
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePosition of gold dictates the photophysical and photocatalytic properties of Cu2O in Cu2O/Au multicomponent nanoparticles†
Dávid
Kovács
 ab,
András
Deák
a,
György Z.
Radnóczi
a,
Zsolt E.
Horváth
a,
Attila
Sulyok
a,
Róbert
Schiller
a,
Ottó
Czömpöly
a and
Dániel
Zámbó
*a
ab,
András
Deák
a,
György Z.
Radnóczi
a,
Zsolt E.
Horváth
a,
Attila
Sulyok
a,
Róbert
Schiller
a,
Ottó
Czömpöly
a and
Dániel
Zámbó
*a
aCentre for Energy Research, Konkoly-Thege M. út 29-33., H-1121 Budapest, Hungary. E-mail: daniel.zambo@ek-cer.hu
bDepartment of Physical Chemistry and Materials Science, Faculty of Chemical Technology and Biotechnology, Budapest University of Technology and Economics, Műegyetem rkp. 3., H-1111 Budapest, Hungary
First published on 12th June 2023
Abstract
Utilizing solar radiation for driving chemical reactions has been of great interest in the last decade. Although Cu2O nanocrystals as a low-cost, abundant, and tailorable photocatalyst are promising candidates to provide a platform for solar-driven applications, their photophysical properties are affected by the limited charge carrier mobility. This paper focuses on the more efficient utilization of the UV light-excited charge carriers in cuprous oxide by preparing different heterostructures with gold. While keeping the size, shape as well as overall composition of the heterostructures identical, the realization of the gold component is varied: either nanograins are created at the surface of the Cu2O nanooctahedra or nanorods are embedded in their interior. The effect of the morphology and the semiconductor-metal contact on the optical and photocatalytic properties is investigated in-detail by spectroscopy, spectrofluorometry, imaging techniques, X-ray diffractometry, and photoelectron spectroscopy extended with optical simulations and single-particle spectroscopy measurements. In terms of particle stability and photocatalytic activity, gold-decorated Cu2O nanooctahedra show superior properties. The comprehensive comparison of the multicomponent nanoparticles underlines the importance of the nanoscale design (including composition, morphology, and surface chemistry), which utilizes the photoexcited carriers in the semiconductor without injecting hot electrons from the metal.
Introduction
Multicomponent plasmonic/semiconductor nanoparticles (NPs) hold great potential to bridge boundless solar energy and various solar-driven applications including photocatalysis, owing to the synergetic properties induced by the interaction of the metal and semiconductor components. Cuprous oxide (Cu2O) is a cheap, abundant and nontoxic p-type semiconductor with a direct bandgap of Eg ∼ 2.17 eV,1 high carrier concentration2 and an extraordinarily high exciton binding energy of around 100–150 meV.3 It has been reported that the overall crystallinity4 and the crystal planes enclosing the Cu2O Mie resonators5 play a central role in controlling the optical properties,1 intrinsic carrier dynamics and therefore the photocatalytic activity.6Advanced wet-chemical synthetic methods enabled the preparation of Cu2O nanocrystals having various morphologies and sizes. It has been demonstrated that the photocatalytic activity shows strong facet-dependency, i.e. an enhanced activity increasing from {100} to {111} facets associated with cubic and octahedral morphologies, respectively, was reported.1 Moreover, surface energies,7 binding of solvent molecules (most importantly ethanol molecules)8,9 as well as the deposition of noble metals on the different crystal planes also show facet-dependent characteristics.7,10,11 Hence, tailoring the morphology of the nanoparticle has opened up new routes towards the synthesis and design of Cu2O-based multicomponent (photo)catalysts.
Although the photocatalytic activity of Cu2O nanoparticles with different morphologies has been investigated thoroughly in the last decade, recent studies implied that size, crystal facets and surface chemistry limit the utilization of the photogenerated carriers at the NPs’ surface. While the used Cu2O nanoparticles often have large dimensions (several hundreds of nanometres in diameter), the diffusion length of the minority carriers (electrons) was found to be less than 200 nm,12,13 which fact significantly shadows their advantageous application in reactions driven on their surfaces. Moreover, trapping of charge carriers plays a crucial role in the photophysical properties of Cu2O: defect states promote the ultrafast relaxation of excited electrons,14,15 hence bulk electrons are unable to reach the surface where the catalytic reaction would take place.16 A vast number of approaches have been implemented to facilitate the separation of electrons and holes in order to enhance their utilization in redox processes, such as preparation of Cu2O/metal heterostructures,17–22 forming heterojunctions with another semiconductor,23–27 metal–organic framework28 or doping.29
In particular, combining Cu2O and Au at the nanoscale was found to be a promising strategy to overcome the above-mentioned limitations rooting in the inefficient charge transport and the lack of charge carrier separation. In these combinations, gold has been introduced to the heterostructures mostly in the form of nanograins (NGs) or nanorods (NRs). A common finding for both types of Cu2O/Au coupled systems is that the charge carrier separation and the photocatalytic activity were found to be significantly enhanced compared to pristine Cu2O nanoparticles. On one hand, the plasmonic properties of gold can be utilized to inject hot electrons into Cu2O through the Schottky barrier formed at the metal/semiconductor interface. On the other hand, photoexcited electrons can be transferred to the Au domain from the conduction band of cuprous oxide. Considering the more advantageous properties of the octahedral morphology, synthetic methods have been explored to reduce the gold salt on the {111} plane,7,10,11,17,30 or overgrowing a CTAB-capped AuNR with an octahedral Cu2O shell in an aqueous medium.18,31–33 While for Cu2O octahedra decorated with small AuNGs (i.e. Cu2O@Au), the enhanced photocatalytic activity was attributed to the improved charge carrier separation, the main contribution of hot electrons was found to be the main reason of the higher activity for Cu2O containing an embedded Au nanorod (i.e. AuR@Cu2O structures).34 Since the hot electron injection from a relatively large AuNR (with a volume above 1000 nm3) has low efficacy,35 a strategy to alter the dynamics of intrinsic carriers generated solely in the semiconductor part would provide a novel platform to utilize low-cost UV LED illumination sources. This inspired us to avoid generating hot carriers and focus instead on the separation of the photoexcited carriers from the semiconductor point of view.
Around an excitation wavelength of 400 nm, the band-to-band transition of the Cu2O part plays the central role in the carrier generation processes in cuprous oxide/gold composites, as an AuNR (buried inside the Cu2O) or small AuNPs (located at the outer surface of the Cu2O) are off-resonant with the incident light. However, the location of gold fundamentally determines the spatial accumulation of the electrons and holes (generated in the Cu2O) at distinct parts of the particles. Furthermore, both the photoexcited electrons and holes seem to be of crucial importance in the degradation mechanism of dye molecules via producing O2− and OH radicals at the nanoparticle-solvent interface, respectively.36,37
These observations motivated us to reveal the impact of the nanoscale design on the photophysical properties of Cu2O in different Cu2O/Au heterostructures. Since both types of heterostructures (i.e. Cu2O@Au and AuR@Cu2O) have implied that the combination drastically improved the photocatalytic properties, understanding the effect of the position of the gold interacting with the octahedral Cu2O (via keeping all parameters identical) on the photophysical properties is of great importance in particular from the semiconductor point of view.
