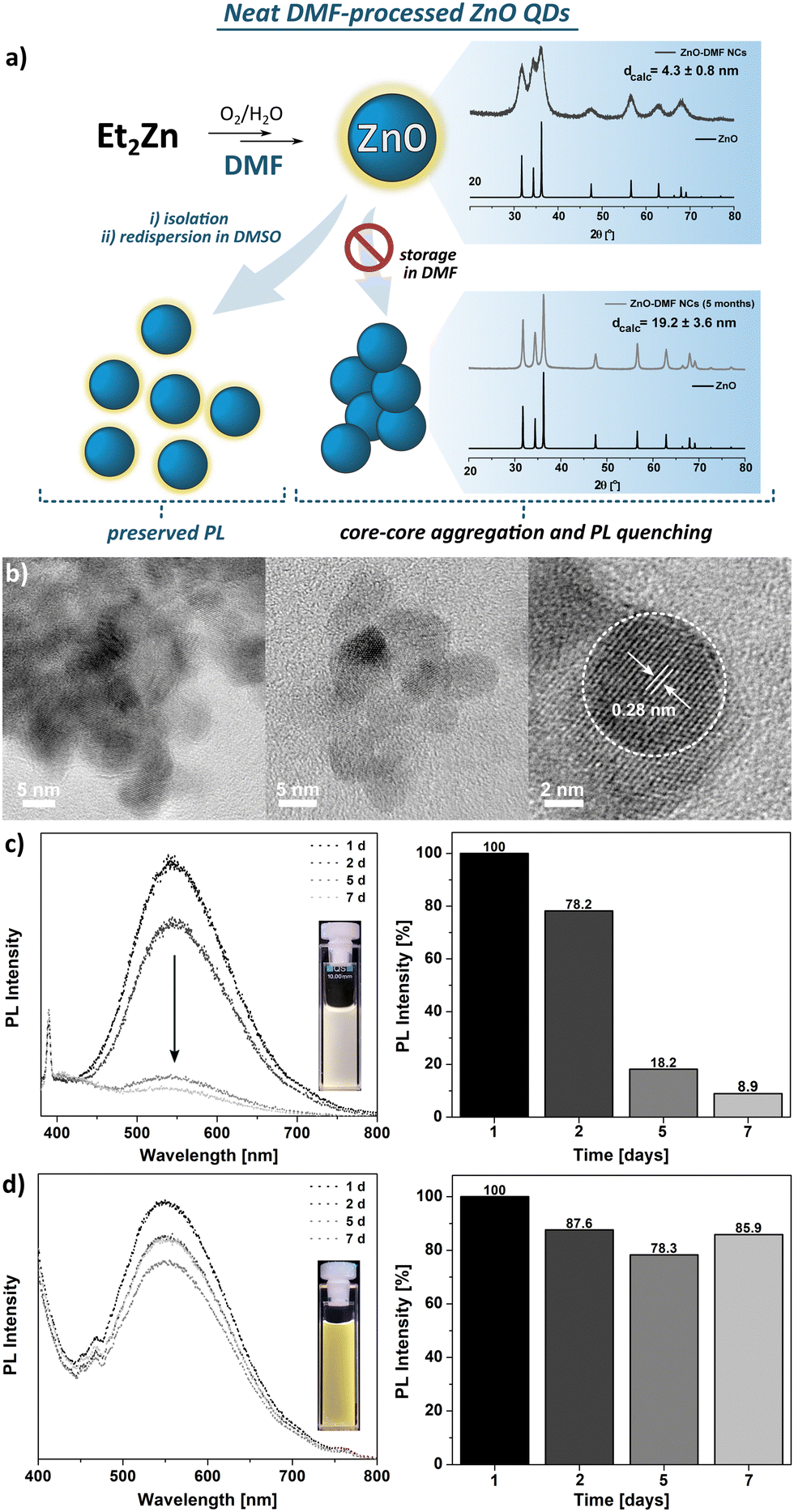 | ||
| Fig. 1 (a) Schematic representation of the organometallic synthesis of ZnO-DMF; (b) HR TEM images of ZnO-DMF, (c) and (d) PL spectra representing the suppresion of PL emission indicating the time-dependent stability of the resulting QDs upon storage in DMF and DMSO, respectively. | ||
The FTIR spectra of the parent DMF ligand-like solvent and ZnO QDs are shown in Fig. S4 (ESI†). The FTIR spectrum of ZnO QDs’ precipitate exhibits a strong band at 1668 cm−1, characteristic for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of free DMF molecules and likely results from residual species of DMF molecules. In turn, two bands of relatively weak intensity at 2928 and 2859 cm−1 are assigned to the N–H vibrational modes of unwashed DMF molecules. This suggests a relatively weak binding affinity of DMF molecule to QD's surface. The TGA profile exhibits multistep decomposition pathways with maximum decomposition rates at 101 °C, 194 °C, 282 °C, and 363 °C (Fig. S5, ESI†). Likely, the first two steps well-match to the boiling points of water and DMF, respectively, and could be associated with the removal of these molecules from the sample along with their thermal decomposition that is observable up to ca. 400 °C with a total weight loss of ca. 12%.
O stretching frequency of free DMF molecules and likely results from residual species of DMF molecules. In turn, two bands of relatively weak intensity at 2928 and 2859 cm−1 are assigned to the N–H vibrational modes of unwashed DMF molecules. This suggests a relatively weak binding affinity of DMF molecule to QD's surface. The TGA profile exhibits multistep decomposition pathways with maximum decomposition rates at 101 °C, 194 °C, 282 °C, and 363 °C (Fig. S5, ESI†). Likely, the first two steps well-match to the boiling points of water and DMF, respectively, and could be associated with the removal of these molecules from the sample along with their thermal decomposition that is observable up to ca. 400 °C with a total weight loss of ca. 12%.
Dispersion of the ZnO-DMF precipitate in DMSO prevents them from irreversible aggregation, as it is noticeable in case of further storage in a DMF solution (Fig. 1d) or in the solid state. Based on the results mentioned above, we can thus suggest that the simple one-step ligand exchange reaction occurred in this system and affords DMSO-protected ZnO QDs. These observations indicate that DMF turns out to be an insufficient L-type QD's surface protector, unable to preserve the primary nanostructure properties (e.g. size, colloidal stability).67,68 The differences between binding affinity to a metal centre of DMSO and DMF arise directly from the chemical character of these molecules, which is reflected in Gutmann number (donor number, DN) as a quantitative measure of Lewis basicity. For DMSO and DMF, the DN equals to 30.0 and 26.5, respectively,69 thus it is reasonable to expect higher binding affinity of DMSO than DMF molecules to the surface Zn sites. The adverse effect of DMF on QD's stability may arise from both a weak biding affinity to the QDs’ surface and restricted abilities to the formation of a ‘swollen’ solvating shell composed of DMF and water molecules, driven by non-covalent interactions, as it was observed for ZnO-DMSO47 (note that ligand-ligand and ligand-water interactions regarding DMSO and DMF have been a subject of numerous spectroscopic and computational studies over the recent years67,68,70).
