
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-fused BODIPY-based acceptor molecules for organic photovoltaics†
Fabien
Ceugniet
*a,
Amina
Labiod
b,
Denis
Jacquemin
 c,
Benoît
Heinrich
d,
Fanny
Richard
e,
Patrick
Lévêque
*b,
Gilles
Ulrich
a and
Nicolas
Leclerc
*a
c,
Benoît
Heinrich
d,
Fanny
Richard
e,
Patrick
Lévêque
*b,
Gilles
Ulrich
a and
Nicolas
Leclerc
*a
aInstitut de Chimie et Procédés pour l’Énergie, l’Environnement et la Santé (ICPEES), UMR CNRS 7515, École Européenne de Chimie, Polymères et Matériaux (ECPM), 25 Rue Becquerel, 67087 Strasbourg Cedex 02, France. E-mail: fabien.ceugniet@chem.ox.ac.uk; leclercn@unistra.fr
bLaboratoire ICube, UMR 7357-CNRS, Université de Strasbourg, 23 rue du Loess, 67037 Strasbourg, France. E-mail: patrick.leveque@unistra.fr
cNantes Université, CNRS, CEISAM UMR 6230, F-44000, Nantes, France & Institut Universitaire de France, 75005, Paris Cedex 5, France
dUniversité de Strasbourg, CNRS, IPCMS UMR 7504, F-67034 Strasbourg, France
eUniversité de Strasbourg, CNRS, Institut de Science et d'Ingénierie Supramoléculaires, 8 allée Gaspard Monge, Strasbourg, 67000, France
First published on 13th July 2023
Abstract
Thanks to the rise of efficient push–pull non-fullerene acceptors (NFAs), bulk-heterojunction organic solar cells have reached a high level of competitiveness with maximum power conversion efficiency over 18% for binary active layers. A common thread of most of these NFAs is the presence of a central unit with several fused aromatic rings. Although this design is very effective in achieving high planarity of the conjugated system, it requires the development of a complex chemistry. In the present work, we designed and synthesized a new series of NFAs, called BTTs, by using the uncommon BODIPY unit as central electron accepting unit and avoiding the intricate chemistry of fused aromatic units. All 6 new BTT molecules exhibit very low optical bandgaps (below 1.5 eV), very high extinction coefficients and a LUMO level deep enough to be used as a NFA with some of the most efficient electron-donor molecules. The appropriate halogenation of the cyanoindanone electron acceptor end units with chlorine or fluorine atoms allows the fine tuning of the energy levels. Despite the unfavorable edge-on orientation of these new BTTs, the most efficient derivative led to one of the best PCEs obtained so far with a BODIPY-based NFA of around 6%.
Introduction
In the last few years, the performance of bulk-heterojunction (BHJ) organic solar cells (OSCs) have dramatically increased to reach a maximum power conversion efficiency over 18% for binary OSCs.1,2 One of the main driving force for this improvement is the important development of non-fullerene acceptors (NFA) having a push–pull design.3 The A–D–A architecture found for instance in ITIC and its derivatives (Fig. 1) was the first one to compete, and then surpass, the traditional fullerene-derivative acceptors.4–6 More recently, Y6 has popularised the A–D–A′–D–A architecture showing outstanding performances as electron-acceptor material.1,7 Due to the exceptional potential of both architectures, a wide range of high-performance materials based on these designs have been developed in recent years.3,8,9 | ||
| Fig. 1 Structures and design of some fused (top) and unfused (bottom) electron acceptor materials. | ||
A common thread of these architectures is the presence of a central unit with several fused aromatic rings. This feature ensures the planarity of the central core and enhances the π-electron delocalisation. These effects allow a diminution of the bandgap of the material, a redshift of its absorption onset and an enhancement of intermolecular interactions. However, a drawback of such molecular design is the synthetic complexity of extended fused aromatic units. To circumvent this issue while ensuring an efficient delocalization of the π-electrons, a new approach, based on non-covalent intramolecular interactions, has started to emerge in the literature.10–15
In 2019, Huang et al. presented a series of NFA with an A–D–π–D–A design in which they considered, based only on basic theoretical calculations, that S⋯O non-covalent interactions were responsible for the central core planarization.16 The efficiency of DOC2C6-2F, the most performing NFA reported in Huang et al. publication, is comparable to ITIC derivatives with a maximum PCE recorded of 13.2%. Later, the combination of non-covalent interactions and A–DA′D–A design improved the maximum PCE of non-fused electron acceptor material up to 14.8% thank to the BTzO/NoCA series.14,17 Very recently, a fully non-fused NFA based on simple A–D–A design, including a very straightforward highly hindered bithiophene central unit has shown a record PCE of 15.2%.18
BODIPYs, due to their large extinction coefficient, good photostability and numerous possibilities of functionalisation,19,20 have been used for different optoelectronic applications including electron donor materials for OSCs.21–27 However, despite the electron deficient character of the BODIPY moiety, examples of BODIPY-based electron acceptor materials are rather scarce in the literature and when we started this project, the maximum PCE achieved with these compounds was under 2%.28,29 Recently, Tao and co-workers reported the first efficient NFA with a BODIPY central unit (tMBCIC) and a maximum efficiency of 9.2%.30 The tetramethyl-BODIPY moiety used in their work induced a dihedral angle over 45° between the central unit and the rest of the molecule. Since then, BODIPY units have also been involved in synthetically more complex 3D-shape efficient NFAs.31,32
In the present work, we investigated a new series A–D–A′–D–A electron acceptor material (BTT series) with a BODIPY as central acceptor unit. For this series a thieno[3,2-b]thiophene unit was used as donating group and three different types of cyanoindanones as end-capping units. To possibly induce intramolecular S⋯O non-covalent interactions, alkoxyphenol side chains were introduced at the 3,5-positions of the BODIPY. This alkoxyphenol unit has been chosen for the two following reasons: (i) it is highly difficult to introduce alkoxy chains on the BODIPY skeleton in 3,5-positions and (ii) it is expected that the benzene ring might disturb the planarity of the central unit in order to avoid over crystallization of the resulting NFAs. By varying the side-chains grafted on this alkoxyphenol unit, six new NFAs were synthesised thanks to a straightforward 4-steps synthetic route (Fig. 2).