In this paper, we aim to enhance the charge carrier separation efficiency of pristine Cu2O nanoparticles enclosed by {111} facets to improve their photocatalytic activity. We report on the synthesis and optical characterization of two colloidal model systems constructed from Cu2O and gold in different arrangements: (i) single gold nanorods were overgrown with octahedral Cu2O shell, and as its inverse counterpart, (ii) the outer surface of Cu2O nanooctahedra were decorated with gold nanograins. To reveal the effect of the position of gold on the photophysical(/optical) and photocatalytic properties, we designed the heterostructured nanoparticles with uniform size, shape and composition (i.e. octahedral shape with 140 nm edge length and identical Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au atomic ratios). Thus, a comprehensive study on the contribution of the same amount (but different appearance) of gold could be carried out using pristine Cu2O nanooctahedra as a reference model system. To understand the optical response of such heteroparticles, optical simulations as well as single particle spectroscopy measurements have been performed. In contrast to the core–shell heterostructure where the gold NR is buried inside the semiconductor, the gold nanograin-decorated system generates more easily accessible charge carriers at the surface of the particles via using a low-cost UV LED source. Our study shows that the sufficient utilization of photoexcited carriers does not necessarily require a high-power (>300 W), broad spectrum Xe source, hence the presented strategy favours the application of Cu2O/Au nanosystems as photocatalysts.
:Au atomic ratios). Thus, a comprehensive study on the contribution of the same amount (but different appearance) of gold could be carried out using pristine Cu2O nanooctahedra as a reference model system. To understand the optical response of such heteroparticles, optical simulations as well as single particle spectroscopy measurements have been performed. In contrast to the core–shell heterostructure where the gold NR is buried inside the semiconductor, the gold nanograin-decorated system generates more easily accessible charge carriers at the surface of the particles via using a low-cost UV LED source. Our study shows that the sufficient utilization of photoexcited carriers does not necessarily require a high-power (>300 W), broad spectrum Xe source, hence the presented strategy favours the application of Cu2O/Au nanosystems as photocatalysts.
Results and discussion
Synthesis and characterization of Cu2O, Cu2O@Au and AuR@Cu2O nanoparticles
:Au ratio can be set in a wide range (Cu:Au atomic ratios from 10 to 50), at the same time retaining the octahedral morphology and ensuring the homogeneous distribution of Au nanograins. Further increase of the applied gold concentration significantly etched the nanooctahedra due to the high local concentration of HCl evolving during the reduction.30Fig. 1d–f show the Cu2O@Au particles being homogeneously decorated with AuNGs preserving the initial size and octahedral shape of the pristine Cu2O. The size of the nanograins was found to be 6.8 ± 1.4 nm. As a third model system, AuNRs were overgrown with Cu2O shells (referred to as AuR@Cu2O) in order to synthesize the counterpart of the Cu2O@Au particles but embedding the gold inside the octahedra. Each octahedron contains a single AuNR (78 × 26 nm in size) located in the middle of the octahedron parallel to the plane of the rectangular base (Fig. 1g–i). Homogeneous and crystalline Cu2O shell forms around the NR which ensures the direct contact between the metal and the semiconductor. Importantly, the shape and size were kept identical for all the three model systems, while the relative amount of gold was equal for Cu2O@Au and AuR@Cu2O nanoparticles (size distribution of the samples is shown in Fig. S2, ESI†). For heteroparticles, the Cu:Au atomic ratios were set to 39.3 ± 0.6.
 | ||
| Fig. 1 Morphology of the synthesized nanoparticles. SEM (a, d and g), TEM (b, e and h) and STEM-EDX elemental maps (c, f and i) of pristine Cu2O (a–c), Cu2O@Au (d–f) and AuR@Cu2O nanooctahedra (g–i). High magnification TEM image of a gold nanorod embedded in a Cu2O octahedron (h). | ||
Crystallinity and surface chemistry were investigated by means of X-ray diffractometry (XRD) and X-ray photoelectron spectroscopy (XPS). Fig. 2a proves that the reflection (corresponding to the planes parallel to the {111} facets facing the substrate surface) is the most dominant reflection in all samples (main peak at 2θ = 36.5°) and the diffraction peaks of the embedded AuNR can also be detected. The absence of the Au lines for Cu2O@Au can be attributed to the small grain size of the metal. XPS provides information about the oxidation state of Cu (Fig. 2b). The particle surface consists of Cu2O, and both the metallic Cu and the overoxidation of the Cu2O to CuO can be excluded. Although the observed peak at 932.4 eV alone is not sufficient to distinguish the Cu oxidation states, the accompanying features support the interpretation. The CuO would yield strong satellites at 940 eV and 943 eV which are absent in the spectra. Moreover, the weak satellite doublet at 944/947 eV is characteristic for Cu2O while metallic Cu has no satellites at all. Beside the dominant peak at 932.4 eV of Cu+, a slight contribution of Cu(OH)2 can be detected at 935.0 eV (Fig. S3, ESI†).39 A further confirmation of the presence of Cu2O phase is the calculated composition. The observed complex shape of oxygen 1s peak (Fig. 2c) includes contributions from: the Cu–O bond in the Cu2O lattice, Cu–OH, Si–O–Si (due to the substrate) and adsorbed ethanol/water, at binding energies 530.5 eV, 531.5 eV, 532.6 eV and 533.1 eV, respectively (Fig. 2c).40 The copper-bound oxygen and copper ratio is near to the stoichiometric 1:2 value. While the surface-attached gold NGs have strong signal in the Au4f regime, embedded AuNRs cannot be detected by the XPS (Fig. 2d), because the mean free path of photoelectrons limits the detection of components to a ca. 10 nm surface region and in this case the covering Cu2O shell was significantly thicker.
 | ||
| Fig. 2 XRD patterns (a) and XPS spectra of the nanoparticles (deposited onto Si substrate from ethanolic solutions) in the binding energy range of Cu2p (b), O1s (c) and Au4f (d). Beside the labeled peaks of Cu2O and Au in panel (a), the peak at 69.2° corresponds to the {400} facet of the applied silicon substrate with {100} orientation. | ||
Taking these considerations into account, after the synthesis, all particles were instantly centrifuged and washed thoroughly with ethanol–water (50:50 V/V%) mixtures to eliminate the unreacted compounds. Finally, the nanoparticles were concentrated and washed with absolute ethanol. These ethanolic stock solutions were stored in sealed glass vials to avoid the oxidation of the sample. Excellent morphological stability and extended shelf life (ambient conditions, daylight) were achieved for Cu2O and Cu2O@Au solutions: the ethanolic samples can be stored for up to 6 months, whilst the octahedral shape and the optical properties can be retained (Fig. S5, ESI†). However, the same storage conditions have deteriorated the morphology of AuR@Cu2O within a month even in ethanolic solutions (Fig. S6, ESI†). It implies that the intrinsic charge carrier separation occurring in these heteroparticles upon ambient conditions partially oxidizes the Cu2O to CuO and the integrity of the particles vanishes in a relatively short period of time. Upon storing the samples in dark and at low temperature (6 °C, fridge), however, the stability of the AuR@Cu2O particles can also be ensured for at least 2 months. Interestingly, for all types of nanoparticles, storing at ambient conditions in a dried form (i.e., deposited particle films) did not lead to the change in the particle morphology (Fig. S7, ESI†) or crystallinity (Fig. S8, ESI†). Consequently, ambient oxygen alone is not sufficient to oxidize the particles; residual water in the solutions might take a central role in the process. Based on these observations, all nanoparticle solutions were stored in ethanol and protected from ambient light in a fridge at 6 °C.
