
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silvanite AuAgTe4: a rare case of gold superconducting material†
Yehezkel
Amiel‡
a,
Gyanu P.
Kafle‡
 b,
Evgenia V.
Komleva
cd,
Eran
Greenberg
ef,
Yuri S.
Ponosov
c,
Stella
Chariton
f,
Barbara
Lavina
fg,
Dongzhou
Zhang
fh,
Alexander
Palevski
a,
Alexey V.
Ushakov
c,
Hitoshi
Mori
b,
Daniel I.
Khomskii
i,
Igor I.
Mazin
j,
Sergey V.
Streltsov
cd,
Elena R.
Margine
b and
Gregory Kh.
Rozenberg
*a
b,
Evgenia V.
Komleva
cd,
Eran
Greenberg
ef,
Yuri S.
Ponosov
c,
Stella
Chariton
f,
Barbara
Lavina
fg,
Dongzhou
Zhang
fh,
Alexander
Palevski
a,
Alexey V.
Ushakov
c,
Hitoshi
Mori
b,
Daniel I.
Khomskii
i,
Igor I.
Mazin
j,
Sergey V.
Streltsov
cd,
Elena R.
Margine
b and
Gregory Kh.
Rozenberg
*a
aSchool of Physics and Astronomy, Tel Aviv University, 69978 Tel Aviv, Israel. E-mail: emtsm@tauex.tau.ac.il; Fax: +972-26785301; Tel: +972-504071266
bDepartment of Physics, Applied Physics, and Astronomy, Binghamton University-SUNY, Binghamton, New York 13902, USA
cM.N. Mikheev Institute of Metal Physics UB RAS, S. Kovalevskaya Str. 18, 620137, Ekaterinburg, Russia
dUral Federal University, Mira St. 19, 620002 Ekaterinburg, Russia
eApplied Physics Division, Soreq NRC, Yavne 81800, Israel
fGSECARS, University of Chicago, Chicago, Illinois 60637, USA
gX-Ray Science Division, Advanced Photon Source, Argonne National Lab, 60439, USA
hSchool of Ocean and Earth Science and Technology, University of Hawai'i at Manoa, Honolulu, HI 96822, USA
iII. Physikalisches Institut, Universität zu Köln, Zülpicher Straße 77, D-50937 Köln, Germany
jDepartment of Physics and Astronomy, George Mason University, Fairfax, USA
First published on 7th July 2023
Abstract
Gold is one of the most inert metals, forming very few compounds, some with rather interesting properties, and only a few of them are currently known to be superconducting under certain conditions. Compounds of another noble element, Ag, are also relatively rare, and very few of them are superconducting. Finding new superconducting materials containing gold (and silver) is a challenge – especially having in mind that the best high-Tc superconductors under normal conditions are based upon their rather close congener, Cu. Here we report combined X-ray diffraction, Raman, and resistivity measurements, as well as first-principles calculations, to explore the effect of hydrostatic pressure on the properties of the sylvanite mineral, AuAgTe4. Our experimental results, supported by density functional theory, reveal a structural phase transition at ∼5 GPa from a monoclinic P2/c to P2/m phase, resulting in almost identical coordinations of Au and Ag ions, with rather uniform interatomic distances. Furthermore, resistivity measurements show the onset of superconductivity at ∼1.5 GPa in the P2/c phase, followed by a linear increase of Tc up to the phase transition, with a maximum in the P2/m phase, and a gradual decrease afterwards. Our calculations indicate phonon-mediated superconductivity, with the electron–phonon coupling coming predominantly from the low-energy phonon modes. Thus, along with the discovery of a new superconducting compound of gold/silver, our results advance the understanding of the mechanism behind superconductivity in Au-containing compounds and dichalcogenides of other transition metals.
1 Introduction
Both gold and silver are known as inert metals, which do not easily react with other chemical elements. Among the gold-containing compounds (which are not very common anyway), only a few are superconducting and the situation with silver-based systems is similar. Famous due to an incommensurate crystal structure, the mineral calaverite, AuTe2,1,2 is a metal, but becomes superconducting3 under applied pressure or when doped with Pd or Pt. Some alloys and intermetallic compounds of gold, e.g., Au2Bi4 and Nb3Au5 are superconducting, but superconducting compounds combining Au and non-metallic elements are in fact extremely rare. While some other systems have been theoretically predicted to be superconducting with rather high critical temperatures,6 we are aware only of one more (besides the above-mentioned calaverite AuTe2) compound SrAuSi3, recently synthesised under high-pressure.7 Therefore, finding new superconducting compounds of gold (and silver) is a challenge.Whereas Cu2+ is quite stable, Ag and even more so Au are rarely seen in +2 oxidation state, so that typically compounds with nominally Ag2+ or Au2+ ions tend to disproportionate into Ag1+ + Ag3+. On the other hand, this very tendency of charge disproportionation could in principle be even more favourable for superconductivity: creating the states with zero and two holes in the d shell (like in Ag3+ or Au3+) reminds us of the tendency to form Cooper pairs, and it is often described theoretically in a “negative-U” Hubbard-like model, corresponding to an effective electron attraction, which could be beneficial for superconductivity.
Keeping all these facts in mind, we undertook a search for novel Au-containing superconductors, and indeed we found superconductivity in a “close relative” of calaverite AuTe2, in sylvanite, AuAgTe4, which can be considered as calaverite where half of the gold ions are replaced by silver. This rather rare mineral has a somewhat simpler crystal structure than AuTe2: it has a layered structure like in typical dichalcogenides MX2, but, in contrast to AuTe2, which exhibits an incommensurate modulation in the triangular layer of, nominally, Au2+ (see the solution to this puzzle in ref. 2), in AuAgTe4 the Au and Ag ions are ordered in a stripy fashion, and they have practically integer valencies Au3+ and Ag1+, though, strictly speaking, these notions may be not fully applicable in this case because the material is a metal. Nevertheless, the structural data under ambient pressure correspond to this valence assignment, and crystal chemistry indeed confirms this: Au and Ag ions sit inside Te octahedra, which are so strongly distorted that they rather resemble square coordination for Au and dumbbells for Ag, very typical for d8 and d10 ions. Due to the Jahn–Teller effect Au3+ with the low-spin d8 configuration and doubly occupied 3z2 − r2 orbital almost always prefers strongly elongated octahedra or even square coordination. The linear coordination of d10 ions such as Ag1+ or Hg2+ is attributed to the second-order Jahn–Teller, which describes mixing of completely filled d and empty s states (see, e.g. ref. 8 and 9).
