
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Centripetal triazine chromophores: towards efficient two-photon absorbers and highly emissive polyimide films†
Pavel
Šimon
 ab,
Milan
Klikar
a,
Zuzana
Burešová
a,
Chrisovalantou
Vourdaki
c,
Alexandros
Katsidas
c,
Jiří
Tydlitát
a,
Jiří
Kulhánek
a,
Jiří
Zelenka
b,
Mihalis
Fakis
*c and
Filip
Bureš
*a
ab,
Milan
Klikar
a,
Zuzana
Burešová
a,
Chrisovalantou
Vourdaki
c,
Alexandros
Katsidas
c,
Jiří
Tydlitát
a,
Jiří
Kulhánek
a,
Jiří
Zelenka
b,
Mihalis
Fakis
*c and
Filip
Bureš
*a
aInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, Pardubice, 53210, Czech Republic. E-mail: filip.bures@upce.cz; Web: https://bures.upce.cz Fax: +420 46 603 7068; Tel: +420 46 603 7099
bToseda s.r.o., U Panasonicu 376, Staré Čivice, Czech Republic
cDepartment of Physics, University of Patras, GR-26504, Patras, Greece
First published on 10th May 2023
Abstract
Four centripetal (D–π–)3–A chromophores based on s-triazine central acceptors and three amino donor-terminated branches were prepared. These chromophores were proved to be thermally stable up to 300 °C with a tuneable HOMO–LUMO gap (3.02–2.51 eV) and longest-wavelength absorption/emission maxima within the range of 390–427/485–551 nm. Relatively large Stokes shift up to 8000 cm−1, quantum yields up to 78% and fluorescence lifetimes ranging between 2 and 4 ns were observed based on the chromophore structure and the used environment. In general, varying the π-linker is proved to be a very useful strategy to tune the fundamental properties of these chromophores. Two-photon absorption measurements showed good to excellent cross-sections reaching 1000 GM. High-quality and well-transparent polyimide films doped with chromophores (0.1%) were further prepared, and showed bright emission in blue and yellow/green regions.
Introduction
Due to their peculiar and tuneable properties, conjugated π-systems end-capped with electron releasing (D) and withdrawing (A) moieties represent a very attractive group of organic materials.1 Besides an ordinary linear D–π–A arrangement, these systems can be arranged in different ways by varying the number and character of D, A and π-parts, while the resulting structure may more or less resemble letters. Based on this alphabet-inspired design, the current portfolio of extraordinary push–pull systems includes Y-, X-, V-, T-, L- and H-shaped molecules.2 Tripodal or star (Y)-shaped push–pull systems that feature three interconnected branches are of particular interest as efficient two-photon absorbing (2PA) molecules. These molecules may adopt two principal arrangements – centrifugal (A–π–)3–D or centripetal (D–π–)3–A.3 The vast majority of centrifugal push–pull molecules were based on triphenylamine central donors.4 On the other hand, (symmetric) centripetal Y-shaped systems with a central electron acceptor are scarce.Aromatic heterocycles such as electron deficient six-membered (di/tri)azines are well-suited as central electron withdrawing moieties.5 Especially 1,3,5-triazine (s-triazine) can be employed for the construction of centripetal systems as it is the only azine scaffold allowing