O stretching vibration. Moreover, the observed slight shift of the abovementioned stretching frequency (Δv = 22 cm−1 compared to the spectrum of pure DBSO) indicates that the DBSO molecules interact with the ZnO surface through an oxygen atom (analogically to DMSO molecules).61 The broad and diffuse band centred at around 3500 cm−1 and the pronounced one at 1586 cm−1 are likely attributed to the stretching vibration modes of residual and surface-attached water molecules. It seems reasonable to assume a complex organic–inorganic interface and a swollen-type shell where water molecules interact with both the surface-associated DBSO molecules and coordinatively unsaturated Zn sites. In the TGA profile, three main decomposition steps are present with a maximum decomposition rate at 119 °C, 240 °C and 402 °C, and a total weight loss of ca. 13% (Fig. S10, ESI†). Notably, the observed 10% of weight loss occurring under 250 °C implies the continuous thermal desorption of the solvating shell composed of water (weight loss ca. 3.7% with maximum decomposition rate at 119 °C) and DBSO molecules (maximum decomposition rate at 240 °C), completed by the degradation of the residual surface-O-bound DBSO molecules at ca. 400 °C (the remaining 3% of weight loss). Moreover, the resulting QDs can be easily re-dispersed in DMSO after the purification process (i.e., precipitation with ‘anti-solvent’, e.g. acetone), which does not affect their optical properties (Fig. S6, ESI†). The presented characteristics allow us to conclude that both DMSO- and DBSO-protected ZnO QDs (previously reported DMSO47 as well as DBSO presented herein) obtained in our one-pot one-step organometallic procedure, hold similar physical properties, thereby neat sulfoxides can play a dual role as effective L-type protectors and solvents for the fabrication of colloidal, quantum-sized ZnO nanocrystals.
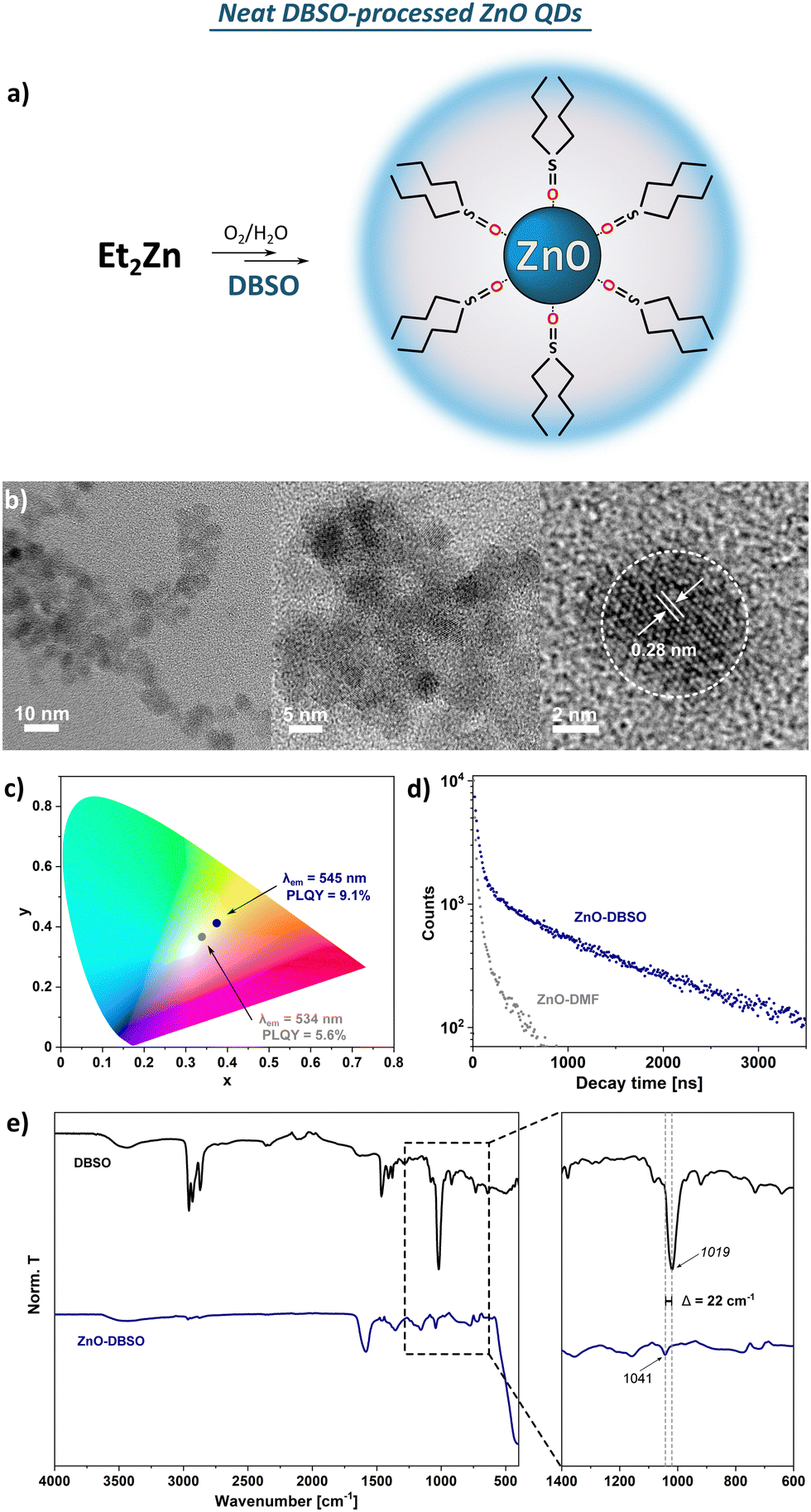 | ||
| Fig. 2 (a) Schematic representation of the organometallic synthesis of ZnO-DBSO; (b) HR TEM images of ZnO-DBSO QDs; (c) chromaticity diagram for ZnO-DBSO and ZnO-DMF excited at 350 nm; (b) photoluminescence decays taken for ZnO-DBSO (blue dots) and ZnO-DMF (grey dots) in the solid state; (d) FTIR spectra of pure DBSO (black line) and with the marked shifts assigned to sulfoxide coordination to QDs’ surface. | ||
One-pot L-type protector-assisted synthesis of ZnO QDs
As mentioned above, the organometallic precursor's character can play a significant role in the nucleation, growth and quality of the final QDs.71,72 Diorganozinc compounds have been known to easily form the Lewis-acid adducts with low-molecular-weight neutral donors73–77 and the adduct formation strongly affects the reactivity of zinc alkyls towards water78 and dioxygen.76 Thus, we were curious to what extent DMF and sulfoxides as Lewis bases could be suitable candidates for tuning the reactivity of dialkylzinc compounds as auxiliary ligands. For this purpose we selected six commercially available low-molecular-weight sulfoxides as potential L-type NC's surface protectors, namely: DMSO and DBSO previously applied for the synthesis in neat L-type ligand-like solvents (vide supra), and additionally tetramethylene sulfoxide (TMSO), methyl phenyl sulfoxide (MPSO), diphenyl sulfoxide (DPSO) and p-tolyl sulfoxide (DTSO) (Fig. 3). In control experiments, we carried out the equimolar reactions between Et2Zn and the selected additive in THF. The as-prepared Et2Zn-based precursors were characterized by liquid-state NMR spectroscopy (Fig. S11–S17, ESI†). The presence of signals distinct for the Et-Zn and the organic additive protons revealed the formation of Lewis acid–base adducts between Et2Zn and the corresponding L-type protector with the general formula [Et2Zn(L)2] (where L is an L-type protector). [Et2Zn(L)]-type adducts were further used as precursors for the fabrication of nanocrystalline ZnO. In the next step, we performed the synthesis of ZnO QDs starting from in situ preparation of an additive-modulated Et2Zn-based precursor in THF followed by the controlled exposition towards air to induce slow transformation to