Some of us have theoretically predicted that this structure with strongly distorted Te octahedra is unstable under pressure10 and now we can confirm this experimentally: at pressures higher than ∼5 GPa the coordination of Ag and Au ions becomes almost identical. And, even more importantly, at ∼1.5 GPa this material becomes superconducting, demonstrating also an abrupt increase of the superconducting critical temperature at the phase transition.
Thus, we have found yet another chemical compound of gold (and silver) that is superconducting. Our theoretical analysis demonstrated that superconductivity here is likely of a conventional type, predominantly due to electron–phonon interactions. Eventual contribution of the “negative-U” mechanism seems to play a minor role here.
2 Experimental and theoretical details
2.1 Samples and characterization
The experiments were performed with high-quality natural single-crystals of silvanite, AuAgTe4.§ Custom diamond anvil cells (DACs) and DACs of symmetric design were used to induce high pressure, with Ne or KCl serving as a pressure-transmitting medium. The pressure was determined using the ruby R1 fluorescence line as a pressure marker, as well as the Ne unit-cell volume in the case of X-ray diffraction (XRD) studies. Single-crystal (SC) XRD experiments were performed at the 13-ID-D beamline (mainly) and the beamline 13-BM-C of the APS synchrotron (Argonne, IL, USA). Electrical resistance measurements were performed as a function of pressure and temperature using the standard four-probe method.2.2 Density functional theory calculations
The Quantum Espresso12 package was used to perform first-principles calculations within the density functional theory (DFT), while the superconducting properties were investigated using the EPW code.13–15For further information on the experimental and theoretical methods see the ESI.†
3 Experimental results
3.1 X-ray diffraction
XRD patterns obtained up to about 10 GPa are shown in Fig. 1(c) (see also Table S1, ESI†). Low pressure (LP) patterns up to ∼5 GPa can be identified as a monoclinic P2/c structure,16 where both Ag and Au are octahedrally coordinated by Te, but these octahedra are so strongly distorted that in fact Ag has a dumb-bell and Au a square planar surrounding, as shown in Fig. 1(a). Each Te atom is surrounded by three Au or Ag atoms and three Te atoms, and it is much closer to one of its three Te neighbors than to the other two. Therefore, there are two types of Te–Te interlayer bonds labeled as short and long in Fig. 1(a). All the Te atoms are thus members of well-defined Te2 clusters.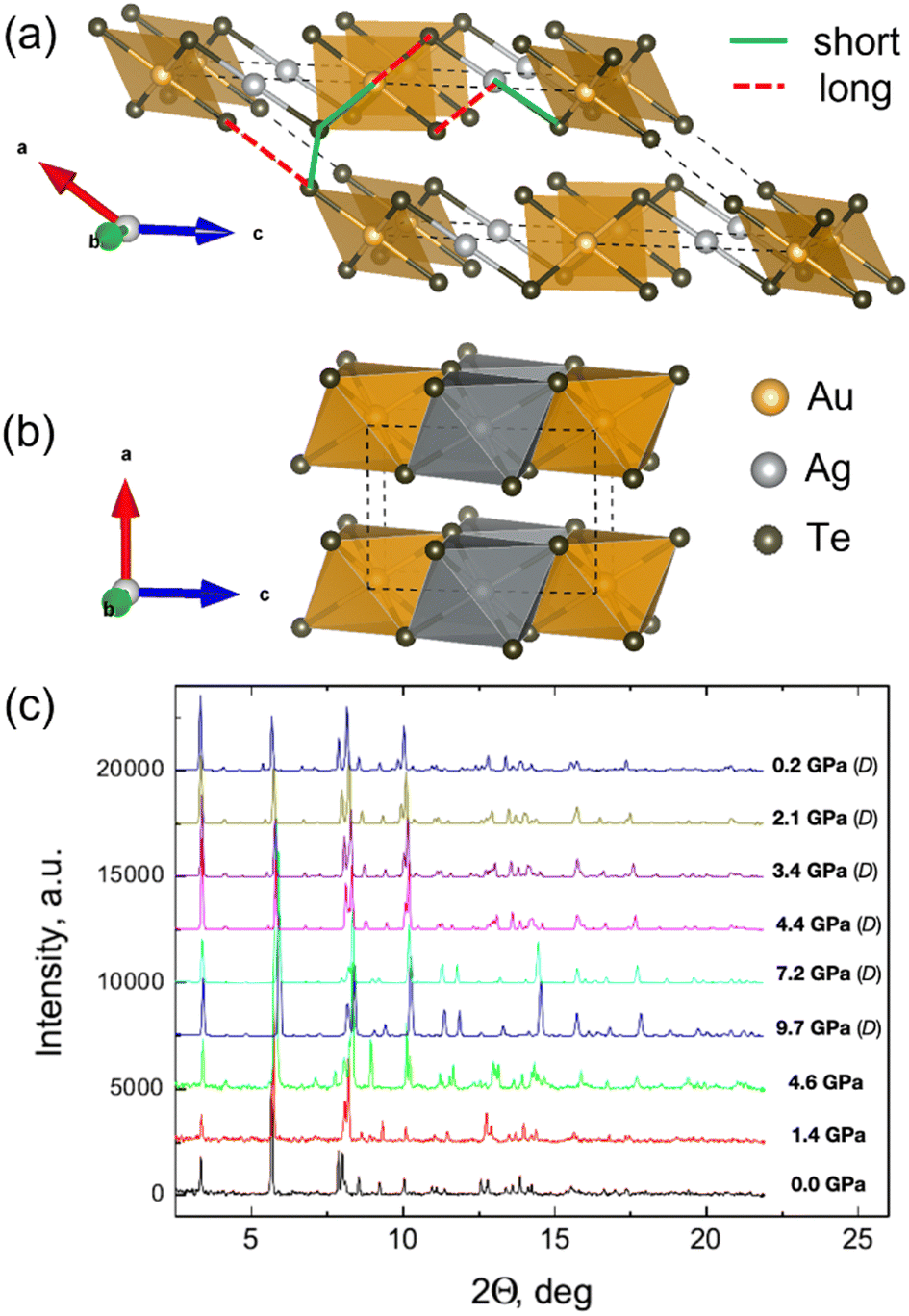 | ||