the construction of symmetrical molecules. Moreover, its synthesis and modification are historically well-explored in the chemistry of triazine dyes. Star-shaped triazine derivatives have been utilized as charge-transfer chromophores6 and fluorophores,7 emissive layers of organic light-emitting diodes (OLEDs),8 light harvesters in dye-sensitized solar cells (DSSCs),9 liquid crystals,10 polymeric cathode materials of Li-ion batteries11 or covalent-organic frameworks (COFs). Second- and third-order nonlinear optical (NLO) properties, especially second-harmonic generation (SHG),12 2PA13 and 2PA-initiated polymerization14 of a few triazine derivatives were also reported.
Aromatic polyimides (PIs) are polymeric materials with high thermal and mechanical robustness utilized across various high-tech areas such as aerospace and aviation, (opto)electronics (PLEDs, SCs, displays, filters, fibres, and lenses), automotive industry, medicine, coatings, membranes, packing, labelling etc.15 Flexible, optically transparent (colourless) and heat-resistant plastic substrates are key materials for these applications. There is a direct relationship between the electronic properties of aromatic diamines and dianhydrides constituting the PI's structure, while embedding alicyclic and perfluorinated components proved to be a suitable strategy bringing transparency.16 Emissive polymeric materials with persisting thermal, chemical and mechanical stability are another important class of organic materials. Due to a variety of additional valuable properties such as (thermally activated delayed) fluorescence, phosphorescence, redox activity and electrochromism, emissive PIs17 evolved into very attractive and burgeoning macromolecules with manifold applications.18
In our previous studies, we focused on star-shaped molecules bearing triphenylamine central donors.19 Subsequently, we also utilized a variety of azines either as peripheral acceptors or π-linkers.20 Herein, we report an opposite strategy using s-triazine as a central scaffold bearing three peripheral triphenylamines (centripetal (D–π–)3–A arrangement). Tripodal chromophores 1–4 (Fig. 1) were synthesized and their fundamental photophysical properties and 2PA activity were examined. These highly emissive fluorophores were further doped into a polyimide made of 1,2,4,5-cyclohexanetetracarboxylic dianhydride and 2,2-bis[4-(4-aminophenoxy)phenyl]propane. In contrast to known emissive PIs, bearing an organic emitter covalently linked to the PI's backbone, our simplified host/guest strategy involves dispersing chromophores 1–4 in the PI matrix.
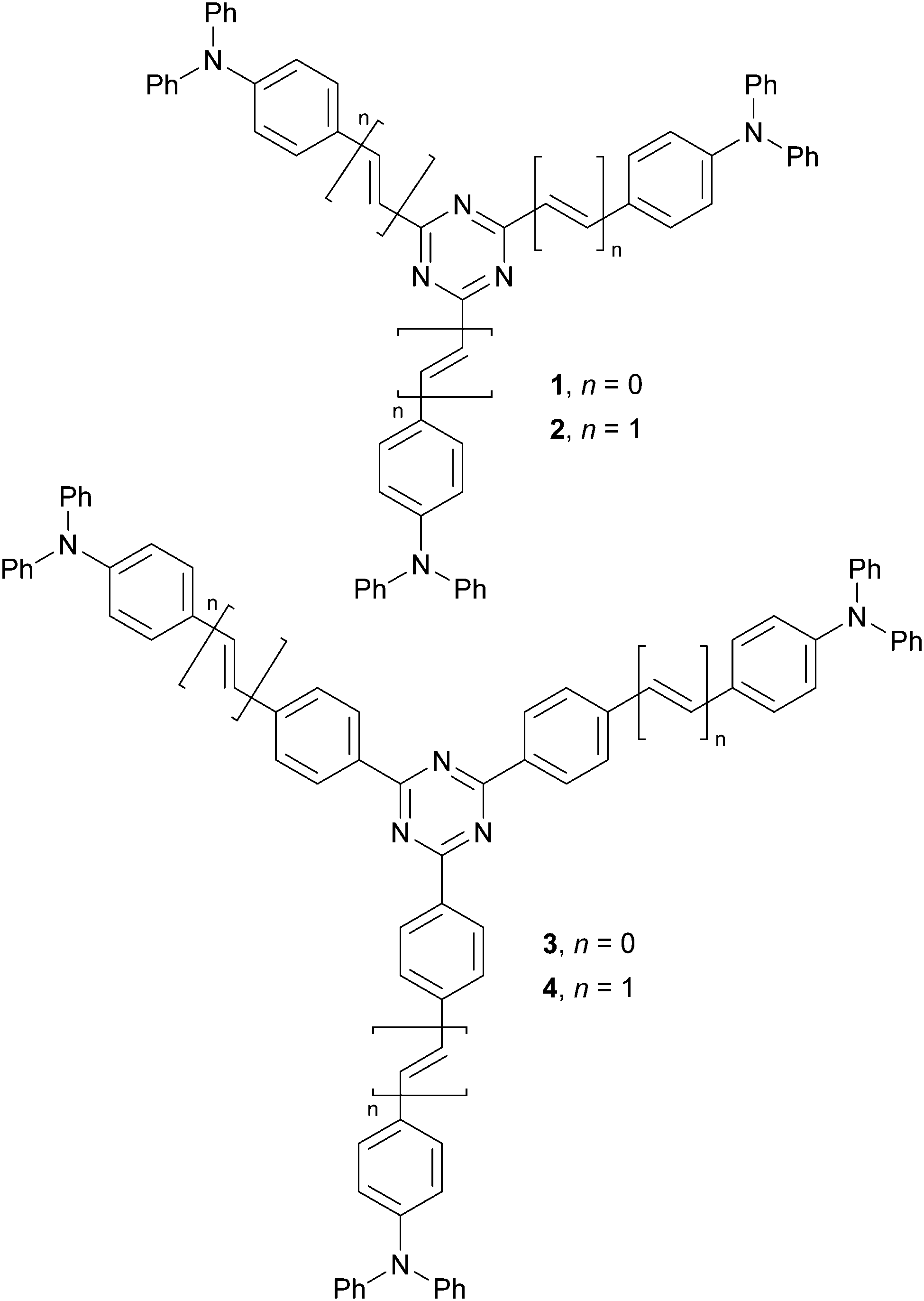 | ||
| Fig. 1 Molecular structure of centripetal triazine chromophores 1–4. | ||
Results and discussion
Centripetal triazines 1–4
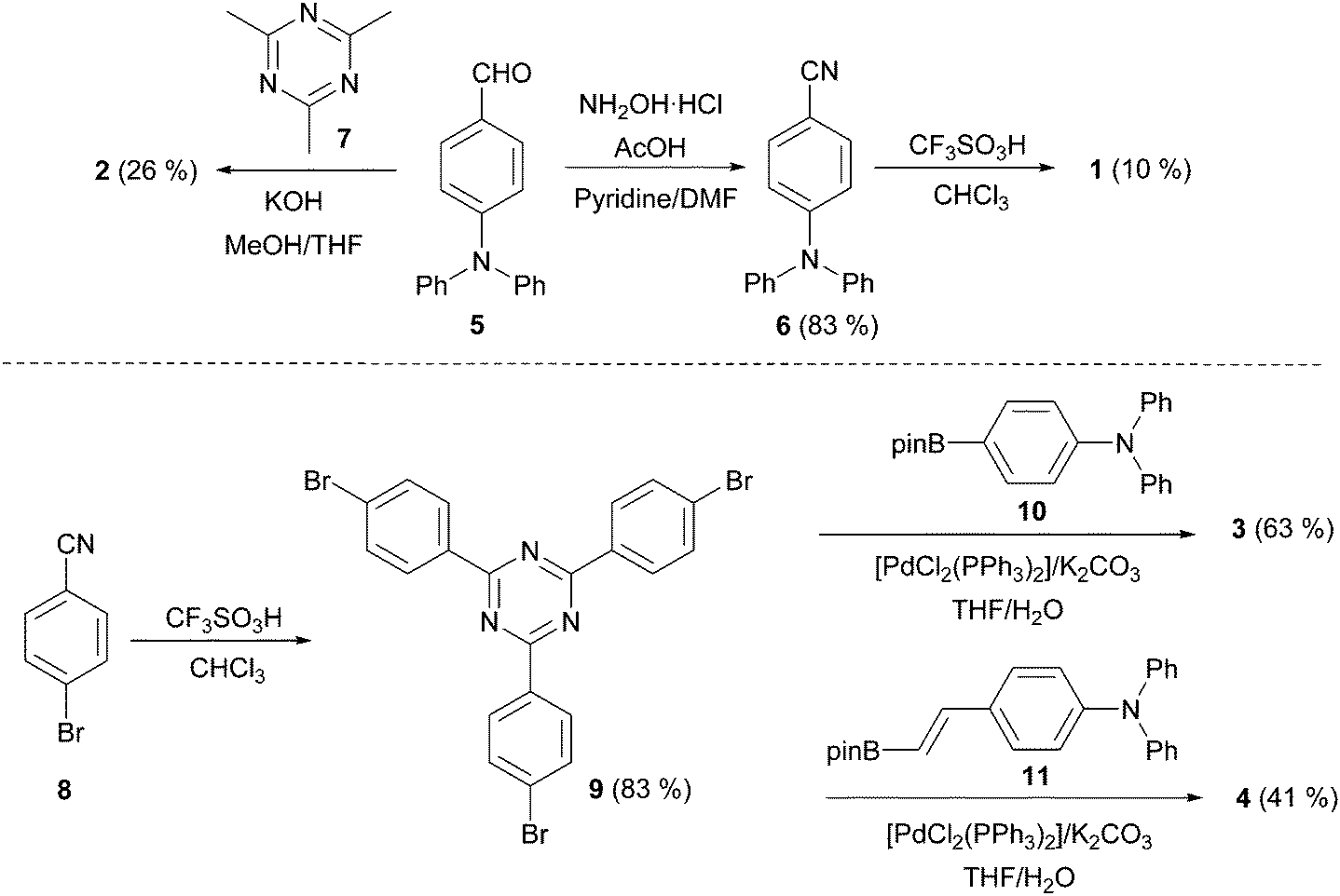 | ||
| Scheme 1 Synthesis of triazine chromophores 1–4. | ||
| Comp. | T m [°C] | T d [°C] | E p(ox1) [V] | E p(red1) [V] | ΔEc [eV] | E HOMO [eV] | E LUMO [eV] |
λ
Amax
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [nm eV−1]
[nm eV−1] |
ε [mol−1 dm3 cm−1] |
λ
Emax
f [nm eV−1] |
Φ
F
[%] |
