
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The challenge with high permittivity acceptors in organic solar cells: a case study with Y-series derivatives†
Peter
Fürk
 a,
Suman
Mallick
a,
Thomas
Rath
*ab,
Matiss
Reinfelds
a,
Mingjian
Wu
c,
Erdmann
Spiecker
c,
Nikola
Simic
d,
Georg
Haberfehlner
d,
Gerald
Kothleitner
d,
Barbara
Ressel
e,
Sarah
Holler
a,
Jana B.
Schaubeder
f,
Philipp
Materna
g,
Heinz
Amenitsch
g and
Gregor
Trimmel
*a
a,
Suman
Mallick
a,
Thomas
Rath
*ab,
Matiss
Reinfelds
a,
Mingjian
Wu
c,
Erdmann
Spiecker
c,
Nikola
Simic
d,
Georg
Haberfehlner
d,
Gerald
Kothleitner
d,
Barbara
Ressel
e,
Sarah
Holler
a,
Jana B.
Schaubeder
f,
Philipp
Materna
g,
Heinz
Amenitsch
g and
Gregor
Trimmel
*a
aInstitute for Chemistry and Technology of Materials (ICTM), NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at; gregor.trimmel@tugraz.at
bJoanneum Research, MATERIALS-Institute for Sensors, Photonics and Manufacturing Technologies, Franz-Pichler Straße 30, 8160 Weiz, Austria
cInstitute of Micro- and Nanostructure Research & Center for Nanoanalysis and Electron Microscopy (CENEM), Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstraße 3, 91058 Erlangen, Germany
dInstitute of Electron Microscopy and Nanoanalysis, Graz University of Technology, Steyrergasse 17, 8010 Graz, Austria
eLaboratory of Quantum Optics, University of Nova Gorica, Vipavska Cesta 11c, 5270 Ajdovščina, Slovenia
fInstitute of Bioproducts and Paper Technology (BPTI), Graz University of Technology, Inffeldgasse 23, 8010 Graz, Austria
gInstitute of Inorganic Chemistry, NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
First published on 26th May 2023
Abstract
Y-series acceptors have brought a paradigm shift in terms of power conversion efficiencies of organic solar cells in the last few years. Despite their high performance, these acceptors still exhibit substantial energy loss, stemming from their low-permittivity nature. To tackle the energy loss situation, we prepared modified Y-series acceptors with improved permittivities via an alternative synthetic route. The route yields a thienothiophene block, which can be easily functionalized further on. That way, two new acceptors (Y-thio-EG and Y-pyr-EG) were synthesized with permittivity-improving glycol side chains in the central pyrrol and outer thiophene substitution site. Both acceptors exhibit a significantly increased relative permittivity (4.73 and 5.24, respectively), compared to Y6 (2.39). When employed in binary bulk heterojunction solar cells with PM6 as polymer, however, both acceptors only reach efficiencies of 8.2% (Y-thio-EG) and 5.3% (Y-pyr-EG). We show by extensive morphology investigation (atomic force microscopy, transmission electron microscopy and X-ray scattering) that the glycol modification and side chain positioning have a strong influence on the crystallization behaviour of these acceptors. A thorough comparison of the acceptors blended with PM6 to PM6:Y6 absorber layers showed that differences in crystallinity and aggregation behaviour are present. However, the degree of the morphological change is likely not the main reason for the large efficiency drops of the solar cells. We hypothesize that the combination of an apolar donor with polar, glycol-modified acceptors yields a much more complex situation in the bulk heterojunction absorber layer, where further effects – such as interface dipole formation, energy level broadening and increased interface-state assisted recombination – are also expected to be highly relevant. Eventually we propose strategies to circumvent these issues in future permittivity modification research.
Introduction
Efficiencies of organic solar cells (OSCs) have increased steadily within the last decades. This quick improvement is in large part due to the continuous synthesis of better-performing conjugated polymers and non-fullerene acceptors.1–3 In terms of the non-fullerene acceptors, Y-series acceptors (YSAs) are the current record-holders. Introduced first in 2019 by the Zou group,4 YSAs have steadily pushed power conversion efficiencies (PCEs) of organic solar cells to record values of up to 19.6% in 2022.5–7Despite their high performance, YSAs, like any other organic semiconductor, still show larger energy loss in OSCs than their inorganic and perovskite counterpart. Typical OSC materials exhibit exciton binding energies of 0.5 eV or larger,8 with an overall energy loss in the range of 0.7 to 1 eV.9–11 A way to greatly reduce the energy loss is to increase the dielectric permittivity of the active layer materials, which would in theory enable OSCs with PCEs beyond the 20% frontier.12,13
To improve the permittivity, different approaches were tried in literature,14 such as adding high-permittivity additives,15–17 expanding the molecular π-system18 or adding polarizable atoms/groups.19 A further effective method is the introduction of polar, flexible side chains, which increase a molecules permittivity by enhancing its polarizability.20–22 This strategy was already used in former work of our research group by modifying perylene monoimide based acceptors with glycol23 and sulfone-ether24 side chains. These modifications resulted in increased relative permittivities (εr) by approx. 15% (from 3.1–3.2 to 3.5–3.7) for the glycol containing acceptors and by 40% to 60% (from 1.9 to 2.8–2.9) for the sulfone containing acceptors. Moreover, Liu et al. modified the popular acceptor ITIC with glycol chains and obtained a significant increase of εr from 4.5 to 7.5–9.5 for the ITIC-OEG acceptor material.22 With the active layer PBDB-T:ITIC-OEG they achieved a PCE of 8.4%, which was slightly decreased compared to the reference PBDB-T:ITIC with a PCE of 10.4%.
The same strategy has also been applied to YSAs. Li et al. have attached a linear glycol chain to the nitrogen atom of the pyrrol in the acceptor core, which increased εr of the acceptor Y6-4O from 3.36 to 5.13.25 That way they achieved a PCE of 15% based on Y6-4O processed from toluene with the donor PM6. Chen et al. introduced two glycol chains to the pyrrol nitrogen position to prepare the symmetric Y-series acceptor BTO.26 Combining BTO with PM6, they achieved a PCE of 11% when processing the active layer from chlorobenzene. When changing to a ternary system with Y6 and 20 wt% of BTO as additive, the PCE was improved to 16%.
Thus, glycol substitution at the center position of YSAs already proved to be an attractive method to yield well-performing OSCs. However, adding linear glycols at the center may be less ideal because it removes the critical branching point, which is known to greatly control the stacking behavior of the molecules.27 Consequently, we rationalized that attaching the glycol chains at the outer thiophene position could be advantageous to the material and solar cell properties. This outer position is, however, more tedious and time-consuming to substitute when following the original synthetic route.4
In this work, we apply an alternative synthetic route towards YSAs bearing OEG side chains, which simplifies the side chain attachment at the outer position while still using inexpensive starting materials. We synthesized two new acceptors (Scheme 1) with permittivity-improving glycol side chains on different positions. The Y-thio-EG contains EG side chains linked via a phenylene unit to the terminal thiophene. Additionally, we prepared the second acceptor Y-pyr-EG with the EG chains on the central pyrrol nitrogen atoms, and alkyl chains on the terminal thiophene. The comparison of both acceptors should shed light on the impact of the EG chain position and the presence of branching points on the photovoltaic performance. We then examined the acceptors’ photovoltaic efficiency in OSCs, combined with a thorough investigation of their morphological behaviour as neat material and in blends with the popular donor polymer PM6, to estimate the magnitude of the glycol chain influence on the solar cell performance.