Optical and photophysical properties of the model systems
To reveal and compare the optical properties of the synthesized nanoparticle solutions, UV-Vis-NIR extinction, absorption and photoluminescence spectra were measured at identical Cu2O concentrations for all samples (0.5 mM). Pristine Cu2O nanooctahedra have a narrow extinction peak centered at 496 nm, which is the superposition of the band-to-band transition in Cu2O and the resonant Mie scattering of the particles (Fig. 3a). Upon decorating the octahedra with small AuNGs, the peak slightly redshifts and broadens due to the absorption of the gold. AuR@Cu2O particles have a complex optical response covering the VIS-NIR region upon utilizing the absorption of the AuNR (transversal, longitudinal plasmon modes), the Cu2O band edge as well as the interaction between Au and Cu2O (detailed discussion below). Eliminating the scattering allows the determination of pure absorption spectra (Fig. 3b), which show the main contribution of the Cu2O band-edge for all samples (below 455 nm), however, the absorption of the NR still remains prominent. From the absorption spectrum, the band gap of the pristine Cu2O was calculated to be 2.15 eV (Fig. S9, ESI†), which agrees well with the earlier reported band gap values for octahedra around 200 nm (tip-to-tip size).1 | ||
| Fig. 3 Extinction (a), absorption (b) and photoluminescence (c) spectra of the nanoparticle solutions in ethanol at identical Cu2O concentrations for each (0.5 mM). Fluorescence decay curves, the fitted biexponential functions and the internal response function (IRF) at the main PL peak ((d), 508 nm, λexc = 320 nm). | ||
To shed light on the photoluminescence properties of the samples, PL spectra as well as PL lifetime decays were measured (Fig. 3c and d) under UV excitation (λexc = 320 nm). At identical Cu2O concentrations, pristine Cu2O shows larger PL intensity, while the radiative recombination is partially suppressed in both Cu2O–Au heterostructures. Beside the main peak corresponding to the radiative recombination near the band edge (at 508 nm), a shoulder centered around 585 nm can be observed for all samples but with different contributions. This transition can be attributed to the vacancies in the Cu2O which act as trap states for the excited carriers. While the contribution of trap states to the total emission is 3.4% and 2.9% for Cu2O and Cu2O@Au, respectively, it increases to 10.0% for the AuR@Cu2O particles. This implies the existence of more crystal defects in the AuR@Cu2O nanooctahedra manifesting in strain and stress-induced lines in the shell (visualized by TEM in Fig. S10, ESI†). Fluorescence lifetime decay curves at the main PL peak have a biexponential nature, containing a fast and a somewhat slower component (Fig. 3d). Upon introducing the gold, the lifetime of the faster component of Cu2O (2.3 ns) further decreases (down to 1.8 ns and 1.6 ns for Cu2O@Au and AuR@Cu2O, respectively). This indicates a more pronounced charge carrier separation in the presence of Au. Moreover, the slower lifetime components show a similar trend (9.2 ns, 7.9 ns and 7.2 ns), however their contribution to the overall lifetime increases from 53.9% (Cu2O) to 64.0% and 80.0% for Cu2O@Au and AuR@Cu2O respectively. Even if an effective electron reservoir is present in the multicomponent nanoparticle, the exciton binding energy might be able to partially overcome the spatial charge carrier separation processes.
Thus, the separation of photoexcited carriers was enhanced due to the presence of the AuNGs and AuNR in the heteroparticles. Both the decreased PL intensity and the shorter lifetime indicate more efficient charge carrier separation for AuR@Cu2O, the increasing contribution of the defect states in the AuR@Cu2O system can play an important role in further suppressing the radiative recombination pathways. Larger concentration of defect states in AuR@Cu2O might be a consequence of the moderate lattice mismatch of Au and Cu2O42 inducing significant strain in the grown thick shell (Fig. S10, ESI†). However, the mismatch of 4.3% does not undermine the formation of a continuous, quasi-epitaxially grown shell in the vicinity of the nanorod.
Revealing the energy landscape of the nanoparticle systems
As previous studies pointed out, electronic properties of Cu2O show a high level of facet-dependency altering the light absorption, electrical conductivity as well as the photocatalytic activity of cuprous oxide nanoparticles.43 From the charge carrier separation point of view, the best choice is the octahedral shape, due to the outstandingly high electrical conductivity of {111} facets resembling that of a metal.44 Thus, photogenerated electrons do not suffer from a hindrance during their transfer towards a gold domain on the Cu2O surface. To untangle the differences in the charge carrier processes for our heterosystems, the relative work functions were determined by single-point Kelvin probe (SKP) measurements. This technique allowed for the determination of the multicomponent nanoparticles’ work function relative to that of the pristine Cu2O nanooctahedra: the work function differences were found to be −150 meV and +145 meV for Cu2O@Au and AuR@Cu2O particles, respectively (Fig. S11a, ESI†). Being in accordance with the SKP measurements, valence band spectra of the heteronanoparticle systems also indicate a prominent difference between the valence band maxima upon changing the position of the gold (around 430 meV, see Fig. S11b, ESI†). Considering that previous studies determined the work function of the {111} facet of Cu2O both via theoretical calculations on thin films (5.03 eV)45 as well as experimentally (4.80 eV),46 the observed effect can be attributed to the contact between the p-type Cu2O and the Au (in different forms). Additionally, heterojunctions containing gold show a size-dependent work function, which is a key factor in the equilibration of the Fermi level (5.3 eV for bulk Au,47 but 3.6–3.8 eV for small Au nanograins with the diameter of 7 nm on Si/SiO2 surfaces).48 Although the work function of AuNGs should decrease upon decreasing their size, at the same time mirror charges would increase it.49 Nevertheless, the contribution of the mirror charges in the NG size regime of 5–10 nm can be neglected,48 hence, it can be assumed that the work function of the Cu2O@Au system becomes smaller (upon contacting) than that of the bulk Au and the {111} facet of the Cu2O. This negative shift in the Fermi level was also reported for TiO2/Au heterojunctions, where the reduction of the Au domains’ size (from 8 to 3 nm) led to the decrease of the apparent work function and resulted in a higher degree of electron accumulation in the metal domains.50The work function differences of the contacting materials induce the formation of metal-semiconductor junctions with different barrier types upon changing the form and the position of the gold within the heteroparticles. Two main contributions are considered: (i) the equilibration of the Fermi levels upon contacting and (ii) the transfer of photoexcited charge carriers upon illumination. During the equilibration of the Fermi levels, electrons flow from Cu2O towards the AuNR and AuNGs due to the larger work function of Au. This process induces an upward band bending in the AuR@Cu2O system which promotes the accumulation of the photogenerated holes and the electrons in the nanorod and the semiconductor, respectively. In contrast, an opposite situation can be observed upon decorating the octahedra with small Au nanograins: after the equilibration of the Fermi levels, a Schottky barrier for the holes is formed, accumulating the holes in the Cu2O, and promoting the electron transfer towards the noble metal domains upon illumination. Based on the SKP measurements and the determined band gap for the Cu2O (Fig. S9, ESI†), energy landscapes are proposed in Fig. 4. This elucidates the different PL properties and stability of the heteroparticles: due to the p-type nature of the Cu2O, the driving force for the intrinsic charge carrier separation (hole transfer upon photoexcitation) is more pronounced if an AuNR is embedded in the Cu2O. This means that the observed particle instability at ambient conditions can be attributed to the extended spatial separation of the charge carriers within the octahedra for AuR@Cu2O heteroparticles. Contrary, Cu2O@Au nanoparticles facilitate the accumulation of both charge carriers at the nanocrystal-solution interface (e− in AuNGs, h+ at the Cu2O surface), which provides room for recombination and temporal relaxation if scavenging processes do not take place. This manifests also in the photoluminescence properties: Cu2O@Au heteroparticles exhibit higher PL intensity and slightly longer PL lifetime compared to their AuR@Cu2O counterparts. Thus, the smaller dimension of the spatial separation ensures the morphological stability of the particles and hinders the overoxidation of the Cu2O@Au heterooctahedra. According to this proposed carrier separation mechanism, the available charge carriers at the outer interface of the heteroparticles are mainly the electrons for AuR@Cu2O, but both holes and electrons for Cu2O@Au implying different photocatalytic activities. Table 1 summarizes the optical properties, crystallinity, and work function differences for the three model systems.