 | ||
| Fig. 2 Chemical structure of tMBCIC and general structure of BTT. | ||
Synthesis of the NFAs
The synthetic pathways towards the six BTTs is shown in Scheme 1. The starting material, tetrabromo BODIPY 1, was synthesized according to the literature.33 Alkoxyphenol substituents were introduced at the 3,5-positions of 1 with excellent yields by SnAr reactions under basic conditions.34 Stille cross-coupling between an excess of 4 (3.5 eq.) and the 2,6-dibromo-3,5-disubstituted BODIPY 2–3 afforded compounds 5–6 in excellent yields (78% and 83%, respectively). The key-step in the synthetic routes is the functionalization of the free α positions of the thieno[3,2-b]thiophene substituents with formyl groups. Good to excellent yields ranging from 68% to 81% were obtained for this step using Vilsmeier–Haack formylation reactions. These intermediates were engaged in Knoevenagel reaction in presence of an excess of terminal acceptor (9a–c depending on the targeted molecule) and β-alanine35,36 to afford the corresponding BTTs with a yield varying from 75% to 90%. It is important to note that primary alcohol such as methanol and ethanol induced slow degradation of intermediates 7–8 and BTT when used as solvents, and therefore must be avoided. All compounds were characterized and confirmed by NMR (1H, 13C, 11B, 19F) and HRMS. | ||
| Scheme 1 Synthetic routes of the BTTs. | ||
Structural analysis and theoretical modelling
All BTTs were first characterized by thermogravimetric analysis (TGA). They all exhibit a degradation temperature (corresponding to a 5% weight loss temperature) in the 280–300 °C range (see Fig. S62, ESI†), well above the temperatures used in organic solar cells elaboration and characterization.Polarizing optical microscopy (POM), differential scanning calorimetry (DSC) and SWAXS on powder were combined to investigate the thermal behavior of these new NFAs. Interestingly, the unsubstituted BTTs (BTTL6 and BTTR8) as well as the chlorinated derivatives (BTTL6-4Cl and BTTR8-4Cl) are molecular crystals at any temperature (see Fig. S63 for DSC and Fig. S64, S65 for SWAXS data, ESI†), as they do not show any thermal transition before degradation. In contrast, the fluorine-substituted derivatives are mesomorphic solids. BTTL6-4F exhibits two mesophases with a transition between the two phases at approximately 145 °C, and BTTR8-4F a single mesophase of different type. Both mesophases of BTTL6-4F have soft-crystalline structures characterized by molten chain zones and a crystallographic lattice defined by the long range positional ordering of the conjugated units, as indicated by the broad scattering from molten chains centered at 1.4 Å−1 and the numerous sharp reflections in the small and medium q-ranges (see SWAXS patterns in Fig. S64, ESI†).37BTTR8-4F forms a frozen liquid crystal like mesophase in which the domains of self-assembled BTT units and domains of molten chains alternate, giving SWAXS patterns composed of broad wide-q scattering signals from the self-assembly inside domains and sharp reflections in the small-q region from the domain alternation (see SWAXS patterns in Fig. S65, ESI†).38 These mesomorphic organizations explain the soft textures of these compounds compared to the chlorinated analogues, where it should be noted that even at temperatures above 200 °C, they stay pasty solids and never flow to fluid liquid crystal phases.
To provide additional insight into these compounds, first-principles calculations on the various molecules were performed using DFT and TD-DFT and modeling the environmental effects with a continuum approach (see the ESI† for computational details). In order to keep calculation time reasonable, we substituted the long alkyl side chains by simple methyl groups, in such a way that there is no longer any distinction between the two sets of molecules and only the changes in the nature of the halogens at the ends are considered. Therefore, the calculated molecules are named BTT, BTT-4F and BTT-4Cl. As detailed in the ESI,† an extensive conformation search was performed for the former (Fig. S66, ESI†). A geometry optimization clearly shows that the conjugated backbone of the molecule is rather flat, with a dihedral angle between the thieno[3,2-b]thiophene and the BODIPY unit in the range of 2–22° for the most stable structures, the flatter structures presenting S⋯O close contacts (smaller than the sum of the VdW radii), but this structure presents a similar energy as the conformers free of these interactions. On the one hand, the thieno[3,2-b]thiophene and lateral acceptor groups are almost perfectly co-planar though amongst the four most stable conformers, there is no direct relationship between relative stability and slight deviations from planarity (Fig. S67 and S68, ESI†). The tolyl substituent in meso-position, exhibits a dihedral angle of 48–50°. The O-Ph-OAlk groups are located out of the chomogen plan for obvious steric reasons (Fig. S68 and S69, ESI†). In addition, we noticed that stronger deviations from planarity result in a large energy cost for the system (of the order of 8 kcal mol−1, see Fig. S66, ESI†). This confirms the high conjugation along the molecules, also obvious from the molecular orbital plots (Fig. S70, ESI†). Besides, it is impossible to precisely predict the orientation of the thieno[3,2-b]thiophene unit with respect to the BODIPY plane, nor the position of the pendant alkoxyphenol substituents, as several conformers show very close total energies, so that there is likely a blend of conformers in the material. Concerning the alkoxyphenol substituents, they are however, and as expected for steric hindrance reasons, positioned approximately perpendicular to the conjugated backbone of the molecule. The occurrence of intramolecular S⋯O non-covalent interactions between these alkoxyphenol substituents and the neighbouring thieno[3,2-b]thiophene units are therefore not sufficiently strong to induce a clear-cut energetic advantage to the structures showing such interactions.