Stokes shift [cm−1 eV−1] | δ 2PA [GM]/λ2PAmax [nm] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a T m = melting point (the point of intersection of a baseline and a tangent of thermal effect = onset). b T d = thermal decomposition (pyrolysis under a N2 atmosphere). c E p(ox1) and Ep(red1) are peak potentials of the first oxidation and reduction, respectively, as measured by CV at a scan rate of 100 mV s−1; all potentials are given vs. SSCE; ΔE = Ep(ox1) − Ep(red1). d Position of the shoulder of the first oxidation peak. e −EHOMO/LUMO = (Ep(ox1) + 0.036) and (Ep(red1) + 0.036) + 4.28 (vs. SCE);24 the increment of +0.036 V corresponds to the difference between the SCE (0.241 vs. SHE) and the SSCE (0.205 vs. SHE).25 f Measured in THF. g Molar absorption coefficient of the longest-wavelength absorption maxima. h Fluorescence quantum yield (± 10%) was determined relative to that of 9,10-bis(phenylethynyl)anthracene in cyclohexane (ΦF = 1.00).26 | |||||||||||||
| 1 | 325 | 380 | 1.33 | −1.69 | 3.02 | −5.65 | −2.63 | 393/3.16 | 62 100 | 485/2.56 | 74 | 4830/0.60 | 205/780 |
| 2 | — | 370 | 1.20d; 1.40 | −1.38 | 2.58 | −5.52 | −2.94 | 427/2.90 | 81 800 | 525/2.36 | 52 | 4370/0.54 | 620/820 |
| 3 | 229 | 350 | 1.25 | −1.53 | 2.78 | −5.57 | −2.79 | 390/3.18 | 70 800 | 519/2.39 | 78 | 6370/0.79 | 600/770 |
| 4 | — | 390 | 1.15d; 1.32 | −1.36 | 2.51 | −5.47 | −2.96 | 418/2.97 | 107 900 | 551/2.25 | 65 | 5770/0.72 | 870/830 |
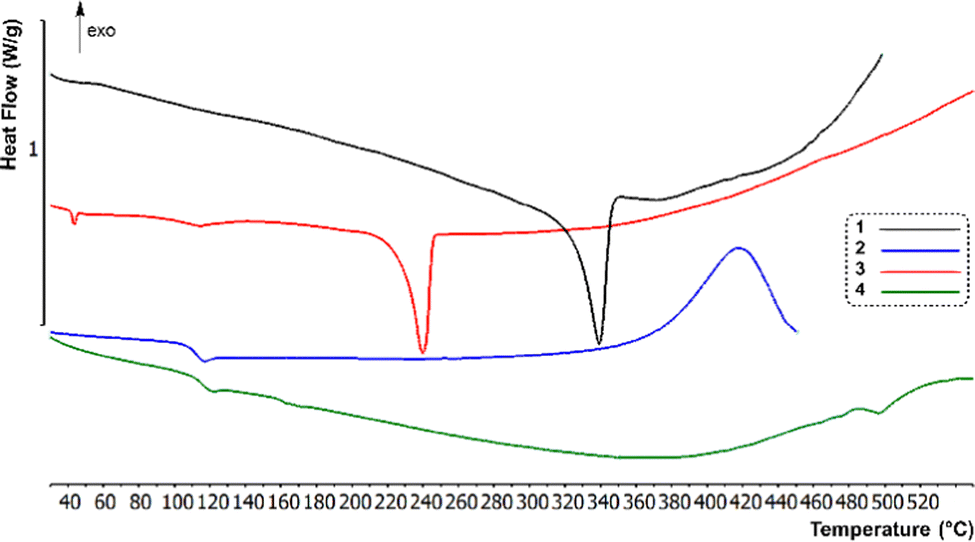 | ||
| Fig. 2 DSC curves of compounds 1–4 obtained using a heating rate of 10 °C min−1 under N2. | ||
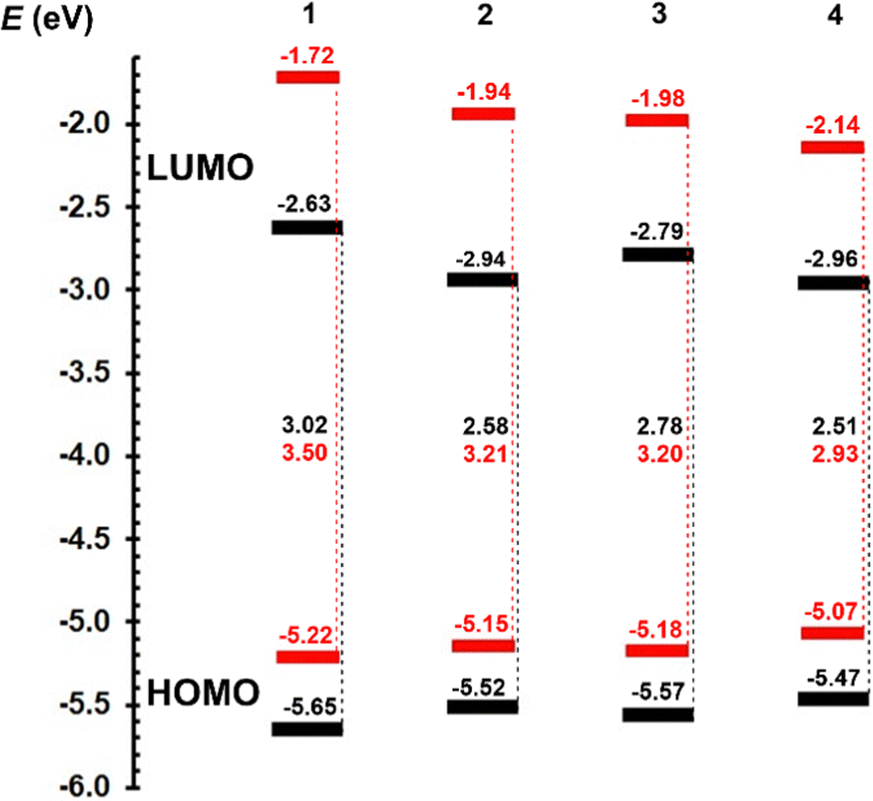 | ||
| Fig. 3 Energy level diagram of electrochemical (black) and DFT-calculated (red) HOMO and LUMO levels of centripetal triazines 1–4. | ||
The HOMO level increases either with planarization (1 → 2 or 3 → 4) or extension (1 → 3 or 2 → 4) of the π-linker by olefinic and 1,4-phenylene units. An opposite but more pronounced trends are seen for the LUMO levels. Thus, chromophore 4 with the most extended and planarized stilbenyl π-linker showed the narrowest HOMO–LUMO gap (2.51 eV), which is, however, not large compared to the gap of 2 bearing a styryl π-system (2.58 eV).
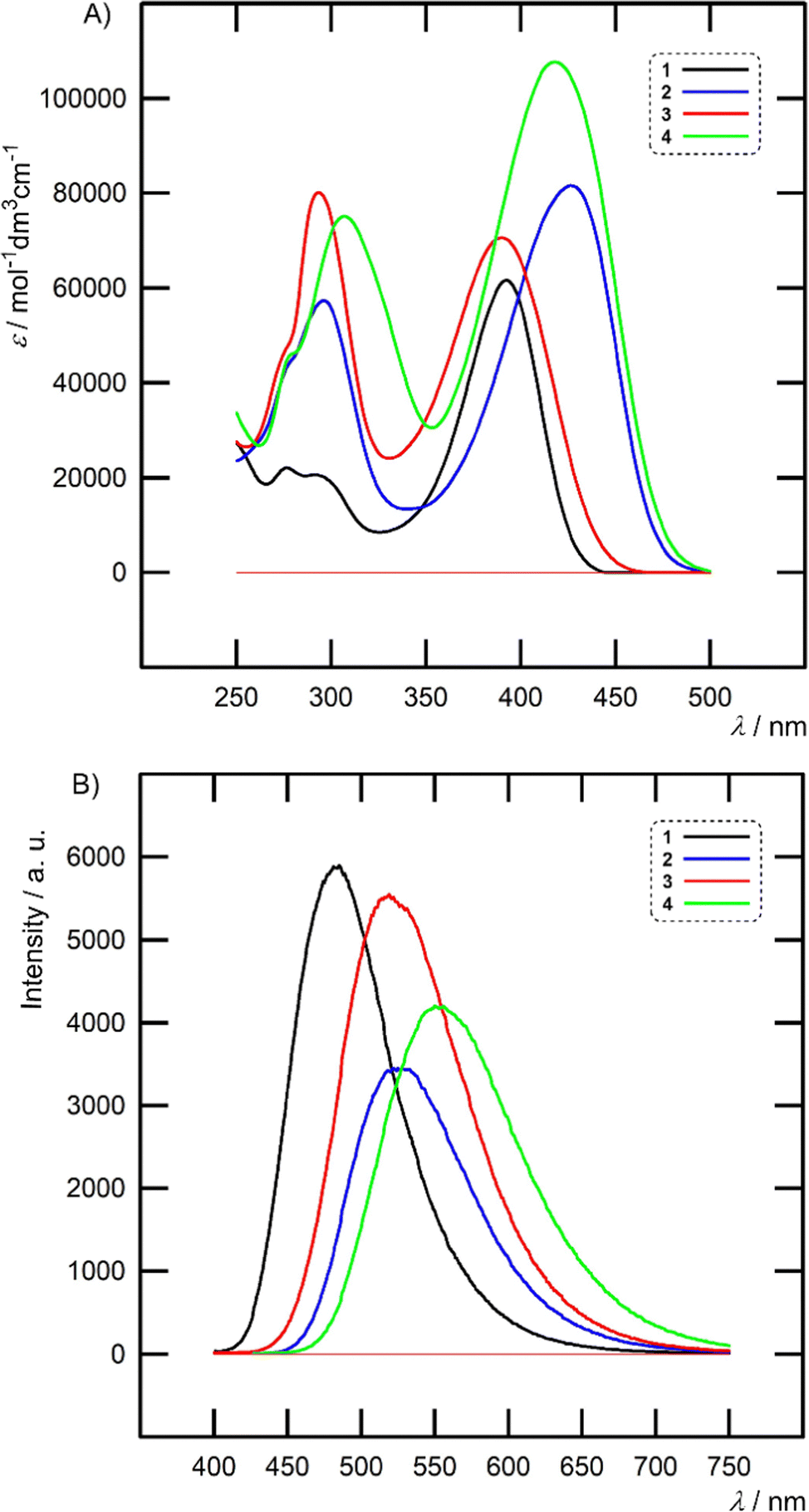 | ||
| Fig. 4 Absorption (A) and emission (B) spectra of chromophores 1–4 measured in THF (c = 1 × 10−5 mol l−1). | ||
Hence, the excited state of 1–4 is generally more stabilized by polar solvents, which is also reflected by a larger Stokes shift measured in THF/acetone (4370–6370/6047–8286 cm−1). The highest quantum yields were measured in toluene and ΦF values decreased steadily when going to THF and acetone. In general, planarized chromophores 2 and 4 with an additional olefinic linker showed lower quantum yields than nonplanar 1 and 3. Their ΦF values diminished to almost zero in acetone (Table S1 in the ESI†). Hence, rigid and twisted interconnection of two (hetero)aryls as presented in 1 and 3 allows more efficient and persisted fluorescence as compared to planarized styryl or stilbenyl π-linkers of 2 and 4. Moreover, concomitant E/Z photoisomerization may further reduce the quantum yield of 2 and 4.