 | ||
| Fig. 4 Schematic contact situations between Cu2O and Au, where the form of the Au is different. Energy landscape of Au and Cu2O before (a and c) and after (b and d) the contact for AuR@Cu2O (a and b) and Cu2O@Au (c and d). Schematics on the right represents the accumulation of the charge carriers (e− and h+) upon illuminating the Cu2O. | ||
| Nanoparticle type | Extinction peak positions (nm) | PL maximum | τ 1 (ns) | τ 2 (ns) | τ average (ns) | Crystallite sizeb (nm) | Δφc (meV) |
|---|---|---|---|---|---|---|---|
|
a Amplitude-weighted average PL lifetime.
b Calculated from XRD measurements.
c Measured by single-point Kelvin probe.
d Set to zero mV and used as reference for the Au-containing heterosystems.
|
|||||||
| Cu2O | 496 | 508 | 2.3 | 9.3 | 4.7 | 26 | —d |
| Cu2O@Au | 503 | 508 | 1.8 | 7.9 | 4.2 | 23 | −150 |
| AuR@Cu2O | 567; 663; 739; 1199 | 508 | 1.6 | 7.2 | 4.1 | 30 | 145 |
Optical simulations and single-particle spectroscopy
The ensemble measurements have shown that combining the anisometric plasmonic metal with the semiconductor results in a complex optical response. To clarify the origin of the different spectral features optical simulations and correlative SEM/scattering measurements have been performed at the single particle level. For pristine Cu2O particles the red-shift of the calculated Mie-scattering peak with increasing particle size is reproduced in our system (Fig. S12a and b, ESI†),51 and a small refractive index dependency is also observed (Fig. S12c and d, ESI†). For gold@Cu2O core@shell structures with other geometries the simultaneous presence of the Mie and plasmon peaks has been demonstrated earlier.52,53 In our system, the peak around 1200 nm can be assigned to the longitudinal mode of the nanorod, whereas the split peak at around 700 nm is related to the transversal plasmon resonance of the rod and its interaction with Cu2O (Fig. 5). This split peak can be also observed in the experimental ensemble spectrum (Fig. 3a) and has been assigned earlier to the octupolar and dipolar resonances of the gold nanorod itself.54,55 It was explained by the presence of a distinct gap between the Cu2O and the NR surface, that – based on simulations – led to the experimentally observed splitting of the extinction peak. It has to be emphasized, however, that due to the different synthetic conditions no gap between the rod and is observed in the present work, hence one can rule out the interface gap as the origin of the observed spectral feature. It can be shown, that for the present AuR@Cu2O system, the hexapolar cavity mode of the Cu2O octahedron and the transversal dipole mode of the NR can couple to generate a binding dark mode (Fig. 5, also see Fig. S13 and S14 (ESI†) and associated text for details). This dark mode cannot be excited by the external electromagnetic field, leading to a Fano resonance dip in the experimentally measured extinction spectrum at around 650 nm. This feature can also be found in the experimental absorption spectrum as a positive peak due to the loss of scattering caused by the Fano resonance (Fig. 3b). It has to be emphasized that increasing the nanorod dimensions to 108 × 43 nm further amplifies the Fano resonance leading to a more pronounced dip and consequently a narrower subpeak at the blue side of the dipolar resonance of the rod (Fig. S15, ESI†). Owing to the nature of Fano resonance, it has a more pronounced impact on the scattering properties of the particles, hence we performed correlative SEM/single nanooctahedron scattering spectroscopy measurements to confirm its presence in the spectra. The correlative single particle scattering spectra shown in Fig. 6 are in-line with the experimental bulk spectrum as well as with the simulated optical responses. Single Cu2O and Cu2O@Au nanooctahedra exhibit a dominant scattering peak at around 480 nm, which is blue-shifted compared to the bulk spectra of ethanolic solutions due to the measurements being carried out in air. The presence of small AuNGs slightly broaden this resonance peak just like for the ensemble measurements. For AuR@Cu2O, the peak splitting at 670 nm can be clearly observed, supporting the findings of the optical simulations. The spectral position of this peak split remains unchanged even for dimers, trimers, or larger oligomers of AuR@Cu2O particles (Fig. S16, ESI†), consequently, it is not emerging due to octahedron–octahedron coupling.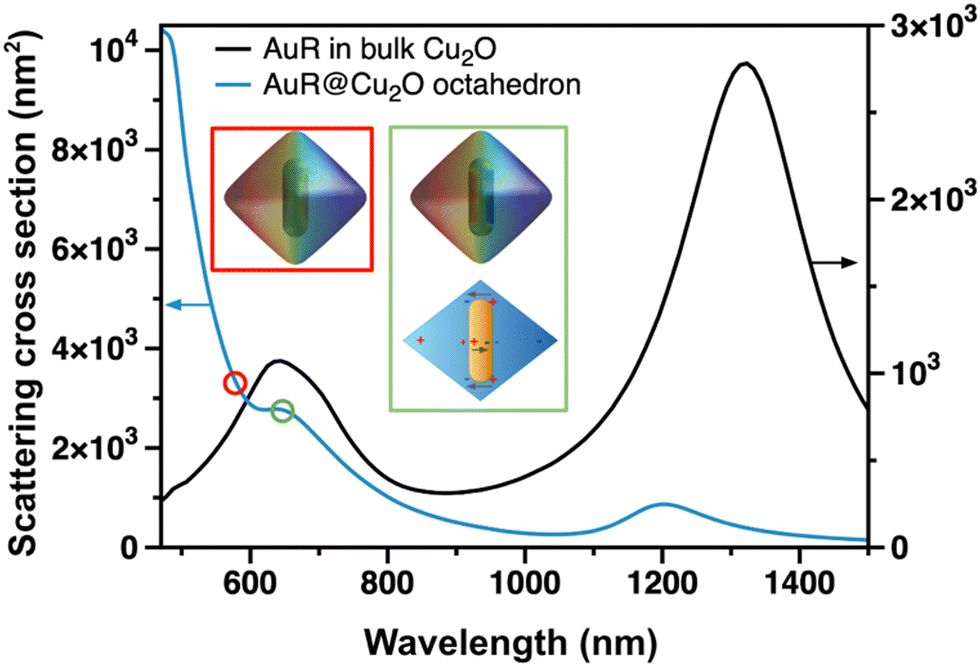 | ||
| Fig. 5 Simulation of scattering cross sections of AuNR in bulk Cu2O and an AuR@Cu2O octahedron. Insets show the surface charge distribution of AuR@Cu2O at 580 nm (red) and 650 nm (green) as well as the schematics of the emerging binding dark mode in AuR@Cu2O. | ||
 | ||
| Fig. 6 Typical correlative single particle scattering spectra of pristine Cu2O (a), Cu2O@Au (b) and AuR@Cu2O (c) on ITO substrate using correlative SEM/single particle spectroscopy. Insets show the SEM images of the measured particles (scale bars represent 100 nm). | ||
Photocatalytic activity and photostability
Photocatalytic activity of the synthesized nanoparticle solutions was investigated in a model reaction to reveal the effect of gold on the utilization of the photogenerated carriers. Fig. 7a summarizes the degradation of methyl orange dye in the absence and presence of the particles in aqueous medium. Importantly, the photodegradation of the dye was tested under UV illumination (λ = 396 nm) using a simple and cheap flashlight equipped with a UV LED. This excitation energy is off-resonant with the localized surface plasmon resonance (LSPR) of the gold (both the transversal and longitudinal modes of the rods as well as the LSPR of the nanograins), thus, it generates photoexcited carriers solely in the Cu2O. Consequently, the contribution of the hot electron injection can be ruled out. Methyl orange (MO) does not degrade upon illumination without the catalyst, nevertheless, pristine Cu2O octahedra were also found to be inactive (owing to the limited charge carrier transfer towards the interface). This inactivity can be attributed to the low power density of the UV flashlight since degradation of the dye can be observed for a higher power source in the presence of pristine octahedra. However, this led to the complete loss of the particle morphology and accompanied with the gradual photocorrosion of the sample (see Fig. S17, ESI† and the associated discussion for details). In contrast, introduction of gold dramatically increases the photodegradation efficiency of MO at low-power illumination: in the presence of AuR@Cu2O and Cu2O@Au particles, 68.2% and 82.2% of the MO degrades in the investigated time window, respectively. Although both multicomponent systems perform superior to the pristine Cu2O, the onset time of the degradation is around 100 minutes after the start of the illumination. To shed light on the origin of this observation, particle morphologies were monitored during the photocatalytic reaction via electron microscopy. While the morphology and Au-loading of Cu2O@Au particles remain intact upon illumination (Fig. 7b), AuR@Cu2O particles suffer from partial photocorrosion within the first 90 minutes which induces the degradation of some of the Cu2O shells and inner NRs appear partly exposed in the SEM images (Fig. 7c). This change in the morphology, however, takes place in the first 30 minutes and does not significantly change upon proceeding further illumination (supported by the change of the extinction spectra in Fig. S18, ESI†). It has to be noted that AuR@Cu2O particles show limited stability even in dark conditions in aqueous solution, while in ethanol, the particles remain intact even under UV illumination (Fig. S19, ESI†). | ||