| Fig. 1 Polyhedral representation of the AuAgTe4 crystal structure of the (a) low-pressure (P < 5 GPa) P2/c phase (the highly distorted octahedra surrounding Au and Ag are shown as square planar and dumb-bell, respectively), and (b) high-pressure (P > 5 GPa) P2/m phase drawn using VESTA11 software. (c) Pressure evolution of the XRD patterns of AuAgTe4 at a compression up to ∼9.7 GPa and following decompression (D) to 0.2 GPa (λX-ray = 0.2952 Å). | ||
We note that AuAgTe4 in its natural occurrences is usually characterized by an excess of Au atoms partially substituting the Ag atoms in the Ag02 sites (see ref. 17–19 and references therein). This substitution may reach 36%, and only a substitution of 50% or more results in the formation of another gold–silver–telluride, krennerite (Au3AgTe8) (see ref. 19 and ref. therein). Taking this into account, structural refinement has been made for refining the occupancy of the Ag site by assuming an Au–Ag solid solution. This refinement at all pressures suggests a partial substitution of Ag by Au resulting in a chemical composition of Au(Ag0.75(3) Au0.25(3))Te4. We note that the experimentally observed Au/Ag–Te intralayer and Te–Te interlayer distances are in good agreement with those calculated using DFT for the fully stoichiometric AuAgTe4 (Fig. 2). Furthermore, the critical pressure values of the onset of the structural phase transition obtained in XRD studies (mixed Ag/Au occupation of Ag02 site), Raman measurements (were performed on very different crystals, see below and Section 2.1 of the paper) and calculated using DFT (the last two-single Ag occupation of the Ag02 site) are almost the same. This suggests that the above mentioned substitution does not have substantial effects on the structural and electronic properties of the sample, which is reasonable taking into account similar chemical and electronic properties of Au and Ag and minor occupancy of Au in the Ag02 site, and also structural stability of sylvanite with a significant substitution of Ag with Au in the Ag02 site (at least up to 36%19).
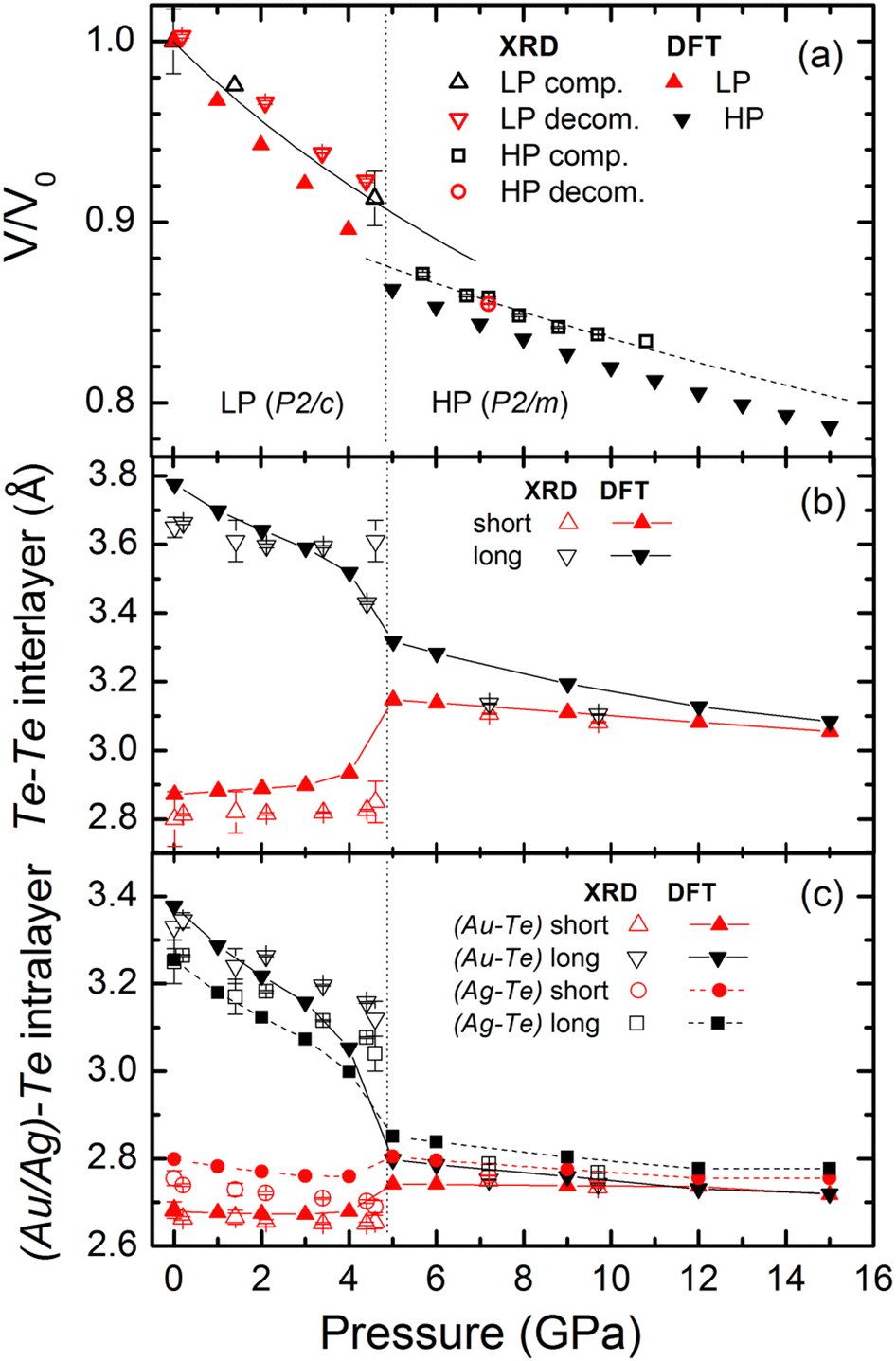 | ||
| Fig. 2 (a) Pressure dependence of the unit-cell volume of AuAgTe4 determined in the single crystal and powder XRD experiments (open symbols), and the fits with the Birch–Murnaghan equation of state20 are shown by lines. DFT results are presented by filled symbols. Experimentally observed (open symbols) and calculated in DFT (lines and filled symbols) pressure dependence of the (b) Te–Te interlayer and (c) (Au/Ag–Te) intralayer distances of AuAgTe4. | ||