Optical and electrochemical properties
The absorption and emission properties of all BTTs and their precursors were studied in DCM or/and CHCl3 at room temperature (see Table S2 and Fig. S72–83, ESI†). In dilute chloroform solution, all six BTTs exhibit a strong and broad absorption band between 600 and 800 nm (Fig. 3a and b). The absorption bands of the six acceptors have a similar shape with an absorption maximum ranging from 720 to 740 nm and a shoulder at lower wavelengths. Along both the BTTL6 and BTTR8 series a bathochromic shift can be observed when going from X = H to X = F and then Cl. This phenomenon has been already observed for the ITIC and its derivative IT-4F and IT-4Cl.6,39 It is worth mentioning that the magnitude of the bathochromic shift is relatively weak in the BTT series with a maximum bathochromic shift of 14 nm between the non-halogenated and its chlorinated counterpart. However, this weak bathochromic shift is in total agreement with the calculated maximum of absorption (Fig. S70 and Table S1, ESI†). In addition, the very high extinction coefficients measured for these six molecules is also confirmed by the very high oscillator strengths given by TD-DFT calculations (Fig. S71, ESI†).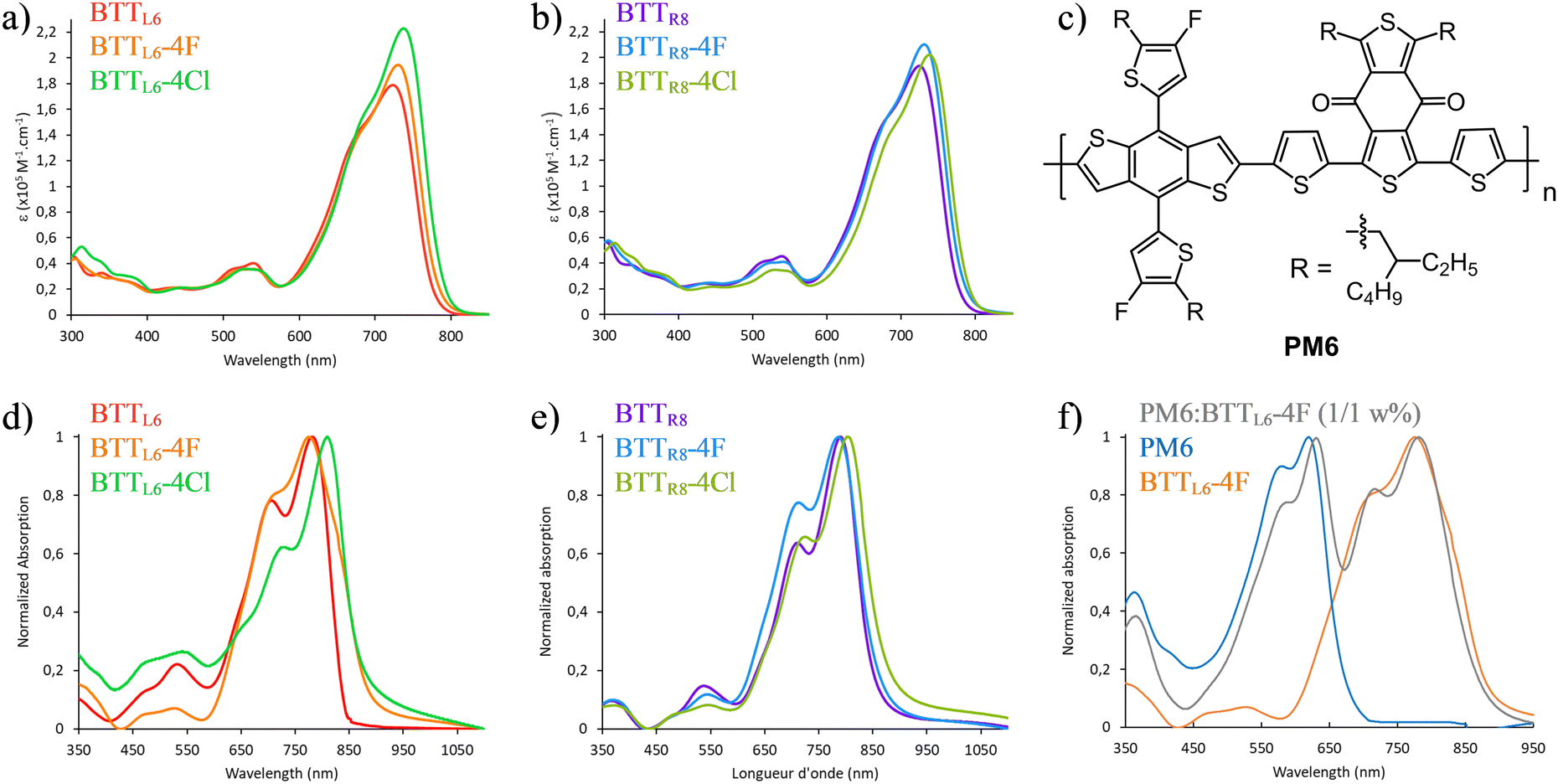 | ||
Fig. 3 (a) absorption spectra of BTTL6, BTTL6-4F and BTTL6-4Cl in CHCl3 (25 °C) (b) absorption spectra of BTTR8, BTTR8-4F and BTTR8-4Cl in CHCl3 (25 °C) (c) molecular structure of PM6 (d) absorption spectra of BTTL6, BTTL6-4F and BTTL6-4Cl in thin film (coated from CHCl3 solution) (e) absorption spectra of BTTR8, BTTR8-4F and BTTR8-4Cl in thin film (coated from CHCl3 solution). (f) Solid state absorption spectra of PM6, BTTL6-4F and PM6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BTTL6-4F (1:1 weight %) blend. :BTTL6-4F (1:1 weight %) blend. | ||
Comparing the UV-vis spectra in solution and in thin-film, the absorption maxima of all acceptors are redshifted. These redshifts ranged from 45 nm for the BTTL6-4F to 72 nm for the BTTL6-4Cl. In the same way, a bathochromic shift of the absorption onset was observed in solid-state with λonset ≈ 900 nm for several BTTs (Fig. 3d and e). The broad absorption spectra for all the BTT acceptors in thin films are relatively similar to those in solution with an absorption maximum around 785 nm and a shoulder at lower wavelengths. Due to their broad redshifted absorption spectra, BTTs are acceptors of interest to complement the absorption of high/medium bandgap donor as PM6 to increase the light-harvesting of the active layer (Fig. 3c and f).40
The energy levels of the frontier orbitals of our materials were estimated via two methods. First, we used the cyclic voltammetry (CV) measurements (Fig. S84 (ESI†), Fig. 4 and Table 1), knowing that our new molecules exhibit clear oxidation and reduction processes. As shown in Fig. 4 (full lines), the energy levels of the six BTTs are quite similar with a variation amplitude of the 130 meV (80 meV) for the LUMO (HOMO) level. For both BTTL6 and BTTR8 series, the introduction of halogen atoms on the end capping units seems to downshift both energy levels of the materials with chlorine atoms inducing a greater effect than fluorine. However, although similar trends have been reported for other NFAs, including the ITIC family,6,35 the variation measured in our molecules are much smaller than the ones usually recorded in A–D–A′–D–A NFAs. Interestingly, this effect and its small amplitude were confirmed by DFT and TD-DFT modelling. Indeed, frontier orbitals of BTT, BTT-4F and BTT-4Cl have been calculated at the PCM(CHCl3)-MN15/6-31+G(d,p)//PCM(CHCl3)-MN15/6-31G(d) level of theory (see Table S1 and Fig. S70, ESI†). Modelling shows very little stabilization of the two energy levels HOMO and LUMO upon halogenation, in the range of 0.06 to 0.12 eV, depending on the halogen. Such a trend is in a very good agreement with experimental values. Moreover, the plot of electronic density in the LUMO level (Fig. S70, ESI†) clearly shows a large electronic delocalization along the molecule, irrespective of the presence of halogens at the extremities, with a strong contribution of the central BODIPY unit. Actually, compared to the thieno- or thienothiophene-fused benzothiadiazole cores, commonly used in state-of-the-art NFAs, the contribution of our electron deficient BODIPY central unit to the electronic delocalization in the LUMO level is higher and could explain the lowest impact of the halogen atoms on the cyanoindanones end-capping units.1
