
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllable and tuneable growth of NaYbF4:Tm(0.5%)Fe(5%)@Na(Yb/Y)F4-core@shell structures and the effect of their geometry on upconversion luminescence†
Elena
Ureña-Horno
 ,
Keqing
Liu
and
Marco
Giardiello
*
,
Keqing
Liu
and
Marco
Giardiello
*
Department of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. E-mail: marco.giardiello@liverpool.ac.uk
First published on 14th August 2023
Abstract
Lanthanide doped upconversion nanoparticles (UCNPs) are a class of nanomaterials with excellent luminescence properties. The practical use of UCNPs, however, has been hindered by their relatively low upconversion (UC) quantum yields. Enhancing their efficiency, and therefore their brightness, is a critical goal for these emerging materials. In this study, a range of novel core@shell structures were synthesized using NaYbF4:Tm(0.5%)Fe(5%) as the core template to regulate the shell growth of Na(Yb/Y)F4. Here, we observed that the size and the morphology of the UCNPs can be fine-tuned by controlling the ratio of the Y3+/Yb3+ ions within the shell material, resulting in small rod-like structures that form when using a high concentration of Y3+ and larger hexagonal plate-like structures when using a high concentration of Yb3+. In terms of the optical properties, the UC luminescence and lifetime of the core-only and core@shell structures were measured. Overall, the emission intensity and lifetime of the UCNPs increased with the nanoparticle size. We observed that, under the same experimental conditions, larger core@shell nanostructures with a hexagonal shape showed brighter UC emission, compared to those with a small nanorod shape and core-only. Based on our findings, we propose a variety of energy transfer pathways arise through geometric alteration of the nanostructures upon varying the Y3+/Yb3+ ion concentration ratios within the shell material. Our results could have important significance for understanding the relationship between the geometry of UCNPs and their UC optical properties.
Introduction
Lanthanide doped upconversion nanoparticles (UCNPs) are a class of luminescence nanomaterials with excellent chemical stability which convert low energy near infrared (NIR) photons into high energy visible (Vis) and ultraviolet (UV) light photons. UCNPs exhibit unique luminescent properties, including sharp excitation and emission bands, long luminescence lifetimes, large anti-Stokes shifts, low autofluorescence, no photobleaching, and high detection sensitivity.1–3 For this reason, UCNPs have attracted considerable interest in recent years in many fields, including 3D display technology,4,5 solar technology,6 photovoltaics,7 photocatalysis,8 optical sensing,9–11 and biomedical imaging applications.12,13 Despite their potential, the practical use of UCNPs has been hindered by their relatively low upconversion (UC) quantum yield, so there is a drive to enhance this efficiency to meet the growing demands of such applications. To this end, various approaches have been employed to tune the UC emission spectra. UCNPs are composed of a host material (typically NaYF4) in which lanthanide (Ln) ions are doped. Structural design is essential for high-efficiency UC emission, which involves the correct choice of the host material, the type of Ln ion used as dopants, their concentration, and their distribution across the UCNP.14,15 Conventionally, in early UCNP development, activator dopant concentration was kept low (typically less than 3% for Er3+ and less than 1% for Tm3+), while the number of sensitiser ions, Yb3+, was high to ensure an appropriate distance between the sensitizer and the activator ions to generate energy transfer and thus a bright emission.16 However, with a high Yb3+ sensitizer content, a concentration quenching effect process could occur, which would be detrimental to the luminescence efficiency.17–19 Recently, various approaches have been developed to alleviate the threshold of concentration quenching in UCNPs. One common approach is to passivate the surface of UCNPs, creating a core@shell structure with one or more shells which can block the energy transfer path to surface quenchers and can also separate sensitizers and activators ions through the use of these multi shells.20–22 Another strategy is to irradiate the UCNPs with high excitation rates so that the majority of dopant ions are at excited states and the number of detrimental ground-state ions is reduced.23–25 Choosing a host material with a large unit cell to maintain a uniform ion–ion distance between the crystal is probably the most common strategy to minimize ion segregation and avoids local concentration quenching.26–30 Another approach is to incorporate another dopant such as Fe3+. Zhang et al. also observed that a high Yb3+ concentration could be significantly enhanced through doping Fe3+ into the host material. They claimed that in the new system, new energy levels form owing to the mixed electron wavefunctions of Yb3+ and Fe3+ It was observed that the UV and blue emissions from the NaYbF4:Tm(0.5%) core doped with Fe3+ (5%) were enhanced by up to 10 and 6 times, respectively, compared to those of the core sample alone.31The relationship between the UCNP geometry with their optical properties is an area of investigation that has been drawing interest since the pathways of UC could be different for different dimensional systems depending on the ions’ distribution across the host material. Therefore, there is still a lot of potential to be explored. Several groups have used synchrotron-based X-ray photoelectron spectroscopy (XPS) to analyse the distributions of Yb3+ sensitizer ions within the NaLnF4 core and have observed that the dopants of sensitizer and activator ions are not uniformly distributed within the host material.32,33 Zhang et al.34 also confirmed that this “heterogeneous doping” can further induce many negative effects on the optical properties of the UCNPs, especially the luminescent efficiency. These findings led us to consider that the sensitizer ions’ distribution across the crystal lattice should play an important role.