Fig. 5 shows the fluorescence decays of chromophores 1–4 in the three solvents while the fitting parameters are summarized in Table S2 (ESI†). The nonplanar molecules 1 and 3 showed simpler decay curves than 2 and 4, which can be fitted adequately by a single exponential function in almost all cases. In toluene, molecules 1 and 3 exhibit single-exponential dynamics with lifetimes of 2.19 and 1.66 ns, respectively. The lifetime increase in THF becomes 3.78 and 3.08 ns, respectively. This elongation of the lifetime is an indication for an emitting state with different nature. In THF, a more relaxed emitting state with an ICT character is responsible for the emission. In acetone, the dynamics become bi-exponential with a fast component of ∼0.2 ns, indicating non-radiative mechanisms and a longer one due to the emission of the relaxed ICT state.
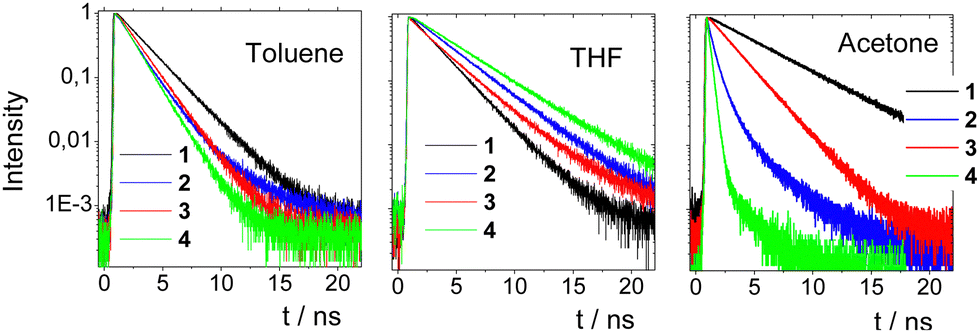 | ||
| Fig. 5 Fluorescence dynamics of chromophores 1–4 on the ns timescale in three solvents. | ||
On the other hand, 2 and 4 show bi-exponential dynamics in all solvents while their dynamics are generally shorter as compared to 1 and 3. In toluene and THF, the first component (0.5–1 ns) has a smaller amplitude and points to a structural relaxation or to emission from an unrelaxed state, while the second component represents the lifetime of the emitting state (1.5–2.9 ns).29a,b The longer component increases from toluene to THF as in the case of 1 and 3 due to the emission from a more relaxed emitting state. In acetone, both molecules exhibit faster dynamics pointing to pronounced non-radiative mechanisms, which is typical in polar solvents.29a,b
In order to shed more light on the fast relaxation phenomena of the excited state as well as on solute–solvent interactions, the fluorescence dynamics have also been examined within the first few tens of ps. Fig. 6 shows the dynamics for all molecules in THF, while the results for toluene are given in Fig. S19 (ESI†). All samples in both solvents exhibited fast decays at short wavelengths accompanied by a slow rise at long ones. This behaviour is associated with a gradual relaxation of the excited state and a red-shift of the emission spectrum. The ps dynamics have been fitted using a global method and the results are given in Table S3 (ESI†). In all cases, two relaxation components have were found, while a third component was also found, which corresponds to the lifetime of the excited state (taken from the TCSPC results) and was fixed. Comparing the non-planar and planarized molecules in toluene, the latter exhibit faster dynamics associated to a more efficient ICT process. For example, compound 2 exhibits relaxation components equal to 0.92 and 5.41 ps vs. 1.86 and 7.69 ps found for compound 1. Besides, in THF, being a more polar solvent, the dynamics are faster than in toluene, which is due to more enhanced solute–solvent interactions meaning that the solutes are facing a stronger solvent reaction field. In THF, a fast relaxation component of ∼1 ps was found for all molecules, which is similar to the average solvation time.30 The second component can be associated with slower solvation mechanisms as well as ICT formation.
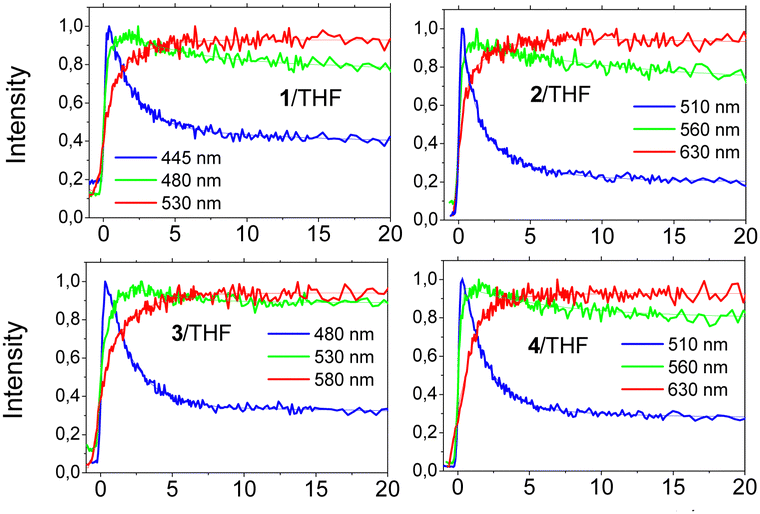 | ||
| Fig. 6 Fluorescence dynamics of chromophores 1–4 on the ps timescale in THF at different emission wavelengths. | ||
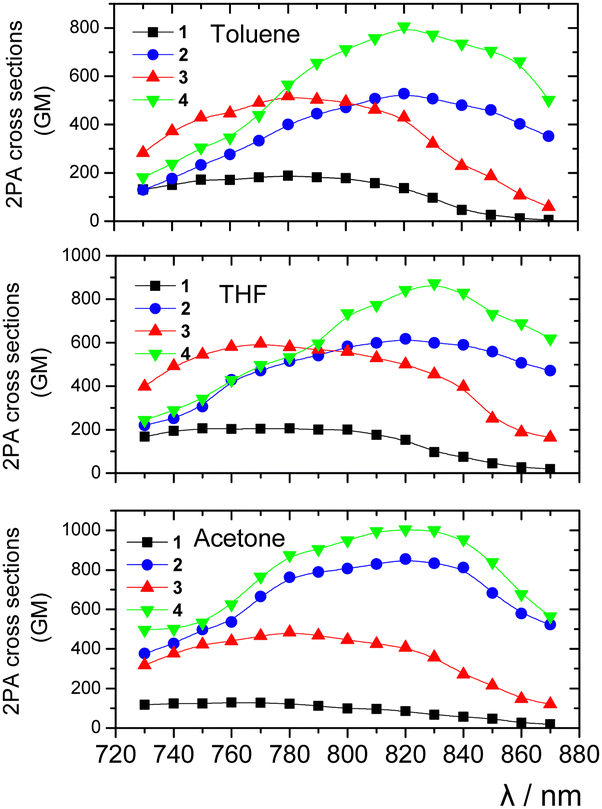 | ||
| Fig. 7 2PA cross-sections of compounds 1–4vs. wavelengths in the three solvents. | ||