 | ||
| Fig. 4 Full lines: energy levels estimated from the onset potential of first oxidation and reduction using the following equation: ELUMO (eV) = − [Eonsetred (vs. SCE) + 4.4] and EHOMO (eV) = − [EonsetOX (vs. SCE) + 4.4], based on an SCE energy level of 4.4 eV relative to the vacuum. Dashed Lines: ionization potentials measured by PESA and electronic affinity estimated by using the IEPESA and the optical bandgap.*PM6 HOMO value from CV measurements is from ref. 40. | ||
| Compound | Absorption spectroscopy | Energy levels | ||||||
|---|---|---|---|---|---|---|---|---|
| Solution | Thin film | CV | PESA | |||||
|
λ
Solmax
(nm) |
ε max (nm) |
λ
filmmax
(nm) |
ΔEoptc (eV) | E HOMO (eV) | E LUMO (eV) | ΔECVd (eV) | E HOMO (eV) | |
| a Done in CHCl3 solution (10−6 M). b Film prepared from CHCl3 solution. c As determined by the absorption onset on UV-visible spectrum in solid-state. d Energy levels estimated from the onset potential of first oxidation and reduction using the following equation: ELUMO (eV) = − [Eredonset (vs. SCE) + 4.4] and EHOMO (eV) = − [EOXonset (vs. SCE) + 4.4], based on an SCE energy level of 4.4 eV relative to the vacuum. | ||||||||
| BTTL6 | 723 | 178700 |
781 | 1.47 | −5.46 | −3.88 | 1.58 | −5.61 |
| BTTL6-4F | 730 | 194000 |
775 | 1.39 | −5.50 | −3.92 | 1.58 | −5.73 |
| BTTL6-4Cl | 737 | 222800 |
809 | 1.42 | −5.52 | −3.95 | 1.57 | −5.72 |
| BTTR8 | 724 | 193600 |
790 | 1.45 | −5.44 | −3.82 | 1.62 | −5.62 |
| BTTR8-4F | 731 | 210300 |
786 | 1.43 | −5.48 | −3.88 | 1.60 | −5.68 |
| BTTR8-4Cl | 738 | 202100 |
803 | 1.41 | −5.49 | −3.91 | 1.58 | −5.49 |
The second method used to estimate the energy levels of the frontier orbitals of our materials is based on the ionization potential (IP) measurement by photoelectron spectroscopy in the air (PESA) to which we add the calculated optical bandgap (ΔEopt) value to extract an electron affinity (EA) value (Fig. 4, dashed lines). First, the same trend can be seen in the evolution of the energy levels, as discussed above. In a series, the IP value is lowered from 0.06 to 0.12 eV upon halogenation. Furthermore, it is worth to note that the IP values measured by PESA are systematically deeper than the ones extracted form CV measurements of about 0.15 to 0.24 eV. Such a variation between these two measurements has been observed recently in different studies.41,42 More interesting is the comparison of IP offsets (ΔIP) when the PM6 polymer, used as electron donor material in this work, is included in the discussion. Indeed, its IP value evolves in the opposite way when comparing the values measured by CV and PESA with its IPPESA being 0.26 eV higher than the IPCV. Here also, such opposite behavior (electron donor vs. electron acceptor materials) has been very recently reported in literature.40 In fact, if we consider only the CV measurements, we end up with a ΔIPCV between PM6 and the BTT acceptors of the order of −0.01 to 0.07 eV. If we consider PESA measurements, ΔIPPESA of the order of 0.42 to 0.54 eV are measured between the electron-donor and the electron-acceptor. In a recent publication, D. Baran et al. investigated and compared the energy levels by CV and photoelectron spectroscopies (Ultraviolet photoelectron spectroscopy (UPS) for IP and low-energy inverse photoelectron spectroscopy (LE-IPES) for EA, respectively).40 They highlighted very similar behaviors as described in the present work, namely, a general upshifting energy levels for electron donor materials when going from CV to UPS and a rather opposite behavior for electron accepting materials. Besides, they characterized several