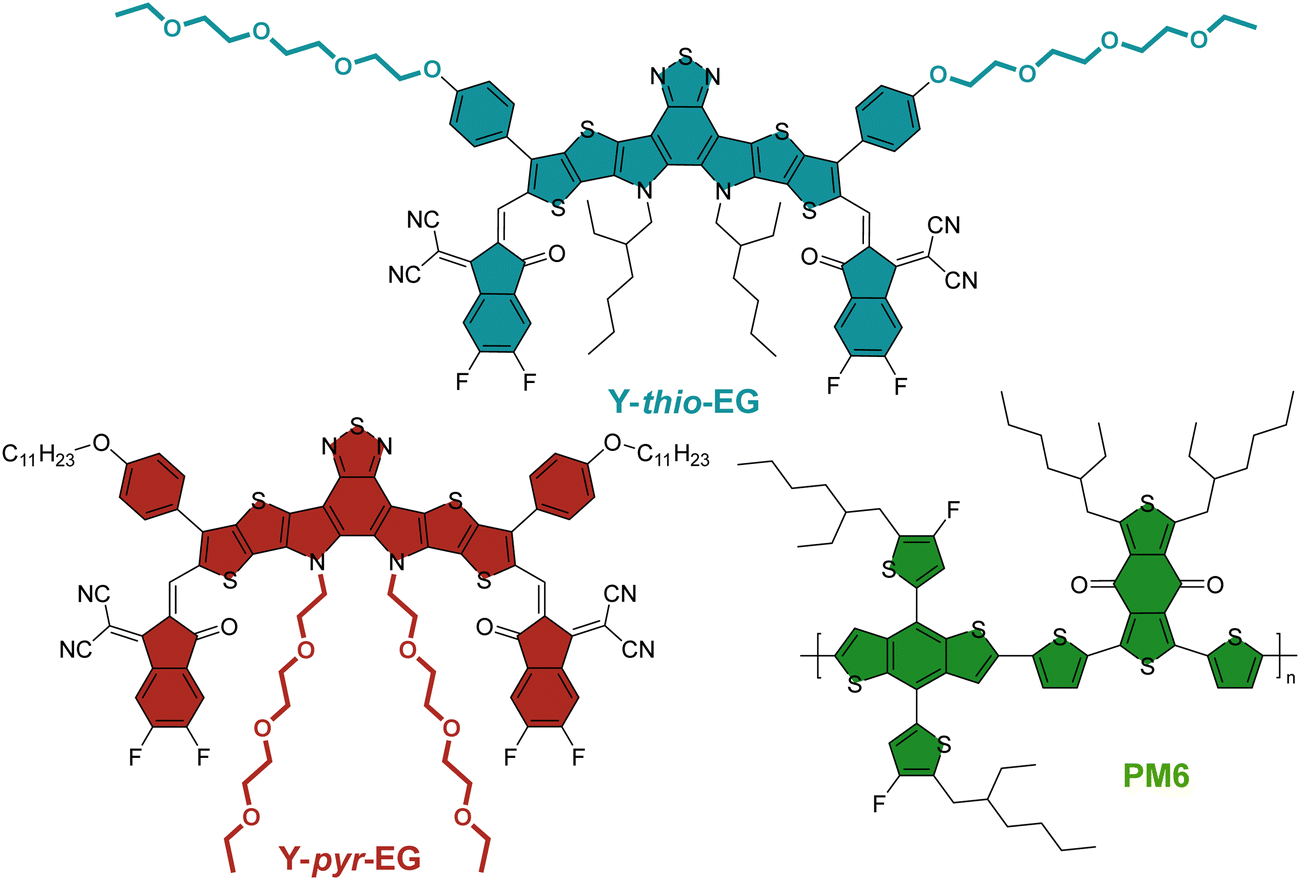 | ||
| Scheme 1 Structure of the novel YSAs Y-thio-EG and Y-pyr-EG and the investigated polymer PM6. | ||
Results and discussion
Synthesis
The preparation of the two YSAs can be divided into two main sections. The first section is the preparation of the functionalized thienothiophene (TT) blocks, and the second section is the subsequent synthesis of the final acceptors. The full synthetic pathway is described in the ESI.†In the first section of the classical route (Scheme 2), the side chain (usually n-undecyl) on the outer thiophene position of the Y-series acceptor is introduced in form of the corresponding alkyloyl chloride to 3-bromothiophene in a Friedel–Crafts acylation. This is followed by a reaction with ethyl thioglycolate, leading to an intramolecular cyclocondensation. Hydrolysis with NaOH, followed by a copper mediated high temperature (260 °C) decarboxylation reaction, yields the substituted TT building block.4 The harsh conditions employed in this route limit the possible variations of the side chains to alkyl chain modifications. Moreover, to introduce such a variation, the side chain has to be, firstly, available as – or converted to – an acid chloride, and secondly, introduced in the very first reaction step. Both factors strongly limit the variability of the thiophene substitution site in YSAs. As a consequence, side chain engineering is mainly applied to the center nitrogen position in the literature.28,29
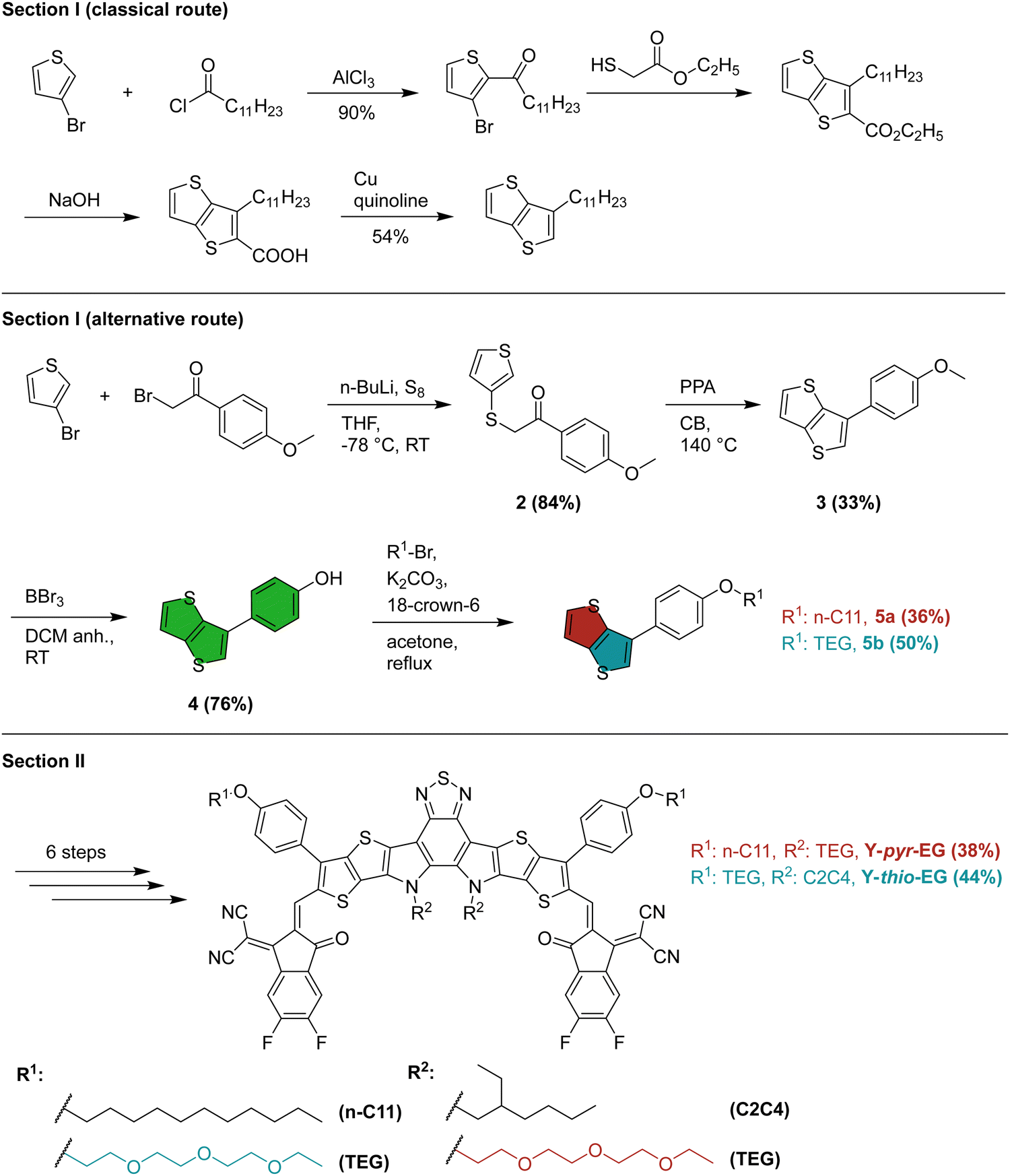 | ||
| Scheme 2 Synthetic route towards functionalized YSAs. The synthesis details and yields for the classical route were taken from the literature.4 | ||
Moreover, we deemed this synthesis strategy not suitable for the introduction of ethylene glycol side chains. Yan et al. demonstrated that Y-series acceptors with an alkyl–phenyl functionalization in the outer thiophenes can reach high efficiencies in OSCs.30,31 To that end, we adopted the synthesis route from the Ozturk group.32,33 In the first step, 3-bromothiophene is activated by a metal-halogen exchange reaction with n-BuLi to give the aryllithium intermediate, followed by quenching with elemental sulfur (Scheme 2). In situ alkylation of the obtained thiol with 2-bromo-4-methoxyacetophenone gives 2 with high yield (84%). Treatment of this intermediate with polyphosphoric acid in refluxing chlorobenzene yields the desired 3-alkylthieno[3,2-b]thiophene derivative in a ring closure reaction. Work up of this reaction proved difficult due to the presence of large excess of acid and we obtained only 33% yield (no yield optimization was performed). However, up to 79% yield can be reached for this step.33 Further deprotection gave the phenol 4, which was alkylated with n-undecyl bromide, and a polar 1-bromo-2-(2-(2-ethoxyethoxy)ethoxy)ethane (TEG-Br), yielding the functionalized TT blocks 5a and 5b, respectively.
Overall, this short synthesis route enables both, a later functionalization of the universal precursor (4), and a wider range of usable side chains. That way, widely functionalized TT blocks can be prepared for YSAs, using only commercially available, inexpensive starting materials. These changes make modification of YSAs in the outer thiophene position simpler and more attractive for future research.