At ∼5 GPa, an onset of a new high-pressure (HP) phase is observed (Fig. 1(c) and Fig. S1, ESI†), whose XRD patterns could be fitted well with the more layered P2/m structure (see Tables S2 and S3, ESI†). In the HP phase the Te6 octahedra around Au and Ag become regular and practically identical, and the Te–Te interlayer distances become almost equal (Fig. 1(b) and 2(b), (c)). We note that this phase transition is in excellent agreement with the recent theoretical prediction and present DFT calculations.10
The LP phase V(P) data can be fit well with a second-order Birch–Murnaghan equation of state (BM2 EOS)20 as shown in Fig. 2(a), resulting in V0 = 335.3(11) Å3 and K0 = 41.1(24) GPa, where K0 and V0 are the bulk modulus and the unit-cell volume at 1 bar and 300 K, respectively, with the bulk modulus first derivative fixed at K′ = 4. For the HP phase, the performed fit using the BM2 EOS results in V0 = 307.8(25) Å3 and K0 = 81(9) GPa (combining both the SC refinements, and the wide images, which were collected during continuous rotation within a single exposure and were analyzed as if they were “powder” data for the HP phase). Close to the transition pressure, at 5 GPa, the unit-cell volume and bulk modulus are V = 303.5(8) Å3 and K = 60(3) GPa, and V = 291.3(8) Å3 and K = 101(9) GPa for the LP and HP phases, respectively. Thus, the phase transition is accompanied by a lattice volume contraction of ∼4% and a significant increase of the bulk modulus.
3.2 Raman spectroscopy
At room temperature and ambient pressure we observe almost all allowed Raman active vibrations for AuAgTe4 (P2/c space group): 7 Ag and 7 weaker Bg phonon modes in the 40–160 cm−1 range. Table 1 shows that the experimental and DFT calculated Raman frequencies are in overall good agreement.| Ag | Expt. | 47 | 61 | 95 | 102 | 121 | 132 | 158 | |
| Calc. | 48 | 60 | 87 | 98 | 112 | 124 | 147 | ||
| Bg | Expt. | 50 | 58 | — | 84 | 88 | 114 | 134 | 147 |
| Calc. | 46 | 52 | 59 | 83 | 112 | 129 | 135 | 147 |
One can see that Raman spectroscopy clearly detects structural changes above 4 GPa, see Fig. 3 and Fig. S4 (ESI†). In particular, new modes are observed at 113 and 142 cm−1 and the intensity of some lines (158 cm−1) changes considerably in the polarized spectra. The polarized spectra in Fig. S3 (ESI†) additionally support a structural transition in the 4–6 GPa region. In addition to the appearance of new lines in the spectrum, the frequencies of a number of lines either increase significantly (61, 133 and 158 cm−1) or decrease (147 cm−1) with increasing pressure, and the energies of others change nonmonotonically (102 cm−1) or increase insignificantly (47 and 121 cm−1) (Fig. S4, ESI†).