L-type additive-capped ZnO QDs (further denoted as ZnO-L QDs). All of the QDs underwent precisely the same purification cycles involving centrifugation and washing with hexane (the isolation procedure is fully described in Experimental section, see Isolation and Purification of ZnO-L QDs), leading to fine powders of ZnO-L QDs. (designated as ZnO-DMF′) or the respective sulfoxide-type ligand shell (designated as ZnO-DMSO′, ZnO-DBSO′, ZnO-TMSO′, ZnO-MPSO′, ZnO-DPSO′, ZnO-DTSO′, respectively). The resulting solids exhibit bright yellow luminescence under UV (λ = 365 nm) irradiation (the representative sample of ZnO-DMSO′ is shown in Fig. 3). It is also worth noting that the sulfoxide-protected ZnO-L QDs are easily dispersible in common solvents independently of the solvent polarity (e.g. DMSO, water, methanol, chloroform, toluene) regardless of long-time storage (over several weeks) of pure powder samples under ambient air conditions. Note that common procedures leading to ligand-free NCs usually require multistep post-synthetic treatment,12,14,79 which significantly decreases their solubility,79 or even causes irreversible aggregation, preventing further redispersion and processability.12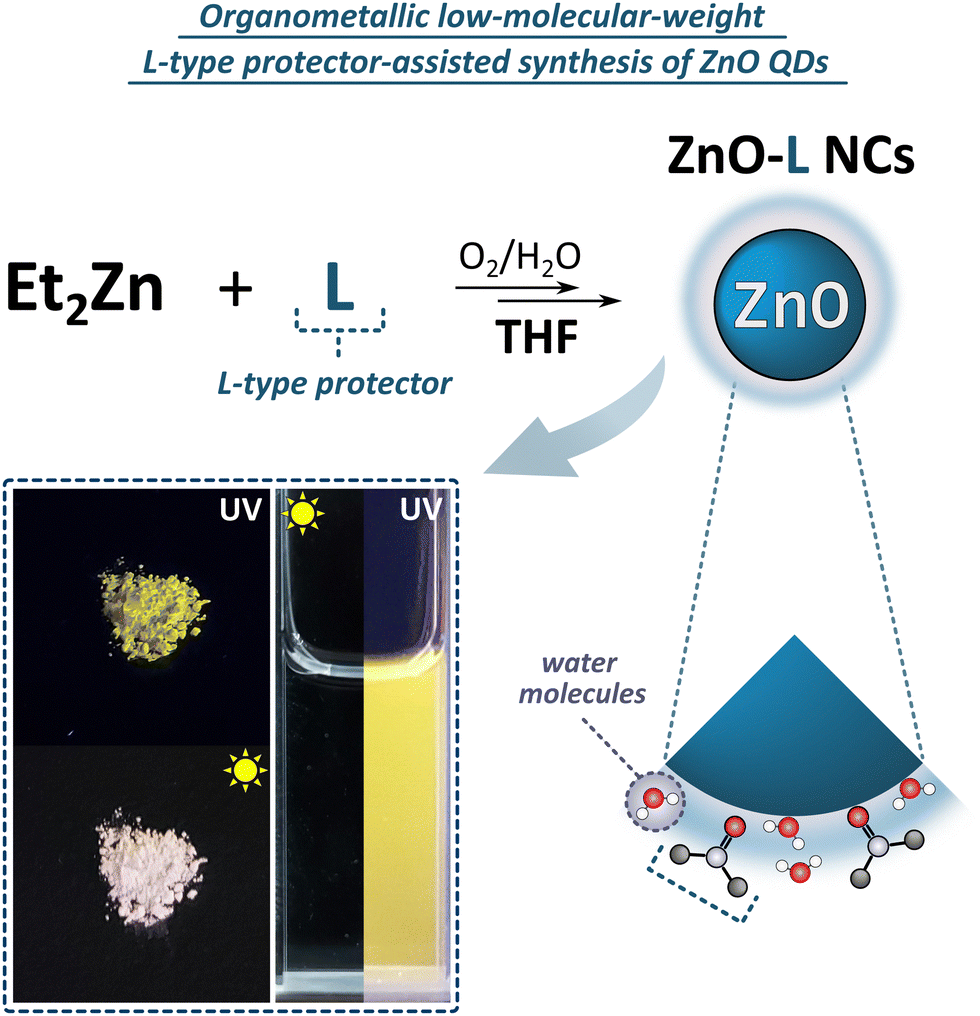 | ||
| Fig. 3 Schematic representation of the L-type-protector-assisted synthetic protocol for the preparation of ZnO QDs with the photographs of the material in the solid state and MeOH solution. | ||
Characterization of sulfoxide-protected ZnO QDs
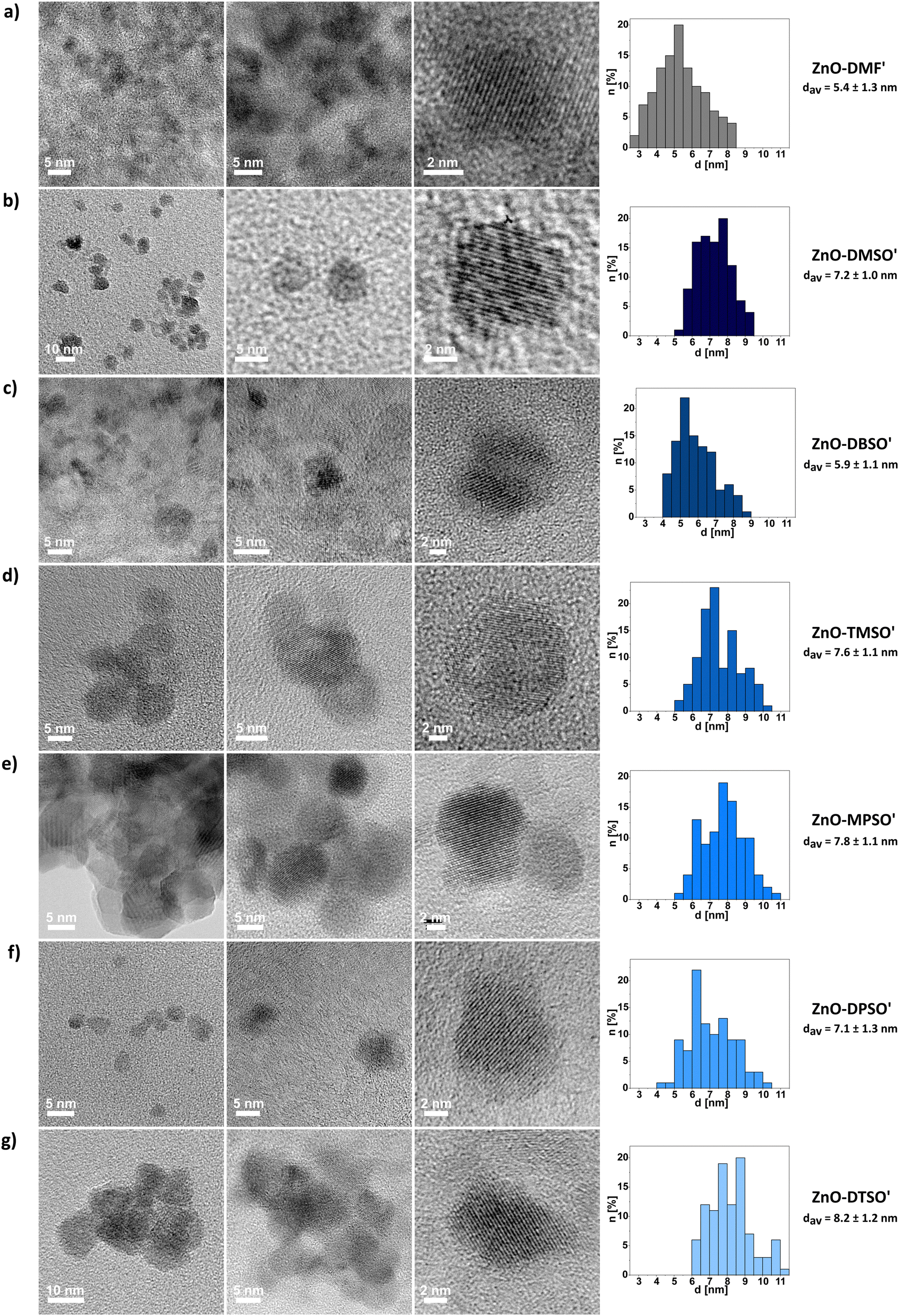 | ||
| Fig. 4 Representative TEM micrographs of (a)–(g) ZnO-DMF′, ZnO-DMSO′, ZnO-DBSO′, ZnO-TMSO′, ZnO-MPSO′, ZnO-DPSO′, ZnO-DTSO′ QDs with their size distributions and average diameters (calculated from at least 100 individual measurements for each sample). | ||
The DLS studies for freshly prepared suspensions of ZnO-DMSO′, ZnO-DBSO′, ZnO-TMSO′, ZnO-MPSO′, ZnO-DPSO′, ZnO-DTSO′ indicate the presence of higher-order soft-type QDs’ associates of 50–80 nm in diameter, appearing in the intensity distribution. In the case of number-based DLS analysis, the results suggest the domination of more separated or individual QDs (solvodynamic diameters within a range of 9–20 nm) in DMSO, confirming their satisfactory colloidal stability. When considering intensity-derived DLS data, the QDs stabilized by aromatic sulfoxides (i.e., ZnO-MPSO′, ZnO-DPSO′ and ZnO-DTSO′) display slightly greater solvodynamic diameters (along with decreasing polydispersity index), which express their propensity to form more stable and higher organized associates (for details, see Fig. S25, ESI†), arising from feasible additional effects involving non-covalent interactions-induced self-assembly.18 The DLS-derived data for ZnO-DMF′ QDs clearly indicate strong aggregation of QDs in solution (with the average solvodynamic diameter of ca. 1 μm, see Fig. S24, ESI†). Despite multiple filtrations, the agglomerate size of ZnO-DMF′ does not decrease, and the solvodynamic diameter remains considerably higher, independent of the distribution type (solvodynamic diameter for number-based distribution equals 254.9 nm). The results may explain the subsequent visible aggregation of ZnO-DMF′ in the solution.
The liquid-phase NMR studies along with FTIR analysis and TGA were performed to better understand the composition of the stabilizing of the L-type protector's coating around the inorganic core. Although the process was conducted in THF solution, all the isolated ZnO QDs are essentially coated only with the corresponding L-type additive. Generally, the 1H NMR spectra of all ZnO-L QDs in DMSO-d6 contain low-intensity proton signals of the characteristic groups in the corresponding sulfoxide ligands, confirming their residual presence on the QDs’ surface (all NMR data is provided in the ESI,† Fig. S26–S32). Moreover, the additional well-resolved singlet at 3.28 ppm, present in all cases, is likely associated with the surface-attached water molecules and strongly supports the concept of a swollen-type organic coating composed of both applied sulfoxide ligands as well as bound and unbound water molecules.47 The FTIR spectra