The second section was kept identical to the typical literature procedure for YSA synthesis. It comprises a stannylation, followed by a Stille coupling of the modified TT blocks to the electron deficient benzothiadiazole (1) center. This gave compound 7a (7b) with 78% (70%) yield. The next step was a ring closure and a subsequent alkylation of the center nitrogen position with the classical 2-ethylhexyl bromide or 2-(2-(2-ethoxyethoxy)ethoxy)ethyl-4-methylbenzenesulfonate (TEG-OTs), respectively, which gave compound 9a (9b) with 14% (31%) yield. Of all steps during the YSA preparation, this ring-closure/alkylation was by far the most problematic in terms of reaction yield. This is likely due to the harsh reaction conditions (180 °C overnight), favoring side reactions. The last steps included a Vilsmeier–Haack formylation to give compound 10a (10b) with 65% (65%) yield, followed by a Knoevenagel-condensation with the end groups 2FIC (2-(5,6-difluoro3-oxo2,3-dihydro-1H-inden-1-ylidene)malononitrile). This gave the final products Y-thio-EG and Y-pyr-EG with 38% and 44% yield in the last reaction step, respectively (Scheme 1). The synthesis was accompanied by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry to confirm the desired compounds (see ESI†).
Physical properties
The absorption and emission properties of the acceptors were measured by UV-Vis absorption and photoluminescence spectroscopy. The results are summarized in Table 1 and Fig. 1. The UV-Vis spectra in chloroform (Fig. 1(a)) and thin film (Fig. 1(b)) show typical absorption behaviour for YSAs (comparison to Y6 see Fig. S1, ESI†). Both acceptors exhibit high absorption coefficients in chloroform solution, with 1.58 × 105 M−1 cm−1 (Y-thio-EG) and 1.61 × 105 M−1 cm−1 (Y-pyr-EG). In thin film, both acceptors reveal bathochromic shifts of 95 nm and 71 nm. They show strong absorption over a wide range of the visible spectrum, reaching into the far red with an absorption onset in the range of 900 to 920 nm. This shows that both acceptors have a strong light harvesting capability for OSCs application.| Acceptor | λ solmax | λ tfmax | ε sol |
E
solg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
E
tfg
|
HOMO/LUMO CV | HOMO UPS | E CVg | T dec | ε r |
|---|---|---|---|---|---|---|---|---|---|---|
| (nm) | (nm) | (105 M−1 cm−1) | (eV) | (eV) | (eV) | (eV) | (eV) | (°C) | (1) | |
| a Optical bandgap, determined from the intersection of absorption and emission spectra in chloroform solution. b Determined from the absorption onset tangent. c Decomposition temperature at 5% weight loss. d Relative permittivity at 103 Hz. e Recalculated from literature4 with Fc/Fc+vs. vac. of 5.39 eV.37,38 | ||||||||||
| Y-thio-EG | 727 | 822 | 1.58 | 1.66 | 1.36 | −6.06/−4.58 | −5.70 | 1.48 | 330 | 4.73 |
| Y-pyr-EG | 728 | 799 | 1.61 | 1.64 | 1.39 | −5.98/−4.50 | −5.72 | 1.48 | 287 | 5.24 |
| Y6 | 732 | 825 | — | — | 1.37 | −6.68/−5.13e | −5.58 | 1.55e | — | 2.39 |
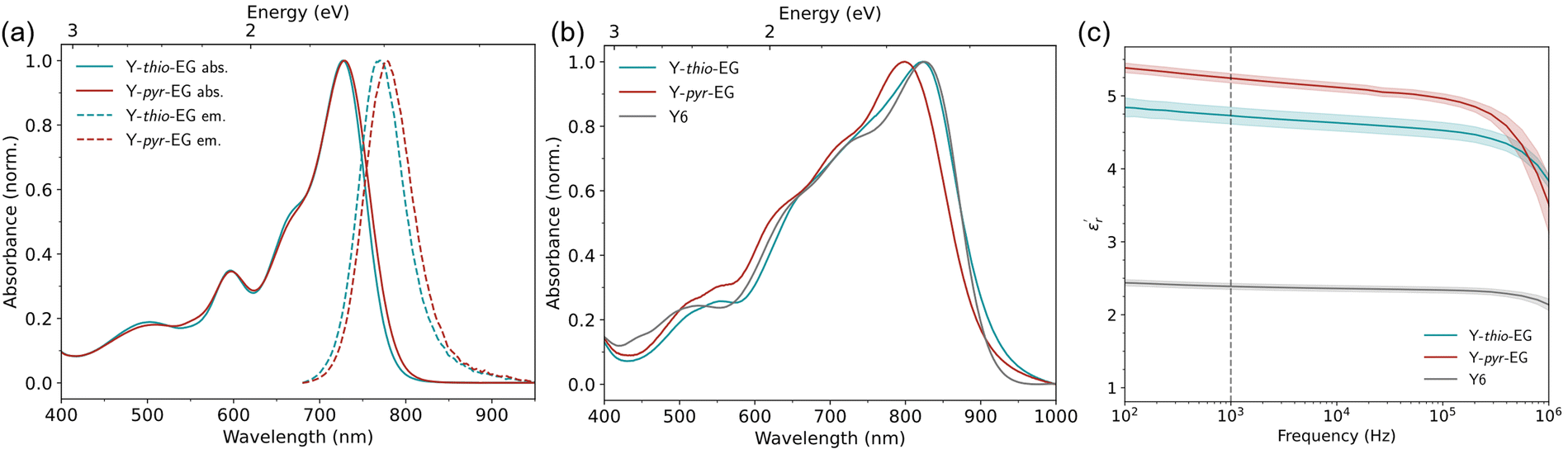 | ||
| Fig. 1 Physical characterization. (a) UV-Vis absorption (solid lines) and photoluminescence (dashed lines) spectra in chloroform. (b) UV-Vis absorption spectra of neat acceptor thin films. (c) Real relative permittivity spectra (solid line: arithmetic mean from 20–30 measurements, shaded area: 95% confidence interval). | ||
Additionally, the thermal properties of both acceptors were investigated by thermogravimetry (Fig. S2, ESI†). Y-thio-EG and Y-pyr-EG exhibit decomposition temperatures (5% weight loss) of 330 °C and 287 °C, respectively. Comparing the values to the similar YSA with p-hexylphenyl groups on the thiophene position, which has a decomposition temperature of 352 °C,30 shows that the TEG chains slightly decrease the thermal stability.
The dielectric permittivity of both acceptors was measured by impedance spectroscopy. For that, parallel-plate capacitors were fabricated with the setup glass/ITO/PEDOT:PSS/acceptor/Ag, with PEDOT:PSS acting as a smoothing layer. The relative permittivities εr as a function of the applied electric field frequency were calculated from the impedance spectra via the capacitor equation
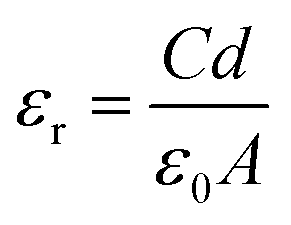 | (1) |
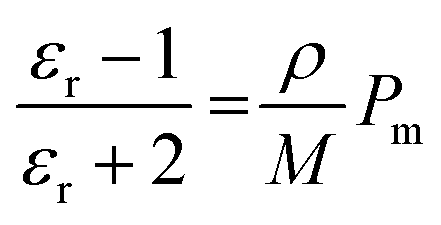 | (2) |
Because of the attached glycol side chains, we expected the acceptors to exhibit altered surface properties. Since the blending behavior of donor and acceptor in the bulk heterojunction are dictated by their surface energy match, we investigated the surface free energy (SFE) of the new acceptors by contact angle measurements (Fig. S3, ESI†). The SFE values were obtained for thin films using water and diiodomethane drops, and calculated by the method of Owens–Wendt–Rabel–Kaelble (OWRK)39 (Table 2). The TEG side chains show a strong influence on the SFE of the materials. Compared to Y6 (SFE 41 ± 6 mN m−1), Y-thio-EG and Y-pyr-EG exhibit increased SFE values of 61 ± 3 mN m−1 and 56 ± 2 mN m−1, respectively. The increase stems from a new, polar contribution of the TEG chains to the SFE. Further, we calculated the Flory–Huggins interaction parameters χ for both acceptors with common donor polymers (Table 3). The values are generally high in the range of 7 to 9, compared to more typical values as seen for Y6 (in the range of 2 to 3). These results indicate, on the one hand, that the new materials could have good solubilities in non-halogenated solvents, and on the other hand, that the interaction with common donor polymers could pose a challenge to get an optimal bulk heterojunction (BHJ) morphology.