| Fig. 7 Degradation kinetics of methyl orange in the absence or presence of the nanoparticles (a). Morphology of the particles after certain times of illumination (b and c). Scale bars represent 100 nm. | ||
These observations imply that another parallel process activates the particles and initiates the degradation of MO. It can be anticipated that the surface-attached ethanol plays a crucial role in the process: within the first 100 minutes, the photogenerated carriers might be consumed for the degradation of the ethanol molecules which hinder the surface attachment and the consequent degradation of the MO molecules in the initial phase. After this prephase, the degradation rate grows rapidly indicating that the surface of the particles becomes available for the dye molecules. This is also supported by the electrophoretic mobility of the particles being positive (+1.3 μm cm V−1 s−1) in aqueous media but becomes zero in ethanol, which suppresses the electrostatic attractive forces between the surface of the nanooctahedra and the dye molecules. The observed difference in the photocatalytic activity of the heterosystems can be attributed to the spatial separation of the photogenerated carriers dictated by the position of the gold.
This also agrees well with the findings based on the particle stability and energy landscape of the Cu2O/Au multicomponent systems enhancing the efficiency of the carrier extraction for Cu2O@Au nanoparticle design. Accumulation of both the holes and electrons at the particle interface improves the activity implying that the efficient degradation of the MO requires the presence of both carriers being spatially separated but directly available. It has to be emphasized that this enhancement in the photocatalytic activity was achieved via improving the separation of the carriers being generated solely in the semiconductor. This underlines that Cu2O combined with Au can be used as an efficient photocatalyst without generating hot electrons or utilizing any plasmonic effect of the noble metal.
Conclusions
The effect of the nanoscale design on the stability, photophysical and photocatalytic properties of Cu2O/Au heteronanoparticles was demonstrated experimentally and their optical response was revealed by optical simulations. While pristine Cu2O nanooctahedra were found to be practically inactive in photocatalysis, decorating the octahedra's facets with small Au nanograins drastically enhanced the activity. This can be attributed to the accumulation of both photoexcited carriers from the Cu2O on the nanoparticle interface, since, their spatial separation boosts the efficiency of their use in light-driven applications. Introducing the same amount of gold as a form of single nanorods embedded inside the octahedron also facilitates the spatial separation of the carriers, however, this arrangement results in the limited availability of the holes. This difference in the charge separation affects the temporal stability of the heteronanoparticles as well: balanced charges on the surface of Cu2O@Au particles endows the systems with superior shelf-life. It was also shown that residual water and ambient light play a crucial role in the reduced stability of the AuR@Cu2O system, thus, storing the samples in ethanol and at a low temperature (6 °C) prolongs the shelf-life of this structure as well. Optical simulations and single particle spectroscopy enabled the identification of a Fano resonance in the optical response of AuR@Cu2O particles in the visible wavelength range. The easy-to-adapt decoration approach makes the Cu2O@Au particles superior in all aspects. We highlighted the application potential of such multicomponent nanostructures by excluding the contribution of the hot-electron injection in the charge carrier separation process providing a new photocatalysis platform where a simple UV LED can be used to trigger reactions at the surface of the particles.Experimental section and methods
Chemicals
Hydrogen tetrachloroaurate (HAuCl4), cetyltrimethylammonium bromide (CTAB), sodium oleate (NaOL), silver nitrate (AgNO3), ascorbic acid (AA), sodium borohydride (NaBH4), copper(II) nitrate (Cu(NO3)2), sodium hydroxide (NaOH), hydrazine hydrate (N2H4 H2O), sodium dodecyl sulfate (SDS), ethanol, methyl orange (MO), ultrapure water. All glassware and stirring bars were cleaned with aqua regia (3:1 V/V ratio of cc. HCl and HNO3), rinsed thoroughly with ultrapure water, and dried at 70 °C for 6 hours. Quartz cuvettes were stored in 2 V/V% Hellmanex® II solutions, rinsed with ultrapure water and 2-propanol followed by drying under N2 flow.
Synthesis of octahedral Cu2O nanoparticles
Pristine Cu2O nanooctahedra were prepared based on Huang et al.,43 but the procedure has been fine-tuned and upscaled to enhance the particle quality and increase the octahedra concentration. The synthesis procedure was as follows: 1 mL of Cu(NO3)2 (0.1 M) solution was added to 91.8 mL of ultrapure water, followed by the addition of 200 μL of NaOH (1 M) solution under vigorous stirring 30 °C (using an aluminum heating block on a hot-plate). The colorless solution turned blue, indicating the formation of Cu(OH)2. Subsequently, 3 mL of freshly prepared hydrazine (0.2 M) solution was swiftly added to the mixture under highly intense stirring. The stirring speed was reduced after 1 minute, meanwhile the color of the blue solution turned into yellow-green, and then gradually to orange, indicating the nucleation and growth of the NPs. After 9 more minutes of gentle stirring, the as-synthesized particle solution was centrifuged (5000 rcf, 10 minutes, 23 °C), the supernatant was removed, and the precipitate was redispersed in a 1:1 mixture of absolute ethanol and ultrapure water. Finally, the product was centrifuged once more, and the precipitate was redispersed in 10 mL of absolute ethanol. The obtained particle solution (containing nanooctahedra with an edge length of 136 ± 12 nm) is stable and can be stored at ambient conditions for months (no spectral changes were observable after 8 months), as well as the process is greatly reproducible. It is to be emphasized that the reducing agent should be used within a minute to ensure the reproducibility of the narrow size distribution and good shape uniformity.