In general, good to excellent 2PA properties have been found with δ2PA reaching 1000 GM. Table 1 summarizes the peak δ2PA for all molecules in THF. More specifically, in all used solvents, the planarized compounds 2 and 4 exhibit larger δ2PA values as compared to those of the non-planarized chromophores 1 and 3. In THF for example, the larger δ2PA for 4 was found 870 GM (830 nm), while for 3 it was 600 GM (770 nm). Comparing the compounds with or without an additional 1,4-phenylene moiety, the former exhibit larger δ2PA values in both planarized and non-planarized series. Overall, the elongation of the conjugated bridge by adding the phenylene moiety and olefinic linkers causes a significant increase in the 2PA activity, rendering compound 4 the most efficient 2PA molecule. Its largest δ2PA was found in acetone (1004 GM at 820 nm). Besides, adding the olefinic subunit also resulted in a red-shift of the 2PA spectra as was the case for the one-photon absorption. Very interesting conclusions can be drawn after comparing the 2PA and one-photon absorption spectra by rescaling them for a better comparison (Fig. S20, ESI†). The 2PA spectra of the non-planarized compounds 1 and 3 almost perfectly match the corresponding one-photon spectra. Such a behaviour is typical for dipolar linear compounds. On the other hand, in tripodal compounds, the excited state splits into three new states, where the two lower-energy states are degenerate. In such case, the lower states can be reached by both 1- and 2-photon transitions, while for the third state, which is higher in energy, only 2-photon transition is allowed.31 In the case of our tripodal compounds, the resemblance of the one-photon and 2PA spectra can be explained by a small excitonic coupling between the branches. A similar behaviour has been reported for tripodal molecules with a central benzene ring.32a For compounds 2 and 4, however, the 2PA peak is obviously blue-shifted as compared to twice the peak wavelength of the one-photon spectrum (vertical lines are shown in Fig. S20 (ESI†) for a better comparison of the spectral shifts). This means that the higher-lying excited state has the dominant contribution to 2PA, as expected for tripodal compounds with a significant excitonic coupling among the branches. Tripodal molecules having N or C+ atoms as the central electron donating or accepting core have also exhibited an enhancement of the 2PA efficiency of the higher energy state due to excitonic coupling.31,32b,c
As a further step, we tried to correlate the 2PA cross sections with the difference in permanent electric dipole moments between the ground state and the excited state, through the equation33
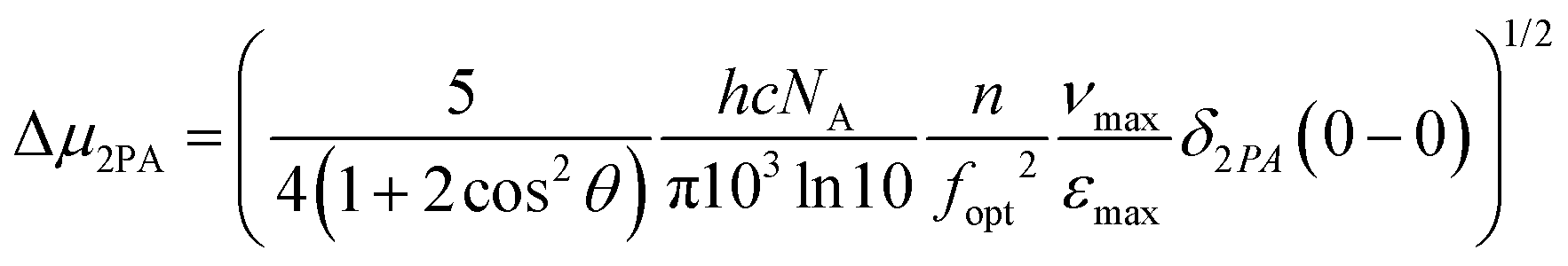 | (1) |
Next, these results are compared with those found using the solvatochromism method. In particular, Δμ is related to the slope of the Lippert–Mataga plot through the equation33c
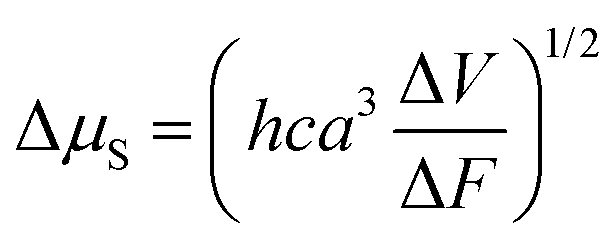 | (2) |
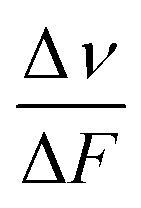 is the slope of the Stokes shift in cm−1versus the polarity function of the solvent
is the slope of the Stokes shift in cm−1versus the polarity function of the solvent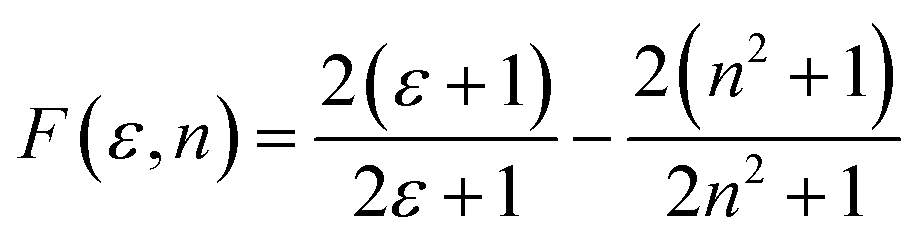 | (3) |
| Comp. | Symmetry group | E HOMO [eV] | E LUMO [eV] | ΔE [eV] | μ [D] | Cavity radius (Å) |
|---|---|---|---|---|---|---|
| 1 | D3 | −5.22 | −1.72 | 3.50 | 0 | 6.23 |
| 2 | C3 | −5.15 | −1.94 | 3.21 | 0.005 | 6.41 |
| 3 | D3 | −5.18 | −1.98 | 3.20 | 0 | 6.58 |
| 4 | C3 | −5.07 | −2.14 | 2.93 | 0.011 | 6.64 |