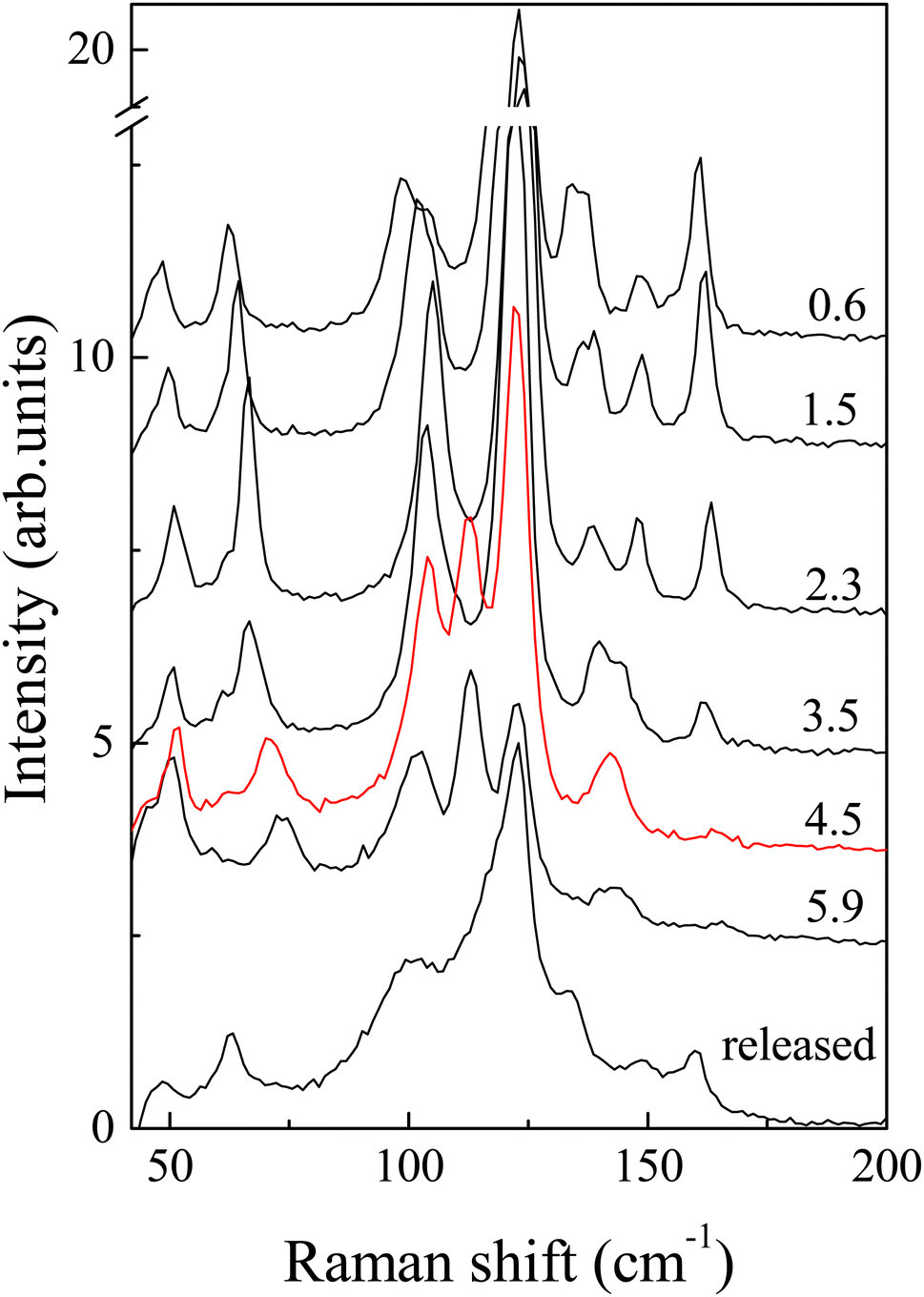 | ||
| Fig. 3 Raman spectra of AuAgTe4 obtained at different pressures with 633 nm excitation in the polarized geometry. Numbers in the figure refer to pressure in GPa. | ||
It is interesting that there are 7 Raman lines above the transition. This number is larger than what follows from the selection rules for the refined HP structure (P2/m space group), where there should only be 6 Raman-active modes: 4Ag + 2Bg. It is well known that Raman spectra provide information not only on the long-range order (since the number of observed lines is determined by the space group of the crystal), but also on the short-range order, being sensitive to local structural distortions. Thus, the appearance of extra lines in the Raman spectra, perhaps, evidences formation of two phases in this pressure range, which is in line with the resistivity measurements discussed below. In addition, one may expect defects in natural crystal, which ensure leakage of a symmetry-forbidden line in the spectrum. In all three experiments, we obtained a somewhat broadened spectrum, compared to the initial crystal, after pressure release (Fig. 3).
3.3 Resistance measurements
The resistivity value of AuAgTe4 under ambient conditions was estimated to be ∼3 × 10−6 Ω m, typical of a bad metal. Under pressure AuAgTe4 shows a significant, about a factor of 30, drop in the resistance when it is compressed up to ∼7 GPa, followed by a slight increase above this (see the inset of Fig. 4).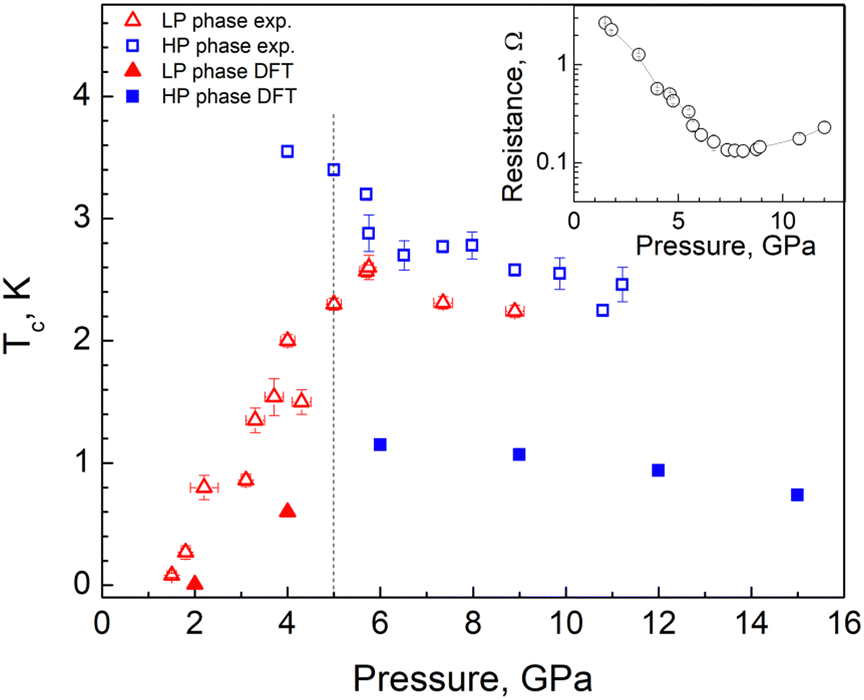 | ||
| Fig. 4 Critical temperature as a function of pressure. The experimental (calculated) results are presented by open (filled) symbols. The vertical line represents the phase separation between the LP and HP phase. The pressure dependence of the room temperature resistance is shown in the inset. | ||
In Fig. 5(a) we show the resistance vs. temperature dependence at various pressures for the most representative run 3 of measurements. One can see that the appreciable drop in resistance coincides with the onset of superconductivity at P ≈ 1.5 GPa with a superconducting critical temperature of Tc ≈ 80 mK. With further pressure increase, Tc increases almost linearly up to ≈2.6 K at 5.7 GPa and then decreases slowly, demonstrating a non-monotonous dome-like shape (Fig. 5(a)). Thus, we can conclude that the onset of superconductivity takes place in the LP phase of AuAgTe4 at pressures above ∼1.5 GPa. With this, starting from P = 4 GPa one can clearly see a drastic change in R(T) behavior: the R(T) curves have two distinct transitions, signifying the appearance of an additional phase with a higher transition temperature (Tc ≈ 3.5 K at P = 4 GPa). As demonstrated by our XRD data, in this pressure range a crystallographic phase transition occurs forming the HP P2/m phase. We can, therefore, interpret the double transition as coexistence of the LP and the HP phases, both being superconducting with a higher Tc for the HP phase. The critical temperature for the HP phase decreases appreciably with pressure, approaching the Tc of the LP phase. Above ∼9 GPa a single transition is observed.