Synthesis of Cu2O@Au nanoparticles
The previously synthesized pristine Cu2O nanooctahedra were used as nucleation centers to fabricate gold nanograin-decorated Cu2O particles at room temperature. First, 2.5 mL of the ethanolic Cu2O nanoparticle dispersion was centrifuged (5600 rcf, 10 minutes) and the precipitate was redispersed in 25 mL of ultrapure water. Afterwards, 25 mL of HAuCl4 solution (0.0116 mM) was added swiftly under highly intense stirring. The stirring bar was removed after 1 minute, and the mixture was kept undisturbed for 19 minutes. Finally, the mixture was centrifuged (5000 rcf, 10 minutes), the precipitate was washed twice as above and redispersed in 5 mL of absolute ethanol. The product was stored in a fridge (6 °C, dark). Under these conditions, the shelf life of the ethanolic solution is at least 6 months.Synthesis of AuNRs
Gold nanorods with (78.3 ± 6.1) nm length and (25.7 ± 1.8) nm width were synthesized according to the seed-mediated protocol of Ye et al. using binary surfactant mixtures applying slight modification.56 To this end, a seed-particle solution and a growth solution were prepared simultaneously.For the seed-particle solution, 5 mL of CTAB (0.2 M) and 5 mL of HAuCl4 (0.5 mM) solution were mixed. Under highly intense stirring, 1 mL of ice-cold, freshly prepared NaBH4 (6 mM) solution was injected in the solution. After 2 minutes of vigorous stirring, the stirring bar was removed, and the solution was kept undisturbed for 30 minutes.
For the growth solution, 1.8 g of CTAB and 0.3086 g of NaOL were completely dissolved in 50 mL of ultrapure water along with a vigorous stirring, assisted with heating the mixture up to 50 °C. After the solution was let cool down to 30 °C, 2.0 mL of AgNO3 (4 mM) solution was introduced and kept the solution undisturbed. Then, during high-speed stirring, 50 mL of HAuCl4 solution (1 mM) was added. After 90 minutes, 420 μL of cc. HCl was injected into the solution. After another 15 minutes of gentle stirring, 250 μL of ascorbic acid (0.064 M) was injected into the solution, followed by the injection of 80 μL of the seed-particle solution after 30 seconds of vigorous stirring. Subsequently, the stirring bar was removed, and the mixture was left undisturbed overnight in a 28 °C bath. For purification, the as-synthesized mixture was centrifuged twice at 5000 rcf for 20 minutes. The supernatant was removed, and the precipitate was redispersed in 50 mL of CTAB (50 mM) solution.
Synthesis of AuR@Cu2O nanoparticles
Core–shell heterostructured nanoparticles with the same dimensions (137 ± 11 nm edge length) as those of the pristine Cu2O NPs were synthesized by growing an octahedral Cu2O shell over the previously synthesized AuNRs at room temperature, based on a procedure reported by Kong et al.31 First, to suppress the CTAB concentration and set an appropriate particle concentration (cAu(0) = 0.6257 mM), the AuNR solution was centrifuged twice, and redispersed in ultrapure water. Next, 17.5 mL of this AuNR seed particle solution was introduced to a growth solution containing 0.0755 g of Cu(NO3)2·3H2O, 0.9013 g of SDS and 25 mL of ultrapure water. Subsequently, 6.25 mL of NaOH solution (2 M) was added to the mixture under vigorous stirring. After 15 minutes, 6.375 mL of N2H4 solution (0.4 M) was added dropwise (ca. 1 droplet per s) over ca. 3 minutes. The ruby-red colour of the mixture gradually changed to dark green, indicating the formation of Cu2O shell. After 40 minutes of gentle stirring, the product was centrifuged (5000 rcf, 10 minutes), and the precipitate was dispersed in a 1:1 mixture of absolute ethanol and ultrapure water. For purification, this step was twice more repeated. Finally, the product was centrifuged once more, the precipitate was redispersed in 5 mL of absolute ethanol, and the product was stored in a refrigerator.
Although the product was not as stable as the pristine and the AuNG decorated Cu2O nanooctahedra, refrigerated storage (excluding the light and providing low temperature) was found to temporarily preserve the particle morphology (i.e., preventing the oxidation of Cu2O to CuO).
Optical characterization
Ethanolic stock solutions of the nanoparticles were diluted with absolute ethanol to reach identical Cu2O concentration (0.5 mM) for each model system. Semi-micro quartz cuvettes were filled with 1–1 mL of these solutions. Extinction spectra were recorded in a Shimadzu UV-3600i UV-Vis-NIR spectrophotometer. Photoluminescence (PL) measurements (emission and excitation spectra) were performed with an Edinburgh FS5 spectrofluorometer ((λexc = 320 nm for emission spectra). Time-correlated single photon counting was carried out using the EPLED-320 (λexc = 320 nm) pulsed source in the Edinburgh FS5. The integrating sphere (SC-30 cassette) was used to record the absorption spectra of the nanoparticle solutions to eliminate the scattering.Electron microscopy
Nanoparticles were deposited from their ethanolic stock solutions onto Si wafers or carbon-coated Cu grids for SEM and TEM investigations, respectively (via drop casting and ambient drying). SEM images were recorded with a Zeiss LEO 1540-XB at an accelerating voltage of 5 keV. Elemental composition of the samples was determined by energy dispersive X-ray spectroscopy using an Oxford Instruments UltimMax 40 detector. TEM, STEM images and STEM-EDX maps were recorded by a ThermoFischer THEMIS 200 spherical aberration corrected transmission electron microscope at 200 keV. EDX data was recorded using a Super-X detector system.XRD and XPS sample preparation and measurements
Ethanolic stock solutions of the nanoparticles were concentrated with a factor of 20 and drop-cast onto Si wafers followed by ambient drying (complete drying occurred within 5 minutes). These samples were transferred into an N2-filled glovebox (cO2 = 1.0–2.0 ppm, cH2O = 0.1–0.2 ppm) to avoid their unwanted overoxidation until they had been measured. XRD measurements were carried out with a Bruker AXS D8 Discover X-ray diffractometer equipped with Göbel-mirror and scintillation detector using Cu Kα (λ = 1.5406 Å) radiation. The X-ray beam dimensions were 1 mm × 5 mm, the 2Θ step size was 0.02°, scan speed 0.2° min−1. XPS measurements were carried out with a ThermoFischer Escalab Xi+ instrument. Optical image as well as XPS identification supported the selection of areas with high particle coverage. Nevertheless, Si substrate was detected beside of Cu2O octahedra (and Au) signal. XPS spectra were measured using Al Kα source focused onto area with 0.5 mm in diameter. Spectra were recorded with 0.5 eV energy resolution at 0.1 eV step size with 0.5 s per point speed. Since applying the built-in charge compensation, the observed spectra showed minimal energy shift from the nominal values. Although carbon contamination was perceived in the samples, the slight organic coverage resulted in no deterioration of the spectra. Binding energy scale was adjusted using the adventitious carbon peak fixed at 284.6 eV.Single-point Kelvin probe