| Acceptor | SFE | Dispersive | Polar |
|---|---|---|---|
| Y6 | 41 ± 6 | 41 ± 6 | 0 |
| Y-thio-EG | 61 ± 3 | 42 ± 1 | 19 ± 2 |
| Y-pyr-EG | 56 ± 2 | 37 ± 1 | 19 ± 2 |
| Acceptor | Polymer | SFE (mN m−1) | χ (1) |
|---|---|---|---|
| Y-thio-EG | PBDB-T | 23 | 9.3 |
| PM6 | 22 | 9.9 | |
| PTQ10 | 22 | 9.5 | |
| Y-pyr-EG | PBDB-T | 23 | 7.4 |
| PM6 | 22 | 7.9 | |
| PTQ10 | 22 | 7.6 | |
| Y6 | PBDB-T | 23 | 2.6 |
| PM6 | 22 | 2.9 | |
| PTQ10 | 22 | 2.8 | |
To identify suitable donor polymers, we determined the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies by cyclic voltammetry (CV) measurements of the neat materials as thin films (Fig. 2(b), (c) and Table 1). Both acceptors show similar energies for the HOMO/LUMO of −6.06/−4.58 eV (Y-thio-EG) and −5.98/−4.50 eV (Y-pyr-EG). Compared to Y6 with a HOMO/LUMO energy of −6.68/−5.13 eV (recalculated from literature4 with Fc/Fc+vs. vac. of 5.39 eV37,38), the modified acceptors show raised HOMO and LUMO energies by approx. 0.6–0.7 eV. The HOMO levels are now close to those of the exemplary measured donor polymers (PBDB-T: −6.01 eV and PM6: −6.07 eV). The small difference could cause inefficient exciton dissociation and hole transfer at the donor–acceptor interface. However, the results might be misleading since energy levels determined from CV, especially of polymers, are known to have a non-neglectable error due to non-reversible redox kinetics.37,40 Therefore, we additionally determined the HOMO energies from ultraviolet photoelectron spectroscopy measurements (UPS, Fig. 2(a), (c) and Table 1). Comparing with the CV results, the HOMO energies of the acceptors are relatively similar with −5.70 eV (Y-thio-EG), −5.72 eV (Y-pyr-EG) and −5.58 eV (Y6). Thus, a comparison with the HOMO energies of both donor polymers with literature values of −5.14 eV (PBDB-T) and −5.25 eV (PM6)41 suggests that there is a sufficient energy offset between both donors and the modified acceptors for efficient charge carrier separation (in the range of 0.4–0.6 eV).
 | ||
| Fig. 2 Electrochemical characterization of neat materials as thin films. (a) Ultraviolet photoelectron spectroscopy measurements. (b) Cyclic voltammetry measurements. (c) Frontier orbital energies, determined by CV (colored rectangles) and UPS (single black bars) measurements. The HOMO values determined via UPS of PBDB-T and PM6 are taken from the literature.41 | ||
Solar cell characterization
For a direct comparison to literature efficiencies, the new acceptors were tested in a setup typically used for Y6-based solar cells, namely the conventional device architecture ITO/PEDOT:PSS/PM6:acceptor/PNDIT-F3N-Br/Ag. Because the high Flory–Huggins interaction parameters with PM6 already indicated a limited miscibility with both acceptors, we refrained from using non-halogenated solvents, which could further complicate the fabrication process. For that reason, we chose the standard solvent system chloroform with 0.5 vol% 1-chloronaphthalene as additive, which is used in most YSA studies.In this device setup, both acceptors achieved substantially lower efficiencies than typical Y6 OSCs (Table 4). Especially limited by the low fill factors (FF), OSCs with Y-thio-EG and Y-pyr-EG merely achieved a PCE of 8.19% (FF 58.7%) and 5.26% (FF 40.3%), respectively. In the identical setup with PBDB-T as donor, similar results were obtained (Fig. S4, ESI†). The shapes of the corresponding JV curves (Fig. 3(a) and (b)) show that the poor FF is mainly caused by low shunt resistances Rsh of both OSCs, indicating high recombination losses in the active layer. The values of the series resistance Rs and shunt resistance Rsh were estimated from the inverse slopes of the JV curves, specifically Rs in the linear region above the VOC, and Rsh around the JSC. The fitted values for the Rsh/Rs are 335/1 Ω cm−2 for the Y-thio-EG based OSCs and 161/5 Ω cm−2 for the Y-pyr-EG based OSCs.
| Acceptor | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) | η diss (%) | η cc (%) |
|---|---|---|---|---|---|---|
| Average values (mean and standard deviation) are calculated from 10 devices. Top values in parentheses. | ||||||
| Y-thio-EG | 0.79 ± 0.01 (0.79) | 17.6 ± 0.2 (17.7) | 59 ± 1 (58.7) | 8.09 ± 0.07 (8.19) | 97.3 | 87.4 |
| Y-pyr-EG | 0.80 ± 0.03 (0.81) | 16.4 ± 0.2 (16.2) | 41 ± 1 (40.3) | 5.23 ± 0.02 (5.26) | 83.4 | 60.5 |
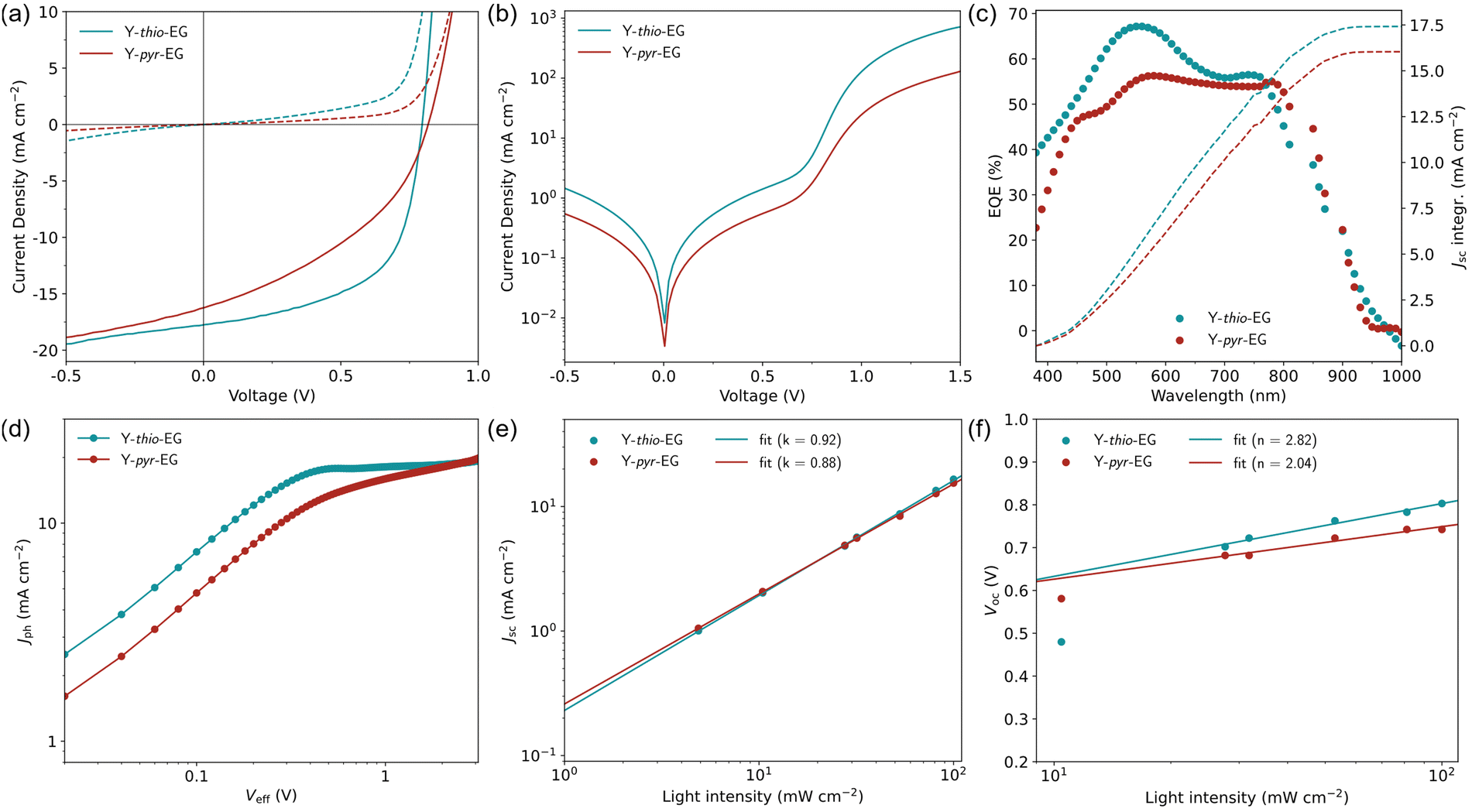 | ||
| Fig. 3 OSC characterization of the new acceptors in the setup ITO/PEDOT:PSS/PM6:A/PNDIT-F3N-Br/Ag (where A is Y-thio-EG, Y-pyr-EG, respectively). (a) JV curves of best OSC in light (solid) and dark (dashed) condition. (b) Dark JV curves in semilogarithmic style. (c) EQE spectra (dots) and integrated JSC curves (dashed lines). (d) Jph–Veff plot. (e) Light intensity dependency measurement of JSC (dots) and fit (solid lines). (f) Light intensity dependency measurement of VOC (dots) and fit (solid lines). | ||
External quantum efficiency (EQE, spectra and corresponding integrated JSC curves see Fig. 3(c)) measurements were conducted. The EQE curves show a wide spectral interval contributing to the current formation, however only moderate EQE values of 50–67% were obtained. The JSC values determined from the EQE spectra (PM6:Y-thio-EG: 17.4 mA cm−2; PM6:Y-pyr-EG: 16.1 mA cm−2) match well with the ones extracted from the JV curves (cf.Table 4).
