
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNegative solvatochromism and sign inversion of circularly polarized luminescence in chiral exciplexes as a function of solvent polarity†
Patthira
Sumsalee
a,
Pierpaolo
Morgante
 b,
Gregory
Pieters
c,
Jeanne
Crassous
a,
Jochen
Autschbach
*b and
Ludovic
Favereau
*c
b,
Gregory
Pieters
c,
Jeanne
Crassous
a,
Jochen
Autschbach
*b and
Ludovic
Favereau
*c
aUniv Rennes, CNRS, ISCR-UMR 6226, F-35000, Rennes, France. E-mail: ludovic.favereau@univ-rennes1.fr
bDepartment of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260, USA. E-mail: jochena@buffalo.edu
cUniversité Paris-Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), SCBM, 91191 Gif-sur-Yvette, France
First published on 18th May 2023
Abstract
The potential control of circularly polarized luminescence (CPL), especially its sign and switching at the molecular level without any chemical modification, is desirable, but remains a considerable challenge owing to the difficulty to finely control the magnitude and relative orientation of the associated electric and magnetic dipole transition moments. To address this challenge, we report the synthesis and chiroptical properties of innovative non-conjugated chiral donor–acceptor systems displaying CPL sign inversion as a function of solvent polarity. Through the formation of a chiral exciplex, it is possible to achieve control of its (non-) radiative deexcitation pathways using solvents of different polarity, resulting in emission from locally excited (LE) or charge-transfer (CT) states (with positive and negative CPL), and thermally activated delayed fluorescence (TADF). Theoretical calculations offer further evidence of the relationship between the nature of the emitting species and the CPL. These results provide original molecular design guidelines to obtain switchable CPL emitters, and new insights into the combination of CPL and TADF, a feature of crucial importance for the development of efficient technologies based on CPL emitters.
Introduction
Designing organic chiral compounds that are able to emit circularly polarized (CP) light has attracted significant interest during the last decades due to the potential applications of CP light in optoelectronics [organic light-emitting diodes (OLEDs), and CP light detectors], as well as in chiral sensing and bioimaging.1–7 For instance, significant efforts are currently directed towards the development of chiral luminophores with thermally activated delayed fluorescence (TADF) for developing OLEDs with polarized electroluminescence, a potential direction for increasing the efficiency of OLED displays.8,9Our recent contributions in this research area investigated CP-TADF emitters based on atropisomer bicarbazole donors functionalized with carbonyl or nitrile accepting units, which afforded promising luminescence dissymmetry factors (glum) up to 2.0 × 10−3.10,11 We also studied intermolecular chiral luminescent exciplexes involving chiral electron donor systems and achiral electron accepting units, introducing a new design of CP-TADF compounds with glum values reaching 7.0 × 10−3.12 During these investigations, we noticed that both the intensity and the sign of the CP luminescence (CPL) differ when comparing the chiral donor alone and the corresponding luminescent exciplex. While this intriguing behaviour has been tentatively rationalized by invoking the different nature of the emitting transition, i.e. locally excited (LE) vs. charge-transfer (CT) states, a more detailed rationalization is still lacking. Indeed, controlling molecular systems to switch the sign of their CPL can bring new opportunities in optoelectronics and bioimaging.13–15 However, examples of chiral compounds with switchable CPL sign remain rare, and rely mostly on dimeric or polymeric structures that require the precise control of both the solvent conditions and the molar concentration of the emitting species.16 For instance, Takaishi, Ema et al. recently reported the switching of the CPL sign in excimeric systems through intermolecular hydrogen bonds formation dependent on the solvent polarity (Fig. 1A).17 At the molecular level, the switching of the CPL sign for a given enantiomer without any chemical modification appears even more challenging and only a few examples have been reported, based mainly on a complex energy dynamics of the lowest unoccupied excited state(s).18,19 This is the case, for instance, in the work of Zhang et al. (Fig. 1B, ref. 19), who reported CPL sign inversion when going from non-polar to polar solvents, associated with a positive solvatochromism of the luminescence spectrum related to an increase of CT character within the emission process.
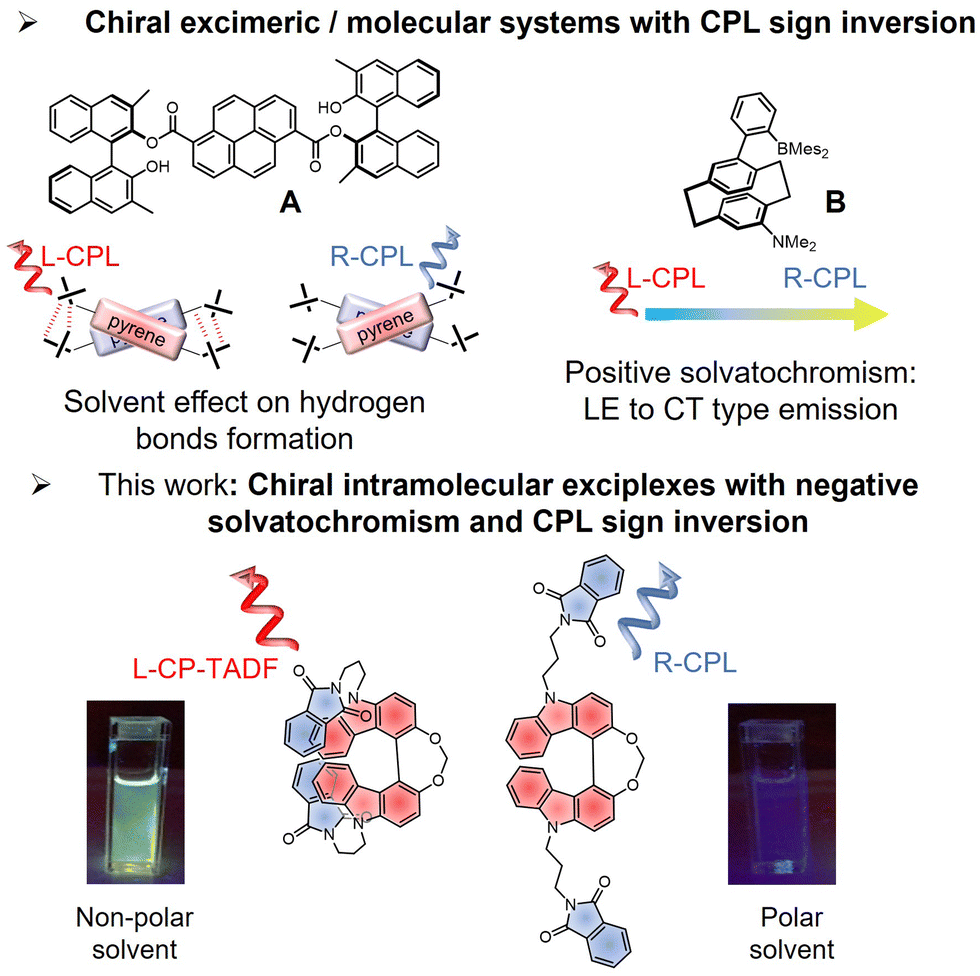 | ||
| Fig. 1 Top: Examples of chiral systems showing CPL sign inversion via excimeric formation (ref. 17) and positive solvatochromism (ref. 19). Bottom: Chemical structures of one the new exciplex emitters, 4, reported in this study with the schematic illustration of their CPL sign (blue and red for right- and left-CPL, respectively), and the picture of their emission colours in non-polar and polar solvents. | ||
Accordingly, innovative synthetic guidelines for designing chiral emitters with switchable CPL are therefore needed to enable further developments in this field. Regarding this aspect, CPL emitters based on excimers are receiving increasing attention because of their peculiar photophysical and chiroptical properties with respect to more classical covalent chiral organic emitters.20,21 Often, the intensity of CPL for these systems is also rather intense, and usually different than the associated ground-state chirality of the related compound.22–25 Surprisingly, their chiral exciplex counterparts are much less investigated in this domain of research, despite their additional potential interest for instance in CP-OLED technology. Indeed, this intermolecular charge-transfer design can afford a new design of CP-TADF emitters because of the spatially separated HOMO and LUMO levels, leading to small singlet–triplet excited states gap, ΔEST, a prerequisite for efficient TADF emitters11,26 While an exciplex can be seen as a simple excited state complex formed between an excited electron donor (or acceptor) molecule and an unexcited electron acceptor (or donor), the photophysics behind this process appears more complex and often includes different excited state intermediates and luminescent responses.27–31 Achieving control of these potential emitters is an interesting strategy to afford different emission profiles and potentially switchable CPL responses with the same enantiopure molecular system.
Herein we report the design of two new non-conjugated chiral donor–acceptor systems, namely 3 and 4, based on a helical bicarbazole unit, functionalized by either one or two phthalimide units. These molecular compounds can form chiral intramolecular exciplexes, and because of the underlying photophysics these excited state complexes show sign inversion of CPL as a function of solvent polarity (Fig. 1). In addition, a negative fluorescence solvatochromism from blue (polar solvent) to green (non-polar solvent) luminescence is observed, which appears related to the radiative vs. non-radiative recombination of the charge-transfer event occurring between the biscarbazole unit and the pendant phthalimide substituents. The CPL sign inversion is further rationalized theoretically through comparison of different conformations of the helical bicarbazole unit functionalized with either one or two phthalimide units, with the former exhibiting a bisignate CPL response in non-polar solvent. Interestingly, luminescence of the two compounds in that medium shows TADF owing to a small singlet–triplet energy gap in the excited state, which provides additional evidence of the exciplex formation. Altogether, these unprecedented results offer a new strategy to design switchable CPL emission at the molecular level, with a precise control of the nature of the transition involved in the CPL.
Results and discussion
Synthesis and characterizations
The synthesis of the two compounds started with the preparation of the helical bicarbazole derivative, Helicol 1, under its enantiopure form (Scheme 1). Following our previous work,32 3-hydroxy carbazole, CBzOH, was engaged in an oxidative homocoupling reaction using a vanadium complex as catalyst, followed by the bridging of the two hydroxyl groups using diiodomethane in 90% yield. At this stage, corresponding enantiomers of Helicol, (P)-(+)- and (M)-(–)-Helicol, were obtained by HPLC separations over chiral stationary phases with ee's up to 99% (see ESI† and ref. 4). Successive nucleophilic substitution reactions were then conducted, first between (P)-(+)- and (M)-(–)-Helicol 1 and an excess of 1,3-dibromopropane to give intermediates (P)-(+)- and (M)-(–)-Helicol 2 in 89% yield, and finally between the latter and the isoindoline-1,3-dione to afford the two mono- and bis-functionalized enantiomers, (P)-(+)- and (M)-(–)- 3 and (P)-(+)- and (M)-(–)-4 respectively, of the Helicol compounds bearing phthalimide unit(s). In fact, it appears that the isolated mono-functionalized Helicol presented a terminal alkene function and not a bromine atom, presumably owing to an elimination reaction, as confirmed by NMR characterizations with the presence of two doublet signals at 5.10 and 5.27 ppm (1H), a singlet at 5.09 ppm (2H) and a multiplet at 6.19 ppm, in addition to the set of three signals of 4 protons found at 2.40, 3.89 and 4.57 ppm, which are assigned to the propyl chain between the nitrogen atoms of the carbazole and phthalimide units in both the mono- and bisfunctionalized compounds. The full experimental conditions and characterization (NMR and mass spectrometry) of all compounds are detailed in the ESI.†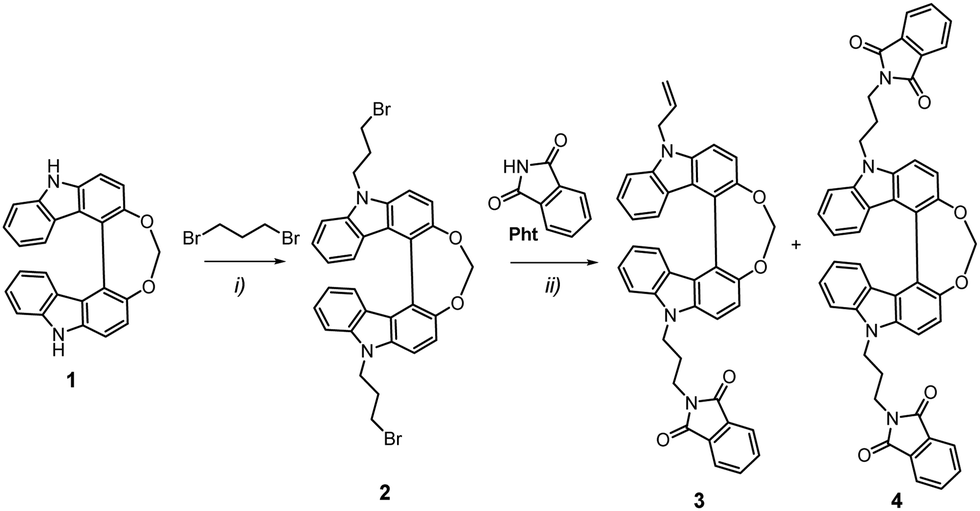 | ||
| Scheme 1 Synthetic pathway leading to chiral emitters 3 and 4. Reaction conditions: (i) 1,3-dibromopropane, NaH, DMF, rt, 89%; (ii) KOtBu, NMP, 60 °C, 42 and 51% for 3 and 4, respectively. Further details are reported in the ESI.† | ||
Computational details
The theoretical investigation of chiroptical properties employed Kohn–Sham density functional theory (KS-DFT) and dynamic response theory via time-dependent (TD) DFT. The range-separated hybrid (RSH) functional ωB97X-D33 and the def2-SV(P) basis set34 were used for geometry optimizations, vibrational frequency calculations, and TD-DFT calculations, which employed the Gaussian 16 (G16) program, version A.03.35 Additional single-point calculations with the def2-TZVP basis34 were performed on the optimized geometries to obtain the energy differences reported in Fig. 5 (vide infra). The conductor-like polarizable continuum model (C-PCM)36 was used to treat effects from the two solvents, dichloromethane (DCM) and methylcyclohexane (MCH).Absorption and electronic circular dichroism (ECD) spectra were obtained considering the 50 lowest-energy excitations calculated using TD-DFT on the ground-state (S0) equilibrium geometry of all compounds. In addition to def2-SV(P), the def2-SVPD and def2-TZVP basis set were also tested, resulting in no significant changes in the calculated excitation energies (see Tables S23–S28 in the ESI†). The transitions were Gaussian broadened with a σ of 0.20 eV to obtain the corresponding spectra. Vibrationally resolved singlet emission and CPL spectra including Franck–Condon (FC) and Herzberg–Teller (HT) effects were calculated as implemented in G16.37 The vibronic transitions were Gaussian broadened with a σ of 0.0248 eV. For the disubstituted compounds, it was not possible to locate the excited state minima for all of the conformers identified in the ground state, despite multiple attempts with different optimization algorithms. For this reason, the vertical gradient (VG) approximation38 was used for the emission and CPL spectra of the conformers of both compounds. For all spectra, a global shift of −0.40 eV was applied to the calculated excitation energies prior to their conversion to wavelengths. RSH functionals such as the one used in this work are known to overestimate experimental excitation energies for systems of the type studied herein,39–42 and therefore the shift was required to align the experimental and calculated spectral bands. In addition, the vertical gradient (VG) approximation used to model the emission spectra is known to require a shift to align with experimental band positions.43 Accordingly, an additional shifting of −0.45 eV, corresponding to the average difference between the 0–0 transitions calculated with the VG and the more reliable adiabatic Hessian approximation for conformers 4a–4c in MCH, was applied to the emission and CPL spectra. For additional computational details, see Section 5 of the ESI.† For an overview of theoretical approaches to calculate natural optical activity parameters, especially with TD-DFT, see ref. 44 and 45.
Ground-state photophysical and chiroptical properties
UV-vis spectra of compounds 3 and 4 were recorded in dichloromethane and show similar features as those of their corresponding phthalimide and helical bicarbazole precursors (Fig. 2 and Fig. S1, ESI†). Both compounds display two maxima of absorption at 270 and 300 nm (ε ≈ 1.3 × 104 and 1.0 × 104 M−1 cm−1), respectively, and a vibronic band between 330 and 390 nm (ε ≈ 0.6 × 104 M−1 cm−1, Fig. 2), mainly assigned to the helicol unit with a significant contribution of the phthalimide group at 270 and 300 nm. This absorption response is not affected by the polarity of the solvent since similar patterns are also recorded in the non-polar methylcyclohexane (Fig. S1, ESI†), which indicates the absence of charge-transfer interaction between the carbazole and the phthalimide units in the ground-state for both 3 and 4.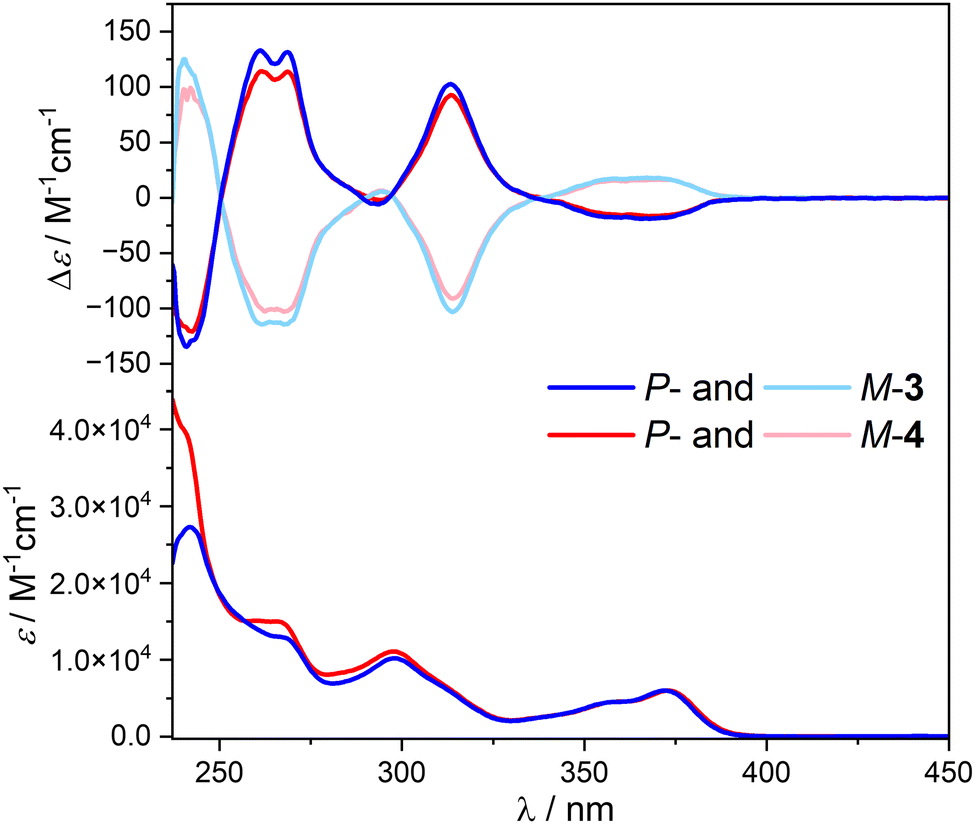 | ||
| Fig. 2 Top: ECD and bottom: UV-vis absorption of 3 (blue) and 4 (red) in dichloromethane at 298 K ([c] ∼10−5 M) | ||
The ECD of the enantiopure compounds P-3 and P-4 shows that the recorded signals are similar to those of the corresponding unsubstituted helicol enantiomers (Fig. 2): a positive exciton couplet around 250 nm (Δe ∼ + 145 M−1 cm−1), a positive band at ca. 310 nm (+100 M−1 cm−1) and a low-energy negative broad band between 340 and 400 nm (−15 M−1 cm−1), with (as expected) no impact of the achiral phthalimide chromophores both in dichloromethane and methylcyclohexane solvents, as also indicated by the corresponding gabs plots (Fig. S2, ESI†).
Excited state photophysical and chiroptical properties
The unpolarized luminescence spectra of compounds 3 and 4 were first recorded in dichloromethane and exhibit a weakly structured fluorescence band centered at 417 nm for both molecules (Fig. 3), characteristic of the Helicol precursor emission (Fig. S3, ESI†). In methylcyclohexane, the two compounds show a negative solvatochromism, with a red-shifted luminescence characterized by a broad structureless band with maxima at 478 and 488 nm for 3 and 4, respectively.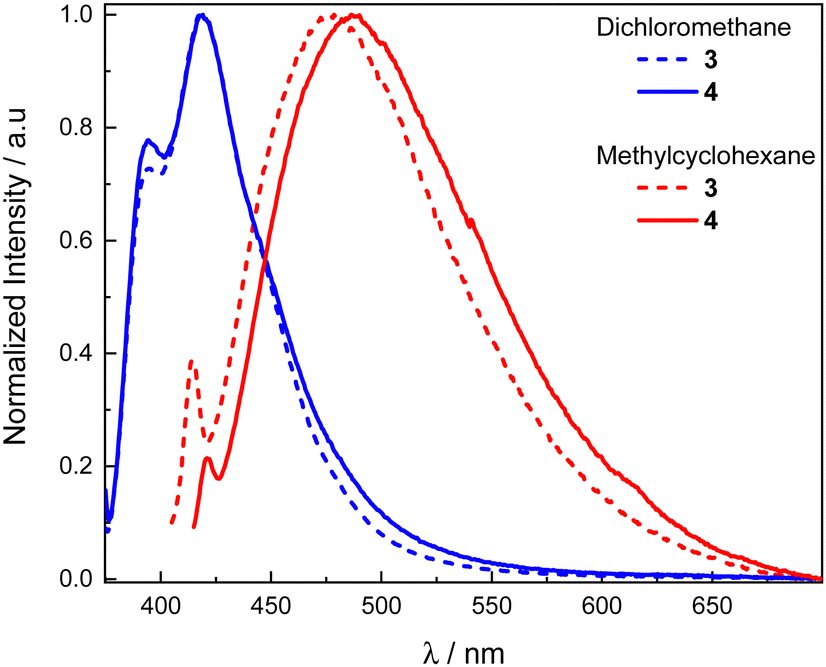 | ||
| Fig. 3 Normalized luminescence spectra in dichloromethane (blue) and methylcyclohexane (red) of 3 (dotted lines) and 4 (solid lines) ([c]∼10−5 M) at 298 K. | ||
Such dramatic change in the luminescence behavior suggests an exciplex-type luminescence in the less polar solvent that results from an intramolecular charge transfer in the excited state between the helicol donor and the electron acceptor phthalimide unit(s). Interestingly, a structured emission signal around 400 nm is present for both compounds, reminiscent of the fluorescence signal obtained in dichloromethane for both 3 and 4. This observation suggests that emission from the LE-state of the biscarbazole system is still present in non-polar solvent. However, the relative intensity between that signal and the one arising from the exciplex is less intense in the case of 4 in comparison to 3, suggesting a more intense exciplex emission for the former presumably due to the presence of the second phthalimide unit.
Further photophysical characterizations of the exciplex emission in methylcyclohexane for 3 and 4 show a decrease of their emission intensity in presence of oxygen, suggesting TADF. Indeed, the luminescence response of 3 and 4 increases by 33 and 45% respectively in degassed methylcyclohexane, and deeper characterization reveals the presence of a delayed fluorescence process, with lifetime of 1.42 and 3.07 μs for 3 and 4, respectively (Table S1, ESI†). The singlet–triplet gap, ΔEST, of these compounds was also measured by recording their luminescence spectra at 77 K. They show a similar structured phosphorescence emission for both compounds with an onset at 455 nm (Fig. S4, ESI†), resulting in corresponding T1 energy levels of 2.71 eV. The onset of the broad luminescence signal in methylcyclohexane (Fig. 3) can be used to estimate the S1 energy levels of both 3 and 4, with values of 3.02 and 2.95 eV, respectively, yielding ΔEST values of 0.31 and 0.24 eV, the classical range found for TADF emitters.46–48
The CPL spectra of compounds 3 and 4 were also recorded in both solvents (Fig. 4). In line with the unpolarized luminescence in dichloromethane, the two emitters show mirror-image CPL signals corresponding to those recorded for the enantiopure helicol (ref. 32 and 49) with identical negative glum factors of –2.0 × 10−3 for P-3 and P-4. The CPL response of the investigated emitters differs significantly in methylcyclohexane, with a sign inversion for both enantiomers of P-3 and P-4.
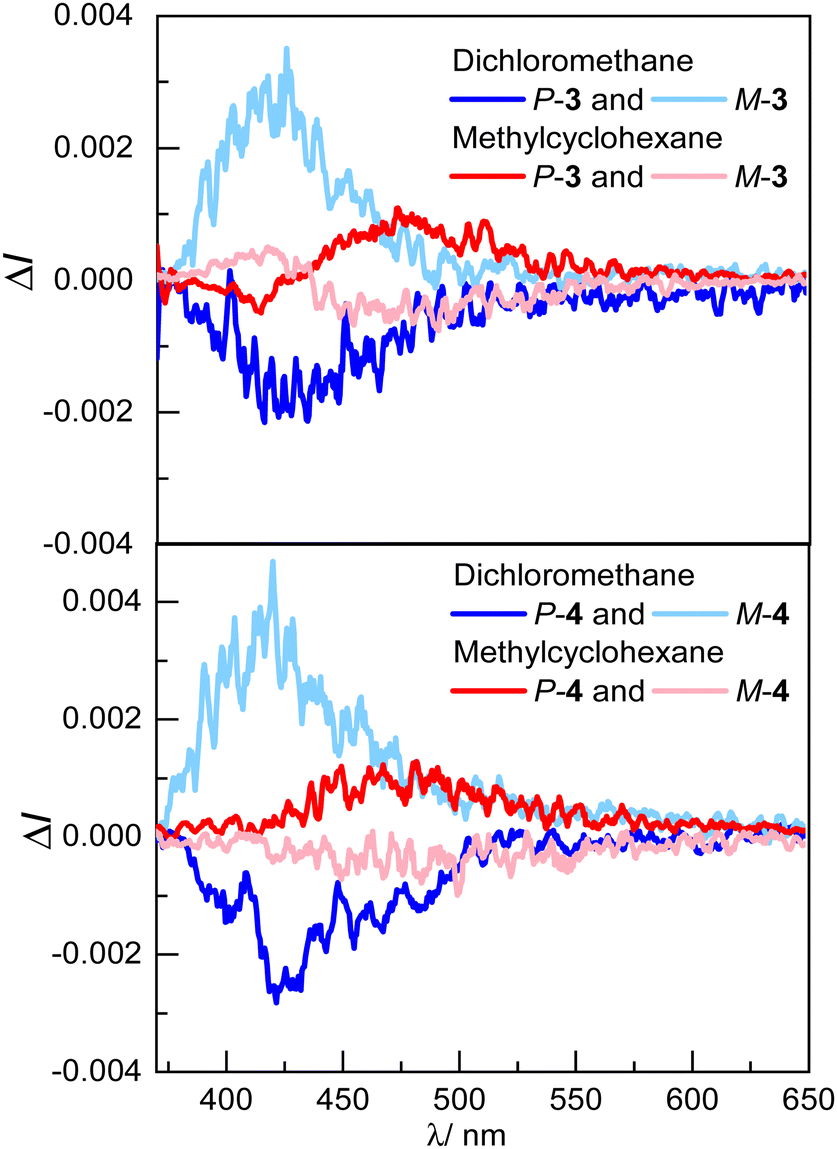 | ||
| Fig. 4 CPL spectra in dichloromethane (blue) and methylcyclohexane (red) of a) P- and M-3 and b) P- and M-4 (∼10−5 M) at 298 K. | ||
Indeed, the CP emissions at around 480 nm for both compounds show a positive signal, with an identical glum factor of ca. +1.0 × 10−3. The sign change observed for the solvents of different polarity, i.e. dichloromethane to methylcyclohexane, is presumably related to the chiral exciplex radiative deexcitation, which is favoured in non-polar solvent. Moreover, a bisignate signal is observed for monophthalimide P-3, with an additional negative band centred at 400 nm, which is however absent for compound P-4. The response for P-3 is presumably arising from radiative deexcitation of both the exciplex and the chiral donor LE-state that is not involved in the exciplex, which may occur to a greater extent than for P-4 because of the presence of only one phthalimide unit (vide infra). To characterize the chiroptical properties of these unprecedented intramolecular chiral exciplexes in more detail, we also estimated the glum/gabs ratio of P-3 and P-4. The ratio is equal to 0.95 and 1.2, respectively, in dichloromethane, and decreases to −0.31 and −0.52, respectively, in methylcylohexane, reflecting the changes observed for chiral emitting excited-states as a function of the polarity of the solvent.50 Since CPL exciplexes are relatively rare in the literature, the brightness for CPL (BCPL) of P-3 and P-4 was also calculated. It is 0.4 and 0.2 M−1 cm−1 in dichloromethane and 0.3 and 0.2 M−1 cm−1 in methylcylohexane, respectively. While these values fall into the low range of what it is usually obtained for organic CPL emitters,51 it is informative to note that the weaker glum obtained for the exciplex emission in methylcyclohexane (compared to the LE-state in dichloromethane) is somehow compensated by a higher photoluminescence quantum yield (PLQY) obtained for the luminescence of the chiral exciplex.
Theoretical analysis
The theoretical investigation first focused on understanding how the solvent polarity affects the relative structural arrangement of the non-conjugated donor and acceptors units. Different conformers of compounds 3 and 4 were identified based on the relative position of the biscarbazole helical core and the flexible phthalimide substituents (Fig. 5). For the mono-/disubstituted compound, three/five different conformers were found. For both molecules, one conformer features the phthalimide chain(s) in an ‘extended’ position, spatially separated from the helical core. This is the case for conformers 4b and 3a. The other structures relate to the extended conformers via rotation of the side chains into a ‘folded’ form with the phthalimide and the helical core in close proximity. This results in additional interactions between the phthalimide and the helix that affect the excited state chiroptical properties of the compounds (vide infra). The relative energy differences for these compounds were found to be sensitive to the choice of the basis set, as shown in Tables S3 and S4 in the ESI.† The energies presented in Fig. 5 were obtained with the def2-TZVP basis. The folded lowest-energy conformers (4d and 3b) benefit from π-stacking interactions between the phthalimide and the terminal benzene rings of the biscarbazole. The partially folded structures such as 4c and 4e, where the stacking of the aromatic rings is not as effective, are higher in energy, and the extended structures represent the highest energy conformers in the ground state (up to 6.0 kcal mol−1 above the lowest-energy conformers). The relative energy of the conformers changes only slightly (within 1.0 kcal mol−1) between the two solvents, as shown by the Gibbs energies reported in Fig. 5. No significant structural changes were observed between the two solvent models, as shown in Fig. S16 in the ESI.†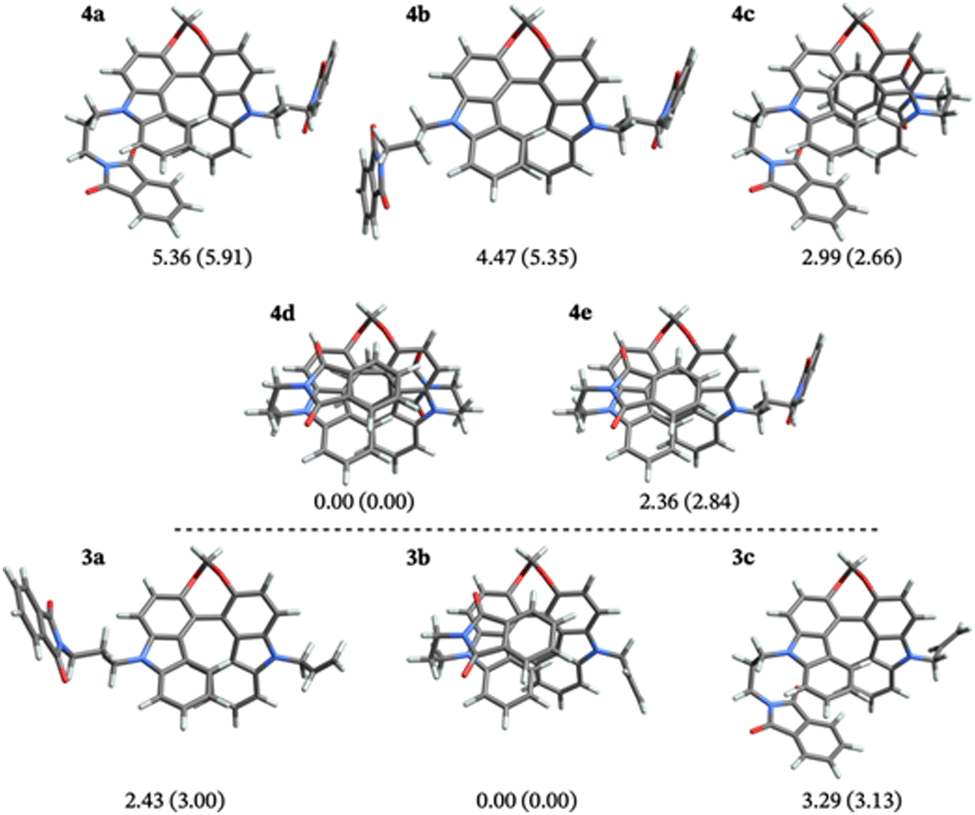 | ||
| Fig. 5 Conformers investigated for the disubstituted bicarbazole unit 4 (top) and the monosubstituted compound 3. The numbers are relative Gibbs energies for DCM (MCH) solvent in kcal mol−1. The structures shown are from the DCM calculations. | ||
The electronic properties were investigated to discern any potential differences between the folded and non-folded donor–acceptor structures. For all conformers and both solvent models, the HOMOs are localized on the biscarbazole core and the LUMOs are localized on the phthalimide substituents, confirming the push–pull characterization of the compounds. HOMO−1, HOMO, LUMO, and LUMO+1 are the orbitals that are dominantly involved in most of the transitions in the calculated ECD spectra, matching previous assignments for similar chromophores.11 HOMO−1 and HOMO are close in energy, independent of the substitution pattern of the helix. Closer inspection reveals that the HOMO and HOMO−1 are best understood as +/− linear combinations of orbitals of the carbazole monomer fragments, with the energetic splitting of HOMO and HOMO−1 indicating a moderate degree of electronic coupling of these fragments in the biscarbazole moiety. Substitution heavily influences the energies and appearance of LUMO and LUMO+1. In the unsubstituted parent compound, LUMO and LUMO+1 represent interacting +/− linear combinations of unoccupied carbazole monomer fragment orbitals. For 3, the LUMO corresponds to the lowest unoccupied phthalimide fragment orbital, which is at lower energy than the carbazole combinations in LUMO+1, with the latter corresponding to the LUMO of the unsubstituted compound. For 4, LUMO and LUMO+1 represent a weakly interacting pair of localized lowest unoccupied orbitals of the phthalimide fragments.
The calculated spectra for compound 4 in DCM and MCH are shown in Fig. 7. The absorption and ECD spectra appear very similar for the different conformers, and they reproduce the spectral features observed in the experiments. Upon shifting the calculated band energies, as explained in the computational details, there is also very good agreement between the positions of the calculated and experimental peaks. The absorption spectra display three prominent peaks, around 350, 300, and 275 nm, with increasing peak height toward lower wavelengths. The ECD peaks exhibit alternating signs, starting for the P stereoisomers with a negative sign around 350 nm. For conformers 4b and 4d this first negative peak is accompanied by a weak positive peak at higher wavelength, from the transitions that cause the corresponding long-wavelength shoulders in the absorption spectra. Very similar features overall are also found for the absorption and ECD spectra of a dipropyl-substituted model compound (the structure on the left of Fig. 6), reported in Fig. S17 of the ESI,† and of the monosubstituted systems (Fig. S18 and S19; see Section 9 in the ESI† for additional details). This confirms the experimental observation that the phthalimide substituents do not strongly influence the absorption properties of these compounds. Based on the relative energies of the conformers in the ground state shown in Fig. 5, and considering that the peaks in the experimental absorption and (especially) ECD spectra do not have prominent shoulders, it is most likely that the folded structure 4d alone is responsible for the spectra observed experimentally.
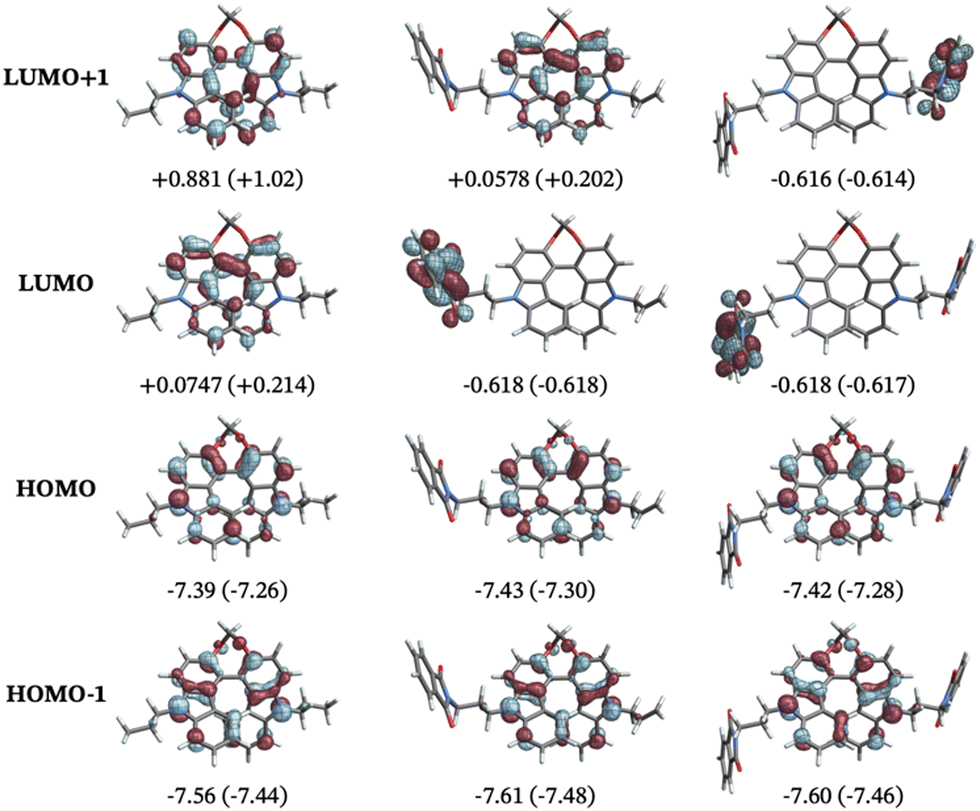 | ||
| Fig. 6 Isosurfaces (±0.04 au) of the relevant frontier molecular orbitals of (left to right) a dipropyl-substituted biscarbazole, 3a, and 4b. The numbers are orbital energies in eV for DCM (MCH) solvent. The orbitals shown are from the DCM calculations. | ||
The CPL spectra, unlike ECD, display a remarkable dependence on the molecular structure. The vibrationally resolved calculated CPL spectrum of the unsubstituted P conformer has a negative sign (Fig. S17 of the ESI†) which is also found for the extended conformer P-4b in both DCM and MCH. The purely electronic rotatory strengths calculated at the excited state minima have the same sign as the vibrationally resolved spectra. We also note that the emission of 4b is blue-shifted relative to the other conformers.
The analysis of the spectra shown in Fig. 7, in Fig. S18 and S19 in the ESI,† with the orbitals shown in Fig. S22–S39 (ESI†), leads to the conclusion that the CPL sign change and wavelength shift in the two solvents is observed most likely because of a different emitting conformer.52 In the excited state, considering that only single-bond rotations are necessary to convert the folded conformers into their extended forms, and considering the fact that the systems contain excess energy following the photon absorption, the energy barriers associated with these bond rotations might be easily overcome,53 and there are likely multiple pathways available for the compounds to reach one of the excited state potential energy minima. A broader exploration of the S1 potential energy surface as well as the explicit treatment of solvent molecules aided by molecular dynamics simulations,31 may clarify the molecular details underlying the mechanism of these processes, but such calculations are beyond the scope of the present study. The calculated dipole moments of the different conformers also reflect the stabilization that benefits the extended and folded structures in the two solvents, especially in the excited state. The extended conformers exhibit the highest dipole moments in the ground state, the folded conformers exhibit the lowest dipole moments, and the partially folded structures have intermediate values (see ESI,† Table S29). Given how energetically close the different conformers are, it is likely that the higher-energy conformers 3a and 4b, responsible for the negative CPL, become accessible in the more polar solvent DCM, whereas the folded conformers that yield a positive CPL remain predominant in the less polar MCH. It should also be noted that the folded structures potentially form non-luminescent exciplexes in the more polar solvent, as sketched schematically in Scheme 2 (vide infra). This statement, and our conclusions derived from calculations are consistent with previous reports,54–57 involving donor–acceptor units linked together by methylene chains [(CH2)n, n = 3 for the molecules studied here], where different conformers of the same compounds were found to be responsible for different emission profiles depending on the solvent polarity. For example, Majima and coworkers found that for linked donor–acceptor units based on naphthalimide and phenothiazine, polar solvents stabilize extended conformations, while non-polar solvents favour folded conformers.54 Therefore, based on our calculations and the cited literature, most likely the two solvents do not modulate the light responsiveness of the molecules directly by, for example, inducing dramatic changes in the electronic structure, but rather by favouring different conformers to dominate the emission and CPL.
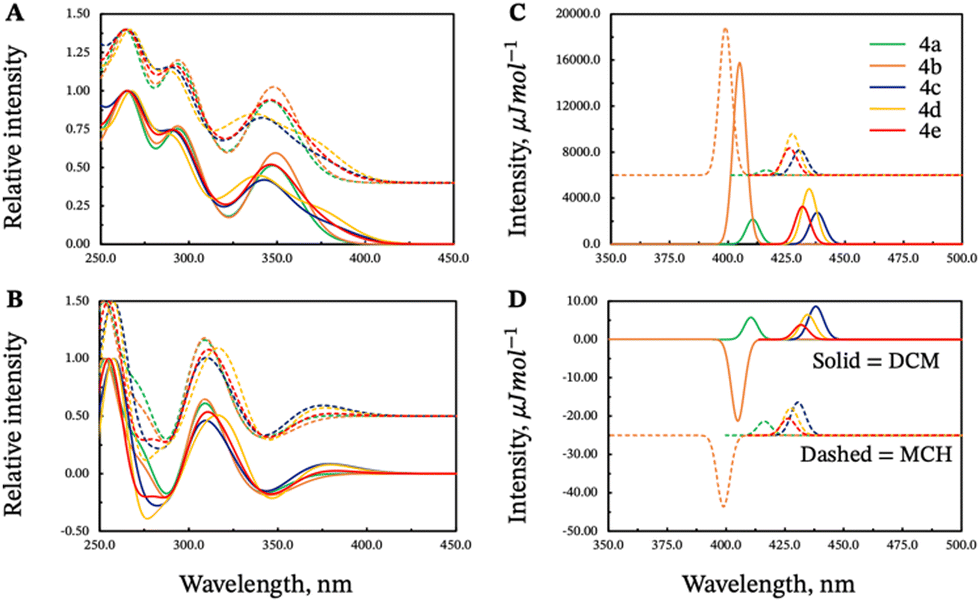 | ||
| Fig. 7 Calculated absorption (panel A), ECD (P configurations, panel B), emission (panel C), and CPL (P configurations, panel D) spectra for the five conformers of compound 4 in DCM (solid lines) and in MCH (dashed lines). The calculated energies were shifted as explained in the text. | ||
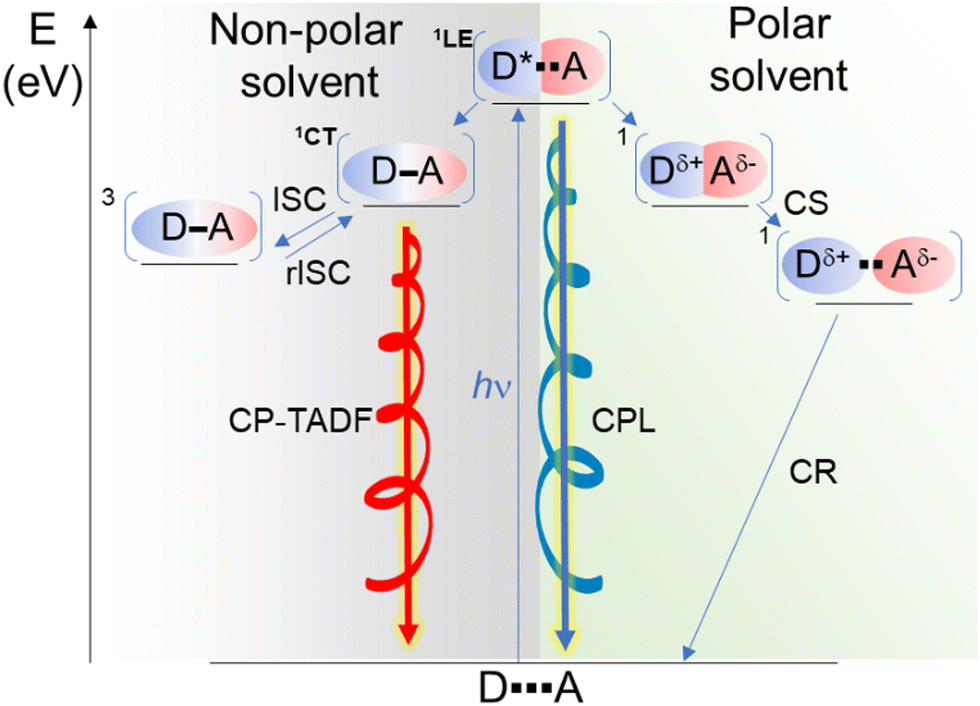 | ||
| Scheme 2 Schematic illustration of the energy-level diagram for 4 in dichloromethane (polar solvent) and methylcyclohexane (non-polar solvent) with two possible pathways for obtaining left- and right-CPL upon excitation. Charge separation (CS), (reverse) intersystem crossing ((r)ISC) and charge recombination (CR). | ||
An exciplex is often described as a result of an incomplete charge-separation upon photoexcitation of either the donor (D) or the acceptor (A) component in a D–A pair. For thermodynamic reasons (notably the enthalpy of bimolecular photoinduced charge separation (CS), see ref. 8), exciplex luminescence is mainly observed in non-polar solvents.27,28 In the present case, the observed behaviour in methylcyclohexane suggests that both 3 and 4 form a [biscarbazole-phthalimide]* complex, and its luminescence is the main deactivation pathway that results in a broad emission band as classically observed for such donor–acceptor association in addition to the presence of TADF (Scheme 2).
In dichloromethane solution, instead, the fluorescence and CPL are due to the LE-state of the biscarbazole. We presume that the polarity of the solvent strongly modifies the energy profile of the [D*A] complex favouring the formation of the radical ion pair (D+–A−) upon excitation. Accordingly, the latter complex does not emit light, and only the radiative deexcitation of the chiral donor LE-state that is not involved in the exciplex is observed, thus explaining the lower observed PLQY of 3 and 4 in dichloromethane (around 7 and 4%, respectively) compared to the reference biscarbazole system (around 20%). Exciton resonance effects between the donor and accepting units could potentially be involved in the emission observed in dichloromethane. However, this hypothesis was rejected because of the similar LE emission observed for compound 3 in methylcyclohexane and the biscarbazole precursor (depicted in the ESI†), and also because if resonance effects were at play the emission of the phthalimide unit would be expected to occur at higher energy with a relatively lower PLQY (reported to be around 380–390 nm and 0.01, respectively).58 Overall, the change of luminescence behaviour, i.e. from LE in dichloromethane to mainly CT state in methylcyclohexane, goes along with a different magnitude and orientation of the corresponding electric and magnetic transition dipole moments for the CPL.
Conclusions
In this work, we described the synthesis of innovative non-conjugated chiral donor–acceptor compounds displaying CPL sign inversion as a function of solvent polarity. The photophysical and chiroptical properties of the investigated compounds show that chiral exciplexes can be formed upon light excitation, and afford either CP-TADF or LE state fluorescence, for non-polar and polar solvents, respectively, with opposite sign of CPL and negative solvatochromism. This different luminescent behaviour is attributed to the presence of molecular folded and non-folded conformers, and the nature of the exciplex deexcitation: non-polar solvent favours the stabilization of the folded structure and CT emission from the chiral exciplex, while more polar solvent such as dichloromethane stabilizes the non-folded conformer in the excited-state, consequently leading to emission from the chiral donor LE-state that is not involved in the exciplex. Aided by theoretical calculations, the results presented in this study further rationalize our previous discoveries on chiral exciplex and bicarbazole donor–acceptor systems regarding the sign inversion of CPL when modifying the nature of the involved radiative transition. While this provides new fundamental insights on the factors governing the CPL process in organic donor–acceptor systems, we also hope that our study may offer new perspectives for the development of new classes of (switchable) CP-TADF materials and their use in optoelectronics or bioimaging.Author contributions
L. F. and P. S. conceived the project. P. S. synthesized the compounds, collected and interpreted the spectral data with the help of J. C., G. P. and L. F. Authors P. M. and J. A. performed the theoretical calculations and accompanying analyses. All authors participated in the writing process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Ministère de l’Education Nationale, de la Recherche et de la Technologie, the Centre National de la Recherche Scientifique (CNRS) and the French National Research Agency (ANR) for financial support (iChiralight project, ANR-19-CE07-0040). J. A. thanks the National Science Foundation (CHE-2152633) for financial support of the theoretical component of this study. P. M. and J. A. thank the Center for Computational Research (CCR) at the University at Buffalo59 for computational resources. Part of this work (ECD measurments) has been performed using the Spectroscopies-CDTP core facility (UMS Biosit, Univ Rennes, Rennes, France).Notes and references
- R. Carr, N. H. Evans and D. Parker, Lanthanide complexes as chiral probes exploiting circularly polarized luminescence, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- J. M. Han, S. Guo, H. Lu, S. J. Liu, Q. Zhao and W. Huang, Recent Progress on Circularly Polarized Luminescent Materials for Organic Optoelectronic Devices, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef.
- B. Kunnen, C. Macdonald, A. Doronin, S. Jacques, M. Eccles and I. Meglinski, Application of circularly polarized light for non-invasive diagnosis of cancerous tissues and turbid tissue-like scattering media, J. Biophotonics, 2015, 8, 317–323 CrossRef PubMed.
- M. Lindemann, G. Xu, T. Pusch, R. Michalzik, M. R. Hofmann, I. Žutić and N. C. Gerhardt, Ultrafast spin-lasers, Nature, 2019, 568, 212–215 CrossRef CAS PubMed.
- L. E. MacKenzie and R. Pal, Circularly polarized lanthanide luminescence for advanced security inks, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS PubMed.
- T. Novikova, A. Pierangelo, S. Manhas, A. Benali, P. Validire, B. Gayet and A. D. Martino, The origins of polarimetric image contrast between healthy and cancerous human colon tissue, Appl. Phys. Lett., 2013, 102, 241103 CrossRef.
- H. Wang, L. Liu and C. Lu, CPLC: Visible Light Communication based on Circularly Polarized Light, Procedia Comput. Sci., 2018, 131, 511–519 CrossRef.
- L. Frederic, A. Desmarchelier, L. Favereau and G. Pieters, Designs and Applications of Circularly Polarized Thermally Activated Delayed Fluorescence Molecules, Adv. Funct. Mater., 2021, 31, 2010281 CrossRef CAS.
- L. Zhou, G. Xie, F. Ni and C. Yang, Emerging circularly polarized thermally activated delayed fluorescence materials and devices, Appl. Phys. Lett., 2020, 117, 130502 CrossRef CAS.
- L. Poulard, S. Kasemthaveechok, M. Coehlo, R. A. Kumar, L. Frédéric, P. Sumsalee, T. d’Anfray, S. Wu, J. Wang, T. Matulaitis, J. Crassous, E. Zysman-Colman, L. Favereau and G. Pieters, Circularly polarized-thermally activated delayed fluorescent materials based on chiral bicarbazole donors, Chem. Commun., 2022, 58, 6554–6557 RSC.
- P. Sumsalee, L. Abella, T. Roisnel, S. Lebrequier, G. Pieters, J. Autschbach, J. Crassous and L. Favereau, Axial and helical thermally activated delayed fluorescence bicarbazole emitters: opposite modulation of circularly polarized luminescence through intramolecular charge-transfer dynamics, J. Mater. Chem. C, 2021, 9, 11905–11914 RSC.
- P. Sumsalee, L. Abella, S. Kasemthaveechok, N. Vanthuyne, M. Cordier, G. Pieters, J. Autschbach, J. Crassous and L. Favereau, Luminescent Chiral Exciplexes with Sky-Blue and Green Circularly Polarized-Thermally Activated Delayed Fluorescence, Chem. – Eur. J., 2021, 27, 16505–16511 CrossRef CAS PubMed.
- Y. Gao, C. Ren, X. Lin and T. He, The Progress and Perspective of Organic Molecules With Switchable Circularly Polarized Luminescence, Front. Chem., 2020, 8, 458 CrossRef CAS PubMed.
- J.-L. Ma, Q. Peng and C.-H. Zhao, Circularly Polarized Luminescence Switching in Small Organic Molecules, Chem. – Eur. J., 2019, 25, 15441–15454 CrossRef CAS PubMed.
- Z.-B. Sun, J.-K. Liu, D.-F. Yuan, Z.-H. Zhao, X.-Z. Zhu, D.-H. Liu, Q. Peng and C.-H. Zhao, 2,2′-Diamino-6,6′-diboryl-1,1′-binaphthyl: A Versatile Building Block for Temperature-Dependent Dual Fluorescence and Switchable Circularly Polarized Luminescence, Angew. Chem., Int. Ed., 2019, 58, 4840–4846 CrossRef CAS PubMed.
- S. Ito, K. Ikeda, S. Nakanishi, Y. Imai and M. Asami, Concentration-dependent circularly polarized luminescence (CPL) of chiral N,N'-dipyrenyldiamines: sign-inverted CPL switching between monomer and excimer regions under retention of the monomer emission for photoluminescence, Chem. Commun., 2017, 53, 6323–6326 RSC.
- K. Takaishi, K. Iwachido and T. Ema, Solvent-Induced Sign Inversion of Circularly Polarized Luminescence: Control of Excimer Chirality by Hydrogen Bonding, J. Am. Chem. Soc., 2020, 142, 1774–1779 CrossRef CAS PubMed.
- W.-B. Lin, D.-Q. He, H.-Y. Lu, Z.-Q. Hu and C.-F. Chen, Sign inversions of circularly polarized luminescence for helical compounds by chemically fine-tuning operations, Chem. Commun., 2020, 56, 1863–1866 RSC.
- M. Y. Zhang, X. Liang, D. N. Ni, D. H. Liu, Q. Peng and C. H. Zhao, 2-(Dimesitylboryl)phenyl-Substituted [2.2]Paracyclophanes Featuring Intense and Sign-Invertible Circularly Polarized Luminescence, Org. Lett., 2021, 23, 2–7 CrossRef CAS PubMed.
- F. Zinna, E. Brun, A. Homberg and J. Lacour, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer Singapore, Singapore, 2020, pp. 273–292 DOI:10.1007/978-981-15-2309-0_12.
- V. Zullo, A. Iuliano, G. Pescitelli and F. Zinna, Tunable Excimer Circularly Polarized Luminescence in Isohexide Derivatives from Renewable Resources, Chem. – Eur. J., 2022, 28, e202104226 CrossRef CAS PubMed.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Tuning symmetry breaking charge separation in perylene bichromophores by conformational control, Chem. Sci., 2019, 10, 10629–10639 RSC.
- A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Gorecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Combined reversible switching of ECD and quenching of CPL with chiral fluorescent macrocycles, Chem. Sci., 2018, 9, 7043–7052 RSC.
- H. Shigemitsu, K. Kawakami, Y. Nagata, R. Kajiwara, S. Yamada, T. Mori and T. Kida, Cyclodextrins with Multiple Pyrenyl Groups: An Approach to Organic Molecules Exhibiting Bright Excimer Circularly Polarized Luminescence, Angew. Chem., Int. Ed., 2022, 61, e202114700 CrossRef CAS PubMed.
- K. Takaishi, K. Iwachido, R. Takehana, M. Uchiyama and T. Ema, Evolving Fluorophores into Circularly Polarized Luminophores with a Chiral Naphthalene Tetramer: Proposal of Excimer Chirality Rule for Circularly Polarized Luminescence, J. Am. Chem. Soc., 2019, 141, 6185–6190 CrossRef CAS PubMed.
- Q. Gu, Z. J. Chen, W. T. Xie, W. D. Qiu, X. M. Peng, Y. H. Jiao, M. K. Li, Z. Liu, G. W. Sun, Y. F. Lu, Y. Y. Gan, K. K. Liu, Z. J. Zhao and S. J. Su, Chiral Exciplex Acceptor Enables Circularly Polarized Electroluminescence with High Dissymmetry Factor Close to 10(-2), Adv. Opt. Mater., 2022, 2201793 CrossRef CAS.
- B. Dereka, M. Koch and E. Vauthey, Looking at Photoinduced Charge Transfer Processes in the IR: Answers to Several Long-Standing Questions, Acc. Chem. Res., 2017, 50, 426–434 CrossRef CAS PubMed.
- J. F. Guo, Y. G. Zhen, H. L. Dong and W. P. Hu, Recent progress on organic exciplex materials with different donor–acceptor contacting modes for luminescent applications, J. Mater. Chem. C, 2021, 9, 16843–16858 RSC.
- D. R. Kattnig, A. Rosspeintner and G. Grampp, Fully Reversible Interconversion between Locally Excited Fluorophore, Exciplex, and Radical Ion Pair Demonstrated by a New Magnetic Field Effect, Angew. Chem., Int. Ed., 2008, 47, 960–962 CrossRef CAS PubMed.
- S. Richert, A. Rosspeintner, S. Landgraf, G. Grampp, E. Vauthey and D. R. Kattnig, Time-Resolved Magnetic Field Effects Distinguish Loose Ion Pairs from Exciplexes, J. Am. Chem. Soc., 2013, 135, 15144–15152 CrossRef CAS PubMed.
- E. Vauthey, Elucidating the Mechanism of Bimolecular Photoinduced Electron Transfer Reactions, J. Phys. Chem. B, 2022, 126, 778–788 CrossRef CAS PubMed.
- S. Kasemthaveechok, L. Abella, M. Jean, M. Cordier, T. Roisnel, N. Vanthuyne, T. Guizouarn, O. Cador, J. Autschbach, J. Crassous and L. Favereau, Axially and Helically Chiral Cationic Radical Bicarbazoles: SOMO–HOMO Level Inversion and Chirality Impact on the Stability of Mono- and Diradical Cations, J. Am. Chem. Soc., 2020, 142, 20409–20418 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, H. N. G. A. Petersson, X. Li, M. Caricato, A. V. Marenich, B. G. J. J. Bloino, R. Gomperts, B. Mennucci, H. P. Hratchian, A. F. I. J. V. Ortiz, J. L. Sonnenberg, D. Williams-Young, F. L. F. Ding, F. Egidi, J. Goings, B. Peng, A. Petrone, D. R. T. Henderson, V. G. Zakrzewski, J. Gao, N. Rega, W. L. G. Zheng, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, K. T. T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, M. J. B. F. Ogliaro, J. J. Heyd, E. N. Brothers, K. N. Kudin, T. A. K. V. N. Staroverov, R. Kobayashi, J. Normand, A. P. R. K. Raghavachari, J. C. Burant, S. S. Iyengar, M. C. J. Tomasi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, R. L. M. J. W. Ochterski, K. Morokuma, O. Farkas and D. J. F. J. B. Foresman, Gaussian 16, Revision B.01, 2016, http://www.gaussian.com Search PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg-Teller effect: the Qx band of porphyrin as a case study, J. Chem. Phys., 2008, 128, 224311 CrossRef PubMed.
- J. Bloino, M. Biczysko, F. Santoro and V. Barone, General Approach to Compute Vibrationally Resolved One-Photon Electronic Spectra, J. Chem. Theory Comput., 2010, 6, 1256–1274 CrossRef CAS.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with Time-Dependent Density Functional Theory, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- B. C. Baciu, P. J. Bronk, T. de Ara, R. Rodriguez, P. Morgante, N. Vanthuyne, C. Sabater, C. Untiedt, J. Autschbach, J. Crassous and A. Guijarro, Dithia[9]helicenes: Molecular design, surface imaging, and circularly polarized luminescence with enhanced dissymmetry factors, J. Mater. Chem. C, 2022, 10, 14306–14318 RSC.
- M. Caricato, G. W. Trucks, M. J. Frisch and K. B. Wiberg, Electronic Transition Energies: A Study of the Performance of a Large Range of Single Reference Density Functional and Wave Function Methods on Valence and Rydberg States Compared to Experiment, J. Chem. Theory Comput., 2010, 6, 370–383 CrossRef CAS PubMed.
- K. Dhbaibi, P. Morgante, N. Vanthuyne, J. Autschbach, L. Favereau and J. Crassous, Low-Temperature Luminescence in Organic Helicenes: Singlet versus Triplet State Circularly Polarized Emission, J. Phys. Chem. Lett., 2023, 14, 1073–1081 CrossRef CAS PubMed.
- Y. Liu, J. Cerezo, G. Mazzeo, N. Lin, X. Zhao, G. Longhi, S. Abbate and F. Santoro, Vibronic Coupling Explains the Different Shape of Electronic Circular Dichroism and of Circularly Polarized Luminescence Spectra of Hexahelicenes, J. Chem. Theory Comput., 2016, 12, 2799–2819 CrossRef CAS PubMed.
- J. Autschbach, L. Nitsch-Velasquez and M. Rudolph, Time-Dependent Density Functional Response Theory for Electronic Chiroptical Properties of Chiral Molecules, Top. Curr. Chem., 2010, 298, 1–98 CrossRef PubMed.
- M. Srebro-Hooper and J. Autschbach, Calculating Natural Optical Activity of Molecules from First Principles, Annu. Rev. Phys. Chem., 2017, 68, 399–420 CrossRef CAS PubMed.
- D. Barman, K. Narang, R. Gogoi, D. Barman and P. K. Iyer, Exceptional class of thermally activated delayed fluorescent emitters that display pure blue, near-IR, circularly polarized luminescence and multifunctional behaviour for highly efficient and stable OLEDs, J. Mater. Chem. C, 2022, 10, 8536–8583 RSC.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, Recent breakthroughs in thermally activated delayed fluorescence organic light emitting diodes containing non-doped emitting layers, J. Mater. Chem. C, 2019, 7, 2172–2198 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- S. Kasemthaveechok, L. Abella, M. Jean, M. Cordier, N. Vanthuyne, T. Guizouarn, O. Cador, J. Autschbach, J. Crassous and L. Favereau, Carbazole Isomerism in Helical Radical Cations: Spin Delocalization and SOMO–HOMO Level Inversion in the Diradical State, J. Am. Chem. Soc., 2022, 144, 7253–7263 CrossRef CAS PubMed.
- H. Tanaka, Y. Inoue and T. Mori, Circularly Polarized Luminescence and Circular Dichroisms in Small Organic Molecules: Correlation between Excitation and Emission Dissymmetry Factors, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- L. Arrico, L. Di Bari and F. Zinna, Quantifying the Overall Efficiency of Circularly Polarized Emitters, Chem. – Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed.
- I. R. Gould, R. H. Young, L. J. Mueller and S. Farid, Mechanisms of Exciplex Formation. Roles of Superexchange, Solvent Polarity, and Driving Force for Electron Transfer, J. Am. Chem. Soc., 1994, 116, 8176–8187 CrossRef CAS.
- Z. Piontkowski and D. W. McCamant, Excited-State Planarization in Donor–Bridge Dye Sensitizers: Phenylene versus Thiophene Bridges, J. Am. Chem. Soc., 2018, 140, 11046–11057 CrossRef CAS PubMed.
- D. W. Cho, M. Fujitsuka, K. H. Choi, M. J. Park, U. C. Yoon and T. Majima, Intramolecular Exciplex and Intermolecular Excimer Formation of 1,8-Naphthalimide−Linker−Phenothiazine Dyads, J. Phys. Chem. B, 2006, 110, 4576–4582 CrossRef CAS PubMed.
- N. Mataga and Y. Murata, Electron donor–acceptor interactions in the fluorescent state of tetracyanobenzene-aromatic hydrocarbon complexes, J. Am. Chem. Soc., 1969, 91, 3144–3152 CrossRef CAS.
- K. Nakatani, T. Okada, N. Mataga and F. C. De Schryver, Photoinduced intramolecular electron transfer and exciplex formation of 1-(1-pyrenyl)-3-(N-skatolyl)propane in polar solvents, Chem. Phys., 1988, 121, 87–92 CrossRef CAS.
- T. Okada, M. Migita, N. Mataga, Y. Sakata and S. Misumi, Picosecond laser spectroscopy of intramolecular heteroexcimer systems. Time-resolved absorption studies of p-(CH3)2NC6H4(CH2)n-1-pyrenyl and 9-anthryl systems, J. Am. Chem. Soc., 1981, 103, 4715–4720 CrossRef CAS.
- V. Wintgens, P. Valat, J. Kossanyi, L. Biczok, A. Demeter and T. Berces, Spectroscopic properties of aromatic dicarboximides. Part1.-N-H and N-methyl-substituted naphthalimides, J. Chem. Soc., Faraday Trans., 1994, 90, 411–421 RSC.
- Center for Computational Research, University at Buffalo, https://hdl.handle.net/10477/79221, accessed 05/2023.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc01528a |
| This journal is © The Royal Society of Chemistry 2023 |