
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One hour road to high-quality arrays of gold nanoparticles coated with organic ligands†
Thibault
Degousée
 *a,
William G.
Neal
b,
Zach
Edwards
c,
Saumya
Singh
c,
Jotham
Selvarajah
a,
Teymour
Talha-Dean
a,
Matteo
Palma
b,
Bob C.
Schroeder
c and
Jan A.
Mol
*a
*a,
William G.
Neal
b,
Zach
Edwards
c,
Saumya
Singh
c,
Jotham
Selvarajah
a,
Teymour
Talha-Dean
a,
Matteo
Palma
b,
Bob C.
Schroeder
c and
Jan A.
Mol
*a
aDepartment of Physics and Astronomy, School of Chemical and Physical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: t.degousee@qmul.ac.uk; j.mol@qmul.ac.uk
bDepartment of Chemistry, School of Chemical and Physical Sciences, Queen Mary University of London, Mile End Road, London, E1 4NS, UK
cDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
First published on 10th November 2023
Abstract
Arrays of gold nanoparticles (AuNP) covered with organic molecules have promising applications in biology, electronics, energy, and fundamental physics, among other areas. Here we consider different approaches to synthesise, coat the AuNP with a ligand and to induce their self-assembly into a network within one hour. Arrays formed from aqueous AuNP are stabilised with small ions or alkane chains of 8 to 18 carbons long as the assembly is induced. Alternatively, the AuNP are exchanged to an organic solvent and covered with alkanethiols or oleylamine before the array formation. Beside the length of the ligand, the network morphology and electronic properties are highly affected by the deposition conditions. By using a solid-state exchange strategy, we alternately replaced the ligand on the AuNP with long and short alkanethiols, for which the change of the network's resistance appears reversible. Using this ligand exchanged strategy, we replaced a commercially available alkanethiol with a bespoke naphthalene diimide, resulting in improved conductivity of the networks and their stability in air. Crucially, this simple, rapid, and versatile AuNP–organic network formation offers a platform to the scientific community to investigate the physical and chemical properties of complex molecules.
Introduction
Gold nanoparticles (AuNP) are versatile materials with a number of potential applications, including sensors,1,2 disease detection from breath,3 surface enhanced Raman spectroscopy (SERS),4 solar cells,5,6 transistors,7 memory devices8 or thermoelectric applications.9,10 These technologies require nanoparticle synthesis,11,12 coating with a ligand,13 and their self-assembly into an AuNP–ligand array (or network).14–17 The opto-electronic properties of the arrays are correlated with their morphology, in which voids and cracks are impediments.18,19 To ease the development of these applications, a fast and reliable route to form homogeneous AuNP–ligand arrays is of high interest.Recently, Martin et al. reported a modified Brust–Schiffrin method to synthesize 3–5 nm AuNP in 10 minutes.20,21 In this method, sodium borohydride (NaBH4) converts gold cations into gold atoms in less than 1 second, forming nanoparticles less than 3 nm in size – in the first instance – that slowly aggregate to form larger nanoparticles.21 The ratio of Au/NaBH4 is the main factor determining the AuNP diameter, so control of the synthesis conditions (pH, temperature, heating time) as in the Turkevich synthesis11,22,23 is not required. The small AuNP synthesized with NaBH4 can rapidly be exchanged to hexane without using surfactant, while at the same time, 1-dodecanethiol (DDT) fully covers the AuNP.20 The excess of ligand remains at the water/organic interface after the phase exchange, thus no rinsing step is necessary. The hexane colloidal solution can be deposited on the surface of a toluene where a floating monolayer of AuNP–DDT self-assembles as hexane evaporated.20,21 Although this self-assembly strategy has been able to cover a 3 inch wafer, only few works24 have used it for opto-electronic applications, possibly because the assembly is highly dependent on the size and charge of the AuNP,21 thus rendering the monolayer formation challenging. Other strategies to self-assemble AuNP from colloidal solutions can be used to complement the fast aqueous AuNP synthesis with NaBH4. Furthermore, the availability of nanoparticles in water or in an organic solvent presents the opportunity to use self-assembly methods for both types of colloidal solutions.
Film formation from aqueous colloidal AuNP can be done by two-phases (liquid–liquid or liquid–air) or three-phases (liquid–liquid–air, 3pD) deposition methods.17,25,26 The injection of ethanol at the water–organic interface destabilizes the AuNP and drives them to the water–organic interface. The film formation at the interface is then guided by the minimization of the free energy of the AuNP in the total system (liquid–liquid or liquid–liquid–air).25,26 Coating of the AuNP is achieved at the interface where a ligand in the organic phase attaches to the surface. Thus, the half-covered AuNP have a Janus-like morphology. Janus materials are a novel class of functional 2D materials with properties useful for opto-electronic, energy storage or catalytic applications.27
For organic colloidal AuNP solutions, the nanoparticles can be assembled at a water–air interface by casting the organic solution on a convex water surface.19,28–31 As the organic solvent evaporates, a monolayer assembles at the surface of water and can be transferred to a substrate using a polydimethylsiloxane (PDMS) stamp in a lift-off manner.
In this work, we used different techniques to self-assembled gold nanoparticles from aqueous or organic colloidal solutions into half-covered or fully covered AuNP–ligand arrays. By using those different assembling strategies, ligands consisting of small ions up to eighteen carbons long alkane chains can be grafted on the nanoparticles. We combined electrical and morphological analysis to study the effects of deposition conditions and ligand length on the AuNP–ligand arrays properties. Using a particular route, all necessary steps in the formation of a homogeneous and uniform AuNP–alkanethiolate monolayers (synthesis, coating, self-assembly and transfer to a substrate) can be done under one hour. Using a solid-state exchange strategy, the initial ligand can be replaced by another ligand in a reversible manner. Therefore, the AuNP–ligand arrays offer a rapid and reliable platform to study the properties of more complex molecules, which we demonstrate with a bespoke naphthalene diimide ligand.
Experimental method
Gold nanoparticle synthesis in water
The synthesis of gold nanoparticles in water follows the work of Martin et al.21 Briefly, a stock solution of gold(III) chloride (HAuCl4·H2O, 50 mM) was made using an aqueous solution of hydrochloric acid (HCl, 50 mM). This AuCl4−/H+ stock solution is stable for months at room temperature. A stock solution of sodium borohydride (NaBH4, 50 mM) was made using an aqueous solution of sodium hydroxide (NaOH, 50 mM). This BH4−/OH− solution is stable for a few hours at room temperature. We used 30 mL soda glass vials from Wheaton. The aqueous colloidal synthesis did not work in borosilicate glass vials with the solution turning black immediately after adding NaBH4 (Fig. S1, ESI†). Deionized water was added to 100 μL of AuCl4−/H+ to reach a total weight of 10 g. While the solution was vigorously mixed with a mechanical mixer, 550 μL of BH4−/OH− solution was added. The solution was further mixed for one minute to allow for the release of hydrogen gas formed during the reaction. Then, the AuNP solution was placed on a hot plate at 100 °C for five minutes. Immediately after, the hot solution was cooled by placing the vial in a beaker of water for several minutes.Monolayer formation from aqueous AuNP
We used the three-phases deposition method – 3pD – as reported by Yang et al.25 Briefly, 4.5 mL of a mixture of hexane and chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v%) containing the organic linker was poured in a glass Petri dish with a substrate. Then, 1 mL of aqueous AuNP solution was added over the substrate with the top of the aqueous droplet exposed to air. The concentration of organic linker was calculated according to the diameter of the nanoparticle. The diameter of one AuNP allowed determination of its volume. Assuming that the volume of one Au unit cell is 0.0679 nm,3 the number of unit cells per AuNP was obtained. As there are four gold atoms per unit cell, the number of gold atoms per AuNP was calculated. Considering that all Au3+ ions are reduced to Au0, the number of Au atoms in the aqueous solution was known, hence the number of AuNP in solution. Furthermore, the AuNP's diameter provided the surface area of an AuNP. Assuming the surface coverage of the Au–S bond to be ∼0.21 nm2,32 we calculated the number of organic linkers needed to cover all the AuNP in solution. Then, using stock solutions of linker in hexane at 1 mg mL−1 we calculated the volume of ligand needed to coat all the AuNP in 1 mL. For all linkers, this theoretical volume was multiplied by five to ensure enough molecules were adsorbed on the AuNP. We used a similar surface coverage for oleylamine as for the alkanethiols despite bonding through the amine group. Note that our calculation overestimates the concentration of ligand as complete coverage of the AuNP is assumed.
:1 v%) containing the organic linker was poured in a glass Petri dish with a substrate. Then, 1 mL of aqueous AuNP solution was added over the substrate with the top of the aqueous droplet exposed to air. The concentration of organic linker was calculated according to the diameter of the nanoparticle. The diameter of one AuNP allowed determination of its volume. Assuming that the volume of one Au unit cell is 0.0679 nm,3 the number of unit cells per AuNP was obtained. As there are four gold atoms per unit cell, the number of gold atoms per AuNP was calculated. Considering that all Au3+ ions are reduced to Au0, the number of Au atoms in the aqueous solution was known, hence the number of AuNP in solution. Furthermore, the AuNP's diameter provided the surface area of an AuNP. Assuming the surface coverage of the Au–S bond to be ∼0.21 nm2,32 we calculated the number of organic linkers needed to cover all the AuNP in solution. Then, using stock solutions of linker in hexane at 1 mg mL−1 we calculated the volume of ligand needed to coat all the AuNP in 1 mL. For all linkers, this theoretical volume was multiplied by five to ensure enough molecules were adsorbed on the AuNP. We used a similar surface coverage for oleylamine as for the alkanethiols despite bonding through the amine group. Note that our calculation overestimates the concentration of ligand as complete coverage of the AuNP is assumed.
The monolayer formation was induced by the addition of ethanol to the organic phase. We carefully added 75 μL of ethanol in 20–30 seconds on top of the organic phase. We repeated this step until a monolayer larger than 1 cm2 formed at the air/water interface (∼650 μL needed). Finally, the excess of organic and aqueous solutions was drained to land the monolayer on the substrate, and the film dried in air. Later, we refer to oleylamine as OA and the alkanethiols as Cx, where x is the number of carbons along their chain.
Phase exchange in organic solvent
Stock solutions of the different organic linkers in hexane were prepared at 1 mg mL−1. The phase exchange of AuNP in water to an organic solvent was done following the recipe by Martin et al.:21 5 grams of acetone were added to the AuNP in water and shaken for 1–2 seconds. To that, 5 grams of organic solvent containing the organic linker were added and the solution vigorously mixed by hand for 30 seconds. Here, the molar ratio of organic molecules to gold nanoparticles was fixed at 10% for all linkers. After a few minutes, the acetone–aqueous phase became transparent, and the organic phase acquired a deep red colour. Although this phase transfer method was first reported for hexane, we successfully transferred the AuNP not only to hexane but also a mixture of hexane–chloroform (1:1 wt%).
Monolayer formation from organic AuNP
For all organic colloidal AuNP solution, we dropped 150 μL on the convex surface of water in a PTFE beaker. The monolayers were formed in ∼1 minute as the organic solvent evaporated. The monolayer was removed from the water surface using a PDMS stamp (Dow, Sylgard 184). After drying the PDMS stamp with a nitrogen flow, the monolayer was transferred to the substrate by gently pressing the stamp on it.For the resistance measurement, we formed a monolayer on the convex water surface and covered 40 devices at a time using a rectangular stamp (1.5 × 0.3 cm). We then repeated this process using the same monolayer to cover the next 40 devices on the interdigitated electrodes (IDE) chip.
UV-Vis spectroscopy
UV-Vis spectroscopy is performed with a Shimadzu UV-Vis 2600 with glass cuvettes (Ossila, path 10 mm). For all measurements, the aqueous AuNP solution was diluted to 0.0125 mM and the organic AuNP solutions were diluted to 0.1:4. The solutions were at room temperature before the measurement. The spectrometer was calibrated with pure solvent (water, pure hexane, or a 1:1 wt% of hexane–chloroform) for the colloidal solutions or a blank ultra-flat quartz coated glass substrate (Ossila). The absorption spectra of all solutions and films were normalized to the surface plasmon resonance peak (SPR) for comparison.
Atomic force microscopy
Atomic force microscopy is performed with a Bruker Dimension Icon in tapping mode.SEM
Scanning electron microscopy (SEM) is performed on a FEI Inspect F under a voltage of 10–20 kV. Depending on the formation method, the arrays were either stamped or drained to silicon substrates (Ossila, resistivity of 0.0005 to 0.001 Ω cm).TEM
Transmission electronic microscopy (TEM) was performed with a JEOL 1240. As for the SEM, the arrays were either stamped or drained to TEM grids (Agar scientific) depending on the formation method. The AuNP diameter was estimated using ImageJ on more than 30000 nanoparticles per type of AuNP–ligand array. The centre-to-centre distance between AuNP was estimated using a nearest neighbour distance algorithm on the same set of nanoparticles used for the diameter estimation.
Resistance measurement
The resistance of each AuNP-molecular array was measured on 80 devices. One device consisted of 100 pairs of interdigitated electrodes (width 1.2 μm, length 35 μm, overlap 34.4 μm) with a gap of ∼600 nm between digits (unless specified). The metal electrodes (10 nm Ti, 50 nm Au) were evaporated on doped Si substrate (10–20 mΩ cm) by ConScience (Sweden). The resistance of the molecular arrays was measured using a Keithley 4200 SCS in a two-probe configuration by applying ±1 V (step 0.01 V) and measuring the current. All measurements were made under vacuum.Freestanding P3HT film
Poly(sodium 4-styrenesulfonate) (NaPSS, Mw ∼ 70000, Sigma-Aldrich) and poly(3-hexylthiophene-2,5-diyl) (P3HT, M1011, 97.6% regioregular, Ossila) and chlorobenzene (CB, Sigma-Aldrich) were used as received. A 10 wt% solution of NaPSS in water was spin coated on glass substrates at 3000 rpm 60 seconds, then the substrates were annealed at 80 °C for 30 minutes. A solution of P3HT in CB (2 mg mL−1) was spin coated at 3000 rpm for 30 seconds. The substrates were immersed vertically in deionised water inducing the dissolution of the NaPSS, leaving a freestanding film of P3HT at the water surface. The P3HT film was then transferred in a lift-off manner to the 3pD layers and dry under vacuum for two hours.
Raman spectroscopy
A Renishaw inVia Raman system with a 633 nm excitation wavelength. All measurements were done using a ×50 objective (spot size of ∼5 μm2), 0.1% of the laser power (∼μW), 10 seconds illumination and three repetitions.Nuclear magnetic resonance spectroscopy
1H-NMR measurements of the coated AuNP were conducted on a Bruker Avance III 600 MHz spectrometer at 298 K. Solids were dissolved in approximately 0.6 mL of D2O (>99.9%). Chemical shifts (δ) are reported in parts per million (ppm), using the residual solvent peak at (δ) 4.79 as internal standard.Results and discussion
Aqueous AuNP and self-assembly by 3pD
The UV-Vis absorption spectra of several aqueous AuNP solutions made with the same molar quantity of NaBH4 indicated high reproducibility of this synthesis route (Fig. 1(a)). The surface plasmon resonance peak (SPR) was observed at 514 nm, as expected for AuNP with a diameter smaller than 10 nm.33 We used the model described by Haiss et al. to estimate the nanoparticles’ diameter.33 From the measured absorbance at the SPR peak and the concentration of AuNP in water, the diameter of our AuNP was estimated at 3.8 ± 0.26 nm, in good agreement with the expected diameter for this molar concentration of NaBH4.21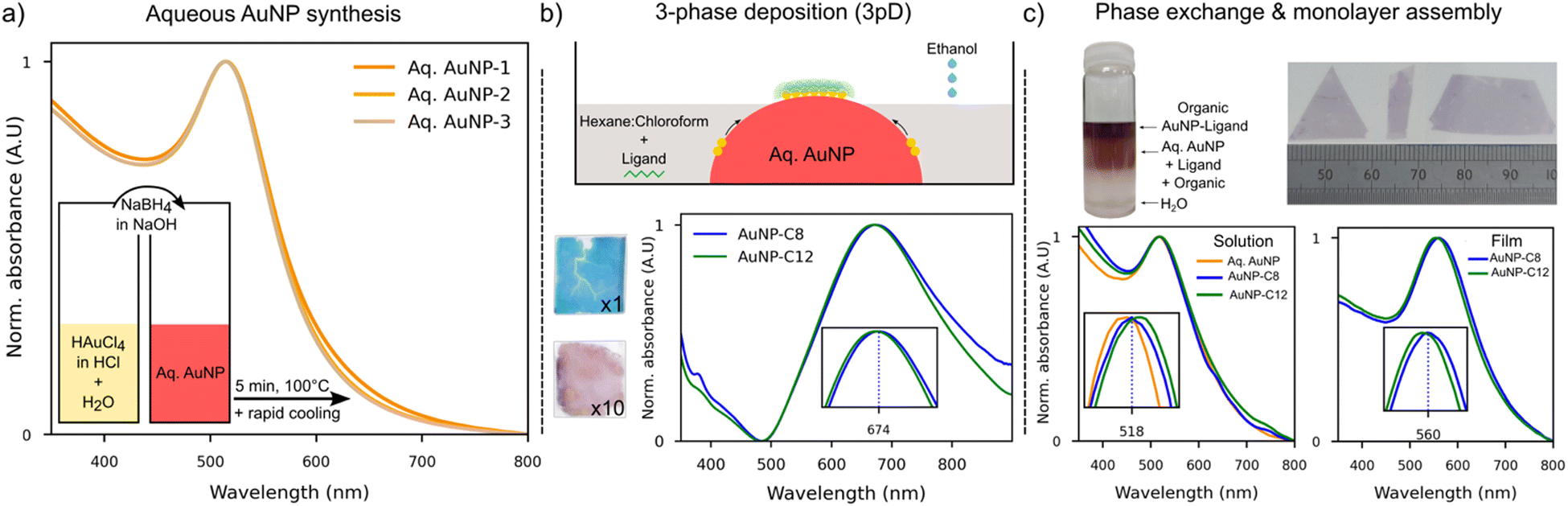 | ||
| Fig. 1 (a) UV-Vis spectra of multiple solutions made with the same volume of NaBH4 showing excellent reproducibility. (b) Schematic of the 3pD assembly, where the injection of ethanol incudes the formation of AuNP islands and their coating with a ligand as they migrate toward the water–air interface. The impact of ligand concentration in the organic phase is shown for the theoretical concentration of ligand (n = 1, blue film) and at a concentration 10 times greater (n = 10, purple film). UV-Vis absorption of AuNP–C8 and AuNP-C12 films made by the 3pD. (c) AuNPs can be fully coated by a ligand during the phase exchange to an organic solvent (vial). The UV-Vis absorption of AuNP–ligand in hexane–chloroform is compared to the aqueous AuNP (bottom left). After assembly on a convex water surface, films of any size and shape can be transferred to a substrate (top right). UV-Vis absorption of AuNP–C8 and AuNP-C12 made from a mixture of hexane–chloroform (bottom right). The dotted line in all insets represents the SPR peak of AuNP–C8 (solution or film). | ||
In the 3pD assembly (Fig. 1(b)), the aqueous AuNP are partially covered with a mixture of hexane–chloroform containing the ligand. The addition of ethanol destabilised the nanoparticles which move toward the water–organic interphase where they are coated by the ligand. It was shown that these nanoparticles acquire a Janus structure, with half of their surface covered by the ligand and the other half remaining stabilised by the reducing agent used in the synthesis. The coated AuNP formed islands that migrated to the water–air interface from the water–organic interface. At the water–air interface, the islands rearranged to form a large layer. As coating of the AuNP is done at the water–organic interface, the concentration of ligand in the organic phase is crucial to ensure enough adsorption of the linker on the AuNP. Monolayers formed with the theoretical concentration of C12 (n = 1), or ten times more (n = 10) had a blue and purple colour, respectively (Fig. 1(b)-top). AuNP monolayers formed with short alkanethiols (10 carbons or less),34 mercaptopropionate or citrate ions35 were of a blue colour comparable to our n = 1 sample, which indicates that the nanoparticles were closely packed. The electrical resistance of AuNP-C12 (n = 1) layer is comparable to that of AuNP stabilized by boron hydrate (Fig. S2, ESI†). Hence, using the theoretical concentration of ligand n = 1 is considered insufficient to coat the AuNP. The reflected colour shifted toward purple as the distance between nanoparticles increased,35,36 like what we observed for our n = 10 film (Fig. 1(b)). However, one can see that this n = 10 film is inhomogeneous and has thick edges. In what follows, all AuNP–ligand networks were formed with a ligand concentration of n = 5.
The absorption spectra of the 3pD arrays (Fig. 1(b)-bottom) show a single and broad band between 500–900 nm centred at 674 nm (3pD–AuNP–C8) and 670 nm (3pD–AuNP-C12). It is known that the SPR peak strongly depends on the interparticle distance and of the dielectric constant on the surrounding media.37 Our results are in good agreement with previous work on 3pD arrays where a broad absorption band blueshifts as the length of the ligand increases.25 However, as seen below, these broad SPR bands centred at ∼670 nm are indicative of stacking of AuNP and not monolayers.
We used a combination of AFM, SEM and TEM to characterise the films made by the 3pD. The AFM showed an average thickness of 8–12 nm, corresponding to 2–3 layers of AuNP (Fig. 2(a) and Fig. S3, ESI†). Whether assembled with a ligand or not, all 3pD layers are inhomogeneous with multiple defects and partial coverage of the substrate. We observed randomly distributed aggregates on the surface or forming long trails of ∼10 nanoparticles thick on some locations. We also noted partial reorganisation of the AuNP islands at the water–air interface, resulting in voids in the films (Fig. 2(a), (b) and Fig. S3, ESI†). Also, the edges of the islands overlap resulting in a thicker film in some locations. A closer look at the islands reveals a multitude of partially connected islets, some being monolayers and others stacks of multiple AuNP (Fig. 2(b)-bottom). These defects might have formed during the 3pD assembly, as seen in the Video S1 (ESI†) where the addition of ethanol destabilises the water–air and water–organic interfaces. Also, the draining step to land the films on the substrates might induce cracks in the films. This is shown in Fig. 1(b) where the hole at the top of the film is caused by the syringe, and the draining step induced a long crack of the entire film’ length.
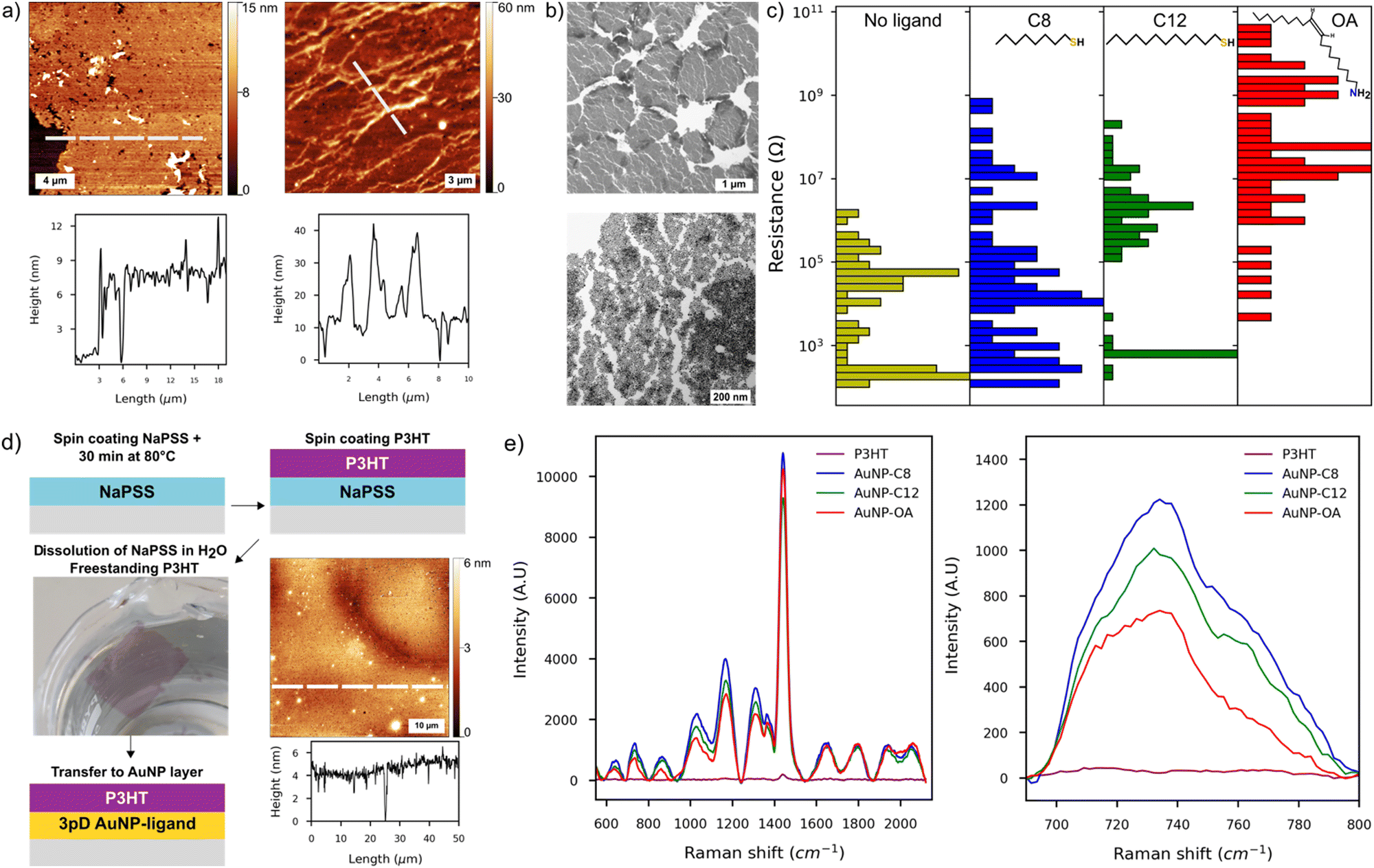 | ||
| Fig. 2 (a) AFM images of 3pD layers assembled without ligand in the organic phase (left) and with dodecanethiol (n = 5, right). The dashed lines indicate the profile position. (b) TEM images of the AuNP islands assembled during the 3pD (top) and higher magnification of an island showing individuals AuNP assembled in 2D arrays with random stacking of AuNP. (c) Distribution of device resistance for 3pD layers assembled with different ligands. (d) Schematic of the deposition of freestanding P3HT thin film. The AFM image show the thin P3HT film transferred to a silicon substrate. (e) SERS activity of P3HT transferred on to the 3pD layers (left) and zoomed in view on the C–S–C vibration of P3HT (right). | ||
For the 3pD assembly, the film inhomogeneity induced the broad distribution of the device resistance spanning several orders of magnitude (Fig. 2(c)). McCold et al. have shown that the network's quality is the key parameter in the charge transport characterization of AuNP–ligand arrays, where defects and voids in the films cause unreliable measurements.19 In our interdigitated electrode design, the resistance of one device corresponded to the equivalent resistance of the 100 sets of electrodes. Hence, defects or partial coverage of a set of electrode will affect the (equivalent) resistance of a device (Fig. S3, ESI†). Devices with resistance less than 10 kΩ have an ohmic behaviour, consistent with multiple layers of AuNP covering the electrodes.38 On the other hand, devices with the highest resistances showed a small charging effect under a bias larger than ±0.5 V. Although observed in monolayers of AuNP–C8,39 this charging effect could be induced partial covering of the electrodes as it is also visible in the AuNP layers without ligand (Fig. S4, ESI†). At room temperature, charge transport in metal nanoparticles arrays follows the sequential tunnelling mechanism.37,40 Hence, by increasing the length of the ligand, the distance between the AuNP increases, which translates to an increase of the resistance of the arrays. Although we noted a resistance increase with the length of the ligand (Fig. 2(c)), charge transport characterization in AuNP–ligand arrays assembled by the 3pD is challenging because of their inhomogeneous morphology.
Nevertheless, the 3pD formation enabled rapid Janus-like coating of AuNP with ligands of any length, which have applications in surface enhanced Raman scattering (SERS).25,41 While previously demonstrated by drop casting rhodamine 6G, we demonstrated the SERS capabilities of the 3pD arrays by transferring a 4 ± 1 nm film of poly(3-hexylthiophene) (P3HT) on top of the arrays (Fig. 2(d) and (e)). P3HT transferred to a plain silicon substrate showed a weak peak at 1440 cm−1. When transferred to a 3pD array, all vibration modes of P3HT can be clearly seen. Because the ligands are carbon chains, their main Raman bands overlap with those of P3HT in the 1200–1600 cm−1 region. Therefore, we used the C–S–C vibration of P3HT at 735 cm−1 to calculate the enhancement factor (EF) by:25
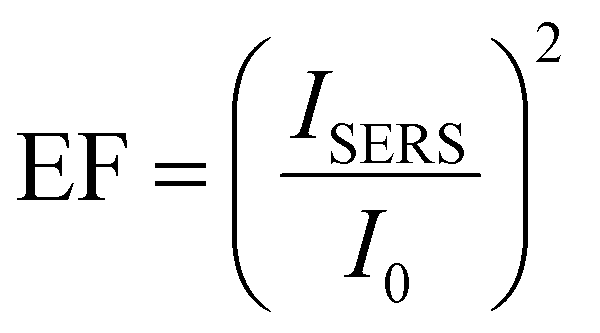
With ISERS and I0 the intensities of the Raman band of interest on the SERS layer and on silicon, respectively. The highest EF measured was for AuNP–C8 (1.6 × 103), followed by AuNP-C12 (6.3 × 102), AuNP–OA (3.1 × 102) and AuNP (1.6 × 102). These EF values are comparable with AuNP monolayers assembled by different strategies.41,42
Organic AuNP and self-assembly on a convex water surface
Starting with a similar aqueous AuNP as in the 3pD formation, we coated the AuNP with C8, C12 or OA during phase exchange in pure hexane or a 1:1 wt% mixture of hexane–chloroform (Fig. 1(c) and Fig. S5, ESI†). After the phase exchange, the SPR peak broadened and redshifted to 518 nm (AuNP–C8) and 520 nm (AuNP-C12) (Fig. 1(c)). This was expected as the different dielectric constant and refractive index of the solvents, and the outer shell of ligand around the AuNP are known to affect the SPR.43 Changing the organic phase to pure hexane had little effect on the SPR peak (Fig. S5, ESI†). Because chloroform is denser than hexane and water, it should remain at the bottom of the vial during the phase exchange to hexane–chloroform, hence transferring the AuNP to hexane only (or half of the organic phase). We performed a phase exchange to pure hexane with twice less organic solvent, and the SPR peak was not affected either (Fig. S5, ESI†). Independent of the organic phase used, phase exchange with OA was more challenging than with C8 or C12 with the AuNP sometimes agglomerating at the aqueous–organic interface. We were not able to identify the parameters influencing the successful phase exchange with OA. As compared to C8 and C12, the UV-Vis spectrum of AuNP–OA was much broader and red-shifted (Fig. S5, ESI†). Upon assembly on a convex water surface, the UV-Vis of the layers showed a single peak centred at 560 nm (AuNP–C8), 554 nm (AuNP-C12) and 569 nm (AuNP–OA) (Fig. 1(c) and Fig. S5, ESI†).
We looked at the topography of the layers to assess the influence of the organic phase on the networks (Fig. 3 and Fig. S6, ESI†). On the top row, the AFM scan of an AuNP-C12 array formed from a colloidal hexane solution, presented a reticulated aspect. The AuNP-C12 seems to assemble into 2–3 μm wide ribbons around circular void areas. Although voids were predominant in the array, a continuous and homogeneous network is observed over the whole scan area. The film thickness was ∼5.4 nm, consistent with the AuNP diameter + ligand length, confirming the self-assembly of the nanoparticles into a 2D array (Fig. 3-middle). On the other hand, the voids induced an irregular coverage of the electrodes. An AFM scan of a set of IDE showed the long continuous network connecting the electrodes so as the inhomogeneous coverage of the gap between them (Fig. S6, ESI†). The reticulated topography was not observed when arrays were cast from a mixture of hexane–chloroform (Fig. 3-bottom and Fig. S6, ESI†). The 50 × 50 μm scan of AuNP-C12 array showed almost complete coverage of the area whereas the voids were greatly reduced. The horizontal profile line indicated similar thickness as for pure hexane monolayer but with homogeneous coverage over tens of micrometres. At the nanoscale, the AuNP were closely packed, forming an almost defect-free monolayer. Although we observed a short-range order over ∼100 nm, we did not observe long-range order in the 2D arrays (Fig. 3(b)). From the TEM images, the AuNP diameter was estimated at 4 ± 0.9 nm in all array-ligands cast with different organic phases. We estimated the length of the ligand between AuNP by subtracting the averaged diameter of the AuNP from the centre-to-centre distance between the AuNPs (Fig. S7, ESI†). Although the large error bars in our analysis do not allow precise measurement of the ligand's length, the trend suggests an increase of AuNP spacing with increasing length of the alkanethiol. Despite being longer than C12, the distance between AuNPs was comparable to or shorter than that observed in AuNP–OA monolayers. Contrary to alkanethiols, OA possesses an unsaturated carbon–carbon double bond forming an angle of 120° with the adjacent single carbon bond.25 As a result, OA allows for closer packing of adjacent AuNP when linear alkanethiols chains form interdigitated structures with the chains on the neighbouring AuNP.
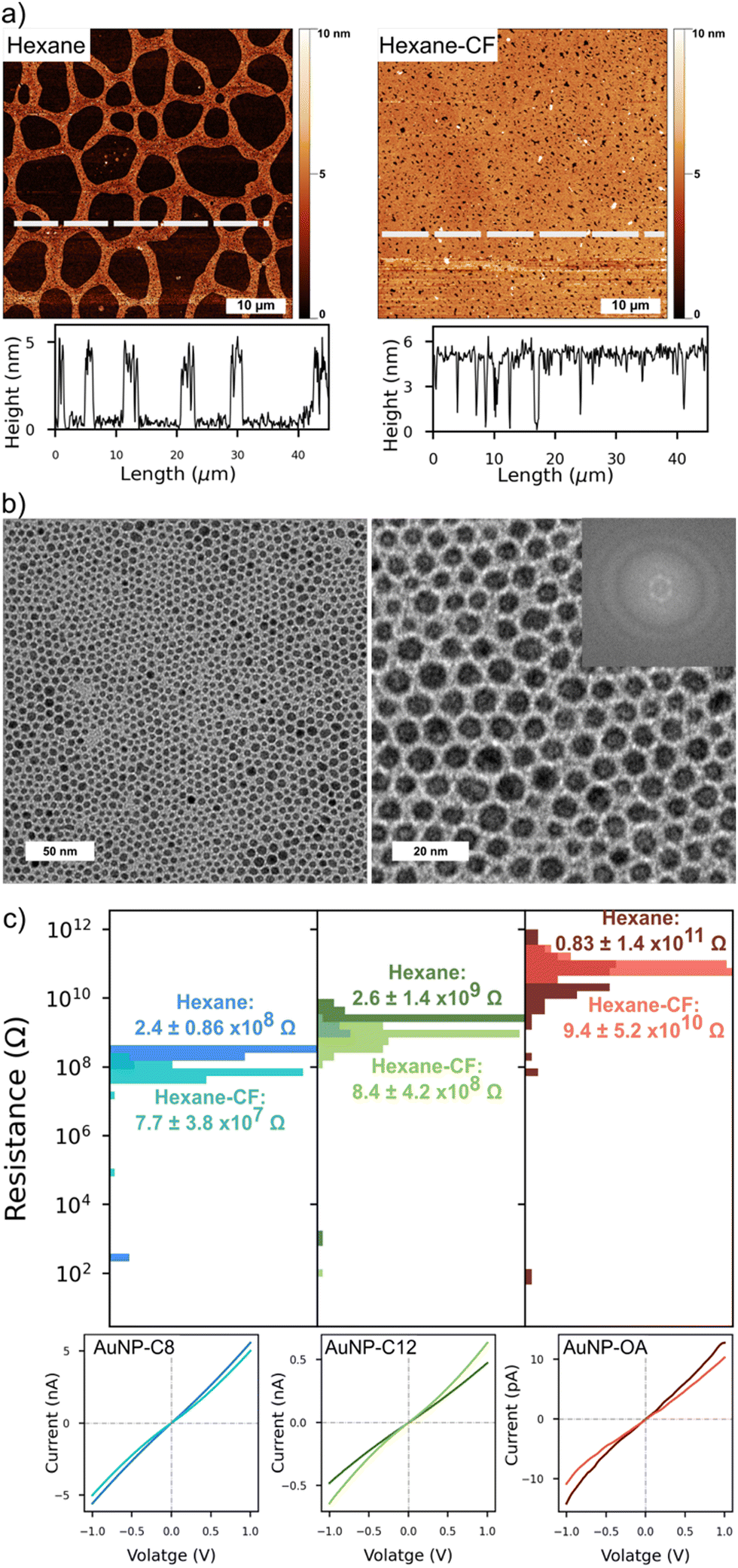 | ||
| Fig. 3 (a) AFM of AuNP-C12 monolayers assembled on a convex water surface from pure hexane (left) and hexane–chloroform (right). The dashed lines indicate where the profile line was taken. (b) TEM images of AuNP-C12 monolayers assembled from hexane–chloroform, long range (left) and short range (right). (c) Distribution of the device resistance for AuNP–ligands monolayers cast from hexane or hexane–chloroform. The IV traces are from devices with the highest resistances for each type of array. | ||
At room temperature, all networks showed Ohmic behaviour under a bias of ±1 V; only a small charging effect was observed for devices in the high end of the resistance distribution (Fig. 3(c)). This charging effect could be due to greater voids in the monolayers resulting in partial coverage of the electrodes. The narrow distribution of device resistances is a consequence of the homogeneous morphology of the monolayers. Arrays cast from pure hexane had a higher average resistance than arrays cast from hexane–chloroform because of their reticulated morphology and partial coverage of the electrodes. Nonetheless, a clear increase of the devices resistance can be seen as the length of the ligand increased. For interdigitated electrodes and a film thickness smaller than the electrodes, the electrical conductivity (σ) of the arrays is given by:44

With L the spacing between the electrodes, N the number of pair of electrodes, l the overlap of the electrodes, t the thickness of the arrays and R its resistance. Using the average resistance of the arrays (Fig. 3(c)), the electrical conductivities of our AuNP–ligand monolayers are in excellent agreement with previous works (Table 1).
| Ligand | Diameter (nm) | σ (S cm−1) | Ref. |
|---|---|---|---|
| C8SH | 4 ± 1 | 1.2 ± 0.4 × 10−7 (hexane) | This work |
| 4 ± 1 | 2.5 ± 1.1 × 10−6 (hexane–chloroform) | This work | |
| 1.2 | 1.8 × 0.8 × 10−5 | 45 | |
| 1.6 ± 0.8 | 9 × 10−6 | 46 | |
| C12SH | 4 ± 1 | 7.2 ± 3.5 × 10−8 (hexane) | This work |
| 4 ± 1 | 2.2 ± 2.0 × 10−7 (hexane–chloroform) | This work | |
| 4.6 | 2 × 10−7 | 47 | |
| 4.8 ± 0.6 | 7.8 × 10−8 | 48 | |
| 1.2 | 2.3 ± 0.8 × 10−7 | 45 | |
| 1.6 ± 0.8 | 5 × 10−7 | 46 | |
| C18NH2 | 4 ± 1 | 0.8 ± 1.4 × 10−8 (hexane) | This work |
| 4 ± 1 | 3.6 ± 1.5 × 10−9 (hexane) | This work | |
The homogeneous and uniform monolayers can be used as platforms for ligand exchange with molecules that are unable to coat AuNP during phase exchange. Previous ligand exchanges in a solid state have been demonstrated with AuNP–C8 and single29 or dithiolated oligo(phenylene ethynylene) (OPE),28,29 oligo(phenylene vinylene) (OPV)49 or perylene tetracarboxylic diimide derivative with pyrroline moieties substituted at the bay positions.50 Here, we replaced C12 with butanethiol (C4), as we failed to coat and exchange the AuNP to hexane–chloroform with this short alkanethiol (Fig. 4). The replacement of C12 by C4 is possible because the space between the AuNP is large enough to accommodate C4. After immersion for 5 h in a 1 mM solution of butanethiol in acetone, the resistance of 80 devices decreased by ∼2 orders of magnitude to 106–108 Ω, consistent with the change of resistance observed with the length of the ligand. The resistance of the devices did not further change upon immersion for 24 h (Fig. 4). Furthermore, the ligand exchange process was reversible, as evidenced by the devices recovering their initial resistance when immersed in 1 mM of dodecanethiol in acetone for 24 hours. This reversible substitution of the organic linker implies that the morphology of the AuNP network is not affected by long immersion in acetone and remains comparable to the initial state when cast. Note that we did not optimize the concentration of ligand, solvent, temperature, or immersion time, all of which are known to affect the solid-state ligand exchange process.51,52
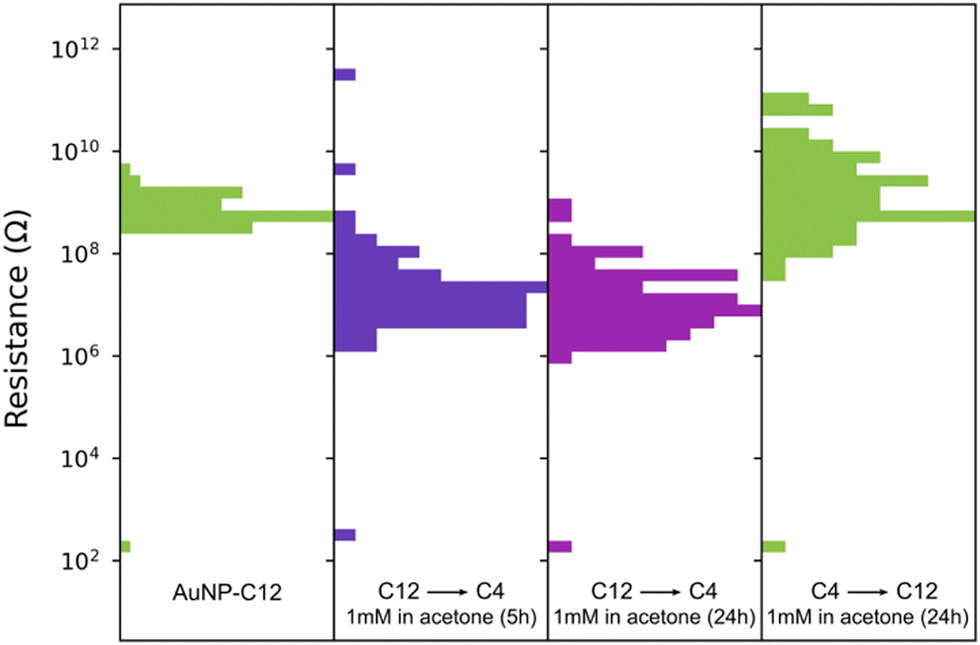 | ||
| Fig. 4 Resistance changes of an AuNP-C12 array when C12 is replaced by butanethiol (C4) and subsequently replaced by C12. The gap between the IDE for this sample was 1 μm. | ||
The solid-state ligand exchange process was also performed with a naphthalene diimide (NDI) derivative, namely 3-(7-(2-(octyldisulfaneyl)ethyl)-1,3,6,8-tetraoxo-3,6,7,8-tetrahydrobenzo[lmn][3,8]phenanthrolin-2(1H)-yl)propanoic acid (7). NDI compounds are known for their air stability which makes them promising n-type electronic materials.53 The detailed synthetic procedure for compound 7 can be found in the ESI† (Fig. S8–S11) and is summarised in Scheme 1. NDI derivative 7 was functionalised with both a disulfide group to facilitate anchoring to the AuNP,54 as well as a carboxylic acid group to enhance solubility in polar solvents. In addition, the polar carbocyclic acid groups facilitate hydrogen bonding between decorated AuNPs or alternatively promote dispersion in solution, especially when deprotonated (pH 8). Two chips of 80 devices were covered with a hexane–chloroform AuNP-C12 monolayer, one of them acting as a control sample.
 | ||
| Scheme 1 Synthetic pathway towards NDI derivative 7. | ||
Initially, the average resistance distribution of both AuNP-C12 monolayers was alike. After immersing one of the arrays for 4 hours in 1 mM of 7 in acetone, the average resistance of each device was reduced by half (Fig. S11, ESI†). After one week in air, the resistance distribution of the control sample spanned over seven orders of magnitude indicating degradation of the monolayer. On the other hand, the resistance distribution of the AuNP–7 layer remained nearly unchanged after one week in air. Furthermore, the UV-Vis spectroscopy of the AuNP-C12 layer before and after ligand exchange with 7 clearly showed the signature of the NDI core, thus confirming that C12 had been replaced (Fig. S11, ESI†). To further evaluate the ligand exchange and verify if it indeed took place via the formation of a chemical Au–S bond, we performed 1H-NMR experiments on the AuNP–7 and compound 7 (Fig. S12, ESI†). Comparing the two spectra, it becomes apparent that numerous new proton environments arise in the AuNP–7 spectrum, further evidence of the successful binding of 7 on the AuNP surface. The spectra can be roughly separated into two regions, downfield (9.0–7.0 ppm) and upfield (4.5–0.5 ppm). The downfield region reflects the aromatic proton environment and multiple new peaks with complex splitting patterns arise once 7 is bound to the AuNP surface. Similarly, a series of additional peaks were recorded for AuNP–7 further upfield in the spectrum resulting from the different chemical environments in the side chains of 7. It is unlikely that these changes are the result of chemical modifications to the structure of 7 when bound to gold, but rather reflect the presence of numerous chemical species bound to the AuNP resulting in a variety of distinct chemical environments.55 Specifically, we could expect dodecanethiolate from the original ligand shell, as well as octanethiolate and 2-(7-(2-carboxyethyl)-1,3,6,8-tetraoxo-3,6,7,8-tetrahydrobenzo[lmn][3,8] phenanthroline-2(1H)-yl)ethane-1-thiolate, the two products resulting from the dissociation of the disulfide bond in 7, to be present on the AuNP surface.
The family of NDI molecules is particularly interesting as multiple side chains or groups can be attached to the core, hence changing the physical and chemical properties of the molecule.56,57 This simple ligand exchange proves that our AuNP–ligand arrays can be used to study the properties of bespoke molecules.
Conclusions
In this work, we combined the advantages of the Martin method for synthesis of aqueous colloidal gold nanoparticles with different AuNP–ligand array formation methods. The aqueous AuNP were assembled by the three-phase deposition method to form Janus-like AuNP–ligand monolayers. This deposition method can form cm2 films with small ions stabilizing the AuNP or with molecular spacers of 8–18 carbons in length; however, 3pD assembly is sensitive to the deposition conditions and an inhomogeneous films consisting of multiple layers of nanoparticles tend to form. The rapid phase exchange and coating of the AuNP developed by Martin et al. was extended to different organic solvents and linkers such as octanethiol, dodecanethiol or oleylamine. By using a convex water surface, we were able to assemble cm2 AuNP–ligand monolayers in a minute. Morphological characterizations showed that phase exchange to a mixture of hexane–chloroform allowed self-assembly of almost defect-free monolayers, whereas phase exchange with pure hexane induced monolayers with a reticulated aspect. The resistance of the devices was dependent of the quality of the network but, the resistance of all monolayers showed an increase of approximately one order of magnitude when the length of the ligand increased by four carbons. Finally, the alkanethiols coating the AuNP could be exchanged by a molecule of interest by soaking the layers in solution. With the present strategy, a reliable platform to investigate the opto-electronic properties of molecules can be synthesized and assembled in less than one hour.Author contributions
T. D.: conceptualization, investigation, formal analysis, visualization, writing – original draft, writing – review & editing. W. G. N. and T. T.-D.: investigation (AFM). J. S.: investigation (AuNP–ligand synthesis and monolayer assembly). Z. E. and S. S.: chemical synthesis and AuNP NMR study. C. S., M. P.: supervision and funding acquisition. J. M.: conceptualization, supervision, and funding acquisition. All authors participated in the writing and correction of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research has been supported by the British Council Newton Fund Institutional Links (ref. 337067) and the UKRI-EPSRC (grant no. 2248056). J. A. M. was supported through the UKRI Future Leaders Fellowship, Grant No. MR/S032541/1. B. C. S. acknowledges the UK Research and Innovation for Future Leaders Fellowship no. MR/S031952/1.Notes and references
- C.-B. Huang, Y. Yao, V. Montes-García, M.-A. Stoeckel, M. Von Holst, A. Ciesielski and P. Samorì, Small, 2021, 17, 2007593 CrossRef CAS PubMed.
- W. H. Steinecker, M. P. Rowe and E. T. Zellers, Anal. Chem., 2007, 79, 4977–4986 CrossRef CAS PubMed.
- M. K. Nakhleh, Y. Y. Broza and H. Haick, Nanomedicine, 2014, 9, 1991–2002 CrossRef CAS PubMed.
- L. He, N.-J. Kim, H. Li, Z. Hu and M. Lin, J. Agric. Food Chem., 2008, 56, 9843–9847 CrossRef CAS PubMed.
- Y.-H. Su, Y.-F. Ke, S.-L. Cai and Q.-Y. Yao, Light: Sci. Appl., 2012, 1, e14–e14 CrossRef.
- X. Chen, X. Yang, W. Fu, M. Xu and H. Chen, Mater. Sci. Eng., B, 2013, 178, 53–59 CrossRef CAS.
- A. Prasad, M. Stoller and R. F. Saraf, ACS Appl. Nano Mater., 2021, 4, 9044–9051 CrossRef CAS.
- S. Paul, C. Pearson, A. Molloy, M. A. Cousins, M. Green, S. Kolliopoulou, P. Dimitrakis, P. Normand, D. Tsoukalas and M. C. Petty, Nano Lett., 2003, 3, 533–536 CrossRef CAS.
- C. E. McCold, L. Domulevicz, Z. Cai, W.-Y. Lo, S. Hihath, K. March, H. M. Mohammad, M. P. Anantram, L. Yu and J. Hihath, J. Phys. Chem. C, 2020, 124, 17–24 CrossRef CAS.
- A. S. Fedorov, P. O. Krasnov, M. A. Visotin, F. N. Tomilin and S. P. Polyutov, Phys. Status Solidi B, 2020, 257, 2000249 CrossRef CAS.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- R. Herizchi, E. Abbasi, M. Milani and A. Akbarzadeh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 596–602 CrossRef CAS PubMed.
- M. Chanana and L. M. Liz-Marzán, Nanophotonics, 2012, 1, 199–220 CAS.
- Q. Shi and W. Cheng, Adv. Funct. Mater., 2020, 30, 1902301 CrossRef CAS.
- M. Singh, N. Kaur and E. Comini, J. Mater. Chem. C, 2020, 8, 3938–3955 RSC.
- L. Song, Y. Huang, Z. Nie and T. Chen, Nanoscale, 2020, 12, 7433–7460 RSC.
- M. D. Scanlon, E. Smirnov, T. J. Stockmann and P. Peljo, Chem. Rev., 2018, 118, 3722–3751 CrossRef CAS PubMed.
- M. A. Mangold, M. A. Niedermeier, M. Rawolle, B. Dirks, J. Perlich, S. V. Roth, A. W. Holleitner and P. Müller-Buschbaum, Phys. Status Solidi RRL, 2011, 5, 16–18 CrossRef CAS.
- C. E. McCold, Q. Fu, J. Y. Howe and J. Hihath, Nanoscale, 2015, 7, 14937–14945 RSC.
- M. N. Martin and S.-K. Eah, MRS Proc., 2009, 1113(1), 301 Search PubMed.
- M. N. Martin, J. I. Basham, P. Chando and S.-K. Eah, Langmuir, 2010, 26, 7410–7417 CrossRef CAS PubMed.
- J. Dong, P. L. Carpinone, G. Pyrgiotakis, P. Demokritou and B. M. Moudgil, KONA, 2020, 37, 224–232 CrossRef CAS PubMed.
- F. Schulz, T. Homolka, N. G. Bastús, V. Puntes, H. Weller and T. Vossmeyer, Langmuir, 2014, 30, 10779–10784 CrossRef CAS PubMed.
- J. Zhou, X. Cao, L. Li, X. Cui and Y. Fu, Nanomaterials, 2019, 9(10), 1468 CrossRef CAS PubMed.
- G. Yang and D. T. Hallinan, Sci. Rep., 2016, 6, 35339 CrossRef CAS PubMed.
- G. Yang and D. T. Hallinan, Nanotechnology, 2016, 27, 225604 CrossRef PubMed.
- V. Montes-García and P. Samorì, Chem. Sci., 2022, 13, 315–328 RSC.
- J. Liao, L. Bernard, M. Langer, C. Schönenberger and M. Calame, Adv. Mater., 2006, 18, 2444–2447 CrossRef CAS.
- J. Liao, M. A. Mangold, S. Grunder, M. Mayor, C. Schönenberger and M. Calame, New J. Phys., 2008, 10, 065019 CrossRef.
- P. A. Reissner, J.-N. Tisserant, A. Sánchez-Ferrer, R. Mezzenga and A. Stemmer, Beilstein J. Nanotechnol., 2016, 7, 2057–2064 CrossRef CAS PubMed.
- V. Santhanam and R. P. Andres, Nano Lett., 2004, 4, 41–44 CrossRef CAS.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lämmerhofer, ACS Nano, 2013, 7, 1129–1136 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- I.-H. Kim, J. H. Kim, J.-Y. Choi, C. H. Shin, J.-H. Kim, G.-T. Bae and K. S. Shin, Chem. Phys. Lett., 2019, 715, 91–99 CrossRef CAS.
- T. Ung, L. M. Liz-Marzán and P. Mulvaney, J. Phys. Chem. B, 2001, 105, 3441–3452 CrossRef CAS.
- C. Kunstmann-Olsen, D. Belić, D. F. Bradley, M. P. Grzelczak and M. Brust, Chem. Mater., 2016, 28, 2970–2980 CrossRef CAS.
- E. J. Devid, P. N. Martinho, M. V. Kamalakar, Ú. Prendergast, C. Kübel, T. Lemma, J.-F. Dayen, T. E. Keyes, B. Doudin, M. Ruben and S. J. van der Molen, Beilstein J. Nanotechnol., 2014, 5, 1664–1674 CrossRef PubMed.
- J. Siegel, O. Lyutakov, V. Rybka, Z. Kolská and V. Švorčík, Nanoscale Res. Lett., 2011, 6, 96 CrossRef PubMed.
- J.-F. Dayen, E. Devid, M. V. Kamalakar, D. Golubev, C. Guédon, V. Faramarzi, B. Doudin and S. J. van der Molen, Adv. Mater., 2013, 25, 400–404 CrossRef CAS PubMed.
- T. B. Tran, I. S. Beloborodov, J. Hu, X. M. Lin, T. F. Rosenbaum and H. M. Jaeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 075437 CrossRef.
- M.-H. Wang, J.-W. Hu, Y.-J. Li and E. S. Yeung, Nanotechnology, 2010, 21, 145608 CrossRef PubMed.
- X. Lu, Y. Huang, B. Liu, L. Zhang, L. Song, J. Zhang, A. Zhang and T. Chen, Chem. Mater., 2018, 30, 1989–1997 CrossRef CAS.
- J. Zhao, A. O. Pinchuk, J. M. McMahon, S. Li, L. K. Ausman, A. L. Atkinson and G. C. Schatz, Acc. Chem. Res., 2008, 41, 1710–1720 CrossRef CAS PubMed.
- A. Zabet-Khosousi and A.-A. Dhirani, Chem. Rev., 2008, 108, 4072–4124 CrossRef CAS PubMed.
- R. H. Terrill, T. A. Postlethwaite, C.-H. Chen, C.-D. Poon, A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark and G. Wignall, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- F. P. Zamborini, M. C. Leopold, J. F. Hicks, P. J. Kulesza, M. A. Malik and R. W. Murray, J. Am. Chem. Soc., 2002, 124, 8958–8964 CrossRef CAS PubMed.
- A. W. Snow and H. Wohltjen, Chem. Mater., 1998, 10, 947–949 CrossRef CAS.
- H. Ahn, A. Chandekar, B. Kang, C. Sung and J. E. Whitten, Chem. Mater., 2004, 16, 3274–3278 CrossRef CAS.
- M. A. Mangold, M. Calame, M. Mayor and A. W. Holleitner, J. Am. Chem. Soc., 2011, 133, 12185–12191 CrossRef CAS PubMed.
- C. E. McCold, Q. Fu, S. Hihath, J.-M. Han, Y. Halfon, R. Faller, K. van Benthem, L. Zang and J. Hihath, Mol. Syst. Des. Eng., 2017, 2, 440–448 RSC.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS PubMed.
- P. Ionita, A. Caragheorgheopol, B. C. Gilbert and V. Chechik, Langmuir, 2004, 20, 11536–11544 CrossRef CAS PubMed.
- S. V. Bhosale, M. Al Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845–9998 RSC.
- H. Grönbeck, A. Curioni and W. Andreoni, J. Am. Chem. Soc., 2000, 122, 3839–3842 CrossRef.
- B. Schuetze, C. Mayer, K. Loza, M. Gocyla, M. Heggen and M. Epple, J. Mater. Chem. B, 2016, 4, 2179–2189 RSC.
- G. Gogoi, L. Bhattacharya, S. R. Sahoo, S. Sahu, N. S. Sarma and S. Sharma, RSC Adv., 2021, 11, 57–70 RSC.
- D. Nava, Y. Shin, M. Massetti, X. Jiao, T. Biskup, M. S. Jagadeesh, A. Calloni, L. Duò, G. Lanzani, C. R. McNeill, M. Sommer and M. Caironi, ACS Appl. Energy Mater., 2018, 1, 4626–4634 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc01497e |
| This journal is © The Royal Society of Chemistry 2023 |