binary blends and related photovoltaic parameters and came to two conclusions. First, the photoelectron spectroscopies are the most accurate techniques to give meaningful IP and EA values, as regards to Voc evolution especially. Second, although still in debate, the required ΔIP for efficient exciton dissociation seems to be of the order of 0.5 eV, as measured by photoelectron spectroscopies. In our case, we assume that the negligible ΔIP measured by CV is purely due to the measurement method and that the energy levels of our new BTT series are in good agreement with an efficient exciton dissociation when blended with PM6, in line with the ΔIPPESA of roughly 0.5 eV.
Charge carrier mobility, morphology and photovoltaic properties
The electron mobility of the BTT series has been investigated using organic field effect transistors (OFETs) in the bottom gate/bottom contacts configuration. The elaboration procedure as well as the output and transfer characteristics (Fig. S85–S92, ESI†) can be found in the ESI† while the charge-carrier mobilities in the saturation regime are summarized in Table 2. Some of the BTT molecules exhibit an ambipolar character after thermal annealing.| Compound | OFET | SCLC | |||
|---|---|---|---|---|---|
| μ e (cm2 V−1 s−1) | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) | |
| a As cast. b Annealed 10 minutes at 100 °C. | |||||
| BTTL6 | (4.9 ± 1.1) × 10−3 | — | (5.7 ± 0.9) × 10−3 | (1.9 ± 0.4) × 10−4 | (3.2 ± 0.4) × 10−3 |
| BTTL6-4F | (2.5 ± 0.7) × 10−3 | — | (3.6 ± 1.0) × 10−3 | — | — |
| BTTL6-4Cl | (5.2 ± 2.0) × 10−3 | — | (6.6 ± 1.9) × 10−3 | — | — |
| BTTR8 | (4.1 ± 1.7) × 10−3 | — | (7.4 ± 1.1) × 10−3 | (5.1 ± 0.7) × 10−4 | (2.2 ± 0.5) × 10−4 |
| BTTR8-4F | (3.4 ± 0.5) × 10−3 | — | (1.4 ± 0.5) × 10−3 | — | (3.4 ± 0.2) × 10−4 |
| BTTR8-4Cl | (2.1 ± 0.6) × 10−2 | — | (3.5 ± 0.5) × 10−2 | — | (7.4 ± 0.6) × 10−4 |
All the electron-mobilities in as-cast OFETs are in the same order of magnitude (ranging from 2.5 × 10−3 cm2 V−1 s−1 to 5.2 × 10−3 cm2 V−1 s−1) except for BTTR8-4Cl whose electron-mobility is an order of magnitude higher. For as-cast materials, there is no sign of ambipolarity in the output characteristics of the transistors.
After thermal annealing at 100 °C for 10 minutes, the general trend is a slight increase in electron mobility, except for BTTR8-4F where a slight decrease was observed. In agreement with their slightly higher HOMO levels, the non-halogenated BTTL6 and BTTR8 materials showed an ambipolar behaviour in OFET after this first annealing step with an extracted moderate hole mobility of roughly 2 × 10−4 cm2 V−1 s−1 and 5 × 10−4 cm2 V−1 s−1 for BTTL6 and BTTR8, respectively.
An extra annealing step at 150 °C for 10 minutes (see Table S3, ESI†) degrades significantly the measured electron mobility in every BTT-based OFET. However, the BTTR8-4Cl OFET showed after this extra annealing an ambipolar behaviour but the extracted hole mobility stays moderate.
The mobility in the direction perpendicular to the substrate was estimated using space-charge limited current (SCLC) diodes. The elaboration conditions of electron–only diodes can be found in the ESI.† Current density (J) times d3 (where d is the organic film thickness) versus the voltage (V) curves for SCLC electron–only diodes have been plotted in Fig. S92 (ESI†). The limited solubility of halogenated BTTL6-4F and BTTL6-4Cl prevented the elaboration of thick and flat films. Therefore, the determination of the film-thickness and consequently of the SCLC electron-mobility was not possible for these two materials. Comparison of OFET electron mobility and SCLC electron mobility is made for as-cast devices in Table 2.
For all BTTs, the SCLC electron mobility is lower than the OFET one. This result could be explained mainly by two considerations: (i) the charge-carrier mobility increases with the charge-carrier density that is much higher in OFET devices compared to SCLC ones, and (ii) the lamella orientation of BTTs (see below) is not suitable for the perpendicular charge transport as probed in SCLC device.