of solid ZnO-L QDs along with the corresponding RZn(L)-type precursors and neat L ligands are shown in Fig. S34–S39 (ESI†). For neat sulfoxides, the bands assigned to high-intensity SO vibration modes are centred within the 1000–1100 cm−1 region) and they are blueshifted to those of the respective sulfoxide-supported QDs (e.g. Δ = 31 cm−1 shift from 1019 cm−1 to 1050 cm−1 frequency for ZnO-DBSO′). Notably, all spectra contain characteristic broad bands around 3300–3500 cm−1 and 1560–1620 cm−1 (e.g. 3340 and 1596 cm−1 for ZnO-DBSO′), designated to stretching vibrations of residual water molecules. The FTIR spectrum of ZnO-DMF′ (Fig. S33, ESI†) reveals a medium-intensity band (at 1558 cm−1) and a broad band around 3300 cm−1 characteristic of the stretching vibrations of water molecules, and a weak band at 1057 cm−1 assigned to the protecting DMF molecules.
Furthermore, the TGA profiles of all sulfoxide-protected ZnO-L QDs showed characteristic ‘crown-like’ derivative curves (Fig. S41, ESI†), with the maximum decomposition rates in the temperature range of 100–400 °C. These complex and multistep decomposition pathways suggest the simultaneous occurrence of various surface-related thermal processes involving the physical desorption of water species and ligand molecules and subsequent decomposition of the residual surface-bound sulfoxide protectors. These results may imply that ZnO-L QDs are solvated by a previously postulated swollen-like shell47 incorporating dissociative water species and ligand molecules, similar to the previously developed ZnO-DMSO.47 It is worth mentioning that for all sulfoxide-supported QDs, the total weight loss does not exceed 15%. For determination of the packing density of organic ligands, the weight loss corresponding to the water content from the TGA profile has been subtracted from the value applied in the equation, showing relatively low organic layer coverage of the QDs’ surface varying from 2 to 5 molecules per nm2 depending on the character of coating ligand (Table S2, ESI†). For example, the packing density for ZnO-DMSO′ and ZnO-DTSO′ is 2 and 5 molecules per nm2, respectively. The difference in separation distances between the sulfoxides can be attributed to the variability of steric demands brought by the alkyl chains and aromatic rings, and the ability of coating ligands to form a ‘swollen’ shell. Despite relatively low grafting density of the organic coating, the prepared QDs possess functionality like luminescence and colloidal stability. Secondly, it seems reasonable that the low grafting density of sulfoxide-coated ZnO QDs substantiates an idealized surface passivation of the resulting QDs47 and corroborates with the presence of aforementioned surface water molecules or hydroxyl groups, which possibly reduces the number of Zn sites available for sulfoxide binding. Notably, as expected, the packing density of sulfoxides is significantly lower to the respective values observed for the OSSOM-derived ZnO QDs with well-passivated organic shell composed of the X-type ligands (e.g. QD coated by carboxylate30 ligands exhibited the packing density of ca. 20 ligands per nm2). In turn, the coating ligand grafting density is within the reported range for sol–gel derived ZnO QDs coated with long-chain-ligand like trioctylphosphine oxide (TOPO) and dodecylamine (DDA) (reaching 2.9 TOPO per nm2,81 and 1.0–3.582 up to 6.781 DDA per nm2). However, such a comparison can be highly misleading. In the case of organometallic-derived QDs the surface is coated solely by an organic ligand and water/hydroxyl molecules, which makes the grafting estimation relatively reliable. In turn, the same cannot be said for ZnO QDs derived from standard sol–gel procedure involving zinc acetate, various alkali hydroxides and alcohols, were the resulting QDs future ZnO QDs with inherently ill-passivated surface and blended coating shell composed of acetate ligands, alcohol and water molecules, and residual OH groups (we would like to emphasize for the benefit of readers that the issue of heterogenous ligand shell in the sol–gel derived QDs is usually neglected in literature).