The ΔμS values range from 19.1 D to 24.6 D (Table S4, ESI†), showing a noticeable difference from the Δμ2PA value for 1 while being in relatively good agreement with the Δμ2PA values for 2–4. In general, the Δμ values found are within the same order of magnitude as those found in the literature and point to a significant increase in the polarity of the molecule in the excited state.33
Visualization of the frontier molecular orbitals of representative chromophore 4 (Fig. 8, see the ESI† for a full list) reveals the s-triazine-centred LUMO including one branch, the LUMO+1 spread over two branches and the LUMO+2 symmetrically localized on the whole π-system. The HOMO is localized on peripheral N,N-diphenylamino donor moieties, whereas the HOMO−1 and HOMO−2 are spread only over two opposite branches. Hence, a significant charge-transfer is revealed that is further pronounced by extension of the π-system. As expected, the calculated ground state dipole moments are zero for chromophores with D3-symmetry (1 and 3) or close to zero for C3-symmetrical molecules 2 and 4.
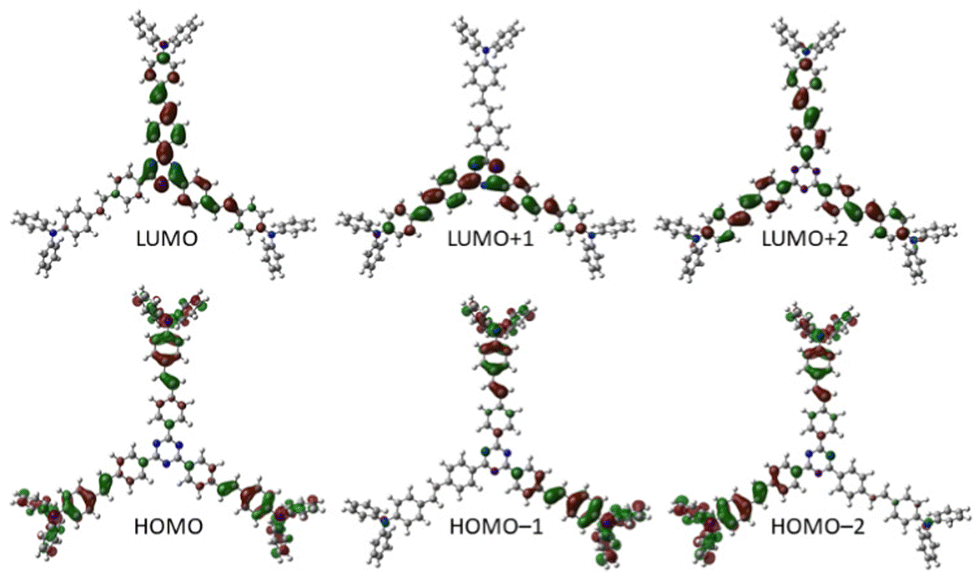 | ||
| Fig. 8 DFT-calculated visualization of frontier molecular orbitals in representative chromophore 4. | ||
Polyimide films
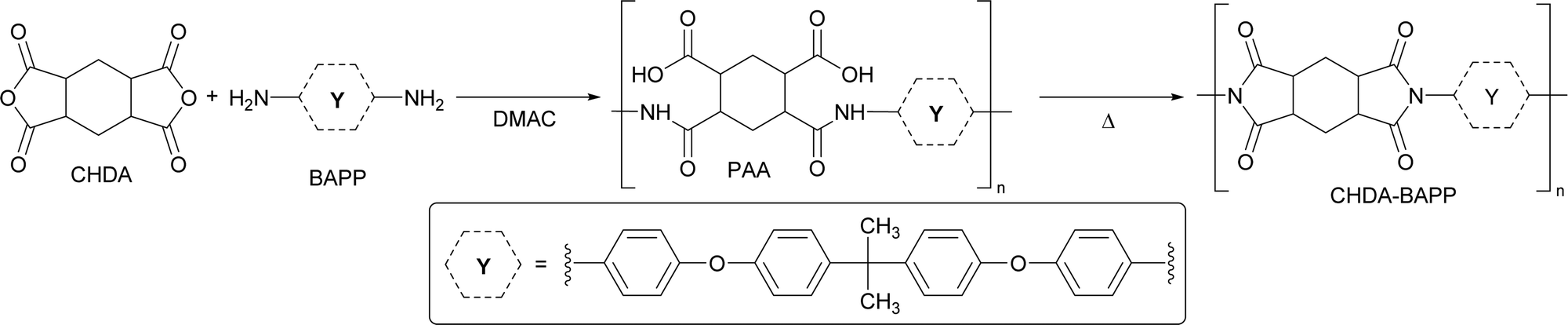 | ||
| Scheme 2 Molecular structure and preparation of polyimide CHDA–BAPP. | ||
| PI film |
T
5%d
[°C] |
T g [°C] | Char yieldc [wt%] | λ cut-off [nm eV−1] | T 400 [%] | T 700 [%] |
λ
Amax
[nm eV−1] |
λ
Emax
[nm eV−1] |
Stokes shift [cm−1 eV−1] | Chromaticity CIEg (x,y) | FWHMh [nm] | 〈τ〉i [ns] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The temperature at which 5% weight loss occurred was measured by TGA. b T g = glass-transition temperature measured by DSC. c Residual weight percentage at 600 °C under the N2 atmosphere as measured by TGA. d Transmittance at 400 and 700 nm, respectively. e The longest-wavelength absorption maxima. f Emission maxima. g CIE coordinates of the emission spectra calculated according to ref. 35. h Full-width at half-maximum of the emission spectra. i Average lifetimes of 1–4 in PI films. | ||||||||||||
| CHDA–BAPP | 454 | 258 | 30 | 295 | 86 | 91 | — | — | — | — | — | — |
| CHDA–BAPP + 1 | 449 | 261 | 32 | 293 | 46 | 89 | 392/3.16 | 461/2.69 | 3820/0.47 | 0.165,0.179 | 72 | 2.61 |
| CHDA–BAPP + 2 | 455 | 263 | 30 | 295 | 18 | 89 | 430/2.88 | 515/2.41 | 3840/0.48 | 0.288,0.531 | 95 | 3.44 |
| CHDA–BAPP + 3 | 460 | 262 | 40 | 296 | 53 | 90 | 390/3.18 | 495/2.51 | 5439/0.67 | 0.220,0.394 | 82 | 2.32 |
| CHDA–BAPP + 4 | 455 | 260 | 37 | 294 | 55 | 90 | 416/2.98 | 526/2.36 | 5027/0.62 | 0.333,0566 | 84 | 2.09 |
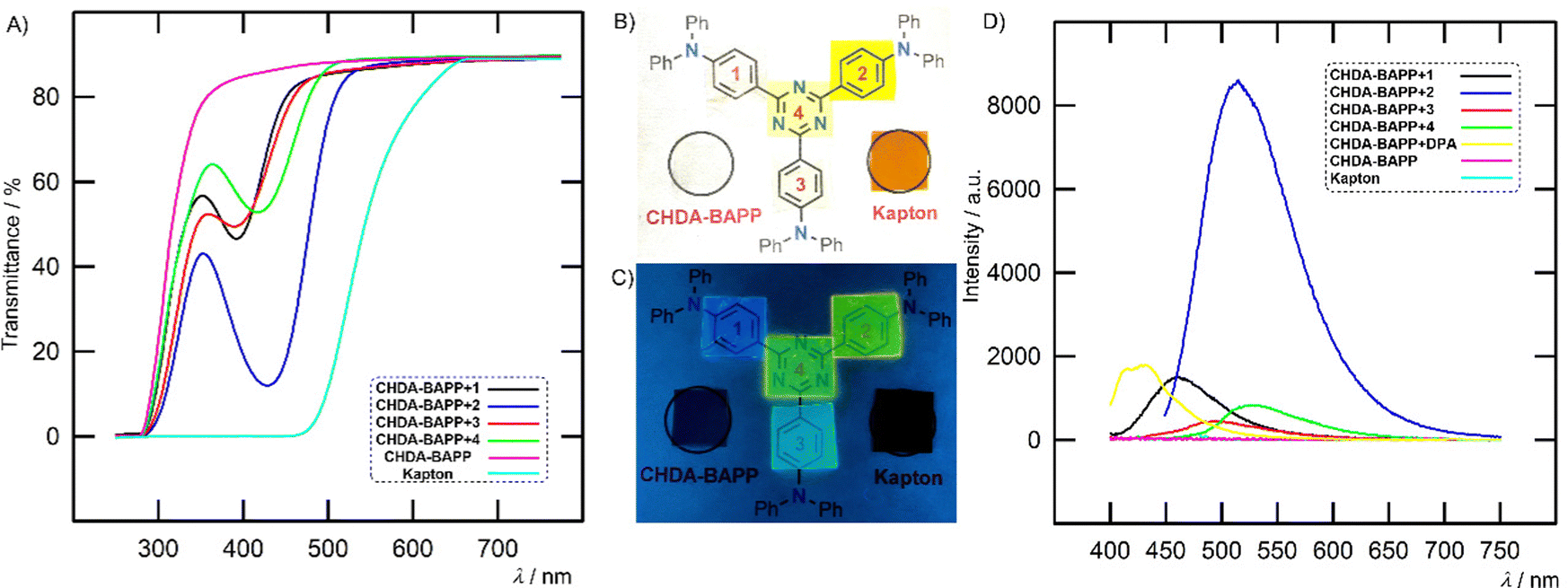 | ||
| Fig. 9 Transmittance of PI films (A), colour of CHDA–BAPP and films doped with 1–4 (numbered in red) under daylight (B) and 254 nm irradiation (C) and emission spectra of films (D). The spectra and native films (CHDA–BAPP doped with 0.1% of 1–4) are shown along with the reference Kapton film. | ||
While the pure polyimides (both CHDA–BAPP and Kapton) are non-emissive, the emission spectra of CHDA–BAPP films show one broad band at 461–526 nm depending on the embedded chromophores 1–4 (Fig. 9(D)). The emission maxima are red/blue-shifted (ΔλEmax ∼ 10–40 nm) as compared to that measured in toluene/THF (ENT = 0.099/0.207).36 Hence, the polarity of the CHDA–BAPP polyimide can be roughly estimated to lie between those of two solvents. Also, the average fluorescence lifetimes of 1–4 embedded in CHDA–BAPP films were found to lie between the values observed in toluene and THF (Table S5 and Fig. S38, ESI†) except for 2, whose lifetime in PI is increased and found to be much longer than that in THF.
The film doped with 2 showed significantly enhanced intensity as compared to other chromophores as well as to a CHDA–BAPP film doped with benchmark diphenylanthracene (DPA). While nonplanar chromophores 1 and 3 can be considered as blue emitters (Fig. 9(C)), planarization of the π-backbone by additional olefinic units red-shifted the emission of 2 and 4 to yellow/green colour. Hence, gradual extension and planarization of the chromophore structure allow tuning the emission colour of the films across the CIE chromaticity diagram (Fig. S32, ESI†). Especially CHDA–BAPP + 1 with CIE coordinates of (0.165, 0.179) can be considered as a promising blue emitter with a relatively narrow emission (FWHM = 72 nm).37
Conclusions
A series of star-shaped centripetal push–pull chromophores based on the s-triazine central acceptor, three N,N-diphenylamino donors and a π-linker of systematically varied length have been synthesized and further investigated. These chromophores exhibited thermal stability exceeding 300 °C, while reversible reduction of the triazine core and kinetically irreversible oxidation of the peripheral amino donors were revealed by electrochemistry. A clear trend in tuning the HOMO–LUMO gap via extension of the π-system and its planarization via an additional olefinic subunit was observed both experimentally and theoretically. Fundamental one- and two-photon absorption and emission properties were screened in various solvents and a distinct intramolecular charge-transfer band was recorded. The emission spectra of chromophores showed one well-resolved band, whose position is strongly influenced by the chromophore structure and the used environment. Single- and bi-exponential fluorescence dynamics were revealed for nonplanar and planar chromophores. Planarity and π-system extension also affect two-photon absorption properties, the latter showed significantly enhanced 2PA activity reaching 1000 GM. The synthesized chromophores were further embedded into polyimide films prepared from 1,2,4,5-cyclohexanetetracarboxylic dianhydride and 2,2-bis[4-(4-aminophenoxy)phenyl]propane. In contrast to benchmark Kapton, these polyimide films are optically well-transparent with thermal robustness persisting up to 450 °C, and can be used as a polymeric matrix for chromophores emitting in the near UV and visible regions. Although the films of nonplanar chromophores emitted in the blue region, planarization and π-linker extension shifted the emission to the yellow/green region. In summary, the developed triazine-derived tripodal chromophores 1–4 proved to be versatile organic materials exhibiting excellent 2PA activity and can be used to prepare emissive polyimide films of high quality.Experimental section
The detailed syntheses and characterizations of target chromophores 1–4, intermediates and polyimide films are given in the ESI.†Author contributions
F. B.: conceptualization. P. Š., M. K., Z. B., C. V., A. K.: investigation. J. T.: software. J. K., J. Z.: supervision. F. B., M. F., P. Š., M. K., Z. B.: writing – original draft. F. B., M. F.: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. Š. and J. Z. acknowledge the financial support from the Ministry of Education, Youth and Sport of the Czech Republic (CZ.02.1.01/0.0/0.0/17_048/0007376, NANOMAT).Notes and references
- F. Bureš, RSC Adv., 2014, 4, 58826 RSC.
- M. Klikar, P. Solanke, J. Tydlitát and F. Bureš, Chem. Rec., 2016, 16, 1886 CrossRef CAS PubMed.
- D. Cvejn, E. Michail, I. Polyzos, N. Almonasy, O. Pytela, M. Klikar, T. Mikysek, V. Giannetas, M. Fakis and F. Bureš, J. Mater. Chem. C, 2015, 3, 7345 RSC.
- (a) X. Lian, Z. Zhao and D. Cheng, Mol. Cryst. Liq. Cryst., 2017, 648, 223 CrossRef CAS; (b) P. Blanchard, C. Malacrida, C. Cabanetos, J. Roncali and S. Ludwigs, Polym. Int., 2019, 68, 589 CrossRef CAS; (c) A. Karak, S. K. Manna and A. K. Mahapatra, Anal. Methods, 2022, 14, 972 RSC; (d) A. Mahmood, Sol. Energy, 2016, 123, 127 CrossRef CAS; (e) P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348 RSC; (f) A. Iwan and D. Sek, Prog. Polym. Sci., 2011, 36, 1277 CrossRef CAS; (g) D. Devadiga, M. Selvakumar, P. Shetty, M. S. Santosh, R. S. Chandrabose and S. Karazhanov, Int. J. Energy Res., 2021, 45, 6584 CrossRef CAS.
- (a) S. Achelle, J. Rodríguez-López, F. Bureš and F. Robin-le Guen, Chem. Rec., 2020, 20, 440 CrossRef CAS PubMed; (b) S. Achelle and N. Plé, Curr. Org. Synth., 2012, 9, 163 CrossRef CAS.
- (a) J. Hu, Y. Li, H. Zhu, S. Qiu, G. He, X. Zhu and A. Xia, ChemPhysChem, 2015, 16, 2357 CrossRef CAS PubMed; (b) H. Meier, E. Karpuk and H. C. Holst, Eur. J. Org. Chem., 2006, 2609 CrossRef CAS; (c) H. Meier, H. C. Holst and A. Oehlhof, Eur. J. Org. Chem., 2003, 4173 CrossRef CAS.
- V. S. Padalkar, V. S. Patil and N. Sekar, Chem. Cent. J., 2011, 5, 77 CrossRef CAS PubMed.
- Y. Zhang, Y. Zheng, B. Wang, H. Ran, X. Wang, J.-Y. Hu and Q. Wang, J. Mater. Chem. C, 2020, 8, 4461 RSC.
- (a) H. Zhang, X.-F. Zang, Y.-P. Hong and Z.-E. Chen, Synth. Met., 2021, 280, 116882 CrossRef CAS; (b) J. Liu, K. Wang, F. Xu, Z. Tang, W. Zheng, J. Zhang, C. Li, T. Yu and X. You, Tetrahedron Lett., 2011, 52, 6492 CrossRef CAS.
- T. Yasuda, T. Shimizu, F. Liu, G. Ungar and T. Kato, J. Am. Chem. Soc., 2011, 133, 13437 CrossRef CAS PubMed.
- Z. Chen, C. Su, X. Zhu, R. Xu, L. Xu and C. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2574 CrossRef CAS.
- Y. Z. Cui, Q. Fang, H. Lei, G. Xue and W. T. Yu, Chem. Phys. Lett., 2003, 377, 507 CrossRef CAS.
- (a) Y. Jiang, Y. Wang, B. Wang, J. Yang, N. He, S. Qian and J. Hua, Chem. – Asian J., 2011, 6, 157 CrossRef CAS PubMed; (b) L. Zou, Z. Liu, X. Yan, Y. Liu, Y. Fu, J. Liu, Z. Huang, X. Chen and J. Qin, Eur. J. Org. Chem., 2009, 5587 CrossRef CAS; (c) Y. Jiang, Y. Wang, J. Hua, J. Tang, B. Li, S. Qian and H. Tian, Chem. Commun., 2010, 46, 4689 RSC; (d) S. Zeng, X. Ouyang, H. Zheng, W. Ji and Z. Ge, Dyes Pigm., 2012, 94, 290 CrossRef CAS; (e) L. Wei, J. P. Shi, Z. Q. Zhou, Y. P. Cui, H. W. Hu and G. Y. Lu, Chin. Chem. Lett., 2012, 23, 867 CrossRef CAS; (f) L. Liu, Z. Q. Zhou, J. P. Shi, C. G. Lu, Y. P. Cui and G. Y. Lu, Chin. Chem. Lett., 2011, 22, 1147 CAS.
- M. Tromayer, P. Gruber, A. Rosspeintner, A. Ajami, W. Husinsky, F. Plasser, L. González, E. Vauthey, A. Ovsiniakov and R. Liska, Sci. Rep., 2018, 8, 17273 CrossRef PubMed.
- (a) M. Hasegawa, Polymers, 2017, 9, 520 CrossRef PubMed; (b) P. K. Tapaswi and C.-S. Ha, Macromol. Chem. Phys., 2019, 220, 1800313 CrossRef; (c) H.-J. Ni, J.-G. Liu, Z.-H. Wang and S.-Y. Yang, J. Ind. Eng. Chem., 2015, 28, 16 CrossRef CAS; (d) A. S. Hicyilmaz and A. C. Bedeloglu, SN Appl. Sci., 2021, 3, 363 CrossRef.
- (a) S. Ando, T. Matsuura and S. Sasaki, Polym. J., 1997, 29, 69 CrossRef CAS; (b) T. Matsumoto, D. Mikami, T. Hashimoto, M. Kaise, R. Takahashi and S. Kawabata, J. Phys.: Conf. Ser., 2009, 187, 012005 CrossRef; (c) H. Yeo, M. Goh, B.-C. Ku and N.-H. You, Polymer, 2015, 76, 280 CrossRef CAS; (d) M. Yamada, M. Kusama, T. Matsumoto and T. Kurosaki, J. Org. Chem., 1992, 57, 6075 CrossRef CAS; (e) C. Cao, L. Liu, X. Ma, X. Zhang and T. Lv, Polim.: Cienc. Tecnol., 2020, 30, e2020017 CrossRef.
- M. Hagesawa and K. Horie, Prog. Polym. Sci., 2001, 26, 259 CrossRef.