In order to investigate this morphological feature, we performed Grazing Incidence Wide-angle X-Ray diffraction (GIWAXS) on BTT's thin-films. All the elaboration and characterization details can be found in the ESI† together with the GIWAXS patterns (Fig. S94, ESI†). BTTL6 and BTTL6-4F self-assemble in a frozen mesomorphic state with a lamellar structure of alternating π-stacked backbones (π-stacking distance from maximum position hπ = 3.55 Å) and molten alkyl chains (hch = 4.5 Å). The lamellar periodicity is about 19 Å (BTTL6) or 20 Å (BTTL6-4F). In both cases, the π-stacking directions is oriented parallel to film in edge-on configuration. BTTL6-4Cl is crystalline with lamellae of periodicity 27 Å lying parallel to film and with a large in-plane periodicity of 38 Å. Unfortunately, similarly to the BTTL6 and BTTL6-4F, the conducting pathways are principally directed in the film plane. For the BTTR8 series, all films display also a frozen mesomorphic state as BTTL6 and BTTL6-4F with characteristic distances hπ = 3.56 Å (BTTR8), 3.54 Å (BTTR8-4F), 3.52 Å (BTTR8-4Cl)) and hch = 4.5 Å. The lamellar periodicity is 19 Å (BTTR8 and BTTR8-4Cl) or 20 Å (BTTR8-4F) and the lamellae orientations are essentially edge-on. The presence of the ramified chains whose crystallization is naturally hampered is probably the reason for structure type change from crystalline to mesomorphic between BTTL6-4Cl and BTTR8-4Cl.
Despite this undesirable orientation, the SCLC mobility values of the BTTs are decent and make it possible to consider using them as electron acceptor compounds in combination with the PM6 electron-donor polymer, with which the absorption complementarity and the ΔIP seem adequate.
Before elaborating the organic solar cells (OSCs) and in order to complete the structural characterisation, the pure PM6 and PM6-BTT blend thin films were also analysed by GIWAXS. The PM6 polymer alone adopts a frozen mesomorphic state with a lamellar structure of alternating π-stacked backbones (π-stacking distance from maximum position hπ = 3.68 Å) and molten alkyl chains (hch = 4.5 Å). The polymer backbones lie always parallel to the substrate, but with mixed edge-on and face-on configurations. Overall, the structure and morphology of PM6 and BTTs domains in the blends are equivalent to those in the neat compounds. There is no sign of a mixed phase with other structural parameters and the BTT lamellas are still mainly oriented edge-on as regards to the substrate plane. It is thus reasonable to assume that the electron mobilities measured for pure BBTs materials in the direction perpendicular to the substrate will be in the same order of magnitude when blended with PM6.
OSCs were elaborated and characterized according to the procedure described in ESI.† The OSC structure was always Glass/ITO/ZnO/active layer/MoO3/Ag and the active layer (AL) was deposited from a solution using CHCl3 as solvent and 1-chloronaphtalene (CN) as additive. The total concentration of the solution and the thermal annealing conditions were optimized for each AL. All the photovoltaic parameters measured in standard illumination conditions can be found in Table 3. The best diode characteristics for each AL are displayed in Fig. 5.
| Compound | [D:A] wt ratio |
C t (mg mL−1) | V oc (mV) | J sc (mA cm−2) | FF (%) | PCEb (%) |
|---|---|---|---|---|---|---|
| All devices were annealed 10 minutes at 110 °C before the top-electrode deposition.a Ct: total concentration. Solvent used was a mixture of 99.5% vol. CHCl3 and of 0.5% vol. CN.b The average value on at least 10 diodes is indicated in parenthesis. | ||||||
| BTTL6 | [1:1.2] |
10 | 880 | 4.0 | 42.2 | 1.5 (1.4) |
| BTTL6-4F | [1:1.5] |
10 | 850 | 11.3 | 59.1 | 5.7 (5.5) |
| BTTL6-4Cl | [1:1.5] |
10 | 840 | 4.8 | 44.4 | 1.8 (1.6) |
| BTTR8 | [1:1.5] |
10 | 900 | 5.1 | 44.4 | 2.0 (1.9) |
| BTTR8-4F | [1:1.5] |
10 | 810 | 9.3 | 49.3 | 3.7 (3.6) |
| BTTR8-4Cl | [1:2] |
12 | 880 | 11.4 | 58.8 | 5.9 (5.7) |
 | ||
| Fig. 5 (J–V) characteristics of the best PM6:BTTs organic solar cells in the dark (close symbols) and under AM1.5G illumination conditions (open symbols). The left figure corresponds to the BTTL6 series and the right one to the BTTR8 series. | ||
When going from non-halogenated BTT molecules to halogenated ones, the open-circuit voltage (Voc) is reduced. This was anticipated as halogenation of the BTT series leads systematically to a deeper LUMO level and it is well known that, at first approximation, Voc increases with the energy offset between the HOMO of the electron-donor and the LUMO of the electron-acceptor. But Voc also depends on a number of interdependent properties of the active layer. The morphology of the active layer can be dependent on the solubilizing chains and optimum [D:A] ratio, and this morphology has a strong influence on the dynamics of carrier recombination and open-circuit voltage (see below). It is therefore risky to base Voc evolution solely on the position of energy levels when comparing the BTTL6 and BTTR8 series, or when comparing the influence of fluorine and chlorine. The main variation on the photovoltaic parameters is however observed on the short-circuit current density (Jsc). Indeed, the optimized BTTL6 OSCs showed a limited Jsc in the 4 mA cm−2 range. This may indicate a strongly limited free charge-carrier photogeneration. A possible explanation could be the too low HOMOs offset between the electron donor PM6 and the non-halogenated BTTs. Indeed, as discussed previously (see also Fig. 4), the ΔIP, as measured by CV is about 0 eV, while it increases to 0.6 eV from PESA measurements. It is thus reasonable to assume that this limited HOMO offset could be one of the reasons for a limited efficiency of the exciton dissociation when a photon is absorbed on BTTL6. By slightly deepening the HOMO level, halogenation of BTTL6 (BTTL6-4F and BTTL6-4Cl) is expected to improve the exciton dissociation. In comparison to BTTL6, the ΔIP between the halogenated BTTs and the PM6 are systematically increased by 0.05 to 0.1 eV. On the one hand, the impact is limited when going from BTTL6 to BTTL6-4Cl with a relative increase of only 20% on Jsc. Further, the FF relative increase is only 5% when going from BTTL6 to BTTL6-4Cl and stays below 50% indicating charge-extraction limitations. On the other hand, Jsc is almost multiplied by a factor of three when going from BTTL6 to BTTL6-4F and the measured FF for BTTL6-4F approached 60%.