Same samples as for XRD and XPS were used for the SKP measurements to investigate the work function, using the Non-Scanning Ambient Kelvin Probe KP 020 (KP Technology Ltd) equipped with a sample positioning system developed by Plósz Engineering Co. Ltd (Hungary). The measurements were carried out at room temperature with a 2 mm tip at 3 different spots on the samples 40 times each.Total-reflection X-ray fluorescence spectroscopy measurements
For determining the Cu concentration of the samples, a compact low-power TXRF configuration was applied comprising a 50 W Mo-anode X-ray tube (operated at 50 kV, 1 mA), a multilayer monochromator (to select Mo-Kα X-ray as excitation source) and a modified WOBI-module attachment. A 50 mm2 silicon drift detector (SDD) (AXAS-D 2.0, Ketek, Germany) was applied to record the X-ray spectra of the samples at ambient conditions (acquisition time was 1800 s). The X-ray spectra were processed with PyMCA software packages.57 50 μL ethanolic stock solution of the nanoparticles, 50 μL HNO3 solution (67% ultrapure), 50 μL Y (as internal standard) (100 ppm) solution were added to 4.85 mL ultrapure water. 5 μL from this solution was drop-cast on a quartz reflector and dried at ambient conditions. Calibration was carried out in the presence of 1 ppm Y on 6 points (0.1, 0.4, 0.6, 1.0, 4.0, 6.0 and 10 ppm) for Cu.Optical simulations
Boundary element method (MNPBEM) was used to calculate the extinction and scattering cross sections as well as the near field plots of the nanoparticles in MATLAB environment.58 The dielectric functions of Cu2O and gold have been taken from the literature.51,59Single particle spectroscopy
ITO-coated glass slides (PGO glass, CES120X) were spin-coated with pristine Cu2O and Cu2O/Au heteroparticles from ethanolic solutions followed by drying under N2 flow. Single particle scattering spectra were taken in an Olympus BX51 upright optical microscope equipped with an XYZ piezo stage (Physik Instrumente, P-545.3R8S) in epi dark field illumination (100 W – Olympus U-LH100IR). The scattered light from the single nanoparticles was collected with a 50X dark field objective (NA = 0.8) and projected on the entrance slit of the imaging spectrograph (Princeton Instruments Isoplane SCT320) equipped with a cooled CCD camera (PIXIS:400BRX). Automatic positioning and autofocusing of the selected individual scatterer in the region of interest were controlled by our lab-built LabView software. The optical transfer function of the system has been corrected by using a dedicated intensity calibration lamp (Princeton Instruments IntelliCal LSVN0518).Electrophoretic mobility
A Malvern ZetaSizer Nano ZS was used to perform dynamic light scattering (λ = 635 nm) measurements on the diluted ethanolic solutions of the particles. Electrophoretic mobilities were determined in folded capillary cells (DTS-1070).Photocatalytic tests
Test reactions were performed in a 25 mL borosilicate beaker placed on a magnetic stirrer. The beaker was covered with a quartz slip, on which the UV flashlight was placed. The UV source was equipped with an Edixeon S series UV LED (λ = 396 nm). The power density of the source was 38 ± 2 mW cm−2. The total volume of the aqueous solution was 25 mL in each case containing 10 mg L−1 Cu2O (from the ethanolic stock solution) and 1.5 mg L−1 MO (from a 100 mg mL−1 aqueous stock). The distance between the LED and the top of the solution was 20 mm. During sampling, 1 mL solution was pipetted into an Eppendorf tube (1.7 mL) and centrifuged quickly at 14100 rcf for 2 minutes. The supernatant was transferred into a PMMA disposable cuvette, and the absorbance spectrum was measured with a fibre-coupled spectrophotometer (Thorlabs CCS200).
Author contributions
D. K.: conceptualization (supporting), data curation (equal), formal analysis (equal), investigation (equal), methodology (supporting), visualization (equal), writing – original draft (equal), writing – review & editing (supporting). A. D.: formal analysis (supporting), funding acquisition (supporting), investigation (supporting), resources (supporting), visualization (supporting), writing – original draft (supporting), writing – review & editing (supporting). R. Gy. Z.: investigation (supporting), visualization (supporting), writing – review & editing (supporting). Zs. E. H.: investigation (supporting), visualization (supporting), writing – review & editing (supporting). A. S.: investigation (supporting), visualization (supporting), writing – review & editing (supporting). R. S.: investigation (supporting), visualization (supporting), writing – review & editing (supporting). O. C.: investigation (supporting), visualization (supporting), writing – review & editing (supporting). D. Z.: conceptualization (lead), data curation (equal), formal analysis (equal), funding acquisition (lead), investigation (equal), methodology (lead), project administration (lead), resources (lead), supervision (lead), visualization (equal), writing – original draft (equal), writing – review & editing (lead).Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. K. acknowledges the support of Pro Progressio and József Varga Foundation. Project no. TKP2021-NKTA-05 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund (NRDI), financed under the TKP2021 funding scheme. Moreover, the work was supported by the NRDI Fund of Hungary under the grants of FK 142148 and FK 128327.References
- M. H. Huang, Small, 2019, 15, 1804726 CrossRef PubMed.
- S. Sun, X. Zhang, Q. Yang, S. Liang, X. Zhang and Z. Yang, Prog. Mater. Sci., 2018, 96, 111–173 CrossRef CAS.
- S. Steinhauer, M. A. M. Versteegh, S. Gyger, A. W. Elshaari, B. Kunert, A. Mysyrowicz and V. Zwiller, Commun. Mater., 2020, 1, 11 CrossRef.
- J. Li, Z. Mei, D. Ye, H. Liang, L. Liu, Y. Liu, A. Galeckas, A. Y. Kuznetsov and X. Du, Opt. Mater. Express, 2013, 3, 2072 CrossRef.
- H. Sugimoto and M. Fujii, Adv. Photonics Res., 2021, 2, 2000111 CrossRef CAS.
- C. Y. Toe, M. Lamers, T. Dittrich, H. A. Tahini, S. C. Smith, J. Scott, R. Amal, R. van de Krol, F. F. Abdi and Y. H. Ng, Mater. Adv., 2022, 3, 2200–2212 RSC.
- H. Zhu, M. Du, D. Yu, Y. Wang, M. Zou, C. Xu and Y. Fu, Dalton Trans., 2012, 41, 13795 RSC.
- K. Marks, Z. Besharat, M. Soldemo, A. Önsten, J. Weissenrieder, J. H. Stenlid, H. Öström and M. Göthelid, J. Phys. Chem. C, 2019, 123, 20384–20392 CrossRef CAS.
- S. Sun, H. You, C. Kong, X. Song, B. Ding and Z. Yang, CrystEngComm, 2011, 13, 2837 RSC.
- H. Zhu, M. Du, D. Yu, Y. Wang, L. Wang, M. Zou, M. Zhang and Y. Fu, J. Mater. Chem. A, 2013, 1, 919–929 RSC.
- X.-W. Liu, Langmuir, 2011, 27, 9100–9104 CrossRef CAS PubMed.
- J. Luo, L. Steier, M.-K. Son, M. Schreier, M. T. Mayer and M. Grätzel, Nano Lett., 2016, 16, 1848–1857 CrossRef CAS PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- L. Grad, Z. Novotny, M. Hengsberger and J. Osterwalder, Sci. Rep., 2020, 10, 10686 CrossRef CAS PubMed.
- S. Dolai, S. Das, S. Hussain, R. Bhar and A. K. Pal, Vacuum, 2017, 141, 296–306 Search PubMed.
- M. Borgwardt, S. T. Omelchenko, M. Favaro, P. Plate, C. Höhn, D. Abou-Ras, K. Schwarzburg, R. van de Krol, H. A. Atwater, N. S. Lewis, R. Eichberger and D. Friedrich, Nat. Commun., 2019, 10, 2106 Search PubMed.