- (a) Y. Long, X. Chen, H. Wu, Z. Zhou, S. S. Babu, M. Wu, J. Zhao, M. P. Aldred, S. Liu, X. Chen, Z. Chi, J. Xu and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7220 CrossRef CAS PubMed; (b) Z. Zhou, Y. Zhang, S. Liu, Z. Chi, X. Chen and J. Xu, J. Mater. Chem. C, 2016, 4, 10509 RSC; (c) K. Kanosue, S. Hirata, M. Vacha, R. Augulis, V. Gulbinas, R. Ishige and S. Ando, Mater. Chem. Front., 2019, 3, 39 RSC; (d) H.-J. Yen, J.-H. Wu, W.-C. Wang and G.-S. Liou, Adv. Opt. Mater., 2013, 1, 668 CrossRef; (e) Z. Zhou, Y. Long, X. Chen, T. Yang, J. Zhao, Y. Meng, Z. Chi, S. Liu, X. Chen, M. P. Aldred, J. Xu and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 34198 CrossRef CAS PubMed; (f) A. Iqbal, H. M. Siddiqi, M. Zubair, T. Akhter, O. O. Park and A. Saeed, High Perform. Polym., 2020, 32, 231 CrossRef CAS; (g) A. Iqbal, S. H. Lee, O. O. Park, H. M. Siddiqi and T. Akhter, New J. Chem., 2016, 40, 5285 RSC; (h) K. Su, N. Sun, X. Tian, S. Guo, Z. Yan, D. Wang, H. Zhou, X. Zhao and C. Chen, Dyes Pigm., 2019, 171, 107668 CrossRef CAS; (i) Y.-W. Liu, L.-S. Tang, L.-J. Qu, S.-W. Liu, Z.-G. Chi, Y. Zhang and J.-R. Xu, Chin. J. Polym. Sci., 2019, 37, 416 CrossRef CAS; (j) T. Xiao, X. Fan, D. Fan and Q. Li, Polym. Bull., 2017, 74, 4561 CrossRef CAS.
- (a) K. Seintis, I.-K. Kalis, M. Klikar, F. Bureš and M. Fakis, Phys. Chem. Chem. Phys., 2020, 22, 16681 RSC; (b) M. Klikar, K. Seintis, I. Polyzos, O. Pytela, T. Mikysek, N. Almonasy, M. Fakis and F. Bureš, ChemPhotoChem, 2018, 2, 465 CrossRef CAS; (c) D. Cvejn, E. Michail, K. Seintis, M. Klikar, O. Pytela, T. Mikysek, N. Almonasy, M. Ludwig, V. Giannetas, M. Fakis and F. Bureš, RSC Adv., 2016, 6, 12819 RSC; (d) K. Seintis, D. Agathangelou, D. Cvejn, N. Almonasy, F. Bureš, V. Giannetas and M. Fakis, Phys. Chem. Chem. Phys., 2017, 19, 16485 RSC; (e) F. Kournoutas, K. Seintis, N. Karakostas, J. Tydlitát, S. Achelle, G. Pistolis, F. Bureš and M. Fakis, J. Phys. Chem. A, 2019, 123, 417 CrossRef CAS PubMed.
- (a) F. Bureš, D. Cvejn, K. Melánová, L. Beneš, J. Svoboda, V. Zima, O. Pytela, T. Mikysek, Z. Růžičková, I. V. Kityk, A. Wojciechowski and N. AlZayed, J. Mater. Chem. C, 2016, 4, 468 RSC; (b) M. Klikar, D. Georgiou, I. Polyzos, M. Fakis, Z. Růžičková, O. Pytela and F. Bureš, Dyes Pigm., 2022, 201, 110230 CrossRef CAS; (c) M. Fecková, I. K. Kalis, T. Roisnel, P. le Poul, O. Pytela, M. Klikar, F. Robin-le Guen, F. Bureš, M. Fakis and S. Achelle, Chem. – Eur. J., 2021, 27, 1145 CrossRef PubMed; (d) M. Fecková, P. le Poul, F. Bureš, F. Robin-le Guen and S. Achelle, Dyes Pigm., 2020, 182, 108659 CrossRef; (e) F. Kournoutas, A. Fihey, J.-P. Malval, A. Spangenberg, M. Fecková, P. le Poul, C. Katan, F. Robin-le Guen, F. Bureš, S. Achelle and M. Fakis, Phys. Chem. Chem. Phys., 2020, 22, 4165 RSC.
- (a) Z. Chen, C. Su, X. Zhu, R. Xu, L. Xu and C. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2574 CrossRef CAS; (b) Y. Zhang, Y. Zheng, B. Wang, H. Ran, X. Wang, J. Y. Hu and Q. Wang, J. Mater. Chem. C, 2020, 8, 4461 RSC.
- W. Li, Y. Dai, C. Zhang and Z. Chen, 2018, CN109053609A.
- G. Kim, T. Shiraki and T. Fujigaya, Bull. Chem. Soc. Jpn., 2020, 93, 414 CrossRef CAS.
- (a) T. J. Carter, R. Mohtadi, T. S. Arthur, F. Mizuno, R. Zhang, S. Shirai and J. W. Kampf, Angew. Chem., Int. Ed., 2014, 53, 3173 CrossRef CAS PubMed; (b) A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894 CrossRef CAS PubMed.
- D. T. Sawyer, A. Sobkowiak and J. L. Roberts, Electrochemistry for Chemists, J. Wiley and Sons Inc., 2nd edn, 1995 Search PubMed.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Chem. Mater., 2013, 25, 3766 CrossRef CAS.
- F. Xu, Z. Wang and Q. Gong, Opt. Mater., 2007, 29, 723 CrossRef CAS.
- (a) M. Fakis, V. Petropoulos, P. Hrobárik, J. Nociarová, P. Osuský, M. Maiuri and G. Cerullo, J. Phys. Chem. B, 2022, 126, 8532 CrossRef CAS PubMed; (b) A. Cesaretti, T. Bianconi, M. Coccimiglio, N. Montegiove, Y. Rout, P. L. Gentili, R. Misra and B. Carlotti, J. Phys. Chem. C, 2022, 126, 10429 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311 CrossRef CAS.
- C. Katan, F. Terenziani, O. Morgin, M. H. V. Werts, L. Porrès, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024 CrossRef CAS PubMed.
- (a) F. Terenziani, C. Le Droumaguet, C. Katan, O. Mongin and M. Blanchard-Desce, ChemPhysChem, 2007, 8, 723 CrossRef CAS PubMed; (b) J. C. Collings, S. Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L. O. Plsson, A. Beeby, J. A. Mosely, H. Martin Kaiser, D. Kaufmann, W. Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem. – Eur. J., 2009, 15, 198 CrossRef CAS PubMed; (c) N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J. L. L. Brédas and J. Perry, J. Phys. Chem. A, 2012, 116, 3781 CrossRef CAS PubMed.
- (a) M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zalesny, W. Bartkowiak, H. Angren, S. Canuto, L. De Boni, E. Ishow and C. R. Mendonca, Sci. Rep., 2014, 4, 4447 CrossRef PubMed; (b) A. Rebane, M. Drobizhev, N. S. Makarov, E. Beuerman, S. Tillo and T. Hughes, J. Lumin., 2010, 130, 1619 CrossRef CAS; (c) A. Rebane, M. Drobizhev, N. S. Makarov, E. Beuerman, J. E. Haley, D. M. Krein, A. R. Burke, J. L. Flikkema and T. M. Cooper, J. Phys. Chem. A, 2011, 115, 4255 CrossRef CAS PubMed; (d) L. H. Zucolotto Cocca, A. Gasparotto Pelosi, L. M. G. Abegao, R. de, Q. Garcia, J.-C. Mulatier, D. Pitrat, C. Barsu, C. Andraud, C. R. Mendonça, M. G. Vivas and L. De Boni, Phys. Chem. Chem. Phys., 2023, 25, 5021 RSC; (e) A. Rebane, M. Drobizhev, N. S. Makarov, G. Wicks, P. Wnuk, Y. Stepanenko, J. E. Haley, D. M. Krein, J. L. Fore, A. R. Burke, J. E. Slagle, D. G. McLean and T. M. Cooper, J. Phys. Chem. A, 2014, 118, 3749 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox,Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- E. H. H. Hasabeldaim, CIE chromaticity diagram 1931, 2021, https://sciapps.sci-sim.com/CIE1931.html.
- F. Bureš, O. Pytela and F. Diederich, J. Phys. Org. Chem., 2009, 22, 155 CrossRef.
- (a) X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913 RSC; (b) M. Xie, M. Sun, S. Xue and W. Yang, Dyes Pigm., 2022, 208, 110799 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, NMR, HR-MS, thermal properties, electrochemistry, 1PA and 2PA data, calculations, and films. See DOI: https://doi.org/10.1039/d3tc00877k |
| This journal is © The Royal Society of Chemistry 2023 |