To gain a deeper insight into the OSC characteristics, we evaluate the individual process efficiencies ηa, ηdiff, ηdiss, ηtr, ηcc, which contribute to the overall device efficiency. These individual terms are the respective efficiencies η of light absorption (a), the exciton diffusion to the donor–acceptor heterojunction (diff), the exciton dissociation (diss), the free charge transport to the electrodes (tr), and the charge collection (cc).42,43 Since both acceptors exhibit strong light absorption and the improved permittivities would suggest efficient exciton dissociation, the limiting factors most likely are the exciton diffusion, the free charge transport, and/or the charge collection. A closer investigation of these efficiencies was done by analyzing the dependency of the photocurrent density Jph on the effective voltage Veff (Fig. 3(d)). From these curves, the exciton dissociation efficiency ηdiss and charge collection efficiency ηcc were calculated (Table 4). Both acceptors Y-thio-EG and Y-pyr-EG exhibit good to moderate values for ηdiss (97.3% and 83.4%), but poor values for ηcc (87.4% and 60.5%). This shows, that the biggest limitation can be attributed to the diffusion and collection of the free charge carriers through the active layer.
For further detail, we investigated the dominating recombination pathways by analyzing the dependency of the short-circuit current density (JSC) and open-circuit voltage (VOC) on the light intensity (Fig. 3(e), (f), JV curves Fig. S5, ESI†). The JSC dependencies reveal slopes of 0.92 and 0.88 for Y-thio-EG and Y-pyr-EG, respectively. Both values show that monomolecular recombination dominates in both OSCs at short-circuit conditions and bimolecular recombination is negligible.44 Further, the ideality factors n of the diode curves can be extracted from the slopes of the VOC dependency on the light intensity.45 In the intensity range of 10–100 mW cm−2, the OSCs based on Y-thio-EG and Y-pyr-EG exhibit an ideality factor of 2.82 and 2.08, respectively, which exceeds the typically discussed interval of 1–2. According to Xiong et al., the deviation to higher values indicates a large energetic disorder in the active layer.46 This high degree of disorder – or non-ideality – might be caused by the suboptimal interaction of the apolar donor and polar acceptor. A poorly defined donor–acceptor interface would make way for an increased charge carrier recombination rate. This assumption is supported by a computational study by Alessandri et al., who concluded that modifying fullerene acceptors with glycol chains results in a broadening of the charge carrier energy levels by electrostatic disorder caused by the glycol dipoles. This would then lead to an increased charge carrier relaxation rate, and consequently, an increased voltage loss.47 Moreover, at lower light intensities, the VOC trends further deviate to even higher values for n, which can be ascribed to the pronounced Shockley–Read–Hall recombination.48
The comparably low efficiencies can be most likely explained by suboptimal interactions of the polar acceptors with PM6 and PBDB-T. The low values for the FF and JSC, as well as the low charge dissociation and transfer efficiencies, suggest problematic BHJ morphologies. This is further supported by the low miscibility between the acceptors and polymer donors, as visible from the Flory–Huggins interaction parameters. Based on these observations, the deterioration of the BHJ morphology seems to overshadow any potential improvements from the higher material permittivities. To get a clear picture of the actual material interplay, a detailed morphological characterization is presented in the following section.
Morphology investigation
For a thorough characterization of the BHJ morphology over the full relevant length scale, we combined atomic force microscopy (AFM), grazing incidence wide-angle X-ray scattering (GIWAXS) and analytical electron microscopic methods.The AFM measurements (Fig. 4 for 2 × 2 μm2 and Fig. S6, ESI† for 5 × 5 μm2 images) show stronger surface aggregation for PM6:Y-thio-EG and especially for PM6:Y-pyr-EG, compared to PM6:Y6. All three films exhibit similar fibrillar microstructures in both the height and phase images. In the PM6:Y6 film, the elongated structures are less pronounced and the film shows an overall more homogeneous picture, which is clearly visible from the smoother structure of the phase image combined with a smaller height contrast. This reveals that the generally desired fibrillar structures are also forming with these acceptors, however the phase separation appears to be more pronounced in the Y-thio-EG and Y-pyr-EG based blends. In addition, the PM6:Y-pyr-EG films show agglomerates of up to 20 nm height (line scans see Fig. S7, ESI†). These aggregates are presumably the acceptor Y-pyr-EG, which likely tends to stronger aggregation due to the lack of the 2-ethylhexyl branching point.
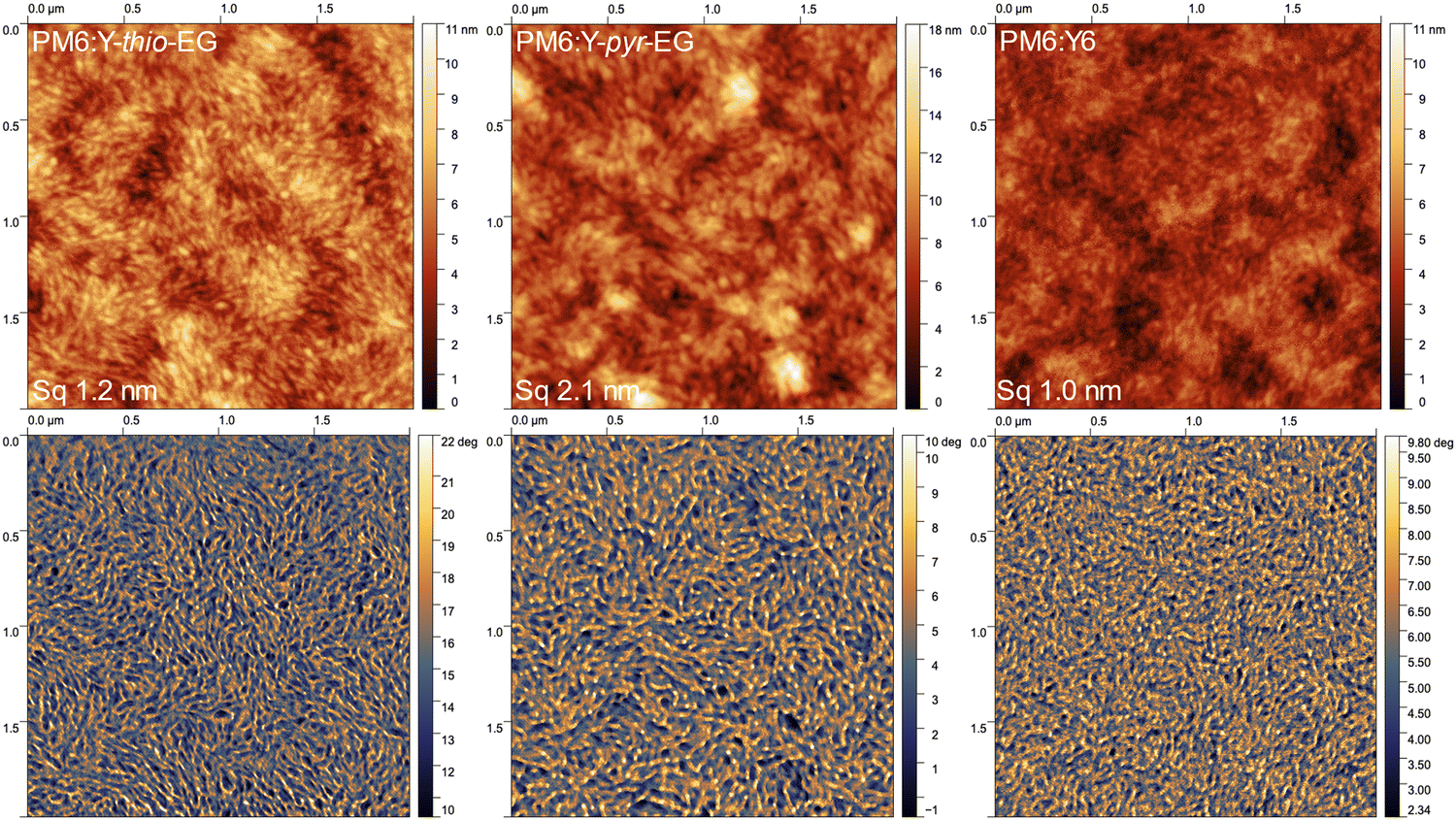 | ||
| Fig. 4 AFM images (2 × 2 μm2) with (top) height and (bottom) phase images of PM6:Y-thio-EG, PM6:Y-pyr-EG, and PM6:Y6 (films deposited onto ITO/PEDOT:PSS substrates). | ||
The impact of the different crystallization levels is also seen in the root-mean-square surface roughnesses (Sq), which increase from Sq 1.0 nm (PM6:Y6) to 1.2 nm (PM6:Y-thio-EG) and 2.1 nm (PM6:Y-pyr-EG). A closer look on the statistical height distributions of the films (Fig. S8, ESI†) gives identical results. The height distribution of PM6:Y6 film is relatively narrow (mean μ = 4.3 nm, standard deviation σ = 1.6 nm), while that of PM6:Y-thio-EG is slightly increased (μ = 6.2 nm, σ = 2.0 nm). For the PM6:Y-pyr-EG sample, a further increase of the height distribution is observed (μ = 8.3 nm, σ = 2.8 nm), with two differently sized populations contributing to the distribution (Fig. S8a, ESI†).
Even though a direct observation of the domain sizes of donor and acceptor is challenging, we can obtain additional hints regarding changes in the domain sizes by STEM-EELS elemental mapping of sulfur and nitrogen (Fig. 5).49,50 The donor PM6 contains 21 wt% sulfur (in the repeating unit), while both acceptors only contain 9–10 wt% sulfur. This difference leads to a usable contrast between donor and acceptor phase in the STEM-EELS elemental mapping images. Additionally, the nitrogen signals origin solely from the acceptor molecules. Generally, the three samples PM6:Y-thio-EG, PM6:Y-pyr-EG and PM6:Y6 show a similar appearance. In more detail, it can be observed that the PM6 regions are slightly larger in the PM6:Y-thio-EG sample. This can be expected from the Flory–Huggins interaction parameter of PM6 with Y-thio-EG, which is the highest among these three samples, indicating a limited miscibility of both compounds (cf.Table 3). In the PM6:Y-pyr-EG sample, the domain sizes of both materials seem smaller, showing a more interwoven network. The reference PM6:Y6 blend exhibits slightly larger structures, however the region contrasts are reduced, which shows that PM6 and Y6 blend better and over larger domain sizes. In any case, however, the differences between the three films seem too nuanced to ascribe the lower PCE of the PM6:Y-thio-EG and PM6:Y-pyr-EG to a too coarse phase separation and too large domain sizes based on the limited miscibility of the two components.
 | ||
| Fig. 5 STEM-EELS sulfur/nitrogen maps (sulfur regions in red, nitrogen regions in green) of the active layer blends PM6:Y-thio-EG, PM6:Y-pyr-EG, and PM6:Y6. | ||
For a closer analysis of the crystallinity and molecular packing, we conducted GIWAXS measurements of the pristine acceptor films as well as for their respective blends with PM6 (Fig. 6). As expected from the lack of branching points at the molecular center, the crystallinity of Y-pyr-EG and its scattering pattern are much more pronounced than that of the Y-thio-EG sample. The pristine Y-pyr-EG sample exhibits a π–π stacking distance of 0.34 nm (qz 18.5 nm−1), which is significantly lower than the one of Y-thio-EG with 0.36 nm (qz 17.5 nm−1). Furthermore, the π–π stacking peak of Y-pyr-EG is distinctly narrower than that of Y-thio-EG, indicating a higher crystalline coherence length (CCL).
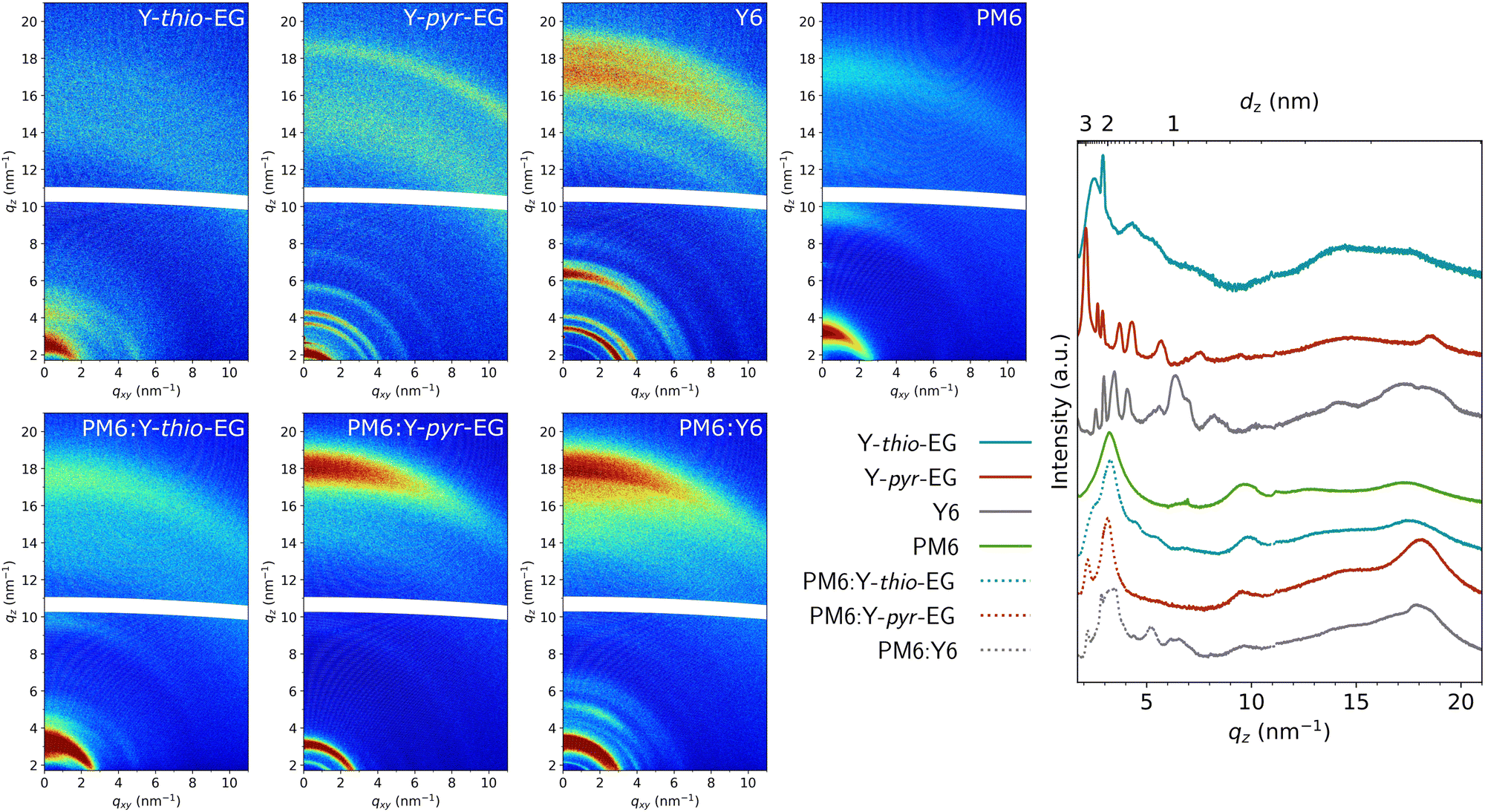 | ||
| Fig. 6 2D GIWAXS patterns of drop-cast films after thermal annealing at 100 °C for 10 min of Y-thio-EG, Y-pyr-EG, Y6, PM6, PM6:Y-thio-EG, PM6:Y-pyr-EG, PM6:Y6, and their corresponding line cuts in qz direction. | ||
Regarding the lamellar stacking, a similar trend is observed. Y-pyr-EG shows narrow peaks, confirming that it forms ordered crystalline domains with a high CCL. The most prominent lamellar stacking peak of Y-pyr-EG located at qz 2.1 nm−1 reveals a lamellar stacking distance of 3.0 nm and a large CCL of 29 nm (full-width-half-maximum (FWHM) of 0.22 nm−1). For Y-thio-EG, the peak is much broader and shifted to qz 2.5 nm−1, corresponding to a stacking distance of 2.5 nm and the broadening indicates crystalline order only over significantly shorter distances.
The blend films of PM6:Y-thio-EG and PM6:Y-pyr-EG exhibit feature contributions of both neat materials, indicating that crystalline domains of donor and acceptor are present in the thin films. The line cut in out-of-plane direction of PM6:Y-pyr-EG indicates more crystalline acceptor domains compared to the PM6:Y-thio-EG sample. This is shown by the wider π–π stacking (0.36 nm, qz 17.5 nm−1) in the PM6:Y-thio-EG film and a much less pronounced lamellar stacking peak, which is only visible as a shoulder in this sample. In the PM6:Y-pyr-EG sample, the CCL of 20 nm (FWHM 0.32 nm−1) of the lamellar stacking peak is slightly smaller compared to the pristine film (29 nm), but still indicates large coherent crystalline acceptor domains in the blend with PM6. Overall, the crystallinities of the pristine Y-pyr-EG film and the blend with PM6 are quite similar to the Y6 analogues, while the Y-thio-EG and PM6:Y-thio-EG films reveal significantly lower crystallinity.
The GIWAXS measurements were performed on drop coated samples with higher film thickness than in the solar cell setup, to obtain a higher signal-to-noise ratio. Therefore, we additionally characterized spin coated donor:acceptor blend films (60–80 nm) with zero-loss-filtered electron diffraction using a tilt of 0° and 75° to observe in-plane and out-of-plane information (Fig. 7). For both the PM6:Y-thio-EG and the PM6:Y-pyr-EG films, the diffraction patterns reveal a preferential face-on orientation of the donor and acceptor in the blend. The backbone signals for PM6 and Y-thio-EG appear both at a q value of 0.47–0.48 nm−1, indicating a lamellar stacking distance of approx. 2.1 nm. In contrast to that, the lamellar stacking peaks of PM6 and Y-pyr-EG appear clearly separated at 0.37 nm−1 and 0.45 nm−1, respectively. Assuming an orthogonal lattice of Y-pyr-EG, the π–π stacking peak will be perpendicular to the backbone peak at high tilt (observed in the 75° tilt image). This means that the inner ring at 0.37 nm−1 corresponds to Y-pyr-EG (real space: 2.7 nm) and the ring at 0.45 nm−1 to PM6 (lamellar stacking distance: 2.2 nm). The π–π stacking of Y-pyr-EG in the blend is measured to be 0.38 nm, while the π–π stacking of Y-thio-EG is slightly larger (0.39 nm). Both observations, the denser π–π stacking of Y-pyr-EG and the larger lamellar stacking distance in Y-thio-EG, match well with the results from the GIWAXS investigations.
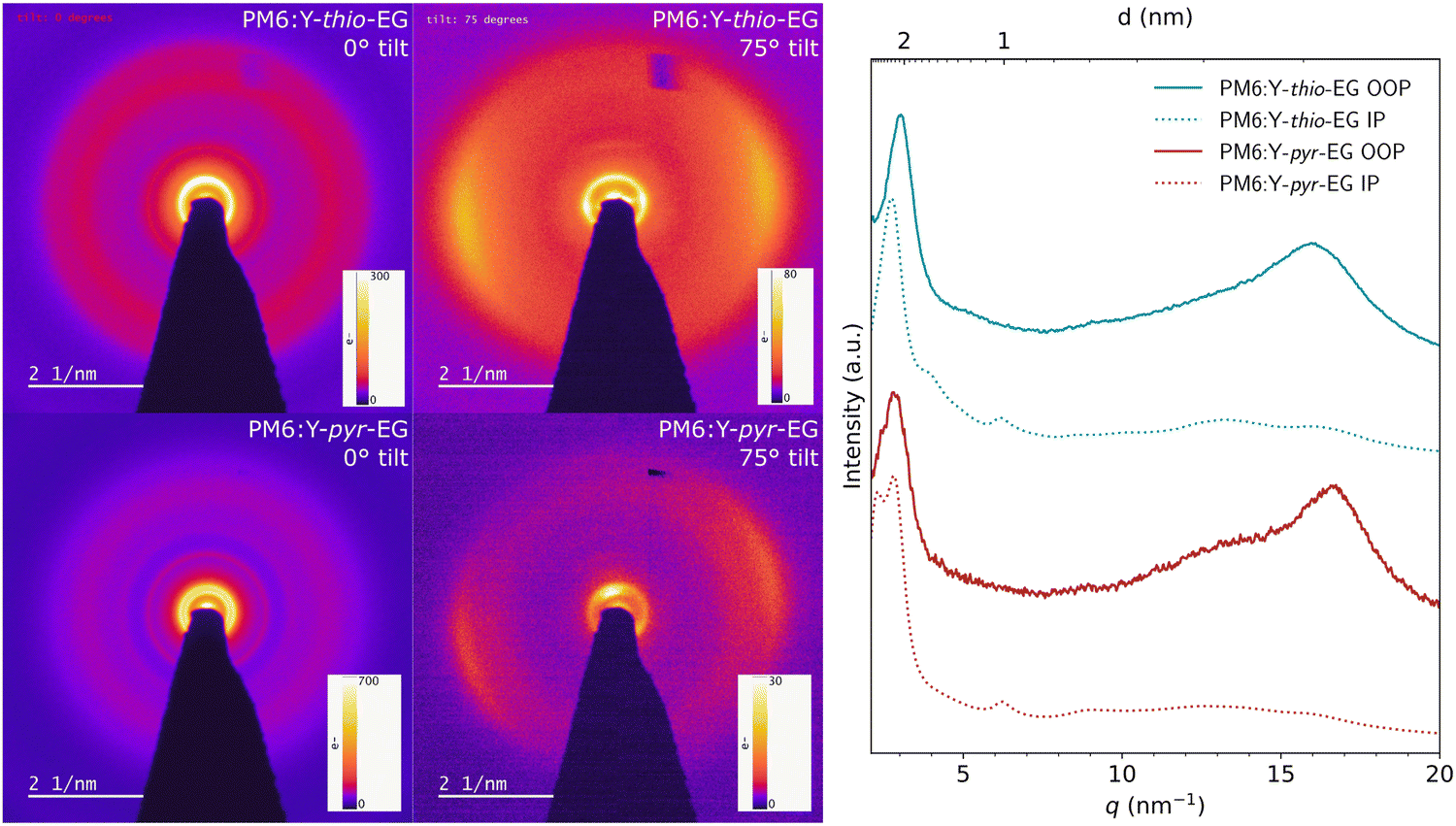 | ||
| Fig. 7 Zero-loss-filtered electron diffraction patterns of thin film blends with 0° and 75° tilt and their extracted line cuts (the q axis was calculated by 2πQ from the original measurement). | ||
Our investigations reveal that the glycol modification, as well as the position of the EG groups have a large influence on the crystallinity of the materials. Moreover, their SFE is affected, which is reflected in changed miscibilities and slight changes in the donor:acceptor morphology, as indicated by AFM images and TEM elemental maps. However, we also conclude that the changes in crystallinity and the domain sizes in the BHJ are most likely not the only reasons that the improved permittivity of the acceptors does not lead to improved efficiencies, and that the situation is much more intricate. In particular, mixed phases between donor and acceptor domains, or the possible formation of interfacial traps, energy level broadening or dipole layer formation between the apolar donor and polar acceptor domains might play a significant role, and thus needs further investigation in future studies. Similar conclusions were drawn by Alessandri et al. from a theoretical study, who stated that energy level broadening, induced by the randomly distributed glycol dipoles, would potentially pose a greater problem than morphological changes.47 Additionally, as indicated by the permittivity measurement (cf.Fig. 1(c)), the contribution of the OEG dipole reorientation to the permittivity might be too small for short-lived processes in the OSC such as exciton dissociation.
Conclusions
In this work, we introduced two Y-series acceptors, Y-thio-EG and Y-pyr-EG, with polar glycol side chains at the outer thiophene, and the inner pyrrol side chain position of the central core. The synthesis was done via an alternative synthetic route, which simplifies the variation of the side chains in the outer thiophene site via a universal thienothiophene building block (compound 4) that can be prepared with good yields from inexpensive starting materials. This alternative route has the potential to make Y-series acceptor functionalization easier and more attractive for future research.Both Y-thio-EG and Y-pyr-EG exhibit greatly improved relative permittivities (4.73 and 5.24) compared to Y6 (2.39), while maintaining the typical attractive physical properties of Y-series acceptors, such as a high thermal stability and strong light absorption over a wide frequency range. The surface free energy of both acceptors is increased (Y-thio-EG 61 mN m−1, Y-pyr-EG 56 mN m−1), which leads to high Flory–Huggins interaction parameters with conventional polymer donors, such as PM6 and PBDB-T.
The new acceptors were tested in a standard setup for Y-series acceptors, namely the conventional setup ITO/PEDOT:PSS/PM6:acceptor/PNDIT-F3N-Br/Ag, processed from chloroform with 0.5 vol% 1-chloronaphthalene. Interestingly, despite their improved permittivities, both Y-thio-EG and Y-pyr-EG based solar cells only achieved efficiencies of 8.09% and 5.23%, respectively.
To pin down the reason for the decreased efficiencies, we conducted a detailed morphological investigation using atomic force microscopy (AFM), grazing-incidence wide-angle X-ray scattering (GIWAXS), and analytic electron microscopy methods. While the glycol modification and the positioning within the acceptor show a large influence on the crystallinity of the pristine acceptor films, the differences in the blend films with PM6 are less pronounced. On a macroscopic scale (AFM), Y-thio-EG and especially Y-pyr-EG exhibit some increased aggregation tendencies in blends with PM6 (compared to PM6:Y6). However, the investigations at smaller lengths scales by analytical electron microscopy revealed only minor differences in the bulk heterojunction morphology between the three systems. These findings led us to the assumption that the morphological differences in the PM6 blends alone do not fully explain the reduced efficiencies of our solar cells. We hypothesize that the combination of apolar polymers with polar glycolated acceptors might yield a much more intricate challenge, with possible adverse interface effects such as dipole formation, energy level broadening, or increased interface-state assisted recombination.
To prevent these numerous issues when employing glycolated molecules as high-permittivity acceptors, an obvious solution is the expansion of the same strategy onto the donor component, to achieve better matching physical properties. A promising example has been recently shown by Neu et al., who modified the popular polymer PTQ10 with glycol side chains to obtain an alcohol-processable donor.51 An alternative solution is to focus on other strategies for the permittivity-modification – such as expanding the π-systems or incorporating functional groups polarizable at higher frequencies (listed in more detail in the Introduction) – which entail less complex system changes.
Experimental
The full methodology, all synthetic routes and synthetic characterizations are included in the ESI.†Author contributions
P. F.: conceptualization, data curation, formal analysis, investigation (synthesis, structural analysis, physical characterization, solar cell characterization, morphological characterization), methodology, project administration, supervision, visualization, writing – original draft, writing – review & editing. S. M.: conceptualization, formal analysis, investigation (synthesis, structural analysis), writing – review & editing. T. R.: conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing. M. R.: conceptualization, writing – original draft, writing – review & editing. M. W.: investigation (STEM-EELS, ZLF-electron diffraction), resources, writing – review & editing. E. S.: investigation (STEM-EELS, ZLF-electron diffraction), resources, writing – review & editing. N. S.: investigation (STEM-EELS, ZLF-electron diffraction), resources, writing – review & editing. G. H.: investigation (STEM-EELS, ZLF-electron diffraction), resources, writing – review & editing. G. K.: investigation (STEM-EELS, ZLF-electron diffraction), resources, writing – review & editing. B. R.: investigation (UPS), resources, writing – review & editing. S. H.: investigation (solar cell characterization), writing – review & editing. J. B. S.: investigation (AFM), writing – review & editing. P. M.: investigation (GIWAXS), writing – review & editing. H. A.: investigation (GIWAXS), resources, writing – review & editing. G. T.: funding acquisition, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support is gratefully acknowledged from the Zukunftsfonds Steiermark and the Klimafonds Graz (project GreenOPVSolutions). Moreover, the authors would like to acknowledge the use of the Somapp Lab, a core facility supported by the Austrian Federal Ministry of Education, Science and Research, the Graz University of Technology, the University of Graz and Anton Paar GmbH. The purchase of the K2-camera for the STEM-EELS measurements was supported by the Zukunftsfonds Steiermark. The authors further thank Ilie Hanzu, Petra Kaschnitz, Marco Sigl, Karin Bartl, and Matthias Schwar for their experimental support.Notes and references
- H. Yao and J. Hou, Angew. Chem., Int. Ed., 2022, 61, e202209021 CAS.