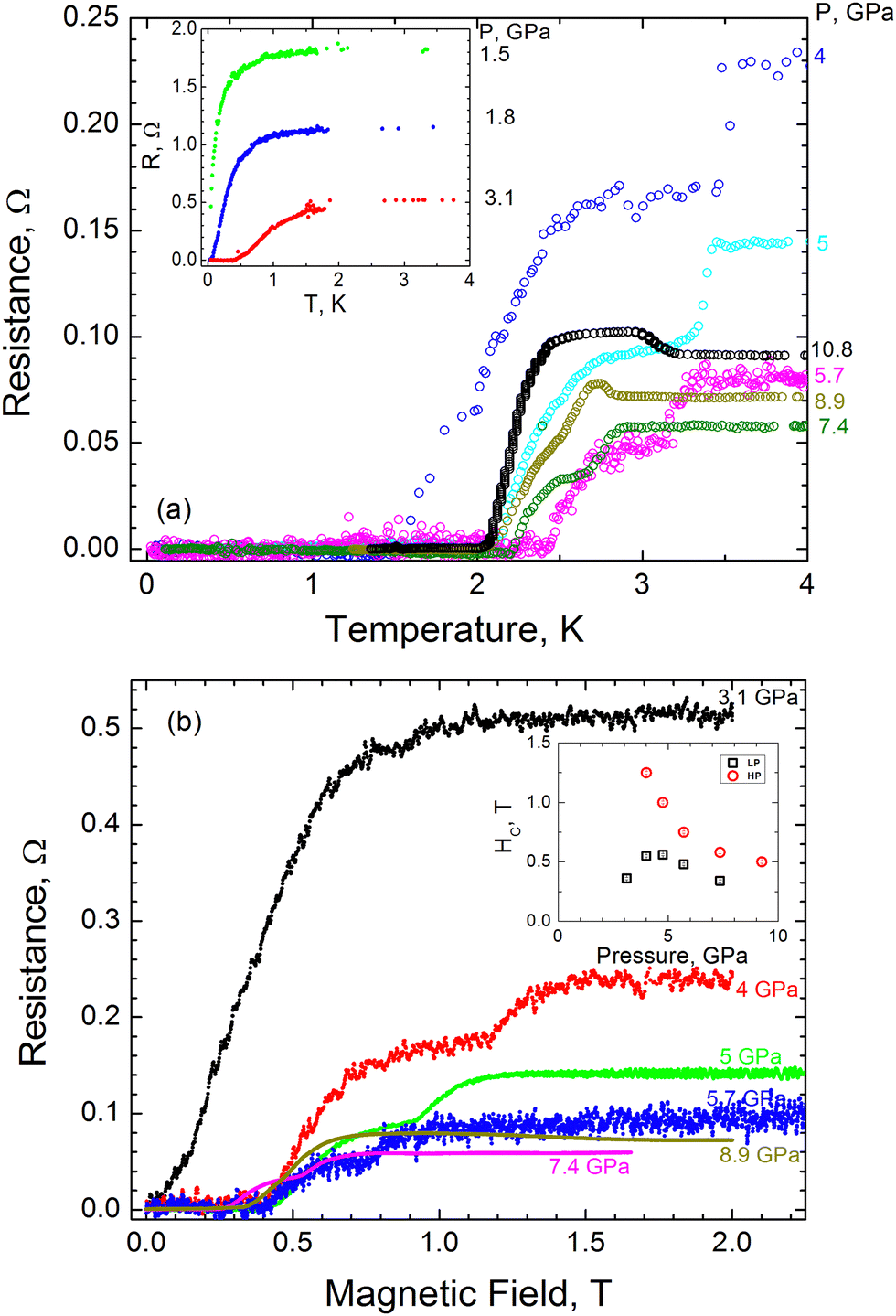 | ||
| Fig. 5 (a) Temperature dependence of the resistance in AuAgTe4. The graph shows complete superconductivity transitions (resistance drops to zero). The double transitions observed in the 4 < P < 9 GPa pressure range are interpreted as a mixture of LP and HP phases. (b) Pressure dependence of the critical field of AuAgTe4. Resistance as a function of magnetic field at different pressures: from 4 to 7.4 GPa two transitions are observed. The inset shows variation in the critical field as a function of pressure. | ||
In Fig. 5(b), we show magnetoresistance measurements at 2 K for run 3. From these curves we can extract the upper critical magnetic field Hc as the field at which the resistance is half of the normal state resistance. In the pressure range from 4 to 7.4 GPa, where the temperature dependence exhibits two transitions, two transitions are observed in the magnetic field. These measurements are consistent with our interpretation that both structural phases coexist within this pressure range. The corresponding upper critical field is plotted in the inset of Fig. 5(b). Our definition of Hc is not appropriate for the pressure range where we observe a double transition (coexistence regime). In the latter case we estimated Hc as the mid-point value of each transition for each phase. The critical magnetic field was found to depend on pressure, as it varies between ∼0.2 and 0.5 T for the LP phase, and between ∼1.2 and 0.5 T for the HP phase. We note that, similar to Tc, the critical field demonstrates a non-monotonous dome-like shape in the LP phase and an appreciable decrease with pressure in the HP phase (see the inset of Fig. 5(b)).