- T. N. Saada, L. Pang, K. Sravan Kumar, A. H. B. Dourado, L. D. Germano, E. D. Vicentini, A. P. L. Batista, A. G. S. de Oliveira-Filho, F. Dumeignil, S. Paul, R. Wojcieszak, S. Melinte, G. Sandu, G. Petretto, G.-M. Rignanese, A. H. Braga, T. F. Rosado, D. Meziane, R. Boukherroub, S. I. C. de Torresi, A. G. M. da Silva and S. Szunerits, Electrochim. Acta, 2021, 390, 138810 Search PubMed.
- L. Guo, Z. Mao, C. Ma, J. Wu, L. Zhu, B. Zhao and Y. M. Jung, ACS Appl. Nano Mater., 2021, 4, 381–388 CrossRef CAS.
- H. Jia, F. Li, T. H. Chow, X. Liu, H. Zhang, Y. Lu, J. Wang and C. Zhang, Nano Lett., 2022, 22, 7268–7274 CrossRef CAS PubMed.
- P. Liu, Z. Cheng, L. Ma, M. Zhang, Y. Qiu, M. Chen and F. Cheng, RSC Adv., 2016, 6, 76684–76690 RSC.
- L. Wang, J. Ge, A. Wang, M. Deng, X. Wang, S. Bai, R. Li, J. Jiang, Q. Zhang, Y. Luo and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 5107–5111 CrossRef CAS PubMed.
- A. Jiao, Q. Cui, S. Li, Y. Tian, H. Ma, C. Wang, M. Zhang, M. Chen, G. Li and X. Liu, Opt. Express, 2022, 30, 588 CrossRef CAS PubMed.
- S.-C. Wu, C.-S. Tan and M. H. Huang, Adv. Funct. Mater., 2017, 27, 1604635 CrossRef.
- S. Raheman AR, B. M. Momin, H. M. Wilson, U. S. Annapure and N. Jha, J. Alloys Compd., 2020, 835, 155262 CrossRef CAS.
- B. Xing, T. Zhang, Q. Han, Q. Wei and D. Wu, Microchim. Acta, 2019, 186, 505 CrossRef PubMed.
- X. Wei, J. Pan, S. Wang, J. Mei, Y. Zheng, C. Cui and C. Li, J. Mater. Sci.: Mater. Electron., 2017, 28, 14079–14084 CrossRef CAS.
- Y. Zhu, Z. Xu, K. Yan, H. Zhao and J. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 40452–40460 CrossRef CAS PubMed.
- Y. Wu, Y. Li, H. Li, H. Guo, Q. Yang and X. Li, Sep. Purif. Technol., 2022, 303, 122106 CrossRef CAS.
- S. Shyamal, P. Hajra, H. Mandal, A. Bera, D. Sariket, A. K. Satpati, M. V. Malashchonak, A. V. Mazanik, O. V. Korolik, A. I. Kulak, E. V. Skorb, A. Maity, E. A. Streltsov and C. Bhattacharya, Chem. Eng. J., 2018, 335, 676–684 CrossRef CAS.
- R. Chen, J. Lu, S. Liu, M. Zheng and Z. Wang, J. Mater. Sci., 2018, 53, 1781–1790 CrossRef CAS.
- L. Kong, W. Chen, D. Ma, Y. Yang, S. Liu and S. Huang, J. Mater. Chem., 2012, 22, 719–724 RSC.
- G. Yuan, M. Lu, J. Fei, J. Guo and Z. Wang, RSC Adv., 2015, 5, 71559–71564 RSC.
- H.-J. Wang, K.-H. Yang, S.-C. Hsu and M. H. Huang, Nanoscale, 2016, 8, 965–972 RSC.
- X. Yu, X. Liu, B. Wang, Q. Meng, S. Sun, Y. Tang and K. Zhao, Nanoscale, 2020, 12, 1912–1920 RSC.
- C. S. Kumarasinghe, M. Premaratne, Q. Bao and G. P. Agrawal, Sci. Rep., 2015, 5, 12140 CrossRef CAS PubMed.
- V. K. Mrunal, A. K. Vishnu, N. Momin and J. Manjanna, Environ. Nanotechnol., Monit. Manage., 2019, 12, 100265 Search PubMed.
- Y. Su, A. Nathan, H. Ma and H. Wang, RSC Adv., 2016, 6, 78181–78186 RSC.
- H. Xu, W. Wang and W. Zhu, J. Phys. Chem. B, 2006, 110, 13829–13834 CrossRef CAS PubMed.
- Z. Dan, Y. Yang, F. Qin, H. Wang and H. Chang, Materials, 2018, 11, 446 CrossRef PubMed.
- J. Han, J. Chang, R. Wei, X. Ning, J. Li, Z. Li, H. Guo and Y. Yang, Int. J. Hydrogen Energy, 2018, 43, 13764–13777 CrossRef CAS.
- S. Zhu, D. Deng, M. T. Nguyen, Y. R. Chau, C.-Y. Wen and T. Yonezawa, Langmuir, 2020, 36, 3386–3392 CrossRef CAS PubMed.
- C. D. Sai and A. B. Ngac, Phys. B, 2018, 532, 216–220 CrossRef CAS.
- J.-Y. Huang, M. Madasu and M. H. Huang, J. Phys. Chem. C, 2018, 122, 13027–13033 CrossRef CAS.
- M. H. Huang and G. Kumar, J. Chin. Chem. Soc., 2022, 69, 1190–1199 CrossRef CAS.
- F. Chiter, D. Costa, V. Maurice and P. Marcus, J. Phys. Chem. C, 2020, 124, 17048–17057 CrossRef CAS.
- C.-S. Tan, S.-C. Hsu, W.-H. Ke, L.-J. Chen and M. H. Huang, Nano Lett., 2015, 15, 2155–2160 CrossRef CAS PubMed.
- G. N. Derry, M. E. Kern and E. H. Worth, J. Vac. Sci. Technol., A, 2015, 33, 060801 CrossRef.
- Y. Zhang, O. Pluchery, L. Caillard, A.-F. Lamic-Humblot, S. Casale, Y. J. Chabal and M. Salmeron, Nano Lett., 2015, 15, 51–55 CrossRef CAS PubMed.
- D. M. Wood, Phys. Rev. Lett., 1981, 46, 749 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- S. Zhang, R. Jiang, Y.-M. Xie, Q. Ruan, B. Yang, J. Wang and H.-Q. Lin, Adv. Mater., 2015, 27, 7432–7439 CrossRef CAS PubMed.
- Y.-C. Yang, H.-J. Wang, J. Whang, J.-S. Huang, L.-M. Lyu, P.-H. Lin, S. Gwo and M. H. Huang, Nanoscale, 2014, 6, 4316 RSC.
- J. Zhu, N. Lu, W. Chen, L. Kong, Y. Yang, D. Ma and S. Huang, J. Nanomater., 2015, 2015, 1–9 Search PubMed.
- X. Shi, Y. Ji, S. Hou, W. Liu, H. Zhang, T. Wen, J. Yan, M. Song, Z. Hu and X. Wu, Langmuir, 2015, 31, 1537–1546 CrossRef CAS PubMed.
- S. Zhang, R. Jiang, Y. Guo, B. Yang, X.-L. Chen, J. Wang and Y. Zhao, Small, 2016, 12, 4264–4276 CrossRef CAS PubMed.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS PubMed.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62, 63–68 CrossRef.
- J. Waxenegger, A. Trügler and U. Hohenester, Comput. Phys. Commun., 2015, 193, 138–150 CrossRef CAS.
- R. L. Olmon, B. Slovick, T. W. Johnson, D. Shelton, S.-H. Oh, G. D. Boreman and M. B. Raschke, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 235147 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc01213a |
| This journal is © The Royal Society of Chemistry 2023 |