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee, T. Wang, H. Ade, T. Heumüller, C. Brabec and P. Meredith, Adv. Energy Mater., 2021, 11, 2003570 CrossRef CAS.
- B. Schweda, M. Reinfelds, P. Hofstadler, G. Trimmel and T. Rath, ACS Appl. Energy Mater., 2021, 4, 11899–11981 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- C. He, Y. Pan, Y. Ouyang, Q. Shen, Y. Gao, K. Yan, J. Fang, Y. Chen, C.-Q. Ma, J. Min, C. Zhang, L. Zuo and H. Chen, Energy Environ. Sci., 2022, 15, 2537–2544 RSC.
- X. Xu, W. Jing, H. Meng, Y. Guo, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2208997 CrossRef CAS PubMed.
- J.-L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef PubMed.
- T. M. Burke, S. Sweetnam, K. Vandewal and M. D. McGehee, Adv. Energy Mater., 2015, 5, 1500123 CrossRef.
- S. M. Menke, N. A. Ran, G. C. Bazan and R. H. Friend, Joule, 2018, 2, 25–35 CrossRef CAS.
- J. Hofinger, C. Putz, F. Mayr, K. Gugujonovic, D. Wielend and M. C. Scharber, Mater. Adv., 2021, 2, 4291–4302 RSC.
- L. J. A. Koster, S. E. Shaheen and J. C. Hummelen, Adv. Energy Mater., 2012, 2, 1246–1253 CrossRef CAS.
- A. Y. Sosorev, D. Y. Godovsky and D. Y. Paraschuk, Phys. Chem. Chem. Phys., 2018, 20, 3658–3671 RSC.
- J. Brebels, J. V. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem. A, 2017, 5, 24037–24050 RSC.
- S. Y. Leblebici, T. L. Chen, P. Olalde-Velasco, W. Yang and B. Ma, ACS Appl. Mater. Interfaces, 2013, 5, 10105–10110 CrossRef CAS PubMed.
- S. Leblebici, J. Lee, A. Weber-Bargioni and B. Ma, J. Phys. Chem. C, 2017, 121, 3279–3285 CrossRef CAS.
- J. Oh, S. Jung, M. Jeong, B. Lee, J. Lee, Y. Cho, S. M. Lee, S. Chen, Z.-G. Zhang, Y. Li and C. Yang, J. Mater. Chem. C, 2019, 7, 4716–4724 RSC.
- K. Mishima, T. Sakai, K. Yokota, M. Taniguchi, Y. Aso, Y. Ie and K. Yamashita, J. Photochem. Photobiol., A, 2020, 401, 112756 CrossRef CAS.
- X. Zhang, D. Zhang, Q. Zhou, R. Wang, J. Zhou, J. Wang, H. Zhou and Y. Zhang, Nano Energy, 2019, 56, 494–501 CrossRef CAS.
- S. Sami, R. Alessandri, R. Broer and R. W. A. Havenith, ACS Appl. Mater. Interfaces, 2020, 12, 17783–17789 CrossRef CAS PubMed.
- J. Brebels, E. Douvogianni, D. Devisscher, R. Thiruvallur Eachambadi, J. Manca, L. Lutsen, D. Vanderzande, J. C. Hummelen and W. Maes, J. Mater. Chem. C, 2018, 6, 500–511 RSC.
- X. Liu, B. Xie, C. Duan, Z. Wang, B. Fan, K. Zhang, B. Lin, F. J. M. Colberts, W. Ma, R. A. J. Janssen, F. Huang and Y. Cao, J. Mater. Chem. A, 2018, 6, 395–403 RSC.
- P. Fürk, J. Hofinger, M. Reinfelds, T. Rath, H. Amenitsch, M. C. Scharber and G. Trimmel, Monatsh. Chem., 2022 DOI:10.1007/s00706-022-02956-2.
- P. Fürk, M. Reinfelds, I. Hanzu, T. Hartl, J. B. Schaubeder, E. Zuccala, H. Amenitsch, T. Rath and G. Trimmel, ACS Appl. Energy Mater., 2023, 6, 1544–1554 CrossRef.
- T. Li, K. Wang, G. Cai, Y. Li, H. Liu, Y. Jia, Z. Zhang, X. Lu, Y. Yang and Y. Lin, JACS Au, 2021, 1, 1733–1742 CrossRef CAS PubMed.
- H. Chen, R. Zhang, X. Chen, G. Zeng, L. Kobera, S. Abbrent, B. Zhang, W. Chen, G. Xu, J. Oh, S.-H. Kang, S. Chen, C. Yang, J. Brus, J. Hou, F. Gao, Y. Li and Y. Li, Nat. Energy, 2021, 6, 1045–1053 CrossRef CAS.
- K. Jiang, Q. Wei, J. Y. L. Lai, Z. Peng, H. K. Kim, J. Yuan, L. Ye, H. Ade, Y. Zou and H. Yan, Joule, 2019, 3, 3020–3033 CrossRef CAS.
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS.
- B. Lu, J. Wang, Z. Zhang, J. Wang, X. Yuan, Y. Ding, Y. Wang and Y. Yao, Nano Select, 2021, 2, 2029–2039 CrossRef CAS.
- G. Chai, Y. Chang, J. Zhang, X. Xu, L. Yu, X. Zou, X. Li, Y. Chen, S. Luo, B. Liu, F. Bai, Z. Luo, H. Yu, J. Liang, T. Liu, K. S. Wong, H. Zhou, Q. Peng and H. Yan, Energy Environ. Sci., 2021, 14, 3469–3479 RSC.
- L. Wang, C. Guo, X. Zhang, S. Cheng, D. Li, J. Cai, C. Chen, Y. Fu, J. Zhou, H. Qin, D. Liu and T. Wang, Chem. Mater., 2021, 33, 8854–8862 CrossRef CAS.
- K. T. Ilhan, S. Topal, M. S. Eroglu and T. Ozturk, RSC Adv., 2019, 9, 38407–38413 RSC.
- T. Celiker, R. İsci, K. Kaya, T. Ozturk and Y. Yagci, J. Polym. Sci., 2020, 58, 2327–2334 CrossRef CAS.
- D. Meschede, Gerthsen Physik, Springer, Berlin, Heidelberg, 2010 Search PubMed.
- P. W. Atkins and J. de Paula, Physical Chemistry, Oxford University Press, Oxford, 2006 Search PubMed.
- E. von Hauff, J. Phys. Chem. C, 2019, 123, 11329–11346 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- T. Nishinaga, Organic redox systems, Synthesis, properties, and applications, Wiley, Hoboken, New Jersey, 2016 Search PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- J. Sworakowski and K. Janus, Org. Electron., 2017, 48, 46–52 CrossRef CAS.
- Y. Xie, W. Wang, W. Huang, F. Lin, T. Li, S. Liu, X. Zhan, Y. Liang, C. Gao, H. Wu and Y. Cao, Energy Environ. Sci., 2019, 12, 3556–3566 RSC.
- A. Moliton and J.-M. Nunzi, Polym. Int., 2006, 55, 583–600 CrossRef CAS.
- J. Yan and B. R. Saunders, RSC Adv., 2014, 4, 43286–43314 RSC.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and P. W. M. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef.
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker and P. W. M. Blom, Appl. Phys. Lett., 2005, 86, 123509 CrossRef.
- C. Xiong, J. Sun, H. Yang and H. Jiang, Sol. Energy, 2019, 178, 193–200 CrossRef CAS.
- R. Alessandri, S. Sami, J. Barnoud, A. H. Vries, S. J. Marrink and R. W. A. Havenith, Adv. Funct. Mater., 2020, 30, 2004799 CrossRef CAS.
- C. Xiong, J. Sun, J. Zhang and Y. Fu, Org. Electron., 2020, 78, 105559 CrossRef CAS.
- G. Haberfehlner, S. F. Hoefler, T. Rath, G. Trimmel, G. Kothleitner and F. Hofer, Micron, 2021, 140, 102981 CrossRef CAS PubMed.
- S. F. Hoefler, G. Haberfehlner, T. Rath, A. Keilbach, M. A. Hobisch, A. Dixon, E. Pavlica, G. Bratina, G. Kothleitner, F. Hofer and G. Trimmel, ACS Appl. Energy Mater., 2019, 2, 7535–7545 CrossRef CAS.
- J. Neu, S. Samson, K. Ding, J. J. Rech, H. Ade and W. You, Macromolecules, 2023, 56, 2092–2103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc01112g |
| This journal is © The Royal Society of Chemistry 2023 |