4 Computational results
As discussed in previous sections, the DFT calculations describe very well the structural transition under pressure (and in fact had predicted this transition10). The calculated pressure dependence of the crystal volume, lattice parameters and interatomic distances are in good agreement with the experimental results (see Fig. 2 and Fig. S2, ESI†).4.1 Electronic properties
The band structure and density of states (DOS) of AuAgTe4 for the LP phase at 2 GPa and the HP phase at 6 GPa are shown in Fig. 6, and those at other pressures are given in the ESI,† Fig. S5. It was shown in ref. 10 that the Te–Te dimerization at ambient pressure opens a pseudogap due to the bonding–antibonding splitting, but this pseudogap gradually closes under pressure. The largest contribution to the DOS at the Fermi level (EF) is provided by the Te p states. As pressure increases to 4 GPa (ESI,† Fig. S5(a)), an electron-like band related to the Te p orbitals lowers in energy along the Γ–Z–D direction, while two hole-like bands along the Γ–Z and Γ–A–E directions increase in energy and cross EF. These changes cause a drastic increase in the total DOS at the Fermi level (NF), as shown in Fig. 7(a) (see also the ESI,† Fig. S5(a)). After the phase transition, the dispersion of all bands is greatly increased, while the major contribution to the DOS remains to be from the states of the Te p character (Fig. 6(b) and Fig. S5(b)–(d), ESI†). At 6 GPa, the NF of the HP phase is 31% of that in the LP phase at 4 GPa. No significant changes are observed in the band structure of the HP phase in the considered 6–15 GPa range, leaving NF nearly constant (see Fig. 7(a)). Overall, we observe that the Te p states provide almost 75% of the total DOS at EF for all pressure points. We also checked the effect of spin–orbit coupling and found that the change in the total DOS near the Fermi level is negligible for both the low- and high-pressure phase (see the ESI,† Fig. S7(a)).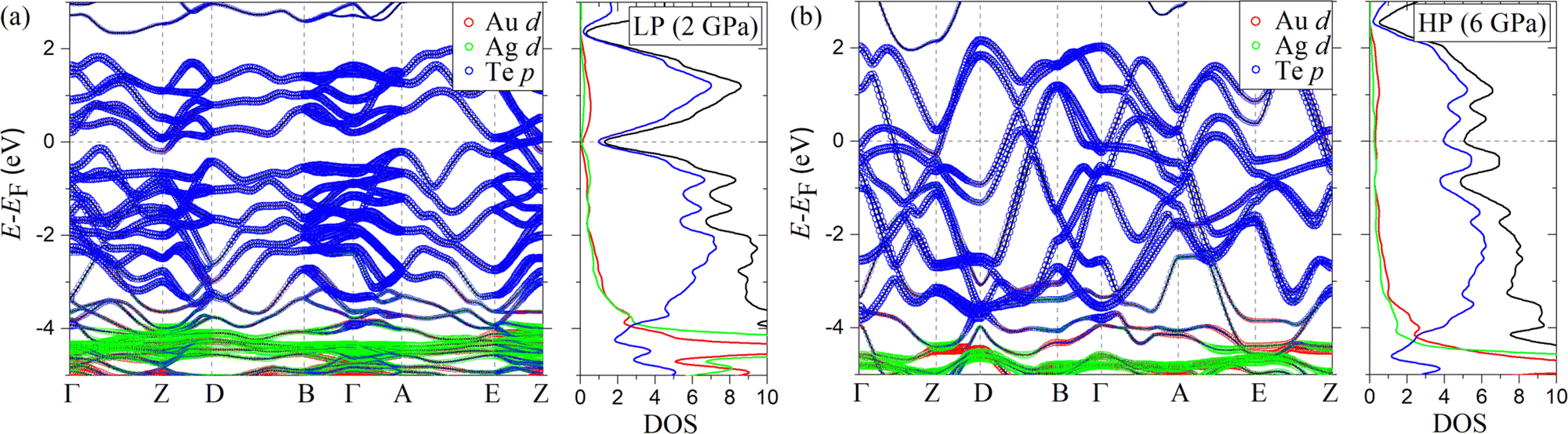 | ||
| Fig. 6 Calculated band structure and density of states (DOS; in states per eV per f.u.) of AuAgTe4 at (a) 2 GPa (low-pressure phase) and (b) 6 GPa (high-pressure phase). The size of the markers is proportional to the contribution of each orbital character. The solid black line in the DOS panel represents the total DOS and the red, green, and blue lines are the contributions to the DOS from the Au, Ag, and Te atoms, respectively. | ||