| Property | Solvent-processed ZnO QDs | L-type-protected ZnO QDs | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ZnO-DMSO47 | ZnO-DMF | ZnO-DBSO | ZnO-DMF′ | ZnO-DMSO′ | ZnO-DBSO′ | ZnO-TMSO′ | ZnO-MPSO′ | ZnO-DPSO′ | ZnO-DTSO | |
| Absorption and emission spectra were collected for ZnO QDs' solutions in DMSO (if not stated otherwise). λab – absorption maximum, λem– emission maximum, FWHM – full width at half maximum of emission peak), PLQY – absolute photoluminescence quantum yield performed in the solid state (excitation wavelength: 350 nm).a Measurement performed in DMF.b Gauss fitting. | ||||||||||
| λ ab [nm] | 330 | 337a/337 | 337 | 337 | 340 | 339 | 342 | 341 | 340 | 341 |
| λ em [nm] | 531 | 534a/541 | 545 | 525 | 540 | 542 | 544 | 544 | 534 | 532 |
| FWHM [nm] | 135 | 112a/115 | 118 | 114 | 135 | 140 | 146 | 133 | 133 | 148b |
| PLQY [%] | ∼10 | 5.6 | 9.1 | 1.7 | 7.8 | 2.8 | 5.1 | 4.7 | 5.2 | 6.2 |
| d av (TEM) [nm] | 4.7 ± 0.8 | 5.4 ± 1.2 | 5.3 ± 0.7 | 5.4 ± 1.3 | 7.2 ± 1.0 | 5.9 ± 1.1 | 7.6 ± 1.1 | 7.8 ± 1.1 | 7.1 ± 1.3 | 8.2 ± 1.2 |
| d av (PXRD) [nm] | 4.41 ± 0.43 | 4.3 ± 0.8 | 5.0 ± 0.6 | 3.6 ± 0.7 | 7.6 ± 0.6 | 6.3 ± 0.5 | 7.4 ± 0.5 | 6.3 ± 0.6 | 6.8 ± 0.7 | 8.2 ± 0.5 |
Photocatalytic activity of ZnO QDs derived from organometallic synthesis in neat DMSO and DMSO-assisted method
At first glance, the developed one-pot organometallic approaches for L-type protector-assisted synthesis of ZnO QDs, i.e. the previously reported synthesis in neat ligand-like solvents47 and the new one based on the finetuning of precursors’ reactivity by Lewis-base chemical additives, appear very similar. They even result in QDs with similar properties regarding the size, optical parameters and solution-stability. However, our control experiments revealed that the resulting QDs dramatically differ in their photocatalytic performance.It is well known that photocatalytic behaviour of nanostructures has been mainly attributed to their method of preparation, and the particle size and the character of surface area.84–86 Thus, we performed a model study on the photodegradation of common organic pollutant, methylene blue (MB), using ZnO QDs as photocatalysts obtained through two different approaches: direct injection of Et2Zn in neat DMSO (previously reported ZnO- DMSO47) or DMSO-assisted method (ZnO-DMSO′). The absorption spectra of photodegraded MB dye are presented in Fig. 5. ZnO-DMSO exhibits excellent photocatalytic activity, promoting almost complete degradation (95%) of MB dye after 90 minutes of UV irradiation (λem = 365 nm) (Fig. 5a). In turn, the photocatalytic ZnO-DMSO′ behaviour is almost negligible and does not exceed 6%. The degradation rates of MB in the presence of ZnO-DMSO and ZnO-DMSO′ were obtained to be 0.0314 and 0.000761 min−1, respectively (Fig. 5c). The photocatalytic performance of ZnO-DMSO is relatively high compared to examples found in literature, including nanostructured ZnO thin films87 calcined85 or doped88 ZnO NCs (note that direct comparison of the results may be misleading due to differences in applied both the methodologies and the character of nanomaterials, e.g. the preparation method, size of nanostructured ZnO, concentration of reagents, light source and its intensity). Therefore, on the basis of our comparison, we may conclude that the applied synthetic procedure affects the both intrinsic and extrinsic properties of the resulting nanomaterials and has great significance in the preparation of QDs with unique characteristics. The above results emphasize the importance of rational design and synthesis of tailored functional nanostructures in terms of future applications.
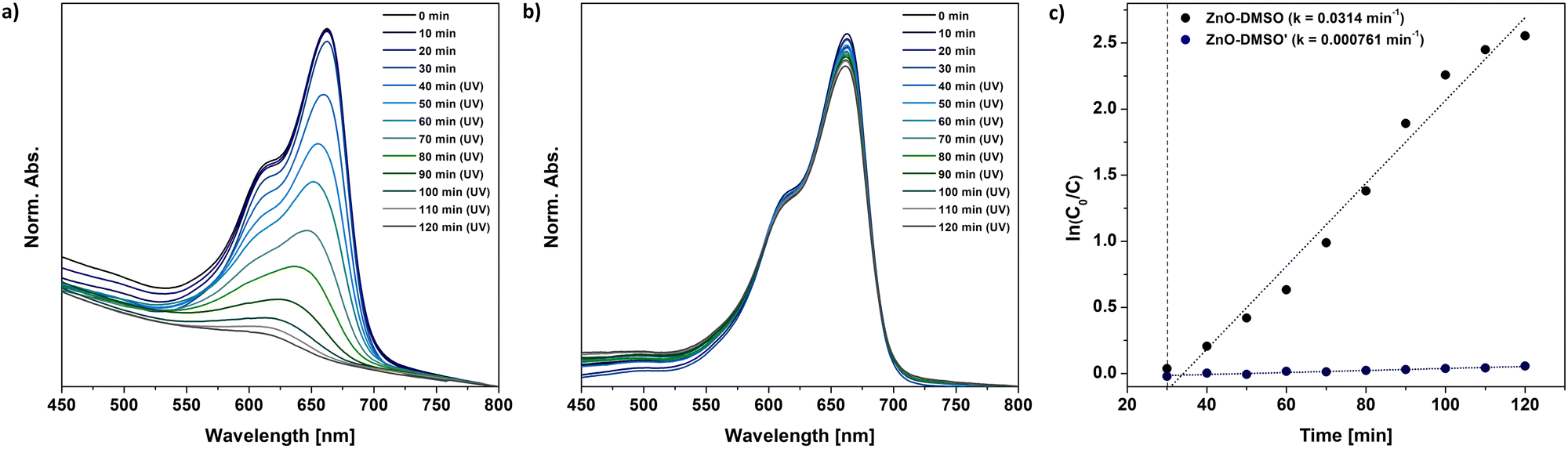 | ||
| Fig. 5 The model photocatalytic degradation of MB under UV light involving (a) – previously reported47 ZnO-DMSO, (b) ZnO-DMSO′ QDs synthesized within the present study, i.e. QDs through the transformation of sulfoxide-modulated Et2Zn-based precursor in THF. (c) First-order kinetics of MB dye degradation under UV light using corresponding ZnO QDs. | ||
Conclusions
The fabrication of colloidal ZnO quantum dots has been a long-standing challenge in the world of modern nanomaterials chemistry and nanoscience. In this perspective, we present a systematic studies on the development of an organometallic route to the preparation of colloidal ZnO QDs coated by small molecules as L-type ligands. The study illustrates the universality of implementing neat liquid sulfoxides for the synthesis of ZnO QDs, providing better stabilization than another type of short-chain L-type ligand (DMF). Moreover, commercially available low-molecular-weight sulfoxides can be implemented as auxiliary ligands (additives) in the in situ generation of Et2Zn-based precursors for the controlled transformation into sulfoxide-capped ZnO QDs under the controlled exposition towards the air. The application of sulfoxides as L-type protectors contributes to the formation of uniform ZnO QDs with low surface grafting density without impeding their colloidal as well as solid-state stability. Small-molecule-coated ZnO QDs exhibit relatively long PL lifetimes (up to 3.2 μs). The study highlights the importance of ligand selection and modulation of precursor reactivity for the rational design of tailored nanostructures. What is more, the established protocols involving small-molecule ligands exclude strongly bonded, interfering stabilizers and do not require multistage post-synthetic treatment. Thus, our systematic studies contribute to the further development of effective approaches to the preparation of insulating ligand-free (and dopant-free) QDs, which do not require multistep post-synthetic treatment and provide stable colloids of QDs. Moreover, preliminary control experiments revealed dramatic differences in the photocatalytic performance of the selected ZnO QDs in the photodegradation of methylene blue, emphasizing the significant impact of the synthetic procedure on the final (intrinsic and extrinsic) properties of the title nanomaterials. Further studies on the functionality and processability of as-prepared ZnO QD as well as the possibility of all-solution device fabrication are in progress.Author contributions
The manuscript was written through the contributions of all authors. All of the authors discussed the results and participated in writing the final version of the manuscript and have given approval to the final version of the manuscript. M. W.-P. proposed the research direction, supervised the project conceptually and experimentally and helped with the interpretation of the results. M. J. participated in the design of the research, performed the syntheses and sample preparation for HR TEM, PXRD, FTIR, DLS, UV-Vis, PL, NMR, TGA characterization as well as performed the PXRD, FTIR, DLS, UV-Vis, PL measurements, including all data collection and further analysis and interpretation. Z. D. performed syntheses of ZnO-DMF and ZnO-DMF′ QDs along with their characterization, including PXRD, FTIR, DLS, UV-Vis, PL, and TGA. J. L. supervised the overall project and the preparation of the manuscript, including the interpretation of the results and participated in finalizing the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the National Science Centre, Grants MAESTRO 11, No. 2019/34/A/ST5/00416 (J. L., M. J., Z. D.) and PRELUDIUM 19, No. 2020/37/N/ST5/01969 (M. J.). Research was cofunded by POB Technologie Materiałowe of Warsaw University of Technology within the Excellence Initiative: Research University (IDUB) programme (M. W.-P., M. J.). We also thank Vadim Szejko for assistance with elemental analysis.References
- M. L. Steigerwald and L. E. Brus, Acc. Chem. Res., 1990, 23, 183–188 CrossRef CAS.
- A. L. Efros and L. E. Brus, ACS Nano, 2021, 15, 6192–6210 CrossRef CAS PubMed.
- H. Morkoç and U. Özgür, Zinc oxide: fundamentals, materials and device technology, Wiley-VCH, 2009 Search PubMed.
- J. Van Embden, S. Gross, K. R. Kittilstved and E. Della Gaspera, Chem. Rev., 2023, 123, 271–326 CrossRef CAS PubMed.
- L. Spanhel and M. A. Anderson, J. Am. Chem. Soc., 1991, 113, 2826–2833 CrossRef CAS.
- E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566–5572 CrossRef CAS.
- T. Schindler, M. Schmiele, T. Schmutzler, T. Kassar, D. Segets, W. Peukert, A. Radulescu, A. Kriele, R. Gilles and T. Unruh, Langmuir, 2015, 31, 10130–10136 CrossRef CAS PubMed.
- B. L. Caetano, V. Briois, S. H. Pulcinelli, F. Meneau and C. V. Santilli, J. Phys. Chem. C, 2017, 121, 886–895 CrossRef CAS.
- P. G. Baranov, S. B. Orlinskii, C. de Mello Donegá and J. Schmidt, Appl. Magn. Reson., 2010, 39, 151–183 CrossRef CAS PubMed.
- A. Asok, M. N. Gandhi and A. R. Kulkarni, Nanoscale, 2012, 4, 4943 RSC.
- A. Grala, M. Wolska-Pietkiewicz, W. Danowski, Z. Wróbel, J. Grzonka and J. Lewiński, Chem. Commun., 2016, 52, 7340–7343 RSC.
- T. Schindler, T. Schmutzler, M. Schmiele, W. Lin, D. Segets, W. Peukert, M. S. Appavou, A. Kriele, R. Gilles and T. Unruh, J. Colloid Interface Sci., 2017, 504, 356–362 CrossRef CAS PubMed.
- D. Lee, M. Wolska-Pietkiewicz, S. Badoni, A. Grala, J. Lewiński and G. De Paëpe, Angew. Chem., Int. Ed., 2019, 58, 17163–17168 CrossRef CAS PubMed.
- M. Gong, Q. Liu, B. Cook, B. Kattel, T. Wang, W.-L. Chan, D. Ewing, M. Casper, A. Stramel and J. Z. Wu, ACS Nano, 2017, 11, 4114–4123 CrossRef CAS PubMed.
- W. Zhang, X. Chen, Y. Ma, Z. Xu, L. Wu, Y. Yang, S. W. Tsang and S. Chen, J. Phys. Chem. Lett., 2020, 11, 5863–5870 CrossRef CAS PubMed.
- Y. Sun, P. D. Donaldson, J. Garcia-Barriocanal and S. L. Swisher, J. Mater. Chem. C, 2018, 6, 9181–9190 RSC.
- N. Li, F. Meng, F. Huang, G. Yu, Z. Wang, J. Yan, Y. Zhang, Y. Ai, C. Shou, Y. Zeng, J. Sheng, B. Yan and J. Ye, ACS Appl. Energy Mater., 2020, 3, 9610–9617 CrossRef CAS.
- O. Lupan, N. Magariu, R. Khaledialidusti, A. K. Mishra, S. Hansen, H. Krüger, V. Postica, H. Heinrich, B. Viana, L. K. Ono, B. R. Cuenya, L. Chow, R. Adelung and T. Pauporté, ACS Appl. Mater. Interfaces, 2021, 13, 10537–10552 CrossRef CAS PubMed.
- M. Shim and P. Guyot-Sionnest, Nature, 2000, 407, 981–983 CrossRef CAS PubMed.
- M. L. Kahn, M. Monge, V. Collière, F. Senocq, A. Maisonnat and B. Chaudret, Adv. Funct. Mater., 2005, 15, 458–468 CrossRef CAS.
- Y. Coppel, G. Spataro, C. Pagès, B. Chaudret, A. Maisonnat and M. L. Kahn, Chem. – Eur. J., 2012, 18, 5384–5393 CrossRef CAS PubMed.
- M. Terlecki, S. Badoni, M. K. Leszczyński, S. Gierlotka, I. Justyniak, H. Okuno, M. Wolska-Pietkiewicz, D. Lee, G. De Paëpe and J. Lewiński, Adv. Funct. Mater., 2021, 31, 2105318 CrossRef CAS.
- E. Chwojnowska, J. Grzonka, I. Justyniak, T. Ratajczyk and J. Lewiński, iScience, 2023, 26(1), 105759 CrossRef CAS PubMed.
- K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Mater., 2012, 24, 2443–2448 CrossRef CAS.
- J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, Z. Wróbel, M. Wadowska, K. Matuła, I. Dzięcielewski, D. Pociecha, J. Smalc-Koziorowska, J. Lewiński and R. Hołyst, Chem. – Eur. J., 2015, 21, 16941–16947 CrossRef CAS PubMed.
- A. M. Cieślak, M. V. Pavliuk, L. D’Amario, M. Abdellah, K. Sokołowski, U. Rybinska, D. L. A. Fernandes, M. K. Leszczyński, F. Mamedov, A. M. El-Zhory, J. Föhlinger, A. Budinská, M. Wolska-Pietkiewicz, L. Hammarström, J. Lewiński and J. Sá, Nano Energy, 2016, 30, 187–192 CrossRef.
- J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, M. Wadowska, Z. Wróbel, K. Matuła, W. Nogala, J. Lewiński and R. Hołyst, ACS Appl. Mater. Interfaces, 2016, 8, 13532–13541 CrossRef CAS PubMed.
- M. Wolska-Pietkiewicz, A. Grala, I. Justyniak, D. Hryciuk, M. Jędrzejewska, J. Grzonka, K. J. Kurzydłowski and J. Lewiński, Chem. – Eur. J., 2017, 23, 11856–11865 CrossRef CAS PubMed.
- E. Chwojnowska, M. Wolska-Pietkiewicz, J. Grzonka and J. Lewiński, Nanoscale, 2017, 9, 14782–14786 RSC.
- M. Wolska-Pietkiewicz, K. Tokarska, A. Grala, A. Wojewódzka, E. Chwojnowska, J. Grzonka, P. J. Cywiński, K. Kruczała, Z. Sojka, M. Chudy and J. Lewiński, Chem. – Eur. J., 2018, 24, 4033–4042 CrossRef CAS PubMed.
- M. Wolska-Pietkiewicz, K. Tokarska, A. Wojewódzka, K. Wójcik, E. Chwojnowska, J. Grzonka, P. J. Cywiński, M. Chudy and J. Lewiński, Sci. Rep., 2019, 9, 18071 CrossRef CAS PubMed.
- M. Monge, M. L. Kahn, A. Maisonnat and B. Chaudret, Angew. Chem., Int. Ed., 2003, 42, 5321–5324 CrossRef CAS PubMed.
- M. L. Kahn, T. Cardinal, B. Bousquet, M. Monge, V. Jubera and B. Chaudret, ChemPhysChem, 2006, 7, 2392–2397 CrossRef CAS PubMed.
- C. Pagès, Y. Coppel, M. L. Kahn, A. Maisonnat and B. Chaudret, ChemPhysChem, 2009, 10, 2334–2344 CrossRef PubMed.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37, 3374 RSC.
- A. Grala, M. Wolska-Pietkiewicz, Z. Wróbel, T. Ratajczyk, J. Kuncewicz and J. Lewiński, Mater. Chem. Front., 2018, 2, 1104–1111 RSC.
- P. Krupiński, A. Grala, M. Wolska-Pietkiewicz, W. Danowski, I. Justyniak and J. Lewiński, ACS Sustainable Chem. Eng., 2021, 9, 1540–1549 CrossRef.
- P. Krupiński, A. Kornowicz, K. Sokołowski, A. M. Cieślak and J. Lewiński, Chem. – Eur. J., 2016, 22, 7817–7823 CrossRef PubMed.
- M. L. Kahn, M. Monge, E. Snoeck, A. Maisonnat and B. Chaudret, Small, 2005, 1, 221–224 CrossRef PubMed.
- D. Bera, L. Qian, S. Sabui, S. Santra and P. H. Holloway, Opt. Mater., 2008, 30, 1233–1239 CrossRef CAS.
- M. V. Kovalenko, M. Scheele and D. V. Talapin, Science, 2009, 324, 1417–1420 CrossRef CAS PubMed.
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Science, 2016, 353, 6302, DOI:10.1126/science.aac5523.
- W. Wang, M. Zhang, Z. Pan, G. M. Biesold, S. Liang, H. Rao, Z. Lin and X. Zhong, Chem. Rev., 2022, 122, 4091–4162 CrossRef CAS PubMed.
- D. Prochowicz, M. M. Tavakoli, M. Wolska-Pietkiewicz, M. Jędrzejewska, S. Trivedi, M. Kumar, S. M. Zakeeruddin, J. Lewiński, M. Graetzel and P. Yadav, Sol. Energy, 2020, 197, 50–57 CrossRef CAS.
- J. Lewiński, M. Wolska-Pietkiewicz and M. Jędrzejewska, PL Pat., 238480, 2021 Search PubMed.
- R. D. Chavan, M. Wolska-Pietkiewicz, D. Prochowicz, M. Jędrzejewska, M. M. Tavakoli, P. Yadav, C. K. Hong and J. Lewiński, Adv. Funct. Mater., 2022, 32, 2205909 CrossRef CAS.
- M. Wolska-Pietkiewicz, M. Jędrzejewska, K. Tokarska, J. Wielgórska, M. Chudy, J. Grzonka and J. Lewiński, Chem. Eng. J., 2023, 455, 140497 CrossRef CAS.
- M. V. Pavliuk, A. M. Cieślak, M. Abdellah, A. Budinská, S. Pullen, K. Sokołowski, D. L. A. Fernandes, J. Szlachetko, E. L. Bastos, S. Ott, L. Hammarström, T. Edvinsson, J. Lewiński and J. Sá, Sustainable Energy Fuels, 2017, 1, 69–73 RSC.
- A. M. Cieślak, E.-R. Janeček, K. Sokołowski, T. Ratajczyk, M. K. Leszczyński, O. A. Scherman and J. Lewiński, Nanoscale, 2017, 9, 16128–16132 RSC.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- J. J. Calvin, A. S. Brewer and A. P. Alivisatos, Nat. Synth., 2022, 1, 127–137 CrossRef.
- Y. Chen, S. Yu, X. B. Fan, L. Z. Wu and Y. Zhou, J. Mater. Chem. A, 2023, 11, 8497–8514 RSC.
- J. A. Clark, A. Murray, J. M. Lee, T. S. Autrey, A. D. Collord and H. W. Hillhouse, J. Am. Chem. Soc., 2019, 141, 298–308 CrossRef CAS PubMed.
- J. Park, A. Jayaraman, A. W. Schrader, G. W. Hwang and H. S. Han, Nat. Commun., 2020, 11, 5748 CrossRef CAS PubMed.
- R. Elbaum, S. Vega and G. Hodes, Chem. Mater., 2001, 13, 2272–2280 CrossRef CAS.
- G. Rodríguez-Gattorno, P. Santiago-Jacinto, L. Rendon-Vázquez, J. Németh, I. Dékány and D. Díaz, J. Phys. Chem. B, 2003, 107, 12597–12604 CrossRef.
- M. E. Wankhede and S. K. Haram, Chem. Mater., 2003, 15, 1296–1301 CrossRef CAS.
- D. Guin, S. V. Manorama, S. Radha and A. Nigam, Bull. Mater. Sci., 2006, 29, 617–621 CrossRef CAS.
- J. Ishida, M. Nakatsuji, T. Nagata, H. Kawasaki, T. Suzuki and Y. Obora, ACS Omega, 2020, 5, 9598–9604 CrossRef CAS PubMed.
- A. E. Raevskaya, Y. V. Panasiuk, O. L. Stroyuk, S. Y. Kuchmy, A. G. Milekhin, L. L. Sveshnikova, T. A. Duda, N. A. Yeryukov and E. E. Rodyakina, Theor. Exp. Chem., 2016, 51, 358–365 CrossRef CAS.
- J. Németh, G. Rodríguez-Gattorno, D. Díaz, A. R. Vázquez-Olmos and I. Dékany, Langmuir, 2004, 20, 2855–2860 CrossRef PubMed.
- T. Nagata and Y. Obora, ACS Omega, 2020, 5, 98–103 CrossRef CAS PubMed.
- S. Repp and E. Erdem, Spectrochim. Acta, Part A, 2016, 152, 637–644 CrossRef CAS PubMed.
- S. Repp, S. Weber and E. Erdem, J. Phys. Chem. C, 2016, 120, 25124–25130 CrossRef CAS.
- S. Nadupalli, S. Repp, S. Weber and E. Erdem, Nanoscale, 2021, 13, 9160–9171 RSC.
- M. H. Aleinawi, A. U. Ammar, M. Buldu-Akturk, N. S. Turhan, S. Nadupalli and E. Erdem, J. Phys. Chem. C, 2022, 126, 4229–4240 CrossRef CAS.
- N. S. Venkataramanan, J. Mol. Model., 2016, 22, 151 CrossRef PubMed.
- K.-I. Oh and C. R. Baiz, J. Phys. Chem. B, 2018, 122, 5984–5990 CrossRef CAS PubMed.
- M. Katayama, M. Shinoda, K. Ozutsumi, S. Funahashi and Y. Inada, Anal. Sci., 2012, 28, 103–106 CrossRef CAS PubMed.
- K. I. Oh, X. You, J. C. Flanagan and C. R. Baiz, J. Phys. Chem. Lett., 2020, 11, 1903–1908 CrossRef CAS PubMed.
- Z. Zhao, Y. Coppel, J. Fitremann, P. Fau, C. Roux, C. Lepetit, P. Lecante, J.-D. Marty, C. Mingotaud and M. L. Kahn, Chem. Mater., 2018, 30, 8959–8967 CrossRef CAS.
- Z. Zhao, Y. Wang, C. Delmas, C. Mingotaud, J. D. Marty and M. L. Kahn, Nanoscale Adv., 2021, 3, 6696–6703 RSC.
- K.-H. Thiele, Z. Anorg. Allg. Chem., 1962, 319, 183–195 CrossRef CAS.
- P. R. Markies, G. Schat, O. S. Akkerman, F. Bickelhaupt and A. L. Spek, J. Organomet. Chem., 1992, 430, 1–13 CrossRef CAS.
- W. Brüser, L. Hilfert, V. Lorenz, C. G. Hrib, K. Bode, A. Adam, J. Vogt and F. T. Edelmann, J. Organomet. Chem., 2016, 806, 77–82 CrossRef.
- J. Lewiński, W. Śliwiński, M. Dranka, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 4826–4829 CrossRef PubMed.
- J. Lewiński, M. Dranka, W. Bury, W. Śliwiński, I. Justyniak and J. Lipkowski, J. Am. Chem. Soc., 2007, 129, 3096–3098 CrossRef PubMed.
- D. Prochowicz, K. Sokołowski and J. Lewiński, Coord. Chem. Rev., 2014, 270–271, 112–126 CrossRef CAS.
- J. A. Pan, H. Wu, A. Gomez, J. C. Ondry, J. Portner, W. Cho, A. Hinkle, D. Wang and D. V. Talapin, ACS Nano, 2022, 16, 16067–16076 CrossRef CAS PubMed.
- Z. Zhao, Z. Zheng, C. Roux, C. Delmas, J.-D. Marty, M. L. Kahn and C. Mingotaud, Chem. – Eur. J., 2016, 22, 12424–12429 CrossRef CAS PubMed.
- N. S. Norberg and D. R. Gamelin, J. Phys. Chem. B, 2005, 109, 20810–20816 CrossRef CAS PubMed.
- C. N. Valdez, A. M. Schimpf, D. R. Gamelin and J. M. Mayer, ACS Nano, 2014, 8, 9463–9470 CrossRef CAS PubMed.
- A. Layek, B. Manna and A. Chowdhury, Chem. Phys. Lett., 2012, 539–540, 133–138 CrossRef CAS.
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei, J. Zhu and Y. Zhu, Inorg. Chem., 2007, 46, 6675–6682 CrossRef CAS PubMed.
- N. S. Ferreira, J. M. Sasaki, R. S. Silva JR, J. M. Attah-Baah and M. A. Macêdo, Inorg. Chem., 2021, 60, 4475–4496 CrossRef CAS PubMed.
- E. Kusiak-Nejman, J. Wojnarowicz, A. W. Morawski, U. Narkiewicz, K. Sobczak, S. Gierlotka and W. Lojkowski, Appl. Surf. Sci., 2021, 541, 148416 CrossRef CAS.
- A. Kulis-Kapuscinska, M. Kwoka, M. A. Borysiewicz, T. Wojciechowski, N. Licciardello, M. Sgarzi and G. Cuniberti, Nanotechnology, 2023, 34(15), 155702 CrossRef PubMed.
- D. G. Ayu, S. Gea, N. Andriayani, D. J. Telaumbanua, A. F. R. Piliang, M. Harahap, Z. Yen, R. Goei and A. I. Y. Tok, ACS Omega, 2023, 8, 14965–14984 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02114a |
| This journal is © The Royal Society of Chemistry 2023 |