The halogenation strategy appears therefore as partly pertinent, even though the morphology is not necessarily always adequate as evidenced in the solubility issues experienced in the elaboration of OSCs as well as of thick films for SCLC devices, using BTTL6-4F and BTTL6-4Cl (see Table 2). Therefore, bulky ramified side-chains were introduced to circumvent the limited solubility of the BTTL6 series. Although slightly increased compared to BTTL6, the rather low BTTR8 PCE supports the hypothesis that the HOMO offset is too limited to efficiently photogenerate free charges. Both halogenated BTTR8-4F and BTTR8-4Cl molecules showed a significant Jsc improvement compared to the non-halogenated BTTR8 molecule. Thanks to a solubility increase, the chlorinated BTTR8-4Cl molecule gave the highest photovoltaic parameters with a Jsc as high as 11.4 mA cm−2, a FF approaching 60% and a Voc of 880 mV. Consequently, the PCE of devices using BTTR8-4Cl as electron acceptor is about 6%. However, BTTs-based blends exhibit moderate PCEs compared to state-of-the-art NFA. Especially the Jsc and the FF are significantly lower than best reported NFA-based solar cells. This is most likely due to a not fully suitable charge transport within the blends. A measurement of the Voc dependence to the incident light power (Pin) in a PM6:BTTL6-4F active layer, is shown in Fig. S96 (ESI†) and confirms this hypothesis. Indeed, a dominant trap-assisted recombination process for the photogenerated free-carriers was evidenced, in-line with charge-extraction limitations and with the moderate Jsc and FF measured for this blend.
Conclusion
In summary, we designed and synthesized a new series of BODIPY-based non-fullerene acceptors by: (i) referring to the most efficient type A–D–A′–D–A design (type Y6) and (ii) avoiding the chemistry of fused aromatic units. A regioselective chemical pathway, discriminating the 3,5- and 2,6-positions of the BODIPY unit, allowed us to synthesize 6 new BTT molecules in good yields. As expected, they exhibit very low optical bandgaps, very high extinction coefficients and deep enough LUMO levels. The appropriate halogenation of the cyanoindanone electron acceptor end units with chlorine or fluorine atoms allows the fine tuning of the energy levels. In particular, the HOMO level adjustment appears to be a key factor in controlling the HOMO offset required for efficient photogeneration of free charges.Although BTTR8-4Cl led to one of the best PCEs obtained from BODIPY-based NFA so far, the maximum PCE obtained, of around 6%, is still limited compared to the state-of-the-art NFA-based OPV devices. The edge-on orientation of these new BTTs, leading to limited perpendicular charge transport, is probably a current limiting key parameter. We believe that increasing the electron donor unit sizes as well as the number, position and length of the solubilizing side-chain are ways to switch to more favorable face-on orientations.43 Such derivatives are currently being developed in our laboratory.
Author contributions
FC contributed to the conceptualization, investigation and writing the original draft. AL contributed to the investigation. DJ contributed to the investigation and review and editing. BH contributed to the investigation, resources and review and editing. FR contributed to the investigation. PL contributed to the funding acquisition, methodology, supervision and writing. GU contributed to the funding acquisition, methodology, supervision and writing. NL contributed to the funding acquisition, methodology, supervision and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. C. thanks the University of Strasbourg and the French Ministry of Research for “contrat doctoral“. D. J. is indebted to the CCIPL/GliciD computational mesocenter installed in Nantes for generous allocation of computational ressources. The authors thank Pohang Accelerator Laboratory (PAL) for giving them the opportunity to perform the GIWAXS measurements, MEST and POSTECH for supporting these experiments, Drs. Hyungju Ahn and Wooseop Lee for adjustments and help, and other colleagues from the 9A USAXS beamline for assistance.References
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, e2102420 CrossRef PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656 CrossRef CAS PubMed.
- S. Dey, Small, 2019, 15, 1900134 CrossRef PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140 CrossRef CAS.
- Q. Wei, W. Liu, M. Leclerc, J. Yuan, H. Chen and Y. Zou, Sci. China: Chem., 2020, 63, 1352 CrossRef CAS.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554 CrossRef CAS.
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356 CrossRef CAS PubMed.
- S. Li, L. Zhan, F. Liu, J. Ren, M. Shi, C.-Z. Li, T. P. Russell and H. Chen, Adv. Mater., 2018, 30, 1705208 CrossRef PubMed.
- J. Zhao, X. Xu, L. Yu, R. Li, Y. Li and Q. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 25214 CrossRef CAS.
- J. Gao, Y. Li, S. Li, X. Xia, X. Lu, M. Shi and H. Chen, Sol. Energy Mater. Sol. Cells, 2021, 225, 111046 CrossRef CAS.
- B.-S. Lu, Y. Zhang, T.-Y. Hu, Y.-F. Ma, Y.-N. Zhu, D.-W. Liu, Z.-Q. Zhang, E. Wang, W. Ma and H.-L. Zhang, Org. Electron., 2021, 93, 106132 CrossRef CAS.
- X. Liu, Y. Wei, X. Zhang, L. Qin, Z. Wei and H. Huang, Sci. China: Chem., 2021, 64, 228 CrossRef CAS.
- H. Huang, Q. Guo, S. Feng, C. Zhang, Z. Bi, W. Xue, J. Yang, J. Song, C. Li, X. Xu, Z. Tang, W. Ma and Z. Bo, Nat. Commun., 2019, 10, 3038 CrossRef.
- X. Zhang, C. Li, L. Qin, H. Chen, J. Yu, Y. Wei, X. Liu, J. Zhang, Z. Wei, F. Gao, Q. Peng and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 17720 CrossRef CAS.
- L. Ma, S. Zhang, J. Zhu, J. Wang, J. Ren, J. Zhang and J. Hou, Nat. Commun., 2021, 12, 5093 CrossRef CAS PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed.
- T. Bura, N. Leclerc, S. Fall, P. Lévêque, T. Heiser, P. Retailleau, S. Rihn, A. Mirloup and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 17404 CrossRef CAS.
- J. Yang, C. H. Devillers, P. Fleurat-Lessard, H. Jiang, S. Wang, C. P. Gros, G. Gupta, G. D. Sharma and H. Xu, Dalton Trans., 2020, 49, 5606 RSC.
- R. Srinivasa Rao, A. Bagui, G. Hanumantha Rao, V. Gupta and S. P. Singh, Chem. Commun., 2017, 53, 6953 RSC.
- T. Li, T. Meyer, Z. Ma, J. Benduhn, C. Körner, O. Zeika, K. Vandewal and K. Leo, J. Am. Chem. Soc., 2017, 139, 13636 CrossRef CAS PubMed.
- T. Jadhav, R. Misra, S. Biswas and G. D. Sharma, Phys. Chem. Chem. Phys., 2015, 17, 26580 RSC.
- B. Liu, Z. Ma, Y. Xu, Y. Guo, F. Yang, D. Xia, C. Li, Z. Tang and W. Li, J. Mater. Chem. C, 2020, 8, 2232 RSC.
- I. Bulut, Q. Huaulmé, A. Mirloup, P. Chávez, S. Fall, A. Hébraud, S. Méry, B. Heinrich, T. Heiser, P. Lévêque and N. Leclerc, ChemSusChem, 2017, 10, 1878 CrossRef CAS.
- A. M. Poe, A. M. Della Pelle, A. V. Subrahmanyam, W. White, G. Wantz and S. Thayumanavan, Chem. Commun., 2014, 50, 2913 RSC.
- W. Liu, J. Yao and C. Zhan, Chin. J. Chem., 2017, 35, 1813 CrossRef CAS.
- X. Song, Y. Xu, X. Tao, X. Gao, Y. Wu, R. Yu, Y. He and Y. Tao, Macromol. Rapid Commun., 2022, 43, 2100828 CrossRef CAS.
- P. Li, F. Wu, Y. Fang, H. Dahiya, M. L. Keshtov, H. Xu, A. Agrawal and G. D. Sharma, ACS Appl. Energy Mater., 2022, 5(2), 2279 CrossRef CAS.
- X. Zong, H. Dahiya, Y. Song, X. Liang, S. K. Pandey, H. Xu and G. D. Sharma, ACS Appl. Energy Mater., 2022, 5, 15624 CrossRef CAS.
- L. Jiao, W. Pang, J. Zhou, Y. Wei, X. Mu, G. Bai and E. Hao, J. Org. Chem., 2011, 76, 9988 CrossRef CAS PubMed.
- J. L. Belmonte-Vázquez, E. Avellanal-Zaballa, E. Enríquez-Palacios, L. Cerdán, I. Esnal, J. Bañuelos, C. Villegas-Gómez, I. L. Arbeloa and E. Peña-Cabrera, J. Org. Chem., 2019, 84, 2523 CrossRef.
- Z. Yao, X. Liao, K. Gao, F. Lin, X. Xu, X. Shi, L. Zuo, F. Liu and Y. Chen, J. Am. Chem. Soc., 2018, 140, 2054 CrossRef CAS.
- Z. Zhang, J. Yu, X. Yin, Z. Hu, Y. Jiang, J. Sun, F. Zhang, T. P. Russell, F. Liu and W. Tang, Adv. Funct. Mater., 2018, 28, 1705095 CrossRef.
- J. Shaya, J.-C. Ribierre, G. Correia, Y. J. Dappe, F. Mathevet, L. Mager, B. Heinrich and S. Méry, Molecules, 2023, 28, 2038 CrossRef CAS.
- C. Tschierske, J. Mater. Chem., 2001, 11, 2647 RSC.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1800613 CrossRef PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655 CrossRef CAS PubMed.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- J. Bertrandie, J. Han, C. S. P. De Castro, E. Yengel, J. Gorenflot, T. Anthopoulos, F. Laquai, A. Sharma and D. Baran, Adv. Mater., 2022, 34, 2202575 CrossRef CAS PubMed.
- O. A. Ibraikulov, B. Heinrich, P. Chávez, I. Bulut, C. Ngov, O. Boyron, N. Brouckaert, S. Swaraj, K. L. Gerasimov, D. A. Ivanov, S. Mery, N. Leclerc, P. Lévêque and T. Heiser, J. Mater. Chem. A, 2018, 6, 12038 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02039h |
| This journal is © The Royal Society of Chemistry 2023 |