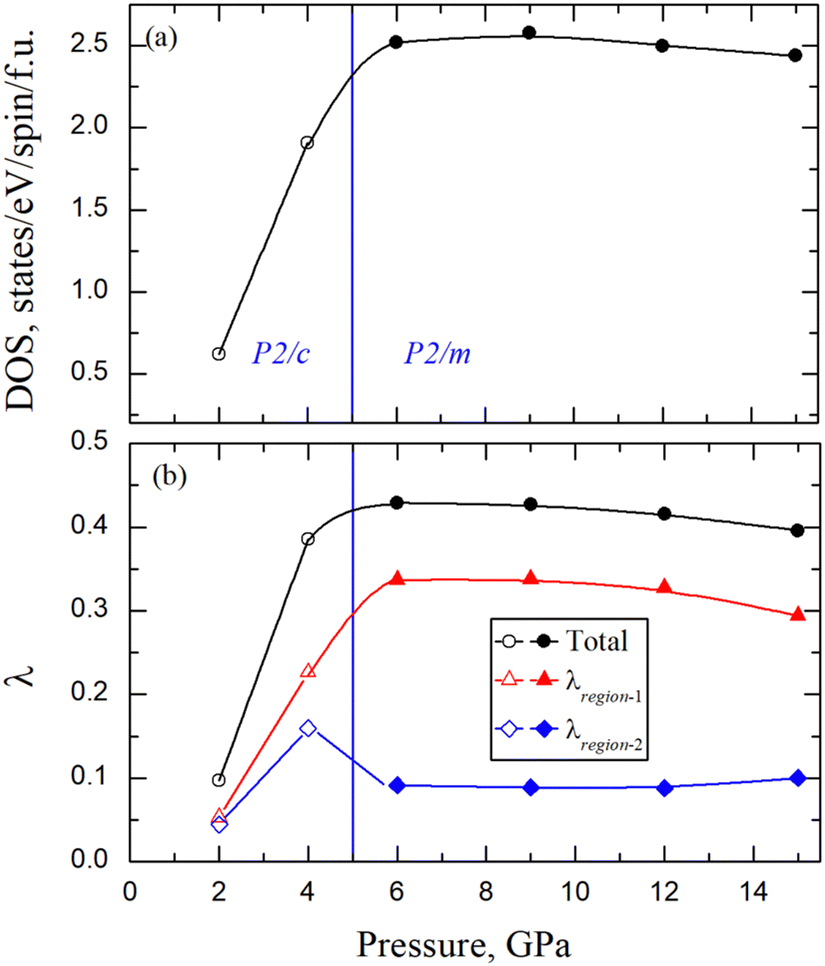 | ||
| Fig. 7 Calculated (a) DOS (states per eV per spin per f.u.) at the EF and (b) total e–ph coupling λ and partitioning of λ for two frequency regions of AuAgTe4 as a function of pressure. The region-1 and region-2 are the separations in the phonon spectrum below and above 11 meV in the LP phase and 15 meV in the HP phase. The vertical blue line represents the phase separation between the LP and HP phases. The open and closed symbols are for the LP and HP phases, respectively. | ||
4.2 Phonons
Fig. 8 shows the phonon dispersion, the phonon density of states (PHDOS), the isotropic Eliashberg spectral function α2F(ω), and the cumulative electron–phonon (e–ph) coupling (λ) of AuAgTe4 for the LP phase at 2 GPa and the HP phase at 6 GPa, and the other pressure points are shown in the ESI,† Fig. S6. We find that the LP phase is dynamically stable at 2 and 4 GPa, but dynamically unstable at 6 GPa. As displayed in Fig. 8(a), the phonon spectrum of the LP phase at 2 GPa is divided into two regions (region-1 and region-2) separated by a large gap around 11 meV. The PHDOS decomposed according to atomic contributions shows that the Te-derived modes extend over the whole spectrum. On the other hand, the Au and Ag contributions to the PHDOS are dominant in region-1, while there is no contribution from either Au or Ag in the 14–18 meV range. In addition, the optical branches above 11 meV are not as dispersed as those below this threshold. At 4 GPa (ESI,† Fig. S6(a)), the frequency gap around 11 meV is closed as one optical branch couples to the modes in the lower region along the B–Γ–A direction. Another noticeable aspect is the softening of the low-energy acoustic branches along the D–B direction.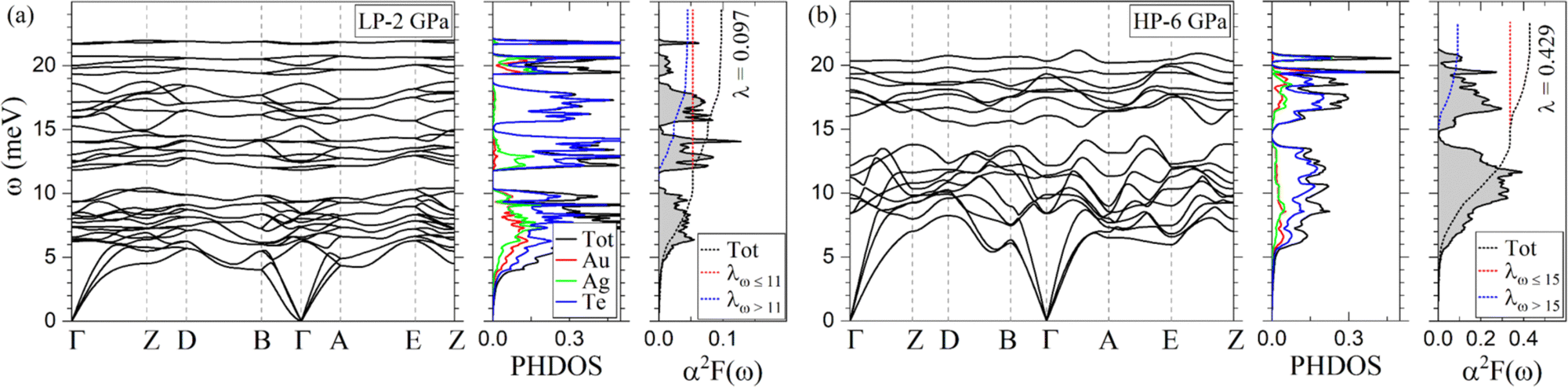 | ||
| Fig. 8 Calculated phonon dispersion, phonon density of states (PHDOS), and Eliashberg spectral function α2F(ω) of AuAgTe4 at (a) 2 GPa (low-pressure phase) and (b) 6 GPa (high-pressure phase). | ||
The HP phase is found to be dynamically stable in the 6–15 GPