
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable electroactive oligothiophene-naphthalimide semiconductors via end-capped engineering: cumulative effects beyond the linker†
Matías J.
Alonso-Navarro‡
 ab,
Alexandra
Harbuzaru‡
c,
Raúl
González-Núñez
c,
M. Mar
Ramos
b,
José L.
Segura
*a and
Rocío
Ponce Ortiz
*c
ab,
Alexandra
Harbuzaru‡
c,
Raúl
González-Núñez
c,
M. Mar
Ramos
b,
José L.
Segura
*a and
Rocío
Ponce Ortiz
*c
aDepartment of Organic Chemistry, Complutense University of Madrid, Faculty of Chemistry, Madrid 28040, Spain. E-mail: segura@ucm.es
bChemical and Environmental Technology Department, Univ. Rey Juan Carlos, Móstoles, 28933, Spain
cDepartment of Physical Chemistry, University of Málaga, Málaga, 29071, Spain. E-mail: rocioponce@uma.es
First published on 21st July 2023
Abstract
Aiming to obtain novel functional semiconducting materials for their use in organic electronics, the combination of strong donor moieties with electron-withdrawing units is one of the most useful strategies to obtain ambipolar semiconductors with tunable properties. Nowadays most of the efforts headed to efficient materials are based on small changes in the alkyl pendant chains or by replacing single atoms. However, a precise design of new functional materials is still challenging. For this reason, in this work we present a new synthetic approach for achieving redox amphoteric organic semiconductors by tuning their opto-electrochemical properties via rational chemical modifications. All these materials present low-lying LUMO levels, lower than −4.00 eV with broad absorption up to 800 nm in the UV-Vis-NIR spectra. In addition, they have been characterized by DFT, absorption and Raman vibrational spectroscopies, while their charge stabilization abilities are studied by means of spectroelectrochemical techniques. The results point out to a quite complex electronic scenario that goes beyond the expected cumulative effects of the independent molecular units constituting the final molecular assembly.
1. Introduction
Organic semiconducting materials have demonstrated to be a good alternative for metal-based semiconductors1 due to their good performance, low-cost, good processability and their tunable structure and properties in optoelectronic2,3 applications like organic light-emitting diodes (OLEDs),4,5 organic solar cells (OSCs),6,7 organic photodetectors (OPD)8 and organic field-effect transistors (OFETs).9–12 One of the key parameters to check the suitability for the application of organic semiconductors in optoelectronic applications is the charge carrier mobility, which is normally measured in an organic field-effect transistor device.13,14 This parameter is affected by different factors such as molecular structure, orbital energy levels or molecular packing.15–17 However, following the advances in the production of new organic π-conjugated hole-conducting (p-type) materials, there is still a need to develop novel n-type (electron-conducting) and ambipolar organic semiconductors.18 For this reason, the design and development of ambient stable and high-performance n-type organic semiconductor materials is still challenging and desirable.19In order to obtain these organic electron transporters, designing π-conjugated electron deficient systems with planar backbones functionalized with solubilizing alkyl chains is one of the best strategies nowadays.10,20–22 This strategy has been used in the last years in order to obtain suitable acceptor–donor–acceptor (A–D–A) fullerene-free architectures with remarkable performances in OFETs23,24 and OSCs.24,25 In this regard, rylenimide derivatives have been described as one of the best choices for efficient n-type organic materials in OFETs, due to their good stability, easy functionalization and tunable optoelectronic properties.26,27 Among them, naphthalimide-based assemblies have been employed as electron acceptors due to their electron-deficient backbone and optimal LUMO levels, tunable absorption range, supramolecular organization properties28,29 and remarkable good transport properties for their single-crystal structures.30
With the aim of obtaining n-type or ambipolar semiconductor materials, Segura and coworkers have developed different series of donor–acceptor molecular assemblies based on oligothiophene-naphthalimide moieties (Fig. 1a) connected through different planar and conjugated linkers.31 This strategy has allowed a precise control over their frontier molecular orbital levels, which was reflected in different optical, electrochemical and also electrical behavior.32–34 In a recent contribution, we have demonstrated that the introduction of different end-capped moieties in pyrazine-based oligothiophene-naphthalimide architectures (Fig. 1b) provides an effectively method to tune the frontier orbital levels and the optoelectronic properties of these novel organic semiconductor materials without modifying the central molecular skeleton.35
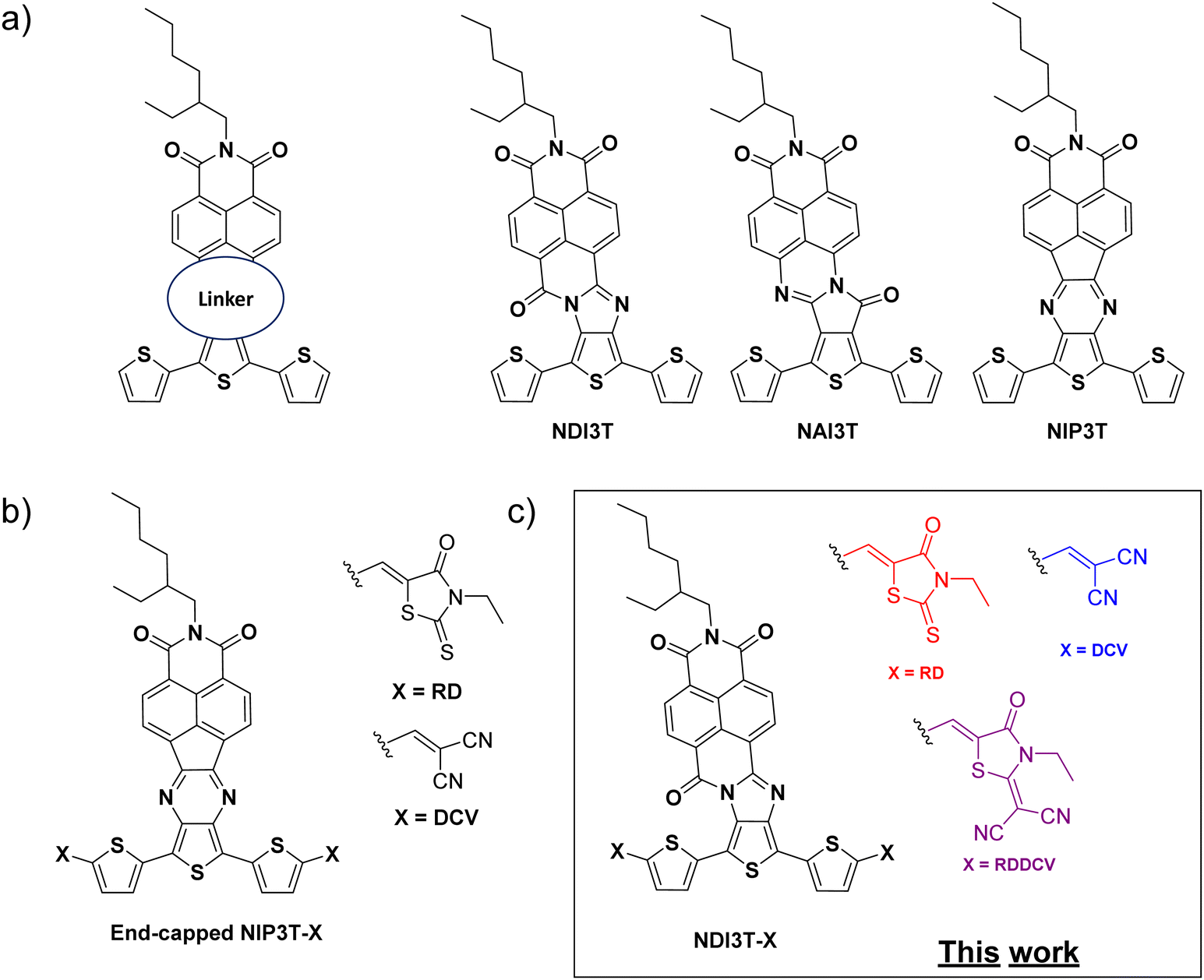 | ||
| Fig. 1 (a) Oligothiophene-naphthalimide assemblies connected through different linkers. (b) Previous published end-capped thienopyrazine derivatives with A2–D–A1–D–A2 architectures and (c) new end-capped oligothiophene-naphthalimide assemblies connected via imidazole linkers reported in this work. | ||
Encouraged by these aforementioned results, we report herein the synthesis of a new family of processable terthiophene–naphthalimide assemblies (Fig. 1c), in which the introduction of an imidazole moiety with a carbonyl group in the alpha position of the central nitrogen of the linker (also known as amidine), makes them a stronger electron–acceptor linker than the previous pyrazine connector (Fig. 1b), and three different strong electron-withdrawing end-capped units yields these novel organic semiconductors with tunable optoelectrochemical properties without modifying the location and topology of the frontier molecular orbitals. The comparison with the previously reported end-capped thienopyrazine derivatives (Fig. 1b) will allow the understanding of the effect of different connector units between the naphthalimide cores and the terthiophene units in the optical and electronic properties of these materials.
2. Results and discussion
The syntheses of D–A assemblies based on oligothiophenes have been widely used for the development of ambipolar organic semiconductors due to their tunable photophysical and electrochemical properties.36,37 In this context, oligothiophene-naphthalimide assemblies are particularly interesting due to the good properties of naphthalimide systems as stable n-type organic semiconductors.38,39In this work, we report the synthesis and characterization of a novel family of organic semiconductors based on 1,8-naphthalimide moieties connected to end-capped oligothiophene fragments through imidazole linkers, as it is depicted in Scheme 1. These chemical modifications pave the way to donor–acceptor materials in which their optical and electrochemical properties can be controlled by tuning the electronic energy levels. These novel assemblies have an A2–D–A1–D–A2 architecture, due to the strong electron acceptor groups introduced in the terminal position of the thienoimidazoline skeleton, being a strategy widely used nowadays to obtain novel functional materials.40–43
 | ||
| Scheme 1 (a) Synthetic route to obtain the novel asymmetric terthiophene–naphthalimide assemblies. (b) Different end-capped A2–D–A1–D–A2 derivatives obtained in this work. | ||
The synthetic route to obtain these end-capped derivatives based on naphthalimide–terthiophene assemblies starts with the condensation of the naphthalimide anhydride derivative NIA44 and the [2,2′:5′,2′′-terthiophene]-3′,4′-diamine 1.45 This condensation reaction allows the formation of the naphthalimide–terthiophene assembly NDI3T with an imidazole linker in its structure.46 The subsequent Vilsmeier–Haack reaction carried out for NDI3T allowed the formylation of the alpha positions of the terthiophene moieties, obtaining NDI3T-CHO in high yields. This formylated derivative can be further functionalized by a Knoevenagel-like reactions with different electron acceptor units such as 3-ethyl-2-thioxothiazolidin-4-one (RD), malononitrile (DCV) and 2-(3-ethyl-4-oxothiazolidin-2-ylidene) malononitrile (RDDCV) to afford the end-capped target molecules NDI3T-RD, NDI3T-DCV and NDI3T-RDDCV.
Due to the relatively good solubility of these oligothiophene-naphthalimide assemblies its structural characterization has been accomplished by nuclear magnetic resonance experiments, Fourier transform infrared spectroscopy analyses and their molecular weights have been determined by matrix-assisted laser desorption/ionization techniques (complete characterization can be found in the ESI,† Fig. S1–S18). However, due to limited solubility of dicyanovinylene-based semiconductors NDI3T-DCV and NDI3T-RDDCV13C-NMR spectra could not be recorded. In addition to this, the thermal stability of these novel assemblies has been analyzed by thermogravimetric analyses, indicating that NDI3T, NDI3T-RD, NDI3T-DCV and NDI3T-RDDCV exhibit favorable thermal stabilities (Fig. S19, ESI†) with decomposition temperatures, Td (weight loss <10%), higher that 300 °C.
Molecular properties
The optimization of the lowest energy molecular structures of the studied semiconductors shows, in contrast to the previously studied and basically planar NIP3T-X systems (Fig. 2 and Fig. S20, ESI†),35 a not coplanar configuration presenting a dihedral angle between two consecutive thiophene rings of approximately 140°. This fact indicates that the election of the central group (pyrazine versus imidazole, see Fig. 1b and c) is crucial from a molecular point of view, in terms of molecular structure. In addition, the modification of the connecting group can actively influence on the tuning of the optoelectronic properties of these organic semiconductors, as it will be seen below.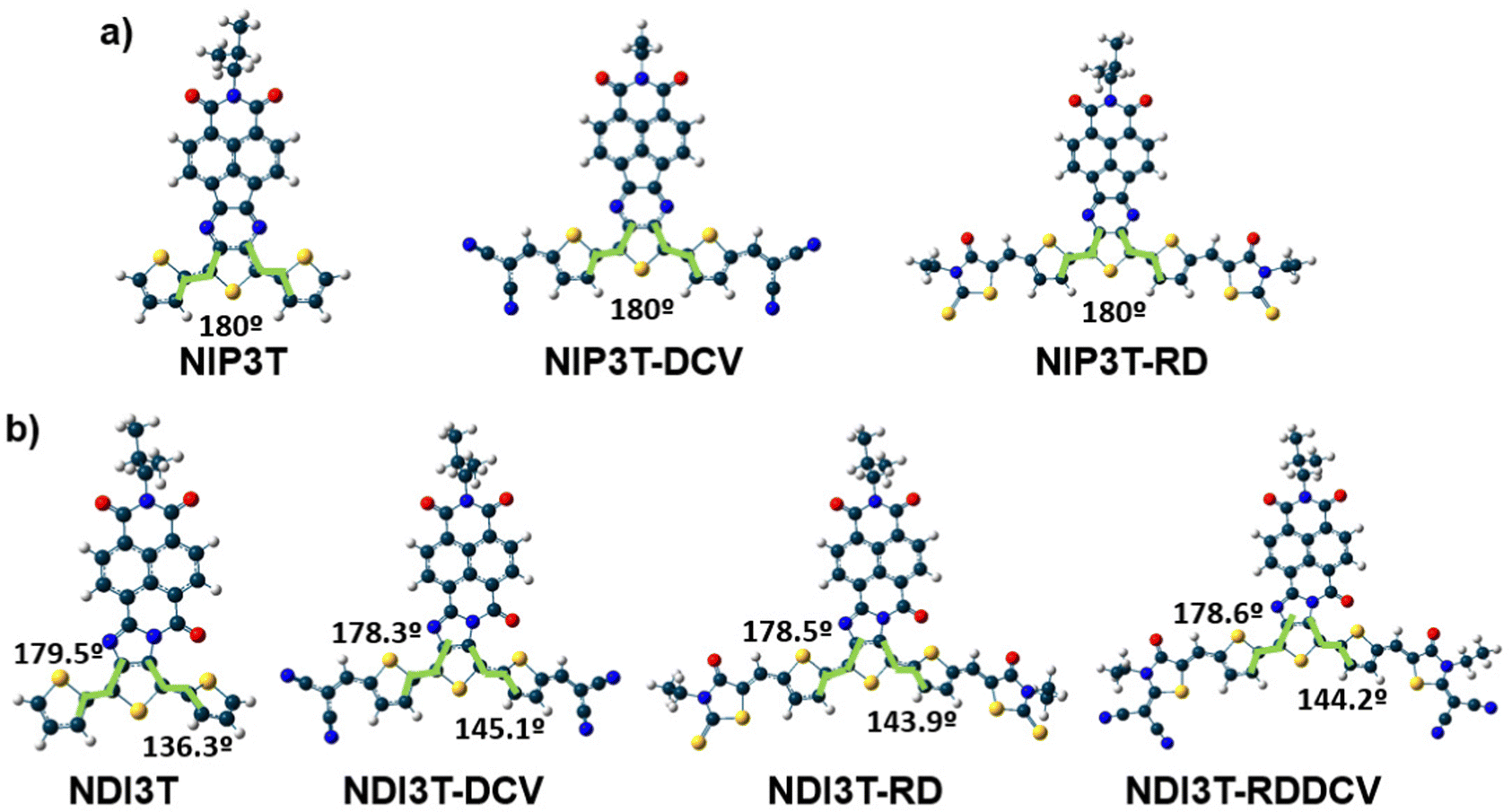 | ||
| Fig. 2 DFT/B3LYP/6-31G** optimized molecular structures for the systems under study: (a) for NIP3T derivatives and (b) for NIP3T derivatives. | ||
FT-Raman spectra were thus recorded to analyze the effect of the lateral substituents on π-conjugation and molecular properties, and the data is shown in Fig. 3. For this end, we make use of the ECC theory,47,48 which has been widely probed to efficiently account for the degree of conjugation of molecular materials in a qualitative way,49 and has been previously applied to naphthalene-modified oligothiophenes.50
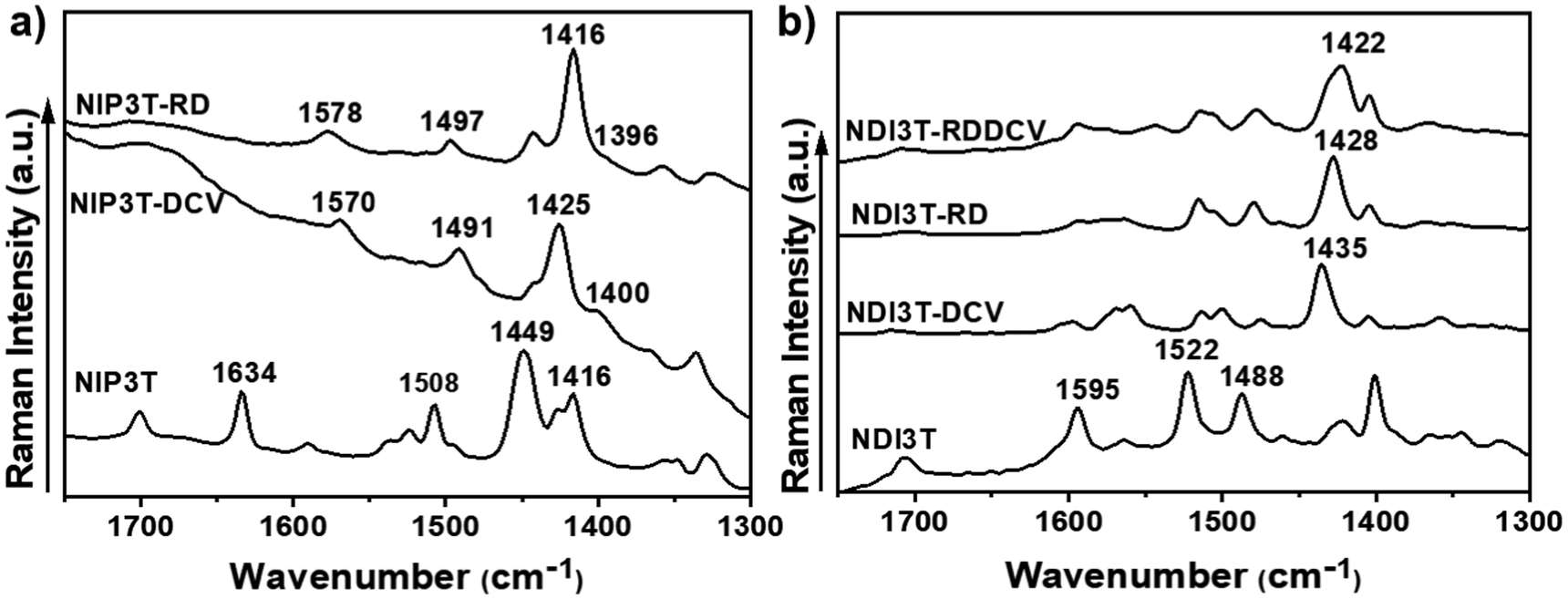 | ||
| Fig. 3 FT-Raman spectra (λ = 1064 nm) of the (a) NIP3T-X and (b) NDI3T-X systems as bulk materials. | ||
Considering the Raman spectra in Fig. 3b and the selected eigenvectors in Fig. S30 (ESI†), we focus on the Raman vibration modes of NDI3T-X systems which are ascribed to the collective totally-symmetric ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) vibration mode of the oligothiophene fragment, whose position can account for the molecular π-conjugation extent in that fragment. This Raman vibration appears at 1488 cm−1 in NDI3T and significantly downshifts upon inclusion of electron-withdrawing groups, being recorded at 1435 cm−1 in NDI3T-DCV, 1428 cm−1 in NDI3T-RD and 1422 cm−1 in NDI3T-RDDCV. This remarkable downshift, up to 66 cm−1 in NDI3T-RDDCV, indicates an effective conjugation extension by the insertion of electron-withdrawing groups.
C) vibration mode of the oligothiophene fragment, whose position can account for the molecular π-conjugation extent in that fragment. This Raman vibration appears at 1488 cm−1 in NDI3T and significantly downshifts upon inclusion of electron-withdrawing groups, being recorded at 1435 cm−1 in NDI3T-DCV, 1428 cm−1 in NDI3T-RD and 1422 cm−1 in NDI3T-RDDCV. This remarkable downshift, up to 66 cm−1 in NDI3T-RDDCV, indicates an effective conjugation extension by the insertion of electron-withdrawing groups.
In addition, the comparison with the previously published NIP3T systems34 indicates a larger π-conjugation extent of the oligothiophene fragment in the NIP derivatives, which is highlighted by the downshift of the aforementioned collective totally-symmetric ν(CC) vibration mode from 1488 in NDI3T to 1449 cm−1 in NIP3T. This extended π-conjugation of the NIP derivative can be explained by the planarization of the thiophenic skeleton on the NIP system due to the absence of repulsive S–O interactions (found in NDI systems), and the appearance of attractive S–N interactions. However, the introduction of the electron-withdrawing lateral substituents on the NIP skeleton does not have such a pronounced effect as in the case of NDI derivatives.35 While a 33 cm−1 downshift is recorded on passing from NIP3T to NIP3T-RD, the same lateral modification entails a 60 cm−1 downshift of the collective totally-symmetric ν(CC) vibration mode for the NDI derivatives. Therefore, the effect of the lateral substituents on conjugational degree is found to be more pronounced for the NDI-based systems, due to the terthiophene planarization, up to 9°, upon substitution with DCV groups. In contrast, in NIP derivatives the terthiophene fragment is totally planar in both, unsubstituted and lateral-substituted derivatives.
Optical and electrochemical properties
The solubility exhibited by the novel assemblies described in this communication allows their proper optical and electrochemical characterization in solution by UV-Vis and cyclic voltammetry techniques. The most relevant optical and electrochemical parameters of both NDI and NIP derivatives are summarized in Tables 1 and 2.| UV-visa | |||||||
|---|---|---|---|---|---|---|---|
| [C] (μM) | λ max (nm) | ε λ max (M−1 cm−1) | λ ICT (nm) | ε ICT (M−1 cm−1) | λ onset (nm) |
E
optg
,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
|
| a UV-Vis absorption in CHCl3 solution. b Absorption maxima in solution. c Molar extinction coefficient to the referred wavelength. d Onset wavelength for the most redshifted absorption band. e Energy band gap derived from the low-energy absorption edge using the equation Eoptg = 1240/λonset. | |||||||
| NDI3T | 0.275 | 305 | 24473 |
599 | 8000 | 745 | 1.66 |
| NDI3T-RD | 0.275 | 516 | 44364 |
647 | 16364 |
758 | 1.64 |
| NDI3T-DCV | 0.275 | 505 | 40364 |
615 | 14909 |
681 | 1.82 |
| NDI3T-RDDCV | 0.275 | 529 | 54545 |
652 | 23273 |
737 | 1.68 |
| NIP3T | 0.275 | 348 | 68000 |
574 | 14910 |
698 | 1.77 |
| NIP3T-RD | 0.275 | 353 | 38180 |
641 | 17820 |
774 | 1.60 |
| NIP3T-DCV | 0.275 | 609 | 40360 |
603 | 11640 |
720 | 1.72 |
| Cyclic voltammetrya | |||||
|---|---|---|---|---|---|
| E 1/2ox I (V) | E 1/2red I (V) | E HOMO (eV) | E LUMO (eV) |
E
elecgap
,
(eV) |
|
| a Cyclic voltammetry recorded in DCM/TBAPF6 (0.1 M) at a scan rate of 0.15 V s−1 using Pt as working and the counter electrode, and Fc/Fc+ as reference. b Estimated from ELUMO = −5.1 eV − E1/2red I. c Estimated from EHOMO = −5.1 eV − E1/2ox I. d Estimated from Eelecgap = EHOMO − ELUMO. e Half wave potential of the reversible wave. f Anodic peak potential of the irreversible wave. | |||||
| NDI3T | 0.59 | −1.09 | −5.69 | −4.00 | 1.69 |
| NDI3T-RD | 0.92 | −0.98 | −6.02 | −4.12 | 1.90 |
| NDI3T-DCV | 0.94 | −1.04 | −6.04 | −4.06 | 1.92 |
| NDI3T-RDDCV | 0.87 | −0.98 | −5.97 | −4.12 | 1.85 |
| NIP3T | 0.50f | −1.30 | −5.60 | −3.80 | 1.80 |
| NIP3T-RD | 0.57f | −1.22 | −5.67 | −3.88 | 1.79 |
| NIP3T-DCV | 0.93f | −1.03 | −6.03 | −4.07 | 1.96 |
Fig. 4a shows the absorption profiles of the previously described NIP3T-X derivatives.35 Here, it is shown that the end-capped functionalization with strong electron–acceptors in the alpha position of the thiophene units have a strong impact in their optical properties, extending their absorption up to 800 nm in comparison with the non-functionalized NIP3T. This is especially evident in the case of NIP3T-RD. On the other hand, as depicted in Fig. 4b, in the case of the NDI3T-X derivatives, the end-capped functionalization with strong electron–acceptors leads to only a slight shift of the absorption tail depending on the electron–acceptor unit introduced. In addition, while the NDI3T-RD and NDI3T-RDDCV systems present a slightly red-shifted absorption tail, the NDI3T-DCV system show a blue-shifted absorption tail compared to that of NDI3T. Note however, that the maximum of the lowest energy absorption bands is consistently blue-shifted for all the NDI3T-X derivatives. This can be explained considering the appearance of two different and competing ICT channels within NDI3T-X molecules (i.e. from the terthiophene to the NDI unit and from the terthiophene to the lateral substituents). These two different ICT pathways were confirmed by TD-DFT calculations, and are related to a one-electron transfer between the HOMO → LUMO (absorption band around 650 nm) and HOMO → LUMO+1 (absorption bands around 516 nm) (Fig. S24–S26, ESI†), phenomena already described for small molecules like o/p-nitrophenols.51,52 Regarding these two different ICT pathways, it is interesting to notice that the more conjugated is the chemical structure, the more red-shifted are both, the HOMO → LUMO transition and the maximum absorption band. For that, NDI3T-DCV shows the most blue-shifted ICT band and absorption maxima, while the NDI3T-RDDCV possesses the most extended and redshifted absorption profile. Note however that, in the case of NDI3T-DCV, the discrete π-conjugation extension imparted by the lateral electron-deficient groups is not sufficient to compensate for the effect of the two competing ICT channels, and thus the blueshifted absorption edge.
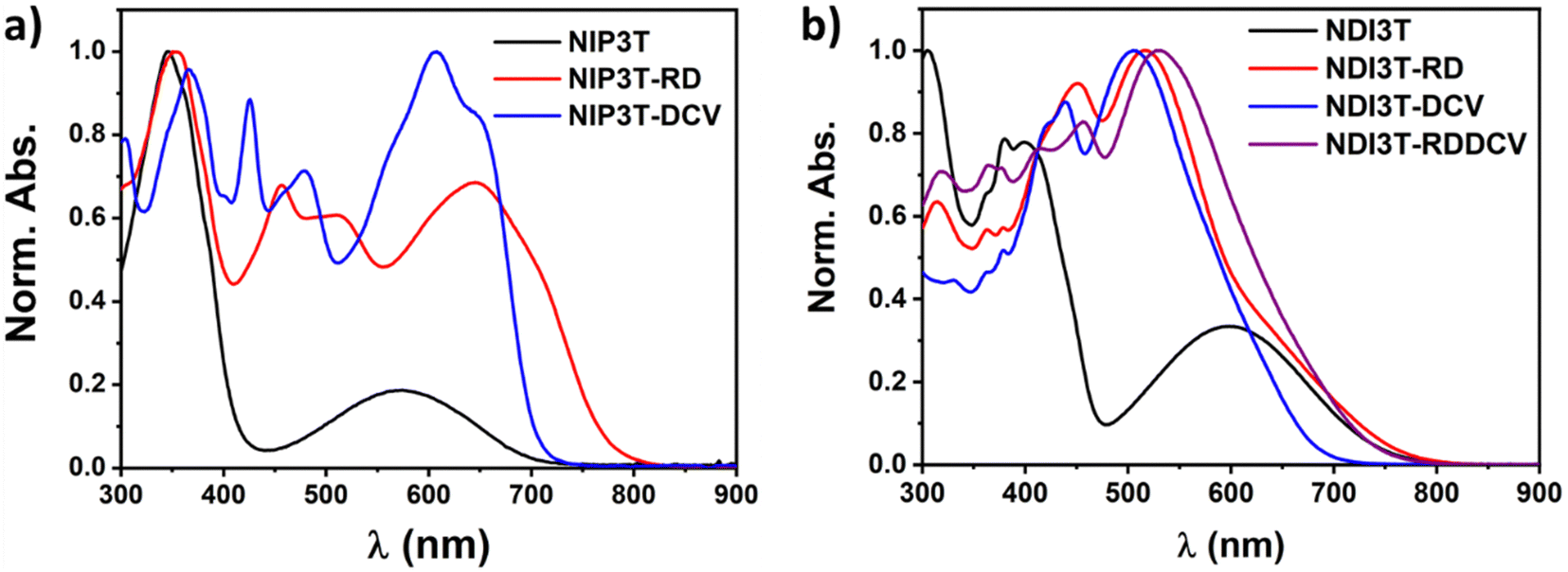 | ||
| Fig. 4 Normalized UV-Vis absorption spectra for (a) NIP3T (black), NIP3T-RD (red) and NIP3T-DCV (blue) and (b) NDI3T (black), NDI3T-RD (red), NDI3T-DCV (blue) and NDI3T-RDDCV (violet) in CHCl3 solutions. | ||
Now, by comparing the pyrazine-based semiconductor NIP3T34 with the imidazole-based NDI3T semiconductor (Fig. 4 and Fig. S40, ESI†), it is observed that the absorption spectrum is blue-shifted in the former, phenomena related with the π-conjugation between the naphthalimide and the oligothiophene units as well as the structural differences between the assemblies. In both cases, the broad absorption bands centered at around 570 nm (NIP3T) and 600 nm (NDI3T) are assigned to intramolecular charge-transfer (ICT) excitations. This is theoretically confirmed by TD-DFT calculations (Fig. S23, S27 and Table S1, ESI†) and can be described as a one-electron HOMO–LUMO excitation consisting of the displacement of the electron density from the HOMO, mainly localized on the oligothiophene fragment, to the LUMO, localized on the naphthalimide unit, in both cases (Fig. 5).33,34
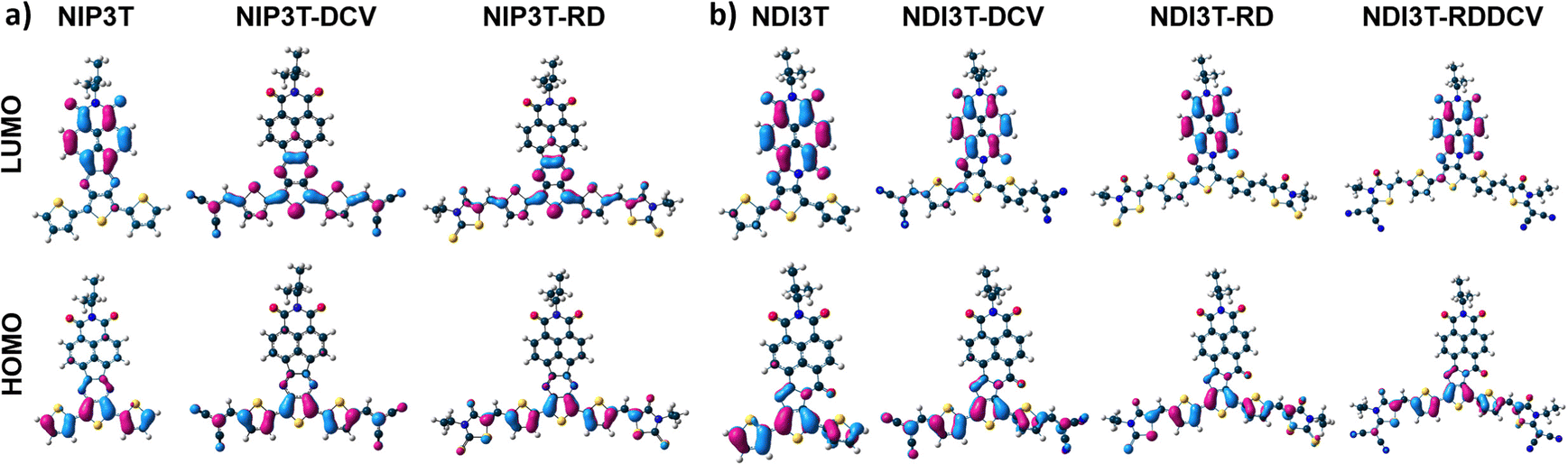 | ||
| Fig. 5 DFT/B3LYP/6-31G** molecular orbital topologies of (a) NIP3T-X and (b) NDI3T-X systems. | ||
In order to further analyze the nature of the lowest-energy absorption band for NIP3T-X and NDI3T-X systems, solvatochromic measurements were carried out by varying solvent polarity (Fig. S41, ESI†). NDI3T systems shows a clear solvatochromic behavior, where the redshift of the ICT absorption band in more polar solvents, implies a positive solvatochromism, which indicates a larger dipole moment on the excited state than on the ground electronic state, being that predicted theoretically (Fig. S22, ESI†). A similar scenario was observed for the NIP3T system, however, for the NIP3T-X end-capped derivatives, no shift of the absorption band was observed when varying the solvent polarity, confirming the absence of any ICT absorption band, as clearly shown by the molecular orbital topologies (Fig. 5 and Fig. S41, ESI†). Note that the HOMO and LUMO orbitals of NIP3T-X are both located on the oligothiophene molecular fragment. On the contrary, the NDI3T-X end-capped derivatives show a modest solvatochromic behavior. For NDI3T-RD and NDI3T-DCV derivatives the maxima absorption bands at around 515 nm experiment a redshift in more polar solvents, namely a positive solvatochromism, which based on TD-DFT calculations, present some extent of intramolecular charge transfer character (Fig. S41, S24 and S25, ESI†). The positive solvatochromism indicates larger dipole moment on the excited state than on the ground electronic state, which was theoretically confirmed (Fig. S22, ESI†). On the other hand, the NDI3T-RDDCV derivative shows a negative solvatochromic behavior, indicating a lower dipole moment on the excited state than on the ground electronic state, which was also theoretically described (Fig. S41 and S22, ESI†). Therefore, in the NDI3T-X end-capped derivatives, it can be demonstrated the ICT nature, albeit modest, of the lowest energy absorption band, as shown in the HOMO and LUMO topologies depicted in Fig. 5. We can assume that the competition between the two different ICT channels due to the presence of different acceptor units within the molecular structure (see above) is behind the modest solvatochromic behavior of the NDI3T-X systems compared to NDI3T.
Concentration-dependent experiments (Fig. S32, S34, S36 and S38, ESI†) do not show the formation of supramolecular aggregates for any of these naphthalimide-based semiconductors NDI3T-X. This contrasts with the previously reported NIP3T-DCV assembly, which allows the formation of aggregates in solution, and it is presumably due to the terthiophene torsion which avoids an optimal π–π interaction.35
UV-vis absorption measurements for this family of oligothiophene-naphthalimide derivatives have been also carried out in thin films (Fig. S39, ESI†). The absorption onsets in thin films are redshifted ca. 100 nm in comparison with that observed in solution, absorbing photons up to 850 nm, thus suggesting stronger π–π interactions in the solid state.53,54
Cyclic voltammetry experiments were carried out under argon atmosphere in an electrochemical set-up at a scan rate of 100 mV s−1 at 20 °C using tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 mol L−1) as supporting electrolyte in dichloromethane. All relevant data are compiled in Table 2 for both NDI and NIP based organic semiconductors.
In Fig. 6 and Table 2 we observe that, on the one hand, for the unsubstituted systems, when a pyrazine connector is substituted by an imidazole linker the reduction potential ascribed to the naphthalimide units shifts around 0.2 V to higher values, due to the stronger electron ability of NDI3T in comparison with the NIP3T analogue. This phenomenon is also observed for the oxidation processes, in this case ascribed to the lack of effective conjugation of the oligothiophene fragment, being less electron-donating unit due to the S⋯O steric interaction. This chemical modification stabilizes both HOMO and LUMO energy values in the NDI3T molecular assembly in comparison with the NIP3T analogue. On the other hand, when we introduce different electron acceptor units in the alpha position of the terthiophene moiety it is observed an impact in the electron acceptor ability of these new materials. Thus, the electrochemical behaviour observed in the NDI3T-X series (Fig. 6b) follows the same trend previously described for the NIP3T-X derivatives, as it is depicted in Fig. 6a where both, NDI3T-RD and NIP3T-RD show an improvement in the electron-accepting ability in comparison with the unsubstituted assemblies NDI3T and NIP3T. Therefore, electrochemical data suggests that those new systems are improved electron–acceptor materials than both the NDI3T skeleton and all the pyrazine-based materials NIP3T-X. On the other hand, the incorporation of these end-capped units has also an impact in the oxidation potential, showing a shift to higher oxidation values when the electron acceptor units are covalently linked to the terthiophene unit (Fig. S42–S45, ESI†), phenomena also observed for the pyrazine-based analogues (Table 2).
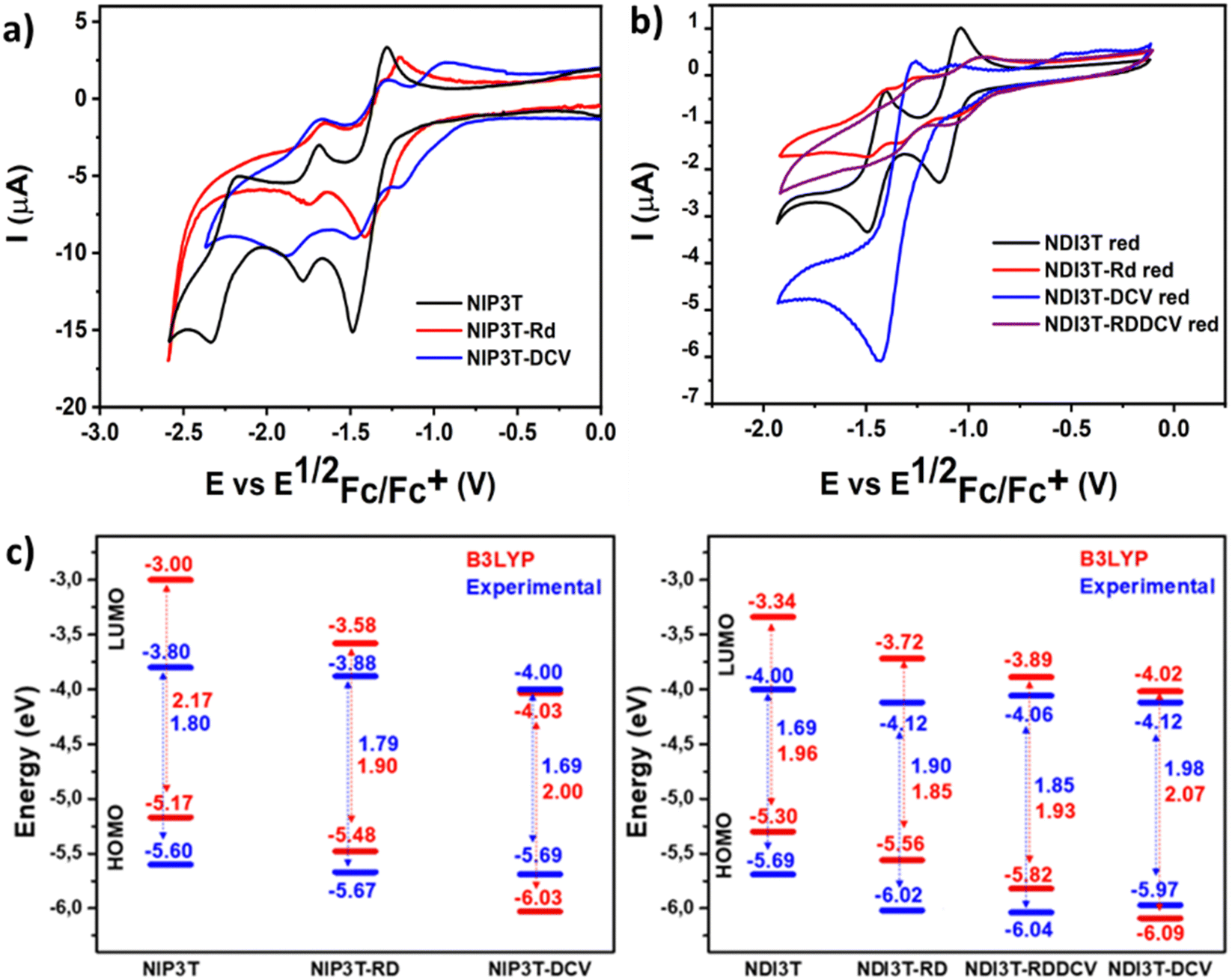 | ||
| Fig. 6 Cyclic voltammetry comparison between (a) NIP3T, NIP3T-RD and NIP3T-DCV (b) NDI3T, NDI3T-RD, NDI3T-DCV and NDI3T-RDDCV reduction processes in dichloromethane solutions and (c) the corresponding energy levels diagram estimated from the experimental electrochemical values for both, NDI and NIP based semiconductors. | ||
The highest occupied molecular orbital (HOMO) levels and the lowest unoccupied orbital (LUMO) levels of these new donor–acceptor materials can be estimated from the first oxidation/reduction potentials obtained by cyclic voltammetry experiments (Fig. 6a, b and Fig. S42–S45, ESI†) in dry dichloromethane (Table 2). As it is depicted in Fig. 6c, the introduction of electron acceptor units at the alpha position of the terminal thiophenes unit allows a precise control over the energy levels of their frontier molecular orbital. These precise modifications in the HOMO/LUMO energy levels present some differences in comparison with the observed values for the NIP3T-X materials, which is related to the different location of the topology of the frontier molecular orbitals in both skeletons, as it is depicted in Fig. 5 and Fig. S21 (ESI†). This effect produces a reduction in the bandgap in the new derivatives and a stabilization of both, HOMO and LUMO in NDI3T-RD, NDI3T-RDDCV and NDI3T-DCV assemblies in comparison with the unfunctionalized NDI3T.
Charged species study by spectroelectrochemistry
In this section, we aim to analyse the structure and stability of the charge carriers present in the electron transport process. As shown in Fig. 7 and 8, both spectroelectrochemical reduction and oxidation processes were recorded for the NDI3T-X systems, as expected considering their amphoteric redox behaviour demonstrated in the CV spectra (Fig. 6a). The evolution of the UV-Vis-NIR spectra of the studied semiconductors were obtained by progressive spectroelectrochemical reduction (Fig. 7) and oxidation (Fig. 8) of low concentration solutions in presence of high excess of Bu4NPF6 supporting electrolyte. The results are supported by TD-DFT theoretical calculations at B3LYP/6-31G** level (Fig. S46–S49, ESI†).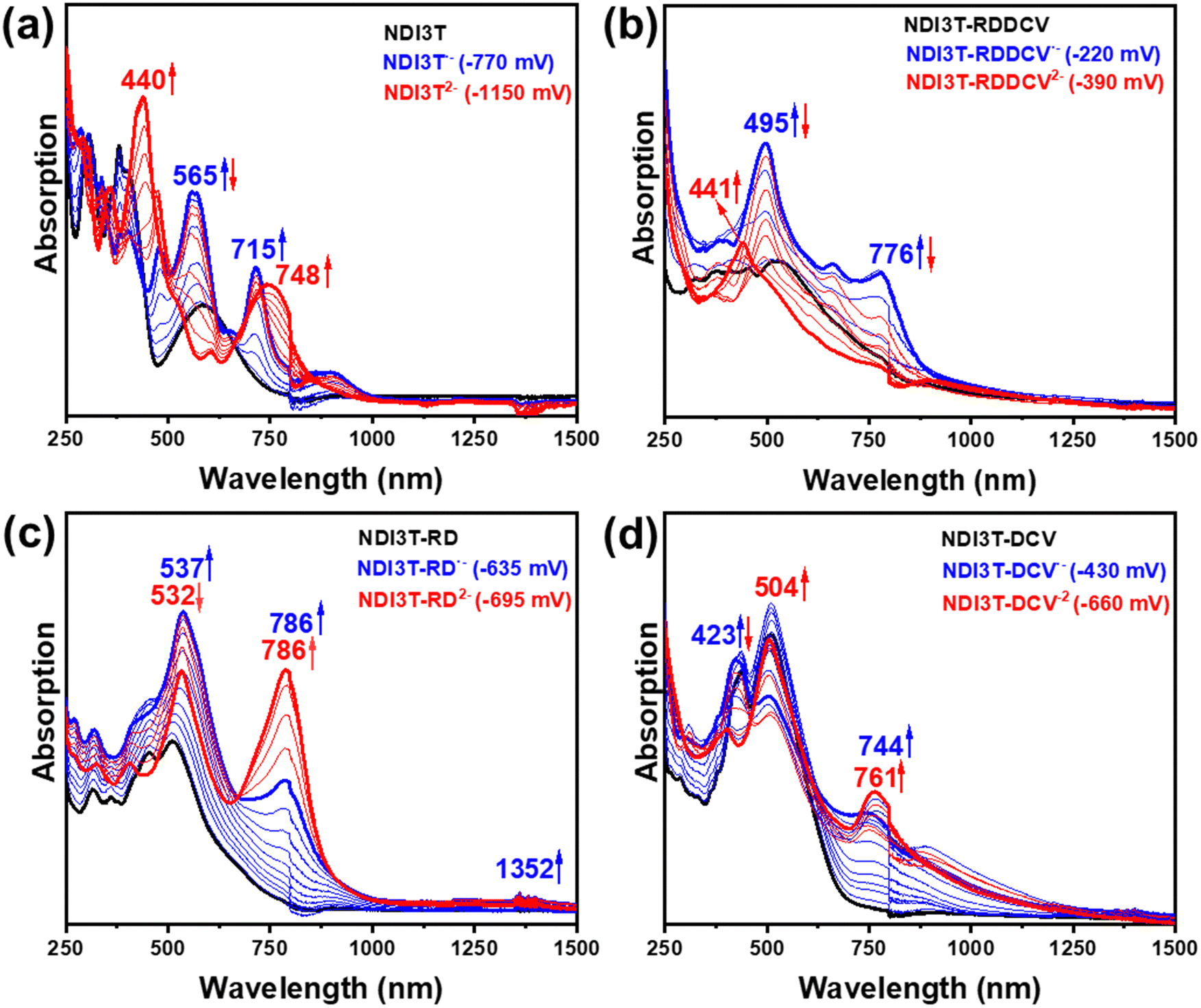 | ||
| Fig. 7 UV-Vis-NIR absorption spectra recorded by electrochemical reduction of (a) NDI3T, (b) NDI3T-RDDCV, (c) NDI3T-RD and (d) NDI3T-DCV in dichloromethane in presence of Bu4NBF4 as supporting electrolyte within an OTTLE cell. | ||
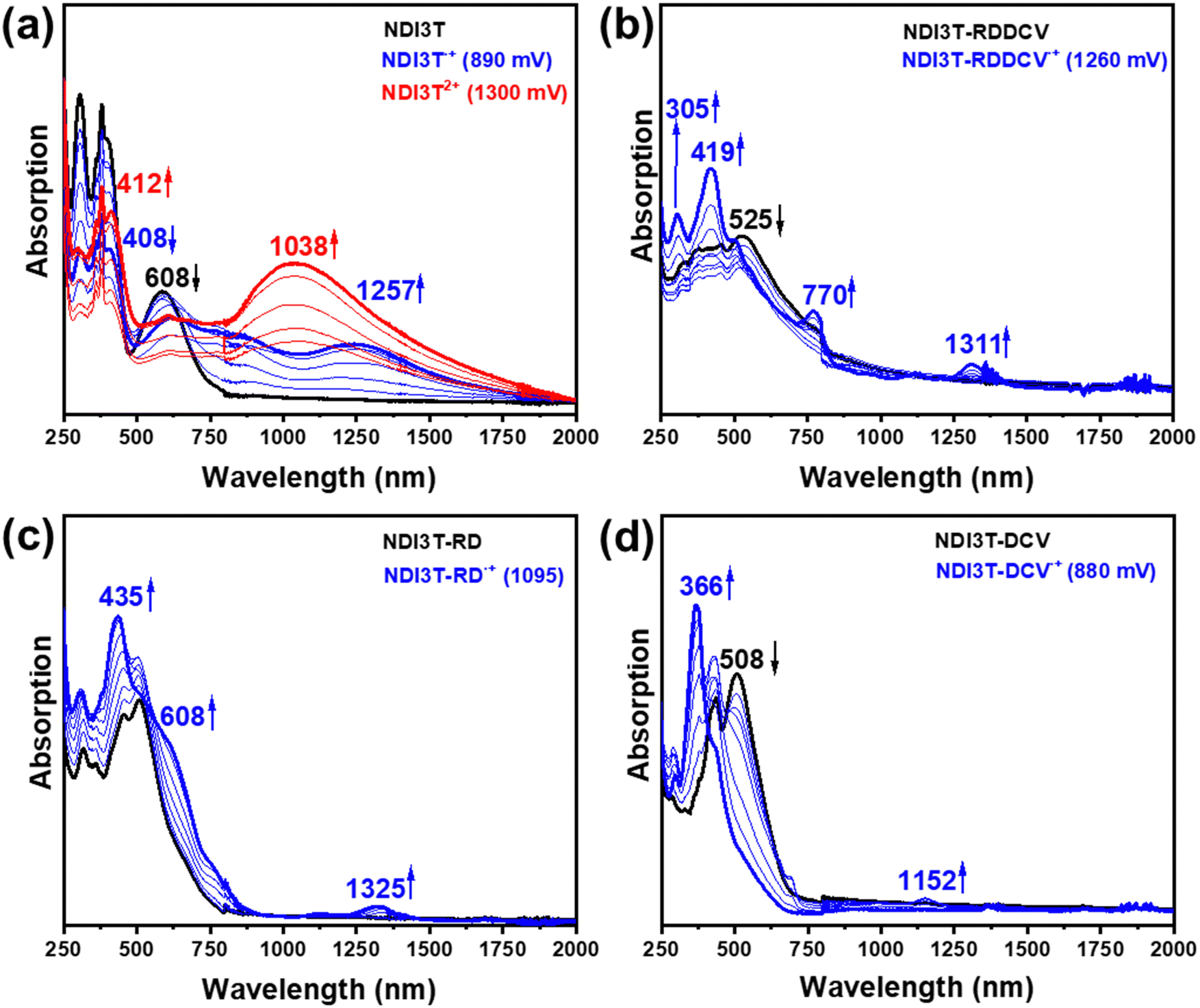 | ||
| Fig. 8 UV-Vis-NIR absorption spectra recorded by electrochemical oxidation of (a) NDI3T, (b) NDI3T-RDDCV, (c) NDI3T-RD and (d) NDI3T-DCV in dichloromethane in presence of Bu4NBF4 as supporting electrolyte within an OTTLE cell. | ||
The UV-Vis-NIR absorption spectra of the NDI3T (Fig. 7a) as neutral species (black curve) progressively evolve to new absorption bands recorded at 565 and 715 nm (blue curve), which are ascribed to the formation of radical anion species (see comparison with theoretical results in Fig. S46, ESI†). Further electrochemical reduction provokes the vanishing of this spectral profile and the appearance of two new absorption peaks, one centred at ca. 440 and a redshifted band at 748 nm.
On the other hand, the UV-Vis-NIR absorption spectra of NDI3T-DCV, NDI3T-RD and NDI3T-RDDCV (Fig. 7b–d), where the terthiophene subunit is laterally substituted with electron-deficient groups, show a similar absorption pattern, where two bands appear at around 500 and 750 nm upon one electron reduction. Further increase of the applied potential is translated in a decrease of these bands while increasing a new absorption band at higher energies, that can be ascribed to the dianion species.
The progressive electrochemical oxidation of NDI3T (Fig. 8a) depresses the absorption of the neutral species while leads to the formation of a red-shifted broad absorption band (blue curve). According to the electrochemical results and TD-DFT calculations, this new absorption can be ascribed to the formation of the radical cation species. Further electrochemical oxidation leads to the appearance of new broad absorption band (red curve), which present two contributions, one centred at ca. 412 and a redshifted band at 1038 nm.
In contrast, the progressive electrochemical oxidation of NDI3T-DCV, NDI3T-RD and NDI3T-RDDCV, leads to only one species (blue curve) which shows similar absorption pattern, with the main absorption band appears at around 400 nm. Further increase of the applied potential does not lead to new species in the compounds with electron-deficient groups at the thiophene subunits.
Therefore, as we can see from Fig. 7 and 8, the NDI3T-X systems present both oxidation and reduction processes, as also found in previously published NIP3T-X systems (Fig. S50 and S51, ESI†). However, the potentials at which both reduction and oxidation take place in NDI3T-X systems are substantially lower than those for NIP3T-X molecules. Note that, in the latter, both HOMO and LUMO are located on the oligothiophene fragment (Fig. 5) and thus the effect of the lateral substituents on the electron-donating properties of the thiophenic chain is much more accentuated, indicating a reduced electron-donating nature of that molecular fragment. Therefore, the ability to stabilize positive charges is diminished in NIP3T-X. Furthermore, the injection of negative charges, which also goes directly to the oligothiophene fragment (in contrast with the injection on the arylene unit in NDI3T-X), is also hindered and thus appears at much higher voltages.
On the contrary, on NDI3T-X systems, similarly to that observed in other naphthalene diimide derivatives, the negative injected charge is mainly stabilized over the naphthalimide fragment, as expected considering the LUMO topologies in Fig. 5. DFT calculations of NDI3T-X charged species indicate that the 70% of the injected charge is stabilized by the NDI units in the NDI3T anion species. This decreases when the terthiophene subunit is laterally substituted with electron-withdrawing groups (Fig. S52–S54, ESI†), which support 20% of the injected charge, indicating the role of the lateral substituents in promoting the stabilization of the negative charged species. In fact, for NDI3T-X systems up to dianion species are recorded at potentials lower than −700 mV, while for NIP3T-X potentials of around −1500 mV are required.
3. Conclusions
We present here a new synthetic approach for achieving redox amphoteric organic semiconductors via rational chemical modifications. For this end, the combination of processable terthiophene–naphthalimide assemblies, having an imidazole connecting group, with three different strong electron-withdrawing end-capped units has been used to design a series of novel organic semiconductors with tunable optoelectrochemical properties. These materials have been compared with previously published terthiophene–naphthalimide semiconductors, having a weaker electron–acceptor pyrazine linker and comparable lateral electron-withdrawing groups. All these materials present low-lying LUMO energy levels and broad absorptions up to 800 nm, being these characteristics ideal for organic electronics.The results indicate a quite complex electronic scenario that goes beyond the expected cumulative effects of the independent molecular units constituting the final molecular assembly. Thus, the modification of the connecting group in unsubstituted derivatives, namely NIP3T and NDI3T, has some impact on the optoelectronic properties but does not modulate the molecular orbital topologies, having the lowest energy absorption band an ICT character. However, when electron-withdrawing lateral substituents are inserted on those molecular platforms, different effects are found on both the energies and, especially, on the topologies of the frontier molecular orbitals. It has been demonstrated here that such effects have profound implications on the electronic properties of the materials, both in the optical absorption properties (energies and nature of the electronic transitions) and on the stabilization of charged species. Thus, when electron-withdrawing groups are inserted on the NIP3T platform (NIP3T-X), the lowest energy absorption band becomes of π–π* character. As a result, the charge distribution on the LUMO becomes localized over the oligothiophene fragment instead on being located on the naphthalimide units, which modulates the electron-donating nature of the oligothiophene chain. Nonetheless, still both positive and negative charges are stabilized, but at high potentials.
On the contrary, the introduction of the electroactive groups on the NDI3T platform (NDI3T-X) provokes the appearance of two different and competing ICT channels within the molecule, which has interesting effects on the electronic absorption spectra. However, the electronic densities on the HOMO and LUMO energies are comparable to those of the unsubstituted systems, and thus both positive and negative charged can be stabilized for the NDI3T-X semiconductors, at much more accessible potentials than in NIP3T-X molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MICINN (PID2019-106268GB-C33, PID2022-139548NB-I00 and TED2021-129886BC43) and the UCM (INV.GR.00.1819.10759). MJAN gratefully acknowledges Universidad Rey Juan Carlos for his postdoctoral contract. R. G.-N. thanks the MICINN for a FPI predoctoral fellowship (PRE2020-092327). Computer resources, technical expertise and assistance provided by the SCBI (Supercomputing and Bioinformatics) center of the University of Málaga are gratefully acknowledged. We also thank the Vibrational spectroscopy (EVI) lab of the Research Central Services (SCAI) of the University of Málaga. Funding for open access charge: University of Malaga/CBUA.References
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3(3), 18003 CrossRef CAS.
- Y. Huang, X. Gong, Y. Meng, Z. Wang, X. Chen, J. Li, D. Ji, Z. Wei, L. Li and W. Hu, Effectively modulating thermal activated charge transport in organic semiconductors by precise potential barrier engineering, Nat. Commun., 2021, 12(1), 21 CrossRef CAS PubMed.
- M. Wang, P. Baek, A. Akbarinejad, D. Barker and J. Travas-Sejdic, Conjugated polymers and composites for stretchable organic electronics, J. Mater. Chem. C, 2019, 7(19), 5534–5552 RSC.
- S. Izumi, H. F. Higginbotham, A. Nyga, P. Stachelek, N. Tohnai, P. D. Silva, P. Data, Y. Takeda and S. Minakata, Thermally Activated Delayed Fluorescent Donor–Acceptor–Donor–Acceptor π-Conjugated Macrocycle for Organic Light-Emitting Diodes, J. Am. Chem. Soc., 2020, 142(3), 1482–1491 CrossRef CAS PubMed.
- A. Minotto, I. Bulut, A. G. Rapidis, G. Carnicella, M. Patrini, E. Lunedei, H. L. Anderson and F. Cacialli, Towards efficient near-infrared fluorescent organic light-emitting diodes, Light: Sci. Appl., 2021, 10(1), 18 CrossRef CAS PubMed.
- D. Liu, T. Wang, Z. Chang, N. Zheng, Z. Xie and Y. Liu, Fused or unfused? Two-dimensional non-fullerene acceptors for efficient organic solar cells, J. Mater. Chem. A, 2021, 9(4), 2319–2324 RSC.
- W. Liu, X. Xu, J. Yuan, M. Leclerc, Y. Zou and Y. Li, Low-Bandgap Non-fullerene Acceptors Enabling High-Performance Organic Solar Cells, ACS Energy Lett., 2021, 6(2), 598–608 CrossRef CAS.
- Z. Zeng, Z. Zhong, W. Zhong, J. Zhang, L. Ying, G. Yu, F. Huang and Y. Cao, High-detectivity organic photodetectors based on a thick-film photoactive layer using a conjugated polymer containing a naphtho[1,2-c:5,6-c]bis[1,2,5]thiadiazole unit, J. Mater. Chem. C, 2019, 7(20), 6070–6076 RSC.
- M. Al-Hashimi, Y. Han, J. Smith, H. S. Bazzi, S. Y. A. Alqaradawi, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Influence of the heteroatom on the optoelectronic properties and transistor performance of soluble thiophene-, selenophene- and tellurophene–vinylene copolymers, Chem. Sci., 2016, 7(2), 1093–1099 RSC.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Donor–Acceptor-Conjugated Polymer for High-Performance Organic Field-Effect Transistors: A Progress Report, Adv. Funct. Mater., 2020, 30(20), 1904545 CrossRef CAS.
- D. Shi, Z. Liu, J. Ma, Z. Zhao, L. Tan, G. Lin, J. Tian, X. Zhang, G. Zhang and D. Zhang, Half-Fused Diketopyrrolopyrrole-Based Conjugated Donor–Acceptor Polymer for Ambipolar Field-Effect Transistors, Adv. Funct. Mater., 2020, 30(21), 1910235 CrossRef CAS.
- A. Velusamy, C.-H. Yu, S. N. Afraj, C.-C. Lin, W.-Y. Lo, C.-J. Yeh, Y.-W. Wu, H.-C. Hsieh, J. Chen, G.-H. Lee, S.-H. Tung, C.-L. Liu, M.-C. Chen and A. Facchetti, Thienoisoindigo (TII)-Based Quinoidal Small Molecules for High-Performance n-Type Organic Field Effect Transistors, Adv. Sci., 2021, 8(1), 2002930 CrossRef CAS.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Charge transport in high-mobility conjugated polymers and molecular semiconductors, Nat. Mater., 2020, 19(5), 491–502 CrossRef CAS.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Recent Progress in High-Mobility Organic Transistors: A Reality Check, Adv. Mater., 2018, 30(36), 1801079 CrossRef.
- S. Prodhan, J. Qiu, M. Ricci, O. M. Roscioni, L. Wang and D. Beljonne, Design Rules to Maximize Charge-Carrier Mobility along Conjugated Polymer Chains, J. Phys. Chem. Lett., 2020, 11(16), 6519–6525 CrossRef CAS.
- Y. Shi, J. Pan, J. Yu, J. Zhang, F. Gao, K. Lu and Z. Wei, Optimizing the Charge Carrier and Light Management of Nonfullerene Acceptors for Efficient Organic Solar Cells with Small Nonradiative Energy Losses, Solar RRL, 2021, 5(4), 2100008 CrossRef CAS.
- H. Usta, D. Kim, R. Ozdemir, Y. Zorlu, S. Kim, M. C. Ruiz Delgado, A. Harbuzaru, S. Kim, G. Demirel, J. Hong, Y.-G. Ha, K. Cho, A. Facchetti and M.-G. Kim, High Electron Mobility in [1]Benzothieno[3,2-b][1]benzothiophene-Based Field-Effect Transistors: Toward n-Type BTBTs, Chem. Mater., 2019, 31(14), 5254–5263 CrossRef CAS.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Insight into High-Performance Conjugated Polymers for Organic Field-Effect Transistors, Chem, 2018, 4(12), 2748–2785 CAS.
- A. Naibi Lakshminarayana, A. Ong and C. Chi, Modification of acenes for n-channel OFET materials, J. Mater. Chem. C, 2018, 6(14), 3551–3563 RSC.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, The role of chemical design in the performance of organic semiconductors, Nat. Rev. Chem., 2020, 4(2), 66–77 CrossRef CAS.
- Y. Ran, Y. Guo and Y. Liu, Organostannane-free polycondensation and eco-friendly processing strategy for the design of semiconducting polymers in transistors, Mater. Horiz., 2020, 7(8), 1955–1970 RSC.
- K. Tajima, K. Matsuo, H. Yamada, S. Seki, N. Fukui and H. Shinokubo, Acridino[2,1,9,8-klmna]acridine Bisimides: An Electron-Deficient π-System for Robust Radical Anions and n-Type Organic Semiconductors, Angew. Chem., Int. Ed., 2021, 60(25), 14060–14067 CrossRef CAS PubMed.
- Y. Park, C. Fuentes-Hernandez, X. Jia, F. A. Larrain, J. Zhang, S. R. Marder and B. Kippelen, Measurements of the field-effect electron mobility of the acceptor ITIC, Org. Electron., 2018, 58, 290–293 CrossRef CAS.
- W. Tang, D. Huang, C. He, Y. Yi, J. Zhang, C. Di, Z. Zhang and Y. Li, Solution-processed small molecules based on indacenodithiophene for high performance thin-film transistors and organic solar cells, Org. Electron., 2014, 15(6), 1155–1165 CrossRef CAS.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, A Facile Planar Fused-Ring Electron Acceptor for As-Cast Polymer Solar Cells with 8.71% Efficiency, J. Am. Chem. Soc., 2016, 138(9), 2973–2976 CrossRef CAS.
- A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Naphthalene and perylene diimides – better alternatives to fullerenes for organic electronics?, Chem. Commun., 2018, 54(98), 13763–13772 RSC.
- A.-J. Payne, N. A. Rice, S. M. McAfee, S. Li, P. Josse, C. Cabanetos, C. Risko, B. H. Lessard and G. C. Welch, Donor or Acceptor? How Selection of the Rylene Imide End Cap Impacts the Polarity of π-Conjugated Molecules for Organic Electronics, ACS Appl. Energy Mater., 2018, 1(9), 4906–4916 CrossRef CAS.
- S. V. Bhosale, Al Kobaisi, M. Jadhav, R. W. Morajkar, P. P. Jones and L. A. George, S., Naphthalene diimides: perspectives and promise, Chem. Soc. Rev., 2021, 50(17), 9845–9998 RSC.
- M. J. Sung, A. Luzio, W.-T. Park, R. Kim, E. Gann, F. Maddalena, G. Pace, Y. Xu, D. Natali, C. de Falco, L. Dang, C. R. McNeill, M. Caironi, Y.-Y. Noh and Y.-H. Kim, High-Mobility Naphthalene Diimide and Selenophene-Vinylene-Selenophene-Based Conjugated Polymer: n-Channel Organic Field-Effect Transistors and Structure–Property Relationship, Adv. Funct. Mater., 2016, 26(27), 4984–4997 CrossRef CAS.
- T. He, M. Stolte, C. Burschka, N. H. Hansen, T. Musiol, D. Kälblein, J. Pflaum, X. Tao, J. Brill and F. Würthner, Single-crystal field-effect transistors of new Cl2-NDI polymorph processed by sublimation in air, Nat. Commun., 2015, 6(1), 5954 CrossRef CAS PubMed.
- M. J. Alonso-Navarro, E. Gala, M. M. Ramos, R. Ponce Ortiz and J. L. Segura, Oligothiophene-Naphthalimide Hybrids Connected through Rigid and Conjugated Linkers in Organic Electronics: An Overview, Electron. Mater., 2021, 2(2), 222–252 CrossRef.
- A. de la Peña, I. Arrechea-Marcos, M. J. Mancheño, M. C. Ruiz Delgado, J. T. López Navarrete, J. L. Segura and R. Ponce Ortiz, Tuning of the Electronic Levels of Oligothiophene-Naphthalimide Assemblies by Chemical Modification, Chem. – Eur. J., 2016, 22(38), 13643–13652 CrossRef.
- R. P. Ortiz, H. Herrera, R. Blanco, H. Huang, A. Facchetti, T. J. Marks, Y. Zheng and J. L. Segura, Organic n-Channel Field-Effect Transistors Based on Arylenediimide-Thiophene Derivatives, J. Am. Chem. Soc., 2010, 132(24), 8440–8452 CrossRef CAS.
- R. Ponce Ortiz, H. Herrera, M. J. Mancheño, C. Seoane, J. L. Segura, P. Mayorga Burrezo, J. Casado, J. T. López Navarrete, A. Facchetti and T. J. Marks, Molecular and Electronic-Structure Basis of the Ambipolar Behavior of Naphthalimide–Terthiophene Derivatives: Implementation in Organic Field-Effect Transistors, Chem. – Eur. J., 2013, 19(37), 12458–12467 CrossRef CAS.
- M. J. Alonso-Navarro, A. Harbuzaru, P. de Echegaray, I. Arrechea-Marcos, A. Harillo-Baños, A. de la Peña, M. M. Ramos, J. T. López Navarrete, M. Campoy-Quiles, R. Ponce Ortiz and J. L. Segura, Effective interplay of donor and acceptor groups for tuning optoelectronic properties in oligothiophene-naphthalimide assemblies, J. Mater. Chem. C, 2020, 8(43), 15277–15289 RSC.
- W. W. A. Koopman, M. Natali, C. Bettini, M. Melucci, M. Muccini and S. Toffanin, Contact Resistance in Ambipolar Organic Field-Effect Transistors Measured by Confocal Photoluminescence Electro-Modulation Microscopy, ACS Appl. Mater. Interfaces, 2018, 10(41), 35411–35419 CrossRef CAS PubMed.
- M. Prosa, S. Moschetto, E. Benvenuti, M. Zambianchi, M. Muccini, M. Melucci and S. Toffanin, 2,3-Thienoimide-ended oligothiophenes as ambipolar semiconductors for multifunctional single-layer light-emitting transistors, J. Mater. Chem. C, 2020, 8(43), 15048–15066 RSC.
- J. L. Segura, H. Herrera and P. Bäuerle, Oligothiophene-functionalized naphthalimides and perylene imides: design, synthesis and applications, J. Mater. Chem., 2012, 22(18), 8717–8733 RSC.
- S.-L. Suraru and F. Würthner, Strategies for the Synthesis of Functional Naphthalene Diimides, Angew. Chem., Int. Ed., 2014, 53(29), 7428–7448 CrossRef CAS.
- U. Ali, A. Javed, A. Tallat, J. Iqbal and A. Raza, Molecular designing of four high performance pyrazine-based non-fullerene acceptor materials with naphthalene diimide-based small organic solar cells, J. Mol. Model., 2019, 25(2), 50 CrossRef PubMed.
- G. Ding, A. Mahmood, A. Tang, F. Chen and E. Zhou, Theoretical and experimental study of electron-deficient core substitution effect of diketopyrrolopyrrole derivatives on optoelectrical and charge transport properties, Chem. Phys., 2018, 500, 67–73 CrossRef CAS.
- K. Do, N. Cho, S. A. Siddiqui, S. P. Singh, G. D. Sharma and J. Ko, New D–A–D–A–D push–pull organic semiconductors with different benzo[1,2-b:4, 5-b′] dithiophene cores for solution processed bulk heterojunction solar cells, Dyes Pigm., 2015, 120, 126–135 CrossRef CAS.
- S. Steinberger, A. Mishra, E. Reinold, C. M. Müller, C. Uhrich, M. Pfeiffer and P. Bäuerle, A–D–A–D–A-Type Oligothiophenes for Vacuum-Deposited Organic Solar Cells, Org. Lett., 2011, 13(1), 90–93 CrossRef CAS.
- E. A. Weiss, M. J. Tauber, M. A. Ratner and M. R. Wasielewski, Electron Spin Dynamics as a Probe of Molecular Dynamics: Temperature-Dependent Magnetic Field Effects on Charge Recombination within a Covalent Radical Ion Pair, J. Am. Chem. Soc., 2005, 127(16), 6052–6061 CrossRef CAS PubMed.
- A. P. Zoombelt, J. Gilot, M. M. Wienk and R. A. J. Janssen, Effect of Extended Thiophene Segments in Small Band Gap Polymers with Thienopyrazine, Chem. Mater., 2009, 21(8), 1663–1669 CrossRef CAS.
- R. P. Ortiz, H. Herrera, C. Seoane, J. L. Segura, A. Facchetti and T. J. Marks, Rational Design of Ambipolar Organic Semiconductors: Is Core Planarity Central to Ambipolarity in Thiophene–Naphthalene Semiconductors?, Chem. – Eur. J., 2012, 18(2), 532–543 CrossRef CAS.
- C. Castiglioni, J. T. Lopez Navarrete, G. Zerbi and M. Gussoni, A simple interpretation of the vibrational spectra of undoped, doped and photoexcited polyacetylene: Amplitude mode theory in the GF formalism, Solid State Commun., 1988, 65(7), 625–630 CrossRef CAS.
- G. Zerbi, M. Veronelli, S. Martina, A.-D. Schlüter and G. Wegner, π-Electron delocalization in conformationally distorted oligopyrroles and ploypyrrole, Adv. Mater., 1994, 6(5), 385–388 CrossRef CAS.
- J. Albero, P. Atienzar, A. Corma and H. Garcia, Efficiency Records in Mesoscopic Dye-Sensitized Solar Cells, Chem. Record, 2015, 15(4), 803–828 CrossRef CAS.
- S. R. González, J. Casado, J. T. L. Navarrete, R. Blanco, J. L. Segura and A. β-Naphthaleneimide-Modified, Terthiophene Exhibiting Charge Transfer and Polarization Through the Short Molecular Axis. Joint Spectroscopic and Theoretical Study, J. Phys. Chem. A, 2008, 112(29), 6732–6740 CrossRef.
- S. K. Panja, N. Dwivedi and S. Saha, Tuning the intramolecular charge transfer (ICT) process in push–pull systems: effect of nitro groups, RSC Adv., 2016, 6(107), 105786–105794 RSC.
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, Tuning the Fluorescence and the Intramolecular Charge Transfer of Phenothiazine Dipolar and Quadrupolar Derivatives by Oxygen Functionalization, J. Am. Chem. Soc., 2021, 143(26), 9933–9943 CrossRef CAS.
- M. J. Alonso-Navarro, A. Harbuzaru, M. Martínez-Fernández, P. Pérez Camero, J. T. López Navarrete, M. M. Ramos, R. Ponce Ortiz and J. L. Segura, Synthesis and electronic properties of nitrogen-doped π-extended polycyclic aromatic dicarboximides with multiple redox processes, J. Mater. Chem. C, 2021, 9(25), 7936–7949 RSC.
- N. Drolet, J.-F. Morin, N. Leclerc, S. Wakim, Y. Tao and M. Leclerc, 2,7-Carbazolenevinylene-Based Oligomer Thin-Film Transistors: High Mobility Through Structural Ordering, Adv. Funct. Mater., 2005, 15(10), 1671–1682 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02099a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |