
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular design of phenazine-5,10-diyl-dibenzonitriles and the impact on their thermally activated delayed fluorescence properties†
Dietrich
Püschel
a,
Julia
Wiefermann
b,
Simon
Hédé
c,
Tobias
Heinen
a,
Leo
Pfeifer
a,
Oliver
Weingart
 *c,
Markus
Suta
*a,
Thomas J. J.
Müller
*b and
Christoph
Janiak
*a
*c,
Markus
Suta
*a,
Thomas J. J.
Müller
*b and
Christoph
Janiak
*a
aInstitute of Inorganic and Structural Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: Markus.Suta@hhu.de; Janiak@uni-duesseldorf.de
bInstitute of Organic and Macromolecular Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@hhu.de
cInstitute for Theoretical Chemistry and Computational Chemistry, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: Oliver.Weingart@hhu.de
First published on 16th May 2023
Abstract
The photoluminescence properties of the compounds 3,3′-(phenazine-5,10-diyl)dibenzonitrile (mBN) and 4′,4′′′-(phenazine-5,10-diyl)bis(([1,1′-biphenyl]-4-carbonitrile)) (BPN) are presented and compared to those of the known fluorophore 4,4′-(phenazine-5,10-diyl)dibenzonitrile (pBN), which has been reported to show thermally activated delayed fluorescence (TADF). In the solid state, pBN shows clear TADF properties. In contrast, TADF is only weakly pronounced in mBN, and BPN is a conventional fluorescent emitter. This is discussed in terms of the provided through-space overlap between donating phenazinediyl and accepting benzonitrile units in these three molecules, which is only effective in pBN. These compounds are only weakly luminescent in toluene, tetrahydrofuran (THF) or dichloromethane (CH2Cl2) solution, most intensively in toluene, with yellow (pBN and mBN) to orange (BPN) colors of the emission. The emission maxima λem,max in toluene differ slightly between pBN (562 nm) and mBN (572 nm) and overlap in THF (634 nm) and CH2Cl2 (∼660 nm), respectively. Their emission is broad-banded and strongly solvent-dependent and thus indicates a CT-type nature of the excited state. BPN shows weak solvent-dependent photoluminescence (604 nm in toluene, 589 nm in THF, and 587 nm in CH2Cl2) and together with an observable vibronic structure in the low temperature spectra of the powder, it can be concluded that emission in BPN occurs from a localized electronic (LE) state non-beneficial for TADF properties. This demonstrates that even tiny modifications in the molecular templating structure of the phenazines can significantly affect their TADF properties.
Introduction
The phenomenon of thermally activated delayed fluorescence (TADF) was first described in detail by Parker and Hatchard in 19611 and discovered even earlier by Perrin in 1929 on uranyl salts.2,3 After a long period of time without application, the concept has been re-introduced by Adachi et al. in the 2010s to motivate the usage of new organic chromophores in organic light-emitting diodes (OLEDs) with the goal to replace common phosphorescent expensive noble-metal complex emitters.4,5 Since then, the field of organic TADF chromophores has received a lot of attention worldwide with vivid development over the last decade.6–13 A key advantage of TADF emitters in OLEDs is the high quantum efficiency based on harvesting of both spin singlet and triplet excitons, which formally allows achieving internal quantum efficiencies of ∼100% and high external quantum efficiencies (EQEs) of up to ∼40%.5,6,14–17 This high efficiency can be achieved because the lowest excited singlet state (S1) and lowest excited triplet state (T1) are energetically sufficiently close that reverse intersystem crossing (rISC) can be thermally triggered.18–23 The energy difference ΔEST of both energy levels is typically less than 0.1 eV for the current highly efficient organic TADF emitters.14,24–27 The smaller the energy gap ΔEST between S1 and T1 the higher the equilibrium population of the higher excited S1 state, which governs up to 25% of the internal quantum efficiency in cases of electric injection of charge carriers.Small organic molecules such as TADF emitters have the advantage that they can be synthesized in high purity or can be optimally purified by recrystallisation and/or sublimation. In addition, the molecules can be modified and optimized in a large variety to lead to high luminous efficacies of light-emitting devices containing these compounds. Ideally, the emitters should have high color purity and narrow emission bands.24,28
It has been shown that the photoluminescence (PL) efficiency of donor–acceptor TADF compounds can be controlled and optimized by the selective choice of specific donor and acceptor moieties.29–31 Furthermore, systems with donor–acceptor–donor (D–A–D)32 and acceptor–donor–acceptor type (A–D–A) topologies have proven to be a beneficial combination.33,34
The phenazine-5,10-diyl molecule with two nitrogen atoms in the central 1,4-dihydrodiazine core is an excellent donor moiety.35 By arylation of the 5 and 10 positions, 5,10-di-aryl derivatives are obtained. The variation of acceptor groups attached to the nitrogen atoms of phenazine-5,10-diyl has been well investigated in this symmetrical A–D–A system. Benzonitrile has been found to be the most effective acceptor in these designed TADF emitters.33,35 Compound 4,4′-(phenazine-5,10-diyl)dibenzonitrile (Fig. 1), which is derived from 5,10-dihydrophenazine (DHPZ) as the strong electron donor and two para-benzonitrile moieties as strong electron acceptor units, has an energy gap of ΔEST = 0.10 eV and a photoluminescence quantum yield of 35.2% with the corresponding EQE of 5–8%. It is an established TADF emitter.33
 | ||
| Fig. 1 Chemical formulae of the investigated phenazine-5,10-diyl dibenzonitriles in this work. | ||
Here, we analyze the effect of a change in the nitrile substitution pattern from a para to meta configuration and an elongation of the phenyl to a biphenyl group on the photophysical properties with special emphasis on potential TADF properties. This offers the possibility of deriving structure–property relationships and to formulate molecular design rules to control desirable TADF parameters such as ΔEST by simple chemical inspection. Thus, we synthesized and investigated the luminescence properties of 3,3′-(phenazine-5,10-diyl)dibenzonitrile (mBN) and 4′,4′′′-(phenazine-5,10-diyl)bis(([1,1′-biphenyl]-4-carbonitrile)) (BPN) and compared them to those of the literature-known TADF emitter 4,4′-(phenazine-5,10-diyl)dibenzonitrile (pBN) (Fig. 1). We analyzed the optical properties both in the solid state and in solution to identify the overall nature of the radiative transition and to verify if aggregation-induced effects in the powder lead to significant changes in the TADF properties. This is usually relevant for applications such as emitting materials in OLEDs, in which thin films are used rather than dissolved dyes. In particular, the presented dyes have high melting points (mBN = 284 °C, pBN and BPN > 300 °C) and can be even processed by sublimation, which is beneficial for applications.
Results and discussion
For the synthesis of the phenazine-5,10-diyl dibenzonitriles, phenazine was first reduced to dihydrophenazine following established literature procedures (see the ESI† for details).33,36 Then, Buchwald–Hartwig coupling with 4-bromobenzonitrile, 3-bromobenzonitrile, or 4′-bromo-[1,1′-biphenyl]-4-carbonitrile forms the products 4,4′-(phenazine-5,10-diyl)dibenzonitrile (pBN), 3,3′-(phenazine-5,10-diyl)dibenzonitrile (mBN) or 4′,4′′′-(phenazine-5,10-diyl)bis(([1,1′-biphenyl]-4-carbonitrile)) (BPN), respectively (Fig. 1). The dihydrophenazine derivatives are obtained in good to high yields of 65–78%. Their purity and identity were confirmed by 1H NMR, mass spectrometry, combustion analysis and single-crystal structures.The crystal structure of pBN was only published recently (α-polymorph, space group P21, no. 4)37 and another β-polymorph (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , no. 2) has been determined in parallel in this work (see the ESI† for details). Both pBN polymorphs consist of two types of molecules, namely a ‘linear’ one and a trans-bent ‘distorted one’ (ratios 1
, no. 2) has been determined in parallel in this work (see the ESI† for details). Both pBN polymorphs consist of two types of molecules, namely a ‘linear’ one and a trans-bent ‘distorted one’ (ratios 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in β- and 1:2 in α-polymorph), with a characteristic non-linearity of the latter associated with the weak, but clearly distinguishable pyramidality at the N atoms of the phenazinediyl core (Fig. 2a). In both molecules of β-pBN and also in crystalline mBN (Fig. 2b), the center of the pyrazine ring coincides with an inversion center; hence, the pyrazine rings are planar by symmetry (see the ESI† for crystallographic details). The dihedral angles between the benzonitrile aryl group and the pyrazine core or phenazine plane are 75.08(5)° or 76.41(4)° in β-pBN molecule 1, 79.37(5)° or 79.93(4)° in β-pBN molecule 2, 86.67(5)° or 85.80(3)° in mBN, respectively. The fused benzo and pyrazine rings are almost coplanar with a small interplanar angle of 2.79(4)°, 1.07(5)° in β-pBN molecule 1,2 and 1.71(4)° in mBN.
:1 in β- and 1:2 in α-polymorph), with a characteristic non-linearity of the latter associated with the weak, but clearly distinguishable pyramidality at the N atoms of the phenazinediyl core (Fig. 2a). In both molecules of β-pBN and also in crystalline mBN (Fig. 2b), the center of the pyrazine ring coincides with an inversion center; hence, the pyrazine rings are planar by symmetry (see the ESI† for crystallographic details). The dihedral angles between the benzonitrile aryl group and the pyrazine core or phenazine plane are 75.08(5)° or 76.41(4)° in β-pBN molecule 1, 79.37(5)° or 79.93(4)° in β-pBN molecule 2, 86.67(5)° or 85.80(3)° in mBN, respectively. The fused benzo and pyrazine rings are almost coplanar with a small interplanar angle of 2.79(4)°, 1.07(5)° in β-pBN molecule 1,2 and 1.71(4)° in mBN.
 | ||
| Fig. 2 Molecular structures of (a) pBN with linear molecule 1 and trans-bent molecule 2, (b) mBN and (c) BPN (CHCl3 solvent molecule omitted) in the crystalline state (50% thermal ellipsoids, H atoms with arbitrary radii). Symmetry transformation in (a) i = −x + 1, −y + 1, −z; (b) i = −x + 1, −y, −z + 2. | ||
Photophysical properties
| Compound | λ max,abs/nm (ε/L mol−1 cm−1) | λ em/nm | τ/ns | Stokes shifta /cm−1 | FWHMb /cm−1 (eV) | CIEc |
|---|---|---|---|---|---|---|
a
 .
b FWHM = full width at half maximum.
c Refers to a transparent solution of the respective compound, not an OLED device.
d In toluene and THF, the absorption measurement starts above 285 nm because of solvent absorption below 285 nm. .
b FWHM = full width at half maximum.
c Refers to a transparent solution of the respective compound, not an OLED device.
d In toluene and THF, the absorption measurement starts above 285 nm because of solvent absorption below 285 nm.
|
||||||
| pBN | ||||||
| Toluene | 427sh (900) | 562 | 5.7 | 5600 | 3624 (0.45) | 0.450, 0.530 |
| 371 (1800) | ||||||
| 318 (2700) | ||||||
| THF | 427sh (1700) | 634 | 2.5 | 7600 | 4018 (0.50) | 0.572, 0.422 |
| 370sh (3800) | ||||||
| 318 (7200) | ||||||
| Dichloromethane | 427sh (2100) | 659 | 1.0 | 8200 | 4436 (0.55) | 0.567, 0.418 |
| 372sh (4200) | ||||||
| 321 (7700) | ||||||
| 255 (42600)d | ||||||
| mBN | ||||||
| Toluene | 381sh (2400) | 572 | 8.9 | 8800 | 3746 (0.47) | 0.468, 0.513 |
| 331 (6800) | ||||||
| THF | 383sh (1900) | 634 | 1.7 | 10300 |
4101 (0.51) | 0.556, 0.422 |
| 329 (5900) | ||||||
| Dichloromethane | 384sh (2600) | 662 | 1.6 | 10900 |
4517 (0.56) | 0.561, 0.432 |
| 332 (7400) | ||||||
| 250 (54200)d | ||||||
| BPN | ||||||
| Toluene | 427sh (2800) | 604 | 6.7 | 6900 | 3564 (0.44) | 0.544, 0.451 |
| 375 (6600) | ||||||
| THF | 428sh (1000) | 589 | 12.0 | 6400 | 3739 (0.46) | 0.525, 0.446 |
| 373 (1900) | ||||||
| Dichloromethane | 427sh (2500) | 6400 | 6442 (0.8) | 0.463, 0.449 | ||
| 373 (6500) | 587 | 5.5 | ||||
| 271sh (71900) | 760 | 0.5 | ||||
| 255 (98000) d | ||||||
 | ||
| Fig. 3 Absorption and emission spectra (λexc = λmax,abs) of pBN, mBN and BPN recorded in (a) toluene, (b) THF, and (c) dichloromethane. c = 10−5 mol L−1, T = 293 K. | ||
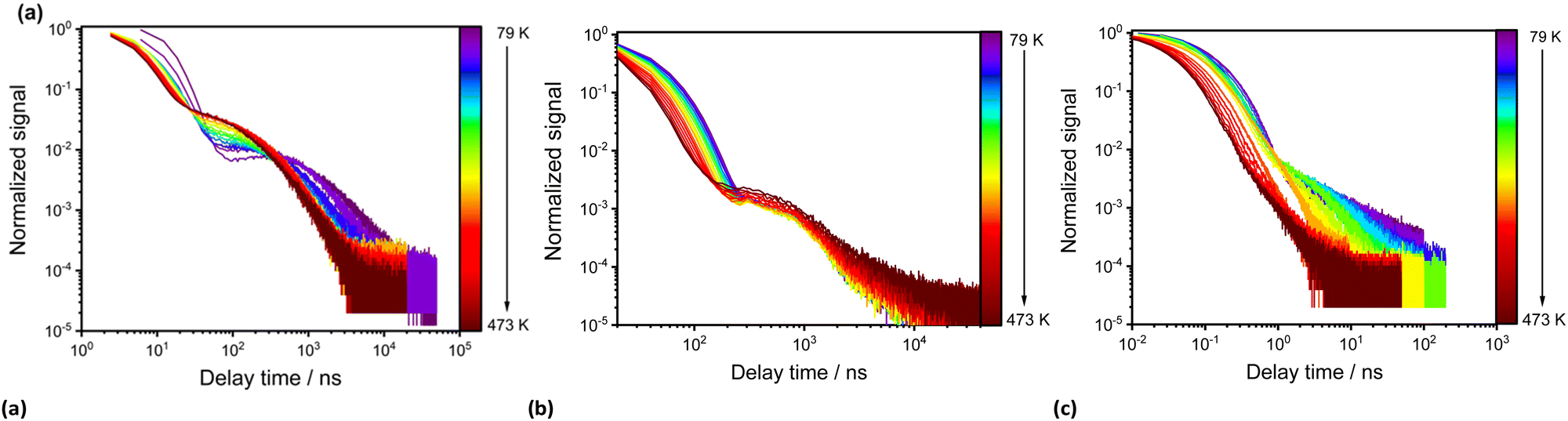
Compound pBN has the strongest absorption maximum at ∼320 nm with a pronounced tailing shoulder at 371 nm and a weak one at 427 nm. In toluene, the absorption at 371 nm is a distinct band (Table 1 and Fig. 3a). In comparison to pBN, for mBN the strongest absorption bands are redshifted to ∼331 nm with a weak shoulder at ∼380 nm. For BPN, an additional redshift of the strongest band to ∼373 nm and the tailing shoulder to 427 nm is observed. This band and shoulder coincide with the two shoulders in pBN. The molar absorption coefficients of the strongest absorptions above 300 nm are relatively low and in the range of 6000–8000 L mol−1 cm−1 (and even lower for pBN in toluene and BPN in THF) (Table 1).
In toluene, the absorption spectra for pBN and BPN are very similar and the longest wavelength absorption maxima appear as incompletely resolved shoulders at 427 nm (Fig. 3a). The comparable lowest energetic absorption of mBN is located at 381 nm (Table 1). This can also be rationalized by DFT/MRCI calculations (see the Theoretical Calculations section).
The compounds are only weakly luminescent in solution, yet, most intensively in toluene. Unlike the absorption spectra, the emission bands of the three compounds recorded upon excitation at each absorption maximum (λexc = λmax,abs) show a pronounced solvatochromicity. For pBN and mBN, the emission maxima differ by 10 nm in toluene with λem = 562 nm and 572 nm, respectively, but lie closely together in THF (λem = 634 nm) and CH2Cl2 (λem = 660 nm) (Table 1 and Fig. 3). The bathochromic shift from toluene to THF and CH2Cl2 of the emission bands indicates a positive emission solvatochromism (see also Fig. S17 and S19, ESI†).38
Compared to the pBN and mBN compounds, the emission maximum for BPN with its biphenyl π-system is bathochromically shifted to 604 nm in toluene but hypsochromically to 589 nm in THF and to 587 nm in CH2Cl2. In CH2Cl2, there is also a second emission band observable at 760 nm. For BPN, the hypsochromic emission shift from toluene to THF and CH2Cl2 represents a negative emission solvatochromism (see also Fig. S21, ESI†).
Compared to the solid-state (at 25 °C), the λmax values for pBN (558 nm) and mBN (551 nm) differ depending on the solvent. In the solid state, the emission maxima of pBN and mBN are located at similar wavelengths. The emission maximum of BPN (596 nm) is at a comparable wavelength to the values in solution and indicates limited solvatochromism.
The emission bands of the chromophores in toluene are narrow, with full width at half maximum (FWHM) values ranging from 0.44 to 0.47 eV (Table 1). The FWHM values only increase slightly for pBN and mBN from toluene to THF and CH2Cl2 (to 0.50–0.56 eV). Emission intensities are low for these molecules in toluene, and the CIE coordinates in toluene comply with the observable yellow (pBN, mBN) to orange (BPN) colors of the emission. In THF the CIE coordinates account for orange emission colors (Table 1 and Fig. S18, S20 and S22, ESI†). In CH2Cl2, the CIE coordinates match the observable orange emission colors of all compounds (Table 1).
Luminescence decay times are in a typical range for fluorescent organic chromophores.39 For pBN and mBN, the decay times decrease from toluene (5.7 ns and 8.9 ns) to THF (2.5 and 1.7 ns, respectively) and further to CH2Cl2 (1.0 and 1.6 ns, respectively) (Table 1). If the decay was purely radiative, the decay time should expectedly increase with redshifted emission wavelength based on the λem3 dependence of the radiative decay time. On the other hand, the increasing refractive index of the solvents from toluene over THF to CH2Cl2 leads to a local field enhancement and thus compensates the pure wavelength dependence of the radiative decay time.40 Finally, redshifted broad-band luminescence is more strongly prone to non-radiative relaxation and consequently lower quantum yields. The general decrease in emission brightness of pBN/mBN from toluene to CH2Cl2 indicates that it is the non-radiative pathway that is most relevant to the observed decrease of the photoluminescence decay time.
For BPN, the decay time first increases from toluene (6.7 ns) to THF (12.0 ns) along with the hypsochromic shift and then decreases again in CH2Cl2 (5.5/0.5 ns) in line with the lower intensity in emission which, however, is not visible anymore. The increase in decay time from toluene to THF is not readily expected given the slightly blue-shifted emission wavelength and similar photophysical properties such as FWHM and Stokes shift of BPN in THF compared to toluene (Table 1). However, the molar absorption coefficients in THF are lowered by a factor of almost 3 compared to those in toluene. If the difference in refractive indices between toluene and THF is considered, this could explain the difference in factor of almost 2 in the luminescence decay times of the luminescence of BPN in these two solvents.
In addition, all chromophores show a higher energy absorption maximum of around 250 nm (Fig. S17, S19, S21, ESI† and Table 1), which might be assigned to π–π* transitions. This absorption is only seen in dichloromethane because toluene and THF are not transparent at this wavelength. While the molar absorption coefficients of the longest wavelength absorption bands were low, the higher energy absorption bands are characterized by molar absorption coefficients of up to 98000 L mol−1 cm−1. The spectral features of the ground state, as reflected by the absorption characteristics, indicate that pBN and its phenylene expanded congener BPN are not only similar, but the ground state is largely insensitive to the change of polarity. Due to steric effects, a significant twist of the N-aryl substituents can be plausibly assumed in the ground state as is also indicated by single crystal structural data (Fig. 2). Yet, the electron-withdrawing nitrile substituent is positioned in conjugation with the phenazine-5,10-diyl nitrogen atoms. For mBN, this conjugative pathway is excluded due to the meta-positioning of the nitrile group, as indicated by the hypsochromic shift with respect to the maxima of the former two chromophores.
The spectral features of the excited states of the constitutional isomers pBN and mBN represented by the spectral ranges of their emission are mutually more similar than for the phenylene expanded system BPN. The positive emission solvatochromicity of the former indicates a polar excited state arising from a charge transfer from the phenazinediyl donor to the benzonitrile acceptor. The expansion of the π-system in BPN leads to negative emission solvatochromicity and hence to a less polar excited state. However, peculiar for the latter system is the occurrence of a second longer wavelength emission band at 760 nm in dichloromethane, which could either indicate a twisted intramolecular charge transfer (TICT) state41–44 or phosphorescence. Especially the latter type of transition should, however, be strongly prone to non-radiative relaxation in solution. BPN can be divided into a donor (phenazinediyl) and an acceptor (biaryl) moiety linked by a single bond. Upon excitation from the ground state, the locally excited (LE) state with a planar conformation can rapidly equilibrate with an intramolecular twisted charge transfer state of lower energy.45 This often results in dual emission with a high energy band through relaxation of the LE state (587 nm) and a low energy band by relaxation of the TICT state (760 nm). In a less polar solvent, like THF, the LE state is slightly energetically increased, but due to lower solvent polarity, the TICT state cannot be stabilized. Only in dichloromethane can the twisted CT state be stabilized and therefore be observed. This might explain the recorded negative emission solvatochromicity. The phenomenon of TICT has previously been observed for biphenyls and terphenyls.46,47
 | ||
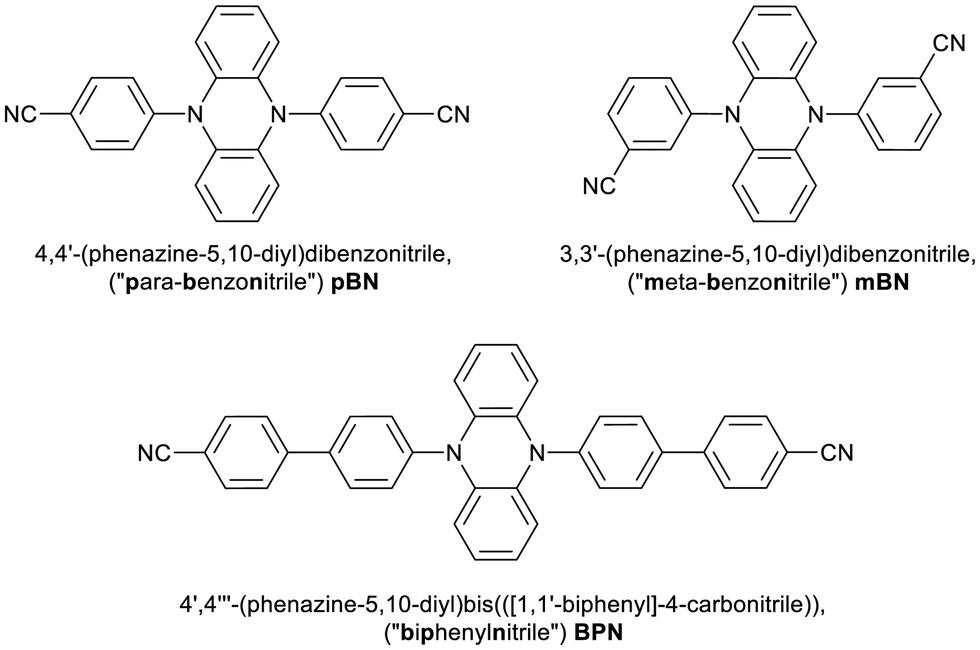
| Fig. 4 Temperature-dependent emission spectra of solid (a) pBN, (b) mBN, and (c) BPN upon excitation at λexc = 420 nm (a, b) and 450 nm (c). Temperature intervals were ΔT = 25 K. | ||
mBN features an emission maximum at λem ≈ 537 nm with a shoulder at 522 nm upon excitation at λexc = 420 nm at 79 K. With increasing temperature, the shoulder is not resolved anymore due to vibronic broadening. The barycenter of the emission band becomes thermally redshifted (Fig. 4b) in the whole regarded temperature range. The location of the emission maximum at shorter wavelengths compared to pBN at 79 K is assigned to the meta position of the nitrile functionalities that limits effective linear charge transfer from the phenazinediyl unit mediated through the phenyl moieties and thus, should lead to expectedly less pronounced TADF properties.
The emission spectra of solid BPN at 79 K show a vibronic fine structure (Fig. 4c). The maximum is located at λem ≈ 583 nm and is also redshifted with increasing temperature accompanied by a loss in the resolution of the vibronic fine structure. Together with the findings of nearly no solvatochromism of the respective emission of BPN in toluene, THF, and CH2Cl2, the observation of vibronic structure in the powder luminescence spectra at low temperatures indicate emission from a LE state. Both this observation and the redshifted luminescence in BPN compared to mBN/pBN can be rationalized by the donor–acceptor distances in the molecule due to the presence of the additional phenylene moiety (see Fig. 1). Charge transfer interaction strength roughly scales inversely exponentially with the donor–acceptor distance, which should be expectedly weakened in the large BPN molecule.48 In addition, the twisted configuration between the intermediate phenyl ring and the benzonitrile moiety additionally minimizes orbital overlap thereby localizing electron density.
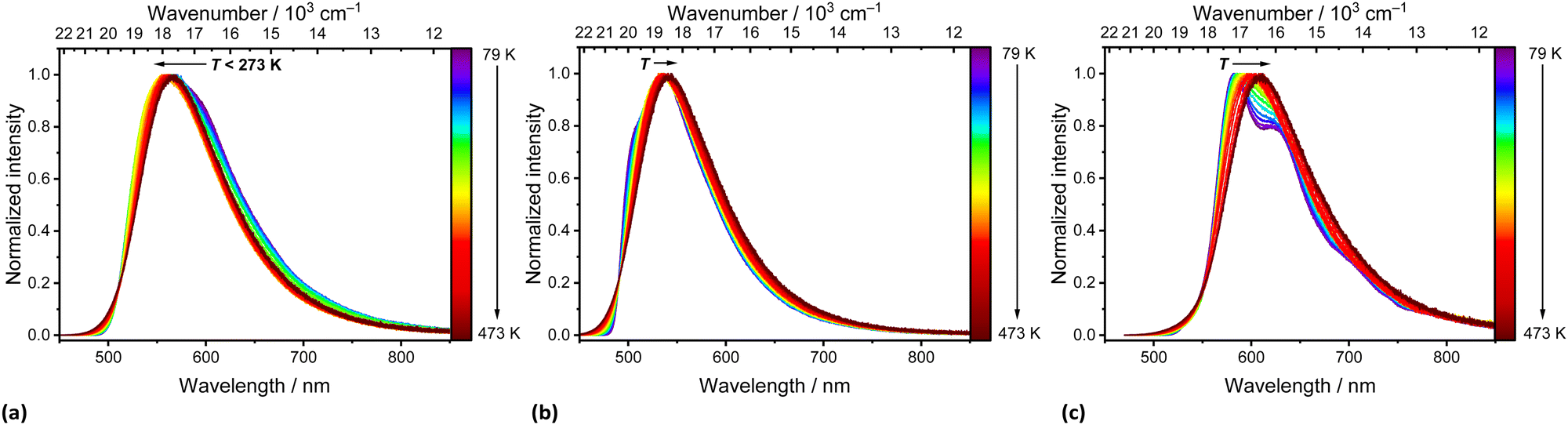
Fig. 5 depicts the temperature-dependent time-resolved luminescence of the three phenazinediyl derivatives. pBN shows clear signatures of TADF such as a prompt component in the ns range (τp(79 K) = 11 ns; τp(273 K) = 6.2 ns) and delayed time component in the μs range (τd(79 K) = 2.0 μs; τd(273 K) = 0.3 μs) as well as a temperature-dependent amplitude ratio between the prompt and delayed component. This is in line with the observable thermally-assisted blueshift of the emission below 272 K (Fig. 4a). From the delayed components at low temperatures (T < 220 K), an effective singlet–triplet gap of ΔEST = (174 ± 3) cm−1 = (21.5 ± 0.4) meV was derived (Fig. S14, ESI†), which is in agreement with the spectroscopically deduced value of ΔEST ≈ 200 cm−1 (25 meV) based on the energy difference of the emission maxima at 79 K and 273 K of pBN (Fig. S16, ESI†). Our derived energy gap is lower than the literature-reported value of ΔEST = 806 cm−1 (100 meV), which refers, however, to a 6 wt% pBN:m-CBP doped thin film and was solely estimated based on the energy difference between noisy spectra.33 In a neat powder, additional intermolecular interactions and aggregate formation expectedly decrease the energy gap consistent with our finding. Another aspect that can affect the energy gap by means of the effective orbital overlap is the relative twisting angle of the benzonitrile moieties to the central dihydrophenazine residue. This angle is fixed in a crystalline solid and may differ from the respective angle upon doping into a thin film. In contrast, mBN (Fig. 4b) shows weak TADF properties with a more dominant prompt (τp(79 K) = 33 ns; τp(273 K) = 24 ns) and weakly defined delayed component (τd(79 K) = 0.9 μs; τd(273 K) = 0.6 μs) with an average singlet–triplet gap of ΔEST = (38 ± 6) cm−1 = (4.7 ± 0.8) meV (Fig. S15, ESI†). No TADF properties are observed for BPN (Fig. 4c) and it shows conventional fluorescence with decay times in the ns range (τp(79 K) = 14 ns; τp(273 K) = 8.6 ns). Unfortunately, no non-radiative intersystem crossing rates are readily accessible with our spectroscopic equipment that would allow a more detailed analysis of the TADF kinetics. It is, however, evident that only pBN is a potent TADF emitter with reasonable photoluminescence quantum yield (ϕPL = 35.8%) if doped into a thin film (as the literature-reported 6 wt% pBN:m-CBP doped film). 33 Its absolute quantum yield in powdered form is much lower (ϕPL = 1.00%), which is understandable given the close contacts between the molecules within the crystalline solid (see section S4 in the ESI†) and agrees with the significant quenching of the luminescence at room temperature (see Fig. S11, ESI†). A similar low quantum yield of only 3.3% was found for pBN in aerated toluene solution with no observation of any delayed fluorescence at all.33 From the time-resolved photoluminescence of pBN at room temperature (see Fig. 5), we derive that the prompt and delayed components have almost equal contributions (ϕp (pBN) = 0.46%, ϕd (pBN) = 0.54%), also in agreement with the findings in a thin film.33 Similarly, the absolute quantum yields of powdered mBN (ϕPL = 3.10%; ϕp (mBN) = 2.97%, ϕd (mBN) = 0.13%) are low and dominantly stem from prompt fluorescence according to the time-resolved luminescence at room temperature (see Fig. 5). This again indicates that mBN is just at the boundary of being a TADF emitter. Finally, also the fluorescence of powdered BPN (ϕPL = 2.80%) is significantly quenched at room temperature, which makes these compounds not readily applicable organic emitters if used as neat powders and requires dilution or doping into thin films.
 | ||
| Fig. 5 Temperature-dependent time-resolved photoluminescence of solid (a) pBN, (b) mBN and (c) BPN upon excitation at λexc = 450 nm. Temperature intervals were ΔT = 25 K. | ||
Theoretical calculations
The DFT/MRCI-computed vertical excitation energies to the HOMO-LUMO S1-state in pBN, BPN and mBN in a vacuum are 402, 415 and 401 nm, respectively. BPN and pBN have zero oscillator strength of the S0 → S1 transition, and the computed value for f in mBN is weak (0.01) (Table S4, ESI†). This results from a lack of overlap between the participating orbitals (Fig. S31, ESI†). The first states with noticeable intensity are S4 in BPN (f = 0.10) and in pBN (f = 0.09) and S6 in mBN (f = 0.14) (Fig. 6). Similar findings hold true for the S1 structures and the corresponding vertical emission from this state. In the S1 geometry, the phenazine moiety flattens and becomes essentially planar (Fig. S26, ESI†). | ||
| Fig. 6 DFT/MRCI computed static absorption spectra of pBN, mBN and BPN in vacuum. | ||
Notably, the lowest triplet state at the S0 geometry is not of HOMO–LUMO character but rather mixed in all models (Table S4, ESI†).
The oscillator strength of both S1 absorption and emission are, however, tuned by motion of the phenyl rings with respect to the phenazine core as depicted in Fig. 7.
 | ||
| Fig. 7 Unrelaxed dihedral scan for phenyl-rotation in pBN showing oscillator strength (same and opposite directions of phenyl ring rotation, the starting point corresponds to the perpendicular arrangement (90°) of phenazine and phenyl rings); see also Fig. S26 (ESI†) for the energy profile. | ||
Hence, low-frequency normal mode vibrations (∼21 cm−1, Fig. S28, ESI†) increase the absorption intensity and slightly shift the corresponding bands, which appear in our zero-point energy (ZPE) simulated absorption spectrum as shoulders at ca. 405 nm (mBN) and 415 nm (pBN) (Fig. 8).
 | ||
| Fig. 8 Combined calculated absorption and emission spectra from vibrational sampling in toluene. | ||
Through ZPE sampling, we note only a slight enhancement in the emission intensity (f < 0.002, Fig. S29, ESI†). Here, the normal mode including torsion of the phenyl rings is shifted towards slightly higher frequencies (34 vs. 21 cm−1). The emission bands gain additional intensity (f ∼ 0.008) after including temperature effects. In our simulation at 300 K the emission bands appear at 547 nm (mBN) and 573 nm (pBN). Thermal sampling at the S0 geometry leads to unphysical distortions indicating the limits of the harmonic approximation in this model.
Variations in phenyl torsions furthermore affect the singlet–triplet gap, this finding is visualized in Fig. S29 (ESI†). The effect is significantly stronger in pBN, where ΔEST decreases from 105 meV to 34 meV by rotation of one phenyl moiety by only −15° (left graph in Fig. S30, ESI†). Fixation in this position through an external force (such as exerted in a rigid crystal structure) may enable efficient tuning of its emission properties and offers an explanation for the difference between the originally reported value of ΔEST = 100 meV in the 6 wt% mBN:m-CBP doped thin film and our slightly smaller value (22 meV).33
Conclusions
The investigated compounds 3,3′-(phenazine-5,10-diyl)dibenzonitrile (mBN) and 4′,4′′′-(phenazine-5,10-diyl)bis(([1,1′-biphenyl]-4-carbonitrile)) (BPN) do not show any evident TADF behavior compared to 4,4′-(phenazine-5,10-diyl)dibenzonitrile (pBN) known from the literature. This indicates that even slight changes in the molecular structure have a significant effect on the photophysical properties of the molecules. Thus, pBN exhibits unique TADF properties both in the solid state and in liquid solution. In the solid state, pBN exhibits a decay time that is in the microsecond range over the entire temperature range measured, along with a blue shift of the emission maximum at temperatures below 220 K. The effective singlet–triplet gap here is ΔEST = (174 ± 3) cm−1 = (21.5 ± 0.4) meV as derived from temperature-dependent time-resolved luminescence data. mBN only exhibits very weak TADF properties with an effective estimated exchange energy gap below 10 meV. We assign this marked difference between pBN and mBN in their TADF behavior to the minimized orbital overlap in the latter case given by the meta-substitution pattern of the nitrile functionalities. Apart from that, the two emitters are strongly related, which is reflected in their similar emission wavelengths and solvatochromism. The emission spectra are broad and featureless, which indicates a CT-like character of the electronic transition in both compounds. In contrast, BPN is a conventional fluorescent emitter and shows a vibronic fine structure indicating an emissive localized state. Their emission maxima are strongly dependent on the solvent, indicating a CT-like character of the excited state. Overall, it can be seen that TADF donor–acceptor-type emitters follow clear electronic guidelines that can be structurally controlled on a molecular scale and within the solid.Materials and methods
The purity of the compounds was determined by NMR spectroscopy, elemental analysis, high-resolution mass spectrometry and single crystal structure analysis (for details, see the ESI†).Solid state optical measurements were performed using an FLS1000 photoluminescence spectrometer from Edinburgh Instruments equipped with a 450 W Xe arc lamp as an excitation source, double excitation and emission monochromators in Czerny–Turner configuration and a thermoelectrically cooled (−20 °C) photomultiplier tube PMT-980 from Hamamatsu. The emission spectra were corrected with respect to the grating efficiency and PMT sensitivity, while excitation spectra were additionally corrected with respect to the lamp intensity. Time-resolved photoluminescence was excited with pulsed laser diodes EPL-450 (Edinburgh Instruments, temporal pulse width: 90 ps, 0.15 mW average incident peak power) or VPL-450 (Edinburgh Instruments, 90 mW average incident peak power in CW mode) with adjustable temporal pulse width (0.1 ms…1 ms) and variable trigger frequency (0.1 Hz…5 MHz) as pulsed excitation sources. The detection mode for the time-resolved measurements was time-correlated single photon counting. Absolute quantum yields at room temperature were measured with a BenFlect®-covered integrating sphere and excitation wavelength of 420 nm in all three regarded compounds.
Solution state absorption spectra were recorded in toluene, tetrahydrofuran and CH2Cl2 of high-performance liquid chromatography (HPLC) grade at 293 K on a PerkinElmer UV/Vis/NIR Lambda 19 spectrometer. For the determination of the molar extinction coefficients ε absorption measurements at five different concentrations were carried out. Emission spectra and fluorescence lifetimes were recorded in toluene, tetrahydrofuran and CH2Cl2 of HPLC grade at 293 K using an Edinburgh FS5 spectrometer. As light sources a 150 W xenon lamp as well as pulsed EPLED-320 (Edinburgh Instruments, 313.5 nm, temporal pulse width: 950 ps, 3 μW average incident peak power) and EPL-375 (Edinburgh Instruments, 372.2 nm, temporal pulse width: 76 ps, 0.15 mW average incident power) laser sources were used.
Computations
Single-molecule structures of pBN, mBN and BPN were geometry-optimized with Gaussian16,49 taking the crystal structure data as input. Calculations were performed with the PBE0 functional,50 the TZVP basis set51 and Grimme D3 dispersion corrections52 in vacuum and in toluene solvent applying the PCM model. TD-DFT was used for the excited S1 and T1 states. Vibrational analysis ensured that the obtained structures were true minima with no imaginary frequencies. For the computation of spectral properties, the optimized structures were recomputed at the BH-LYP/TZVP level of theory using Turbomole 7.5.53 Solvation was considered with the COSMO model.54 A DFT/MRCI computation was then performed with the R2022 Hamiltonian,55 the tight parameter set and an energy selector of 0.8Eh. The starting wavefunction was computed including eight orbitals and eight electrons with single and double excitations. An updated reference space was obtained by performing a second run with the same parameters to yield the final values for energies and oscillator strengths. Combined unrelaxed scans along the dihedral angles involving the phenyl rings were applied to document changes in ΔEST and oscillator strengths f.Vibrational effects in emission and absorption data were considered by sampling the vibrational modes (zero-point-energy (ZPE) sampling) generating 200 structures for each molecule and state in pBN and mBN. Thermal sampling at a temperature of 300 K was performed in addition to the S1 structures. After orbital computation with BH-LYP and the SV(P) basis, the resulting geometries were processed with DFT/MRCI and the R2016 Hamiltonian56 using the same selector choices stated above. The final spectra with vibrational contributions were obtained by Gaussian broadening using a FWHM of 0.3 eV.
Author contributions
Conceptualization: D. P and M. S.; methodology: D. P., J. W., T. H. and M. S.; validation: D. P.; formal analysis: D. P., J. W., T. H., L. P. and M. S.; investigation: D. P., J. W., T. H., L. P. and M. S.; resources: M. S., T. J. J. M. and C. J.; data curation: D. P., J. W., T. H. and M. S.; writing – original draft preparation: D. P. and J. W.; writing – review and editing: D. P., M. S., T. J. J. M. and C. J.; visualization: D. P., J. W. and C. J.; supervision: C. J.; project administration: C. J.; funding acquisition: M. S., T. J. J. M. and C. J.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationship that could have appeared to influence the work reported in this paper.Acknowledgements
D. P., J. W., T. J. J. M and C. J. thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for grant 396890929/GRK 2482 and grant Mu 1088/9-1. The Rigaku X-ray diffractometer was funded by the DFG through grant 440366605. M. S. gratefully acknowledges funding from a materials cost allowance of the Fonds der Chemischen Industrie e. V. and the “Junges Kolleg” of the North Rhine-Westphalian Academy of Sciences and Arts. The authors thank Dr István Boldog for his support in the supramolecular analysis of the single crystal structures of pBN. We also thank the Center for Molecular and Structural Analytics at Heinrich Heine University (CeMSA@HHU) for recording the mass spectrometric and NMR-spectrometric data.References
- C. A. Parker and G. G. Hatchard, Trans. Faraday Soc., 1961, 57, 1894–1904, 10.1039/TF9615701894.
- R. Delorme and F. Perrin, J. Phys. Radium, 1929, 10, 177–186, DOI:10.1051/jphysrad:01929001005017700; J. Perrin and N. Choucroun, Fluorescence sensibilisée en milieu liquide (transfert d’activation par induction moléculaire), Comptes Rendus, 1929, 189, 1213–1216 Search PubMed; F. Perrin and J. Perrin, Activation et deactivation par induction moléculaire, Activation et structure des molécules: Rapports et discussions, 1929, pp. 354–382 Search PubMed.
- M. N. Berberan-Santos, Pioneering Contributions of Jean and Francis Perrin to Molecular Luminescence, in New Trends in Fluorescence Spectroscopy, ed. B. Valeur and J. C. Brochon, Springer Series on Fluorescence, Springer, Berlin, Heidelberg, 2001, vol. 1 DOI:10.1007/978-3-642-56853-4_2.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238, DOI:10.1038/nature11687.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 42, DOI:10.1063/1.3558906.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444, DOI:10.1002/adma.201605444.
- M. K. Manna, S. Shokri, G. P. Wiederrecht, D. J. Gosztola and A. J.-L. Ayitou, Chem. Commun., 2018, 54, 5809–5818, 10.1039/C8CC01553H.
- T. Chatterjee and K.-T. Wong, Adv. Opt. Mater., 2018, 7, 1800565, DOI:10.1002/adom.201800565.
- X. Yin, Y. He, X. Wang, Z. Wu, E. Pang, J. Xu and J. Wang, Front. Chem., 2020, 8, 725, DOI:10.3389/fchem.2020.00725.
- S. Achelle, M. Hodée, J. Massue, A. Fihey and C. Katan, Dyes Pigm., 2022, 200, 110157, DOI:10.1016/j.dyepig.2022.110157.
- X. Wang, S. Gao, M. Zhao and N. Marom, Phys. Rev. Res., 2022, 4, 033147, DOI:10.1103/PhysRevResearch.4.033147.
- C. Li, A. K. Harrison, Y. Liu, Z. Zhao, C. Zeng, F. B. Dias, Z. Ren, S. Yan and M. R. Bryce, Angew. Chem. Int. Ed., 2022, 61, e20211540, DOI:10.1002/anie.202115140.
- L. Hua, Y. Liu, B. Liu, Z. Zhao, L. Zhang, S. Yan and Z. Ren, Nat. Commun., 2022, 13, 7828, DOI:10.1038/s41467-022-35591-w.
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282, DOI:10.1126/sciadv.1603282.
- Y. Yu, L. Ma, Z. Feng, B. Liu, H. Zhou, H. Qin, H. Li, J. Song, G. Zhou and Z. Wu, J. Mater. Chem. C, 2019, 7, 5604–5614, 10.1039/C9TC00789J.
- T. B. Nguyen, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Mater., 2020, 32, 1906614, DOI:10.1002/adma.201906614.
- J. Wiefermann, P. Schmeinck, C. Ganter and T. J. J. Müller, Chem. – Eur. J., 2022, 28, e202200576, DOI:10.1002/chem.202200576.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025, DOI:10.1021/acs.chemrev.7b00617.
- D. Hu, L. Yao, B. Yang and Y. Ma, Philos. Trans. R. Soc., A, 2015, 373, 20140318, DOI:10.1098/rsta.2014.0318.
- I. Lyskov and C. M. Marian, J. Phys. Chem. C, 2017, 121, 21145–21153, DOI:10.1021/acs.jpcc.7b06187.
- J. Gibson, A. Monkman and T. Penfold, Chem. Phys. Chem., 2016, 17, 2956–2961, DOI:10.1002/cphc.201600662.
- M. Inoue, T. Serevicius, H. Nakanotani, K. Yoshida, T. Matsushima, S. Jursenas and C. Adachi, Chem. Phys. Lett., 2016, 644, 62–67, DOI:10.1016/j.cplett.2015.11.042.
- J. Wiefermann, J. M. Kaminski, E. Pankert, D. Hertel, K. Meerholz, C. M. Marian and T. J. J. Müller, ChemPhotoChem, 2023, 7, e202200265, DOI:10.1002/cptc.202200265.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020, DOI:10.1038/natrevmats.2018.20.
- I. Marghad, D. H. Kim, X. Tian, F. Mathevet, C. Gosmini, J.-C. Ribierre and C. Adachi, ACS Omega, 2018, 3, 2254–2260, DOI:10.1021/acsomega.7b01570.
- R. K. Konidena, J. Lim and J. Y. Lee, Chem. Eng. J., 2021, 416, 129097, DOI:10.1016/j.cej.2021.129097.
- Z. Zhao, C. Zeng, X. Peng, Y. Liu, H. Zhao, L. Hua, S.-J. Su, S. Yan and Z. Ren, Angew. Chem. Int. Ed., 2022, 61, e202210864, DOI:10.1002/anie.202210864.
- F. Rodella, S. Bagnich, E. Duda, T. Meier, J. Kahle, S. Athanasopoulos, A. Köhler and P. Strohriegl, Front. Chem., 2020, 8, 657, DOI:10.3389/fchem.2020.00657.
- T. Matulaitis, P. Imbrasas, N. A. Kukhta, P. Baronas, T. Bučiūnas, D. Banevičius, K. Kazlauskas, J. V. Gražulevičius and S. Juršėnas, J. Phys. Chem. C, 2017, 121, 23618–23625, DOI:10.1021/acs.jpcc.7b08034.
- E. V. Verbitskiy, R. M. Gadirov, L. G. Samsonova, K. M. Degtyarenko, A. E. Kurtcevich, E. V. Sapozhnikova, M. V. Medvedeva, T. S. Svalova, A. N. Kozitsina, G. L. Rusinov and V. N. Charushin, Dyes Pigm., 2022, 207, 110716, DOI:10.1016/j.dyepig.2022.110716.
- D. Gudeikaa, O. Bezvikonnyia, D. Volyniuka and J. V. Grazuleviciusa, Dyes Pigm., 2020, 172, 107789, DOI:10.1016/j.dyepig.2019.107789.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599–4604, 10.1039/C3TC30699B.
- J. Lee, K. Shizu, H. Tanaka, H. Nakanotani, T. Yasuda, H. Kaji and C. Adachi, J. Mater. Chem. C, 2015, 3, 2175–2181, 10.1039/c4tc02530j.
- S.-J. Woo, Y. Kim, M.-J. Kim, J. Y. Baek, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, Chem. Mater., 2018, 30, 857–863, DOI:10.1021/acs.chemmater.7b04437.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963, DOI:10.1021/acs.chemmater.6b05324.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086, DOI:10.1126/science.aaf3935.
- Q. Wan, Y. Li, K. Ding, Y. Xie, J. Fan, J. Tong, Z. Zeng, Y. Li, C. Zhao, Z. Wang and B. Z. Tang, J. Am. Chem. Soc., 2023, 145, 1607–1616, DOI:10.1021/jacs.2c09210.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038, DOI:10.1021/ja00460a031.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006 Search PubMed.
- D. Toptygin, J. Fluoresc., 2003, 13, 201–219, DOI:10.1023/A:1025033731377.
- W. Rettig and M. Zander, Chem. Phys. Lett., 1982, 87, 229–234, DOI:10.1016/0009-2614(82)83131-X.
- Z. R. Grabowski and J. Dobkowski, Pure Appl. Chem., 1983, 55, 245–252, DOI:10.1351/pac198855020245.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Chem. Soc. Rev., 2021, 50, 12656–12678, 10.1039/D1CS00239B.
- A. M. El-Zohry, E. A. Orabi, M. Karlsson and B. Zietz, J. Phys. Chem. A, 2021, 125, 2885–2894, DOI:10.1021/acs.jpca.1c00629.
- S. Sasaki, G. P. C. Drummen and G. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743, 10.1039/C5TC03933A.
- S. Delmond, J.-F. Létard, R. Lapouyade and W. Rettig, J. Photochem. Photobiol., A, 1997, 105, 135–148, DOI:10.1016/S1010-6030(96)04544-3.
- S. Sharm, Z. Wei, T. C. Grozema and S. Sengupta, Phys. Chem. Chem. Phys., 2020, 22, 25514–25521, 10.1039/d0cp04579a.
- D. L. Dexter, A Theory of Sensitized Luminescence in Solids, J. Chem. Phys., 1953, 21, 836–850, DOI:10.1063/1.1699044.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and G. E. Scuseria, J. Chem. Phys., 1999, 111, 2889–2899, DOI:10.1063/1.479571.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305, 10.1039/B508541A.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, Turbomole: Modular program suitefor ab initio quantum-chemical and condensed-matter simulations, J. Chem. Phys., 2020, 152, 184107, DOI:10.1063/5.0004635.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805, 10.1039/P29930000799.
- D. R. Dombrowski, T. Schulz, M. Kleinschmidt and C. M. Marian, J. Chem. Phys. A, 2023, 127, 2011–2025, DOI:10.1021/acs.jpca.2c07951.
- I. Lyskov, M. Kleinschmidt and C. M. Marian, J. Chem. Phys., 2016, 144, 034104, DOI:10.1063/1.4940036.
Footnote |
| † Electronic supplementary information (ESI) available: Sources of chemicals, synthesis, crystal structure details, supramolecular interaction analysis, photophysical properties, and quantum chemical calculations. CCDC 2238744, 2222499 and 2222500. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc01228j |
| This journal is © The Royal Society of Chemistry 2023 |