The aim of this study was to evaluate a new tuneable growth mechanism and set of shapes for NaYbF4:Tm. To this end, a range of novel core@shell structures were synthesized by growing various NaYF4:Yb(x%) shell materials in which the Yb3+ doping concentration was set at x = 0%, 20%, 40%, 60%, 80% and 100% around a core of NaYbF4:Tm(0.5%), with 5% Fe3+ incorporated following the interesting findings of Zhang et al. Here, we have first observed that the nanoparticle size and the morphology can be tuned by precisely controlling the sensitizer concentration of Yb3+ within the shell material. The resulting core@shell nanoparticles exhibited a pure hexagonal phase and increasing sizes with increasing Yb3+ concentrations. Additionally, we correlated the structure with their optical properties for the range of the prepared UCNPs and have observed that the Yb3+ concentration is not limited by the concentration quenching effect. We saw that a high dopant concentration of sensitizer ions enhanced the overall brightness of UCNPs. It was found that the UC intensities of the UV and blue emissions for the core@shell with 100% Yb3+ were enhanced by ∼75 and ∼55-fold, respectively, compared to that of the core sample alone, showing also longer-lived emission lifetimes. Due to the core@shell formation, we hypothesize a new distribution for the pairs Yb3+–Tm3+ and Tm3+–Tm3+ within each UCNP from the core to the UCNP surface, forming distinct types of energy transfer network within the systems, depending on the introduction of Y3+/Yb3+ ions. We believe that these results would complement and extend the findings on the distribution of Y3+/Yb3+ ion dopants around a core shell. We have investigated this using a NaYbF4:Tm(0.5%)Fe(5%) core, however, this could be further applied to other UCNP cores with different dopant ions.
Experimental
Materials
All the reagents for the synthesis of UCNPs were purchased form Sigma-Aldrich. These are: yttrium(III) chloride hexahydrate (YCl3·6H2O), ytterbium(III) chloride hexahydrate (YbCl3·6H2O), thulium(III) chloride hexahydrate (TmCl3·6H2O), iron(III) chloride hexahydrate (FeCl3·6H2O), oleic acid (OA, 90%), 1-octadecene (ODE, 90%), sodium hydroxide (NaOH, ≥98%) and ammonium fluoride (NH4F, ≥98%).Synthesis of NaYbF4:Tm(0.5%)Fe(5%) core UCNPs
NaYbF4:Tm(0.5%)Fe(5%) core UCNPs were synthesized using a Schlenk-line and following a previously reported protocol with slight modification to increase the scale.35 In a 250 mL three-neck round-bottomed flask, YbCl3·6H2O (4.75 mmol, 1.83 g), TmCl3·6H2O (0.025 mmol, 9.60 mg), and FeCl3·6H2O (0.25 mmol, 67.58 mg) were mixed with 30 mL of OA (90%) and 60 mL of ODE (90%). The solution was then heated to 150 °C under the presence of N2 gas for 2 h. After that, the solution was cooled down to room temperature, and a mixture of NaOH (12.5 mmol, 500 mg) and NH4F (20 mmol, 740.80 mg) dissolved in 30 mL of methanol was added dropwise under vigorous stirring. The mixture was stirred for 60 min at room temperature. After evaporating methanol, a vacuum was applied for 45 min at 100 °C. Finally, the solution was heated up to 305 °C and maintained for 30 min under a N2 atmosphere. The solution was cooled down naturally, and UCNPs were precipitated by centrifugation and washed with ethanol three times (3000 rpm, 15 min). The as-prepared UCNPs were re-dispersed in hexane for further use.Synthesis of NaYbF4:Tm(0.5%)Fe(5%)@NaYF4-core@shell UCNPs
The as-synthesised UCNPs served as a core to grow an epitaxial shell of NaYF4. The synthesis was again performed using a standard Schlenk-line following a method described in the literature with some adjustments.36,37 In a 100 mL three-neck round bottom flask, YCl3·6H2O (0.5 mmol, 151.68 mg) was mixed with 6 mL of OA and 15 mL of ODE under a N2 gas flow. The solution was heated to 150 °C to form a homogenous mixture. After that, the solution was cooled down to 80 °C under a steady flow of N2, and the previously prepared core UCNPs (125 mg) dispersed in hexane (20 mg mL−1) were added. After removal of the hexane, the temperature of the solution was reduced to room temperature and a mixture of NaOH (1.25 mmol, 50 mg) and NH4F (2 mmol, 74.08 mg) dissolved in 5 mL of methanol was added dropwise under vigorous stirring. The mixture was stirred for 45 min at room temperature. After evaporation of the methanol, the solution was heated up to 305 °C and maintained for 1 h and 30 min under a N2 atmosphere. The as-prepared UCNPs were cooled down naturally and were precipitated by centrifugation, and washed with ethanol three times (8000 rpm, 15 min). Finally, UCNPs were re-dispersed in hexane, and stored for further use.Synthesis of NaYbF4:Tm(0.5%)Fe(5%)@NaYF4:Yb (x%)-core@shell
NaYF4:Yb (x%), where x = 20%, 40%, 60%, 80% and 100% were created around the core structure by using a similar procedure as described above. (Note, where x = 0%, the shell is denoted NaYF4 and where x = 100% the shell is referred to as NaYbF4). Here, both YCl3·6H2O and YbCl3·6H2O were incorporated into the synthesis at various ratios, as stated in Table 1.| Shell material | YCl3·6H2O (mmol) | YCl3·6H2O (mg) | YbCl3·6H2O (mmol) | YbCl3·6H2O (mg) |
|---|---|---|---|---|
| NaYF4 | 0.5 | 151.68 | — | — |
| NaYF4:Yb(20%) | 0.4 | 121.34 | 0.1 | 38.75 |
| NaYF4:Yb(40%) | 0.3 | 91.01 | 0.2 | 77.50 |
| NaYF4:Yb(60%) | 0.2 | 60.67 | 0.3 | 116.25 |
| NaYF4:Yb(80%) | 0.1 | 30.34 | 0.4 | 155.00 |
| NaYbF4 | — | — | 0.5 | 193.75 |
Characterization
Transmission electron microscopy (TEM) was performed on a FEI Tecnai with an operating voltage of 120 kV. TEM grids were Carbon Films on 300 Mesh Grids Copper (from Agar Scientific). The nanoparticle size distribution was analysed with ImageJ (National Institutes of Health, USA) software considering over 150 nanoparticles for the statistical analysis. XRD analysis was performed using a Bruker X-ray diffractometer equipped with a CuKα source (λ = 1.54059 nm) in transmission mode and using a stainless-steel plate. Fourier transform infrared (FTIR) spectra were recorded in the spectral range of 525 and 4000 cm−1 on a Vertex 70 Fourier transform infrared spectrometer (FT-IR) spectrometer at room temperature. For the sample preparation, nanoparticles were dried and deposited directly on the glass (approximately 1 mg required for analysis). The UC luminescence spectra and lifetime decay were obtained using a FLS1000 Photoluminescence Spectrometer equipped with an external 980 nm CW laser with 0.001 to 2 W power. The UCNPs were dispersed in hexane to the concentration of 10 mg mL−1. All samples were measured using a quartz cuvette with a path length of 10 mm. Best fit for the life curve decay was obtained using the Advanced Analysis of Fluorescence Kinetics software FAST.Results and discussion
Effect of epitaxial shelling on NaYbF4:Tm(0.5%)Fe(5%) UCNPs
Fig. 1a shows the transmission electron microscopy (TEM) image of the synthesized NaYbF4:Tm(0.5%) Fe(5%) core UCNPs showing uniformity in size with a mean diameter of 11.5 nm ± 3.6 nm. Here, we used 5 mol% of Fe3+ ions which, according to previous reports, is optimal for the UC enhancement in UCNPs.31 Core@shell structures were achieved through epitaxial growth, in which NaYbF4:Tm(0.5%)Fe(5%) served as the core template for the shell growth.38–40 Different lanthanide (Ln) doped shell materials of NaYF4:Yb (x%) where used, where x refers to the Yb3+ doping concentration incorporated into the shell material. For this study, six samples were prepared: NaYbF4:Tm(0.5%)Fe(5%)@NaYF4:Yb(x%)-core@shell structures, where x was set at 0%, 20%, 40%, 60%, 80% and 100%. Note, where x = 0%, the shell is denoted NaYF4 and where x = 100% the shell is referred to as NaYbF4. | ||
| Fig. 1 TEM images of core and core@shell structures using different Yb3+ concentrations during shell growth. (a) NaYbF4:Tm(0.5%) Fe(5%) core and (b)–(g) NaYbF4:Tm(0.5%) Fe(5%)@NaYF4:Yb(x%)-core@shell where x = 0%, 20%, 40%, 60%, 80%, and 100%. Size histograms are shown in Fig. S1 (ESI†). All scale bars show 100 nm. | ||
Overall, it was observed that the deposition of the shell material on the core led to highly unbalanced epitaxy in different crystallographic directions when different shell materials were used. As one can see from Fig. 1(b)–(g) core@shell nanostructure synthesized with a low concentration of Yb3+ tended to form small rod-shaped structures while a higher concentration of Yb3+ favoured the larger hexagonal plate nanostructures. This result is expected due to the smaller ionic radius of Yb3+vs. Y3+ and its higher relative electronegativity allowing for a greater binding energy to F− ions to occur, which results in the formation of a larger structure.41 The size and morphology are summarized in Table 2.
| Shell material | Core@shell length (L) (nm) | Core@shell width (W) (nm) | AR (L/W) | Shape |
|---|---|---|---|---|
| NaYF4 | 34.3 ± 1.7 | 26 ± 1 | 1.32 |

|
| NaYF4:Yb(20%) | 43.6 ± 1.7 | 35.8 ± 2.8 | 1.22 |

|
| NaYF4:Yb(40%) | 52.4 ± 1.9 | 60.7 ± 3.7 | 0.86 |

|
| NaYF4:Yb(60%) | 69.1 ± 5.7 | 115.2 ± 6.8 | 0.60 |

|
| NaYF4:Yb(80%) | 103.0 ± 8.1 | 200.7 ± 11.8 | 0.51 |

|
| NaYbF4 | 119.1 ± 9.7 | 255.9 ± 9.6 | 0.47 |

|
Fourier-transform infrared (FTIR) spectroscopy was used to characterize the functional groups on the surface of the nanoparticles (see Fig. S2, ESI†). The spectra show two main vibrations at 2924 cm−1 and 2853 cm−1, which were attributed respectively to the asymmetric and symmetric stretching modes of the methylene (−CH2) groups of the OA molecule. The characteristic bands at ∼1700 cm−1 corresponding to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from OA, were practically absent in all the spectra. Instead, two bands at 1559 cm−1 and 1459 cm−1 appeared and were attributed to the asymmetric and symmetric stretching vibrations of the carboxylate group [(–COO–)3Ln3+] of the NaLnF4, respectively.
O from OA, were practically absent in all the spectra. Instead, two bands at 1559 cm−1 and 1459 cm−1 appeared and were attributed to the asymmetric and symmetric stretching vibrations of the carboxylate group [(–COO–)3Ln3+] of the NaLnF4, respectively.
To obtain information about the crystal structures, we carried out X-ray diffraction (XRD) measurements (see Fig. S3, ESI†). It shows the XRD patterns of the core and the different core@shell structures. Our experiments revealed that the initial core had a pure cubic phase (α) while all core@shell structures had a pure hexagonal phase (β), without other impurity peaks. In addition, we saw that some peaks, such as the (101) and (201) reflections, were shifted to lower 2 theta angles when NaYF4 was grown as the shell material and slightly shifted to higher values when NaYbF4 was used instead (see Fig. S4, ESI†). This is consistent with the Bragg equation λ = 2dsin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ; Y3+ ions, which have a larger ionic radius compared to Yb3+ ions, will form unit cells with larger d-spacing, and therefore they will need to be compressed to fit around the NaYbF4 core.42,43
θ; Y3+ ions, which have a larger ionic radius compared to Yb3+ ions, will form unit cells with larger d-spacing, and therefore they will need to be compressed to fit around the NaYbF4 core.42,43
It is known that β-NaLnF4 generally features the (0001) crystal planes for the top and the bottom and six equivalents ±(10–10)/±(1–100)/±(01–10) axial planes, usually simplified as (10–10).38,44,45 The rate of overall crystal growth, according to general principles, is related to the relative growth rate of the individual crystal planes within the crystal structure.46,47 The major cause of anisotropy during shell growth is the lattice mismatch between the core and shell material. Another important factor is the competing effect of the selective adsorption of the OA ligand vs. the deposition of the shell material on specific faces of the core.48–50Fig. 2 shows the proposed mechanism of the epitaxial growth of the NaLnF4 (Ln = Y and Yb) shell around the NaYbF4:Tm(0.5%) Fe(5%) core.
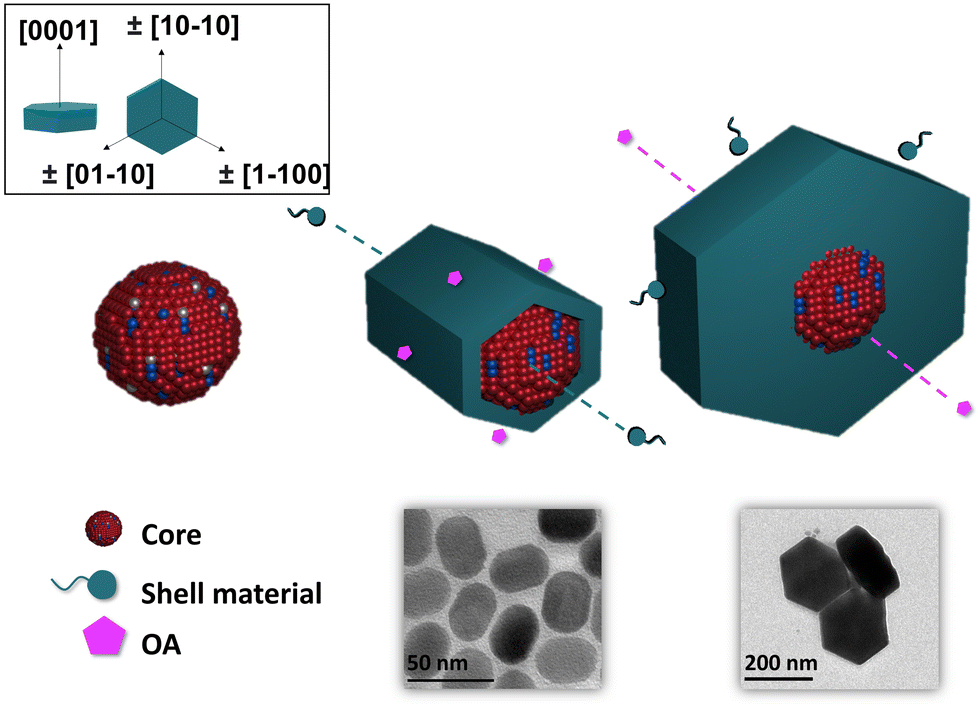 | ||
| Fig. 2 Proposed mechanism of epitaxial growth of NaLnF4 shells around the NaYbF4:Tm(0.5%) Fe(5%) core. | ||
When the core@shell structure was created with 0% and 20% Yb3+, it is postulated that OA adsorbed preferentially on the axial planes, thereby slowing the growth in this direction. Consequently, the shell grew preferentially on the [0001] lateral direction, and thus, produced structures with a rod-like shape and high AR. By increasing the Yb3+ content to 40%, the shell material could adhere to the (0001) facets as well as to the less preferred (10–10) axial facets, and as a result, the growth rates would be comparable along both directions. Here, the ratio between the length and width was very close to 1, more precisely AR = 0.86. When the concentration of Yb3+ was above 60%, we reasoned that the material would adhere selectively more on the prismatic axial facets of the core. This way the shell grew faster in the axial direction, leading to the formation of hexagonal shaped core@shell. When the core@shell structure was created with 80% and 100% Yb3+, the shell grew preferentially on the (10–10) axial facets, and thus, produced mostly flattened hexagonal plate-shaped structures and low AR (0.51 and 0.47). These arguments can explain the change of morphology we observed in our experiments.
Upconversion luminescence properties of nanoparticles with different core@shell structures as a function of Yb3+ content in the shell
To systematically correlate the optical properties to the structure of both the core and the different core@shell structures, the UC luminescence properties were investigated under 980 nm irradiation.One can observe from the luminescent spectra several characteristic enhanced UC emissions, where the UV emissions at 344 nm and 362 nm can be assigned to the 1I6 → 3F4 and 1D2 → 3H6 transitions, and the blue emissions at 450 and 475 nm correspond to the 1D2 → 3F4 and the 1G4 → 3H6 transitions of the Tm3+ ion, respectively (see Fig. S5, ESI† for the full spectra).
As a reminder to the reader, all the samples were prepared from the same core material used throughout, i.e. with 94.5% Yb3+ as the sensitizer and 0.5%Tm3+ as the activator. Therefore, the only synthetic modification was the incorporation of Y3+ and Yb3+, in different concentrations for the core@shell structures. With the core-only structures, nanoparticles had a high surface area to volume (S/V) ratio, therefore, most of the Ln3+ dopants were exposed to surface deactivations arising from surface defects (i.e., disorder, vacancy, and interstitial defects).20 Overall, it was observed that upon introduction of any shell material, the recovery from the core's surface defects enhanced the UC luminescence. Compared to the UC emission of the core, the emission from the core@shell prepared with 100% of Y3+ ions was enhanced by ∼4 fold and ∼2.6 fold in the UV and blue range, respectively, as can be seen in Fig. 3a. As the same core material is used throughout, we can assume the same distribution and concentration of Tm3+ and Yb3+ ions in both the core and core@NaYF4 shell structures. As such, we attribute the change in the emission intensity to the surface quenching effect, which depends primarily on the particle size. When the shell was created using 100% of Yb+3 ions as the material, the UC emission was enhanced by ∼75 and ∼55 -fold with respect to the core-only structure, shown in Fig. 3b. In general, for a thin surface coating in the 0–2.5 nm range, a linear dependence of UC emission intensity and shell thickness has been observed.51 However, it has been demonstrated that a steady enhancement in UC emission is generally unexpected by using very thick shells and the UC emission intensity reaches a plateau at a shell thickness of ∼3 nm.40,52 Based on this finding, we propose that the variation in the UC efficiency could depend on the final size but also on the different pathways of UC in the new geometry for the NaYbF4@NaYbF4 core@ shell.
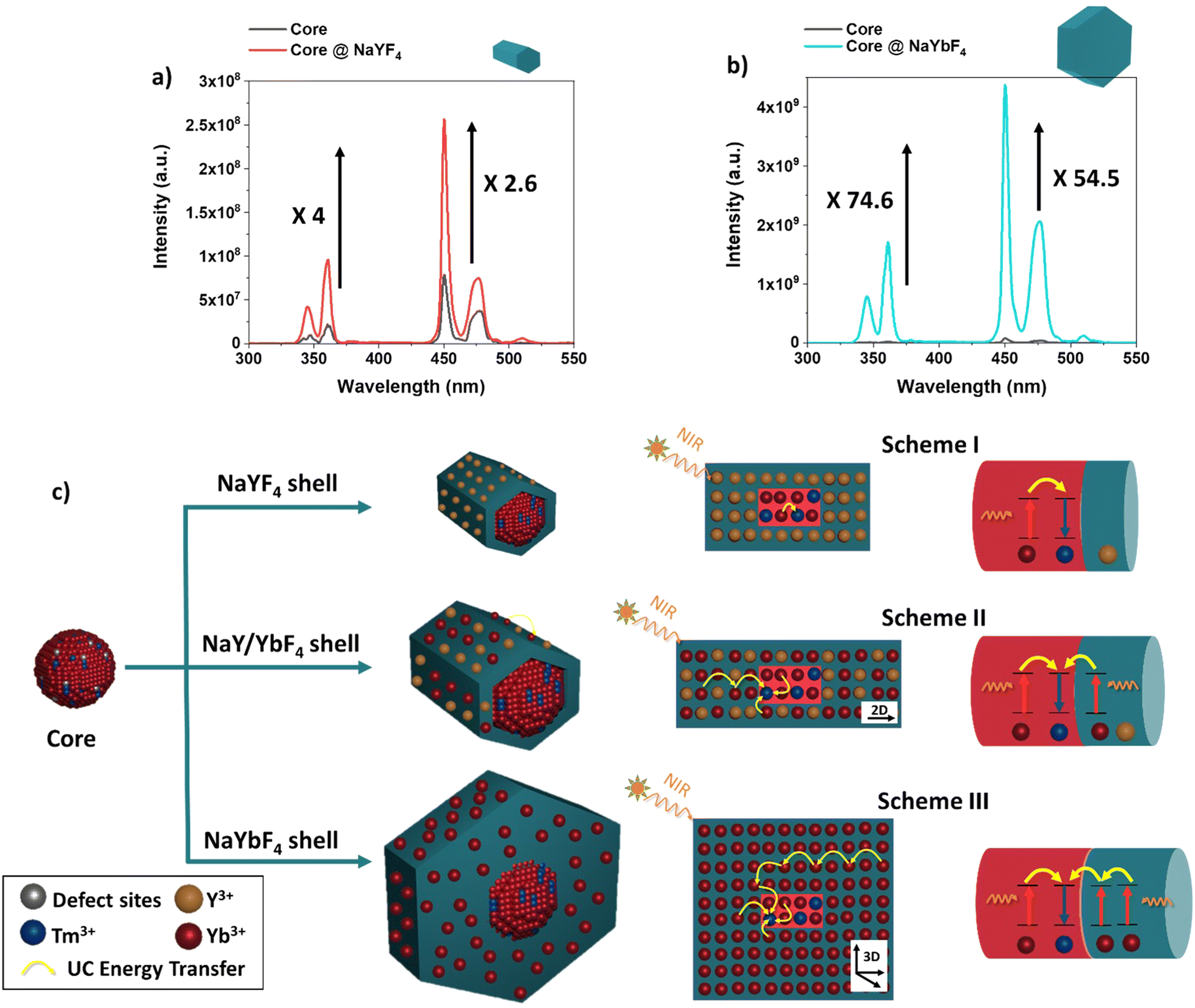 | ||
| Fig. 3 Comparison of UC emission spectra of (a) NaYbF4:Tm(0.5%)Fe(5%) core vs. NaYbF4:Tm(0.5%)Fe(5%)@NaYF4-core shell, and (b) NaYbF4:Tm(0.5%) Fe(5%) core vs. NaYbF4:Tm(0.5%) Fe(5%)@NaYbF4-core shell. For the measurements, nanoparticles were dispersed in hexane and measured with a 980 nm CW laser excitation. (c) Schematic illustration of the UC luminescence mechanism for the core@NaYF4, core@NaY/YbF4 and core@NaYbF4 structures, when measured with a 980 nm CW laser excitation. Scheme I represent a core composed of a sensitizer and activator coated with an optical inert outer shell. Scheme II illustrates the mechanism when the core is coated with a partially active shell of Y3+ and Yb3+, demonstrating the suggested 2D energy transfer network along the length of the rod like nanostructure. Scheme III illustrates the mechanism when the core is coated with an active shell of 100 Yb3+, demonstrating the suggested 3D energy transfer network throughout the entirety of the hexagonal platelike structure. | ||
As known, when UCNPs are excited at 980 nm there is an absorption by sensitizers Yb3+, producing an efficient energy transfer to the activator Tm3+ ions, which occurs because each Tm3+ ion is surrounded by many excited Yb3+ ions.53 In general, the energy transfer depends on the strength of the ion–ion interaction and on the concentration of Yb3+–Tm3+ and Tm3+–Tm3+ ions within the crystal.54 Therefore, as the relative concentration of Yb3+ and Tm3+ is expected to be constant in the core of all samples, we can assume that the gradual incorporation of Yb3+ ions and their new arrangement within the shell crystal structure will favour this energy transfer process, to a greater or lesser extent.
When Yb3+ was incorporated into the shell material, as shown in Fig. 3c (Scheme II and III), we can assume that the variation in particle shapes leads to a different pathway for energy transfer for the pair Yb3+–Tm3+ in the crystal structure. The Yb3+ sensitizer located at the shell would act as a bridge and allow for energy migration between Yb3+ ions across the shell, resulting in the excitation of core Tm3+ ions that are ultimately responsible for emitting UC light. This is more prominent where the shell is composed of 100% Yb3+ (Fig. 3c (Scheme III)) vs. shells containing Y3+ (Fig. 3c (Scheme II)). The rod like structures that form when Y3+ is present in the core encourage more 2D energy transfer networks through the length of the structure, while the hexagonal plate like nanostructures that form upon less to no Y3+ present in the shell allow for greater opportunities for 3D energy transfer networks across the overall crystal structure. Emission enhancement is further demonstrated when looking at the integrated intensities, which shows the enhancement of UC luminescence as a function of Yb3+ concentration incorporated in the shell growth (see Fig. S6, ESI†).
The pump power dependence of the UV and blue emission for the samples was also studied, with the results displayed on a double logarithmic plot (see Fig. S7, ESI†). It is known that the relationship between the UC emission intensity (IUC) and laser power (P) can be approximately expressed as: IUC ∼ Pn, where (n) refers to the number of photons absorbed per photon emitted, and its value can be derived from the slope of the fitted line of the plot log (IUC) vs. log(P).55 As expected, the UC emission changed gradually when increasing the pump power. The values of the exponent n were extracted from a linear fit of the experimental data. In detail, the values of the parameter n (photons absorbed per photon emitted) for 345 nm, 362 nm, 450 nm and 475 nm were found to be 2.00, 1.85, 1.94 and 1.69 for the core, 2.18, 1.95, 1.88 and 1.98 for the core@shell with 0% Yb3+, and 2.21, 1.94, 1.901 and 1.85 for the core@shell with 100% Yb3+. Overall, the n-values of the 4 bands are much lower than the theoretical value (n = 5 for 345 nm and n = 4 for 362 nm, 450 nm, and 475 nm) that could be strongly ascribed to the saturation effect, which has been seen previously by other groups.56–60
Lifetime measurements
The upconversion luminescent (UCL) decay behaviours for the pair Yb3+–Tm3+ were subsequently studied. As shown in (Fig. S8(a)–(c), ESI†) it was found that the emission of Tm3+ (1D2 → 3H6 transition 362 nm) had a decay lifetime of τ = 49 μs for the core, while the decay of Tm3+ emission when coated with a layer of NaYF4 and NaYbF4 registered a lifetime of 198 μs and 234 μs, respectively.When the content of Yb3+ in the shell increased from 0% to 100%, the emission lifetime of Tm3+ prolonged noticeably by 36 μs, indicating that the Yb3+ concentration is a crucial factor affecting the energy transfer from the Yb3+ to Tm3. As a result, the longer lifetime at the 362 nm emission peak observed in the core@shell structured UCNPs indicates the lower non-radiative relaxation rate which agrees with the result shown in Fig. 3. For the 1D2 → 3F4 transition 450 nm (see Fig. S8(d)–(f), ESI†) a similar trend was observed; a decay lifetime of τ = 60 μs for the core, 201 μs when coated with a NaYF4 and 205 μs when coated with NaYbF4.
Conclusion
In this study we have developed novel NaYbF4:Tm(0.5%)Fe(5%)@Na(Y/Yb)F4 core@shell UCNP structures, possessing varied Y3+/Yb3+ concentration ratios. We have demonstrated that the incorporation of Yb3+ ions into the shell material further enhanced the emission in both the UV and visible regions, while also offering the ability to incrementally tune particle size and morphology. Based on our XRD observations and aligned with known theory, we propose a shift in the shell growth mechanism from a preferential growth in the [0001] lateral direction when low Yb3+ is present in the shell material, producing structures with a rod-like shape, to a preferential growth on the (10–10) axial facets when higher Yb3+ is present in the shell material, producing flattened hexagonal plate-shaped structures. An increase in the emission intensity was also observed at higher Yb3+ concentrations in the shell material. We hypothesize that the enhancement in the UC efficiency is attributed to a combination of an increase in particle size, but also variation of the energy transfer pathways arising through geometric alteration.We propose that a reduction in the Y3+/Yb3+ ratio encourages a 3D energy transfer network throughout the entirety of the plate-like structures, providing greater opportunities for energy transfer where the Yb3+ concentration is high within the shell. In contrast, at higher Y3+/Yb3+ ratios fewer opportunities for energy transfer are present, resulting in more 2D energy transfer networks along the length of the formed rod-like structures where the Y3+ concentration is high. These findings are useful in designing core@shell nanostructures, which could be exploited further in creating accurate models of core@shell designs expanded to other cores, highlighting new findings for multifunctionality and optimal performance in luminescent applications.
Author contributions
EUH and MG designed the experiments and wrote the manuscript. EUH and KL conducted all laboratory work, including the synthesis, characterisation and data interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge funding from the UKRI Future Leaders Fellowship (MR/T021306/1), from the EPSRC for a vacation bursary (KL). The authors also thank the UoL Materials Innovation Factory (MIF) for access to laboratories, the UoL Biomedical Electron Microscopy Unit (EM Unit) for access to TEM and Amy Lunt of the Chong group (UoL) for access to analytical equipment (XRD).References
- K. Du, J. Feng, X. Gao and H. Zhang, Light: Sci. Appl., 2022, 11, 222 CrossRef CAS PubMed.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed.
- E. Downing, L. Hesselink, J. Ralston and R. Macfarlane, Science, 1996, 273, 1185 CrossRef CAS.
- R. Deng, F. Qin, R. Chen, W. Huang, M. Hong and X. Liu, Nat. Nanotechnol., 2015, 10, 237 CrossRef CAS PubMed.
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173 RSC.
- P. Zhang, L. Liang and X. Liu, J. Mater. Chem. C, 2021, 9, 16110 RSC.
- Q. Tian, W. Yao, W. Wu and C. Jiang, Nanoscale Horiz., 2019, 4, 10 RSC.
- H. Rabie, Y. Zhang, N. Pasquale, M. J. Lagos, P. E. Batson and K. B. Lee, Adv. Mater., 2019, 31, 1806991 CrossRef PubMed.
- T. Wang, F. Sun, Z. Jiang, J. Wang, Y. Ma, H. Lin, H. Lv, S. Xu, J. Li and G. Zhou, J. Mater. Chem. C, 2021, 9, 17451 RSC.
- T. Zhang, L. Liu, R. Wang, W. Zhang, X. Liu, C. Yuan and R. Hua, RSC Adv., 2023, 13, 9273 RSC.
- S. Chen, A. Z. Weitemier, X. Zeng, L. He, X. Wang, Y. Tao, A. J. Y. Huang, Y. Hashimotodani, M. Kano, H. Iwasaki, L. K. Parajuli, S. Okabe, D. B. Loong Teh, A. H. All, I. Tsutsui-Kimura, K. F. Tanaka, X. Liu and T. J. Mchugh, Science, 2018, 359, 679 CrossRef CAS PubMed.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed.
- S. Wen, J. Zhou, K. Zheng, A. Bednarkiewicz, X. Liu and D. Jin, Nat. Commun., 2018, 9, 2415 CrossRef PubMed.
- D. L. Dexter and J. H. Schulman, J. Chem. Phys., 1954, 22, 1063 CrossRef CAS.
- B. Chen and F. Wang, Acc. Chem. Res., 2020, 53, 358 CrossRef CAS PubMed.
- L. Sun, R. Gao, T. Pan, X. C. Ai, L. Fu and J. P. Zhang, Nanoscale, 2019, 11, 18150 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed.
- F. Wang, J. Wang and X. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456 CrossRef CAS PubMed.
- A. Pilch, C. Würth, M. Kaiser, D. Wawrzyńczyk, M. Kurnatowska, S. Arabasz, K. Prorok, M. Samoć, W. Strek, U. Resch-Genger and A. Bednarkiewicz, Small, 2017, 13, 1701635 CrossRef PubMed.
- X. Liu, X. Kong, Y. Zhang, L. Tu, Y. Wang, Q. Zeng, C. Li, Z. Shi and H. Zhang, Chem. Commun., 2011, 47, 11957 RSC.
- F. Benz and H. P. Strunk, AIP Adv., 2012, 2, 2 Search PubMed.
- D. J. Gargas, E. M. Chan, A. D. Ostrowski, S. Aloni, M. V. P. Altoe, E. S. Barnard, B. Sanii, J. J. Urban, D. J. Milliron, B. E. Cohen and P. J. Schuck, Nat. Nanotechnol., 2014, 9, 300 CrossRef CAS PubMed.
- J. Zhao, D. Jin, E. P. Schartner, Y. Lu, Y. Liu, A. V. Zvyagin, L. Zhang, J. M. Dawes, P. Xi, J. A. Piper, E. M. Goldys and T. M. Monro, Nat. Nanotechnol., 2013, 8, 729 CrossRef CAS PubMed.
- C. Ma, X. Xu, F. Wang, Z. Zhou, D. Liu, J. Zhao, M. Guan, C. I. Lang and D. Jin, Nano Lett., 2017, 17, 2858 CrossRef CAS PubMed.
- L. Marciniak, W. Strek, A. Bednarkiewicz, A. Lukowiak and D. Hreniak, Opt. Mater., 2011, 33, 1492 CrossRef CAS.
- J. Wang, R. Deng, M. A. Macdonald, B. Chen, J. Yuan, F. Wang, D. Chi, T. S. Andy Hor, P. Zhang, G. Liu, Y. Han and X. Liu, Nat. Mater., 2014, 13, 157 CrossRef CAS PubMed.
- J. Zuo, Q. Li, B. Xue, C. Li, Y. Chang, Y. Zhang, X. Liu, L. Tu, H. Zhang and X. Kong, Nanoscale, 2017, 9, 7941 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed.
- Y. Zhang, Y. Shen, M. Liu, Y. Han, X. Mo, R. Jiang, Z. Lei, Z. Liu, F. Shi and W. Qin, CrystEngComm, 2017, 19, 1304 RSC.
- K. A. Abel, J. C. Boyer and F. C. J. M. Van Veggel, J. Am. Chem. Soc., 2009, 131, 14644 CrossRef CAS PubMed.
- X. Xu, C. Clarke, C. Ma, G. Casillas, M. Das, M. Guan, D. Liu, L. Wang, A. Tadich, Y. Du, C. Ton-That and D. Jin, Nanoscale, 2017, 9, 7719 RSC.
- X. Li, R. Wang, F. Zhang and D. Zhao, Nano Lett., 2014, 14, 3634 CrossRef CAS PubMed.
- Z. Li and Y. Zhang, Nanotechnology, 2008, 19, 345606 CrossRef PubMed.
- X. Wu, Y. Zhang, K. Takle, O. Bilsel, Z. Li, H. Lee, Z. Zhang, D. Li, W. Fan, C. Duan, E. M. Chan, C. Lois, Y. Xiang and G. Han, ACS Nano, 2016, 10, 1060 CrossRef CAS PubMed.
- G. Jiang, J. Pichaandi, N. J. J. Johnson, R. D. Burke and F. C. J. M. van Veggel, Langmuir, 2012, 28, 3239 CrossRef CAS PubMed.
- H. S. Qian and Y. Zhang, Langmuir, 2008, 24, 12123 CrossRef CAS PubMed.
- S. Liu, L. Yan, J. Huang, Q. Zhang and B. Zhou, Chem. Soc. Rev., 2022, 51, 1729 RSC.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318 RSC.
- G. Chen, T. Y. Ohulchanskyy, W. C. Law, H. Ågren and P. N. Prasad, Nanoscale, 2011, 3, 2003 RSC.
- A. Lay, C. Siefe, S. Fischer, R. D. Mehlenbacher, F. Ke, W. L. Mao, A. P. Alivisatos, M. B. Goodman and J. A. Dionne, Nano Lett., 2018, 18, 4454 CrossRef CAS PubMed.
- C. Zhang and J. Y. Lee, ACS Nano, 2013, 7, 4393 CrossRef CAS PubMed.
- C. Li, J. Yang, Z. Quan, P. Yang, D. Kong and J. Lin, Chem. Mater., 2007, 19, 4933 CrossRef CAS.
- X. Liang, X. Wang, J. Zhuang, Q. Peng and Y. Li, Adv. Funct. Mater., 2007, 17, 2757 CrossRef CAS.
- M. Ding, D. Chen, S. Yin, Z. Ji, J. Zhong, Y. Ni, C. Lu and Z. Xu, Sci. Rep., 2015, 5, 12745 CrossRef CAS PubMed.
- C. Li, J. Yang, Z. Quan, P. Yang, D. Kong and J. Lin, Chem. Mater., 2007, 19, 4933 CrossRef CAS.
- N. Kang, J. Zhao, Y. Zhou, C. Ai, X. Wang and L. Ren, Nanotechnology, 2019, 30, 105701 CrossRef CAS PubMed.
- J. L. Fenton, B. C. Steimle and R. E. Schaak, Science, 2018, 360, 513 CrossRef CAS PubMed.
- J. Zhao, X. Chen, B. Chen, X. Luo, T. Sun, W. Zhang, C. Wang, J. Lin, D. Su, X. Qiao and F. Wang, Adv. Funct. Mater., 2019, 29, 1903295 CrossRef CAS.
- F. Zhang, R. Che, X. Li, C. Yao, J. Yang, D. Shen, P. Hu, W. Li and D. Zhao, Nano Lett., 2012, 12, 2852 CrossRef CAS PubMed.
- Q. Su, S. Han, X. Xie, H. Zhu, H. Chen, C. Chen, R. Liu, X. Chen, F. Wang and X. Liu, J. Am. Chem. Soc., 2012, 134, 20849 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- D. Yu, T. Yu, A. J. van Bunningen, Q. Zhang, A. Meijerink and F. T. Rabouw, Light: Sci. Appl., 2020, 9, 107 CrossRef CAS PubMed.
- Y. Yang, J. Liao, Z. Yang, S. Lai, B. Shao, J. Li, J. Qiu and Z. Song, J. Phys. Chem. C, 2014, 118, 17992 CrossRef.
- S. Hu, X. Wu, Z. Chen, P. Hu, H. Yan, Z. Tang, Z. Xi and Y. Liu, Mater. Res. Bull., 2016, 73, 6 CrossRef CAS.
- F. Pandozzi, F. Vetrone, J. C. Boyer, R. Naccache, J. A. Capobianco, A. Speghini and M. Bettinelli, J. Phys. Chem. B, 2005, 109, 17400 CrossRef CAS PubMed.
- S. Xu, H. Ma, D. Fang, Z. Zhang and Z. Jiang, J. Lumin., 2006, 117, 135 CrossRef CAS.
- H. Sun, Z. Duan, G. Zhou, C. Yu, M. Liao, L. Hu, J. Zhang and Z. Jiang, Spectrochim. Acta, Part A, 2006, 63, 149 CrossRef PubMed.
- J. Grube, Appl. Spectrosc., 2022, 76, 189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM histograms, FTIR, XRD, luminescence and lifetime decay data. See DOI: https://doi.org/10.1039/d3tc01215h |
| This journal is © The Royal Society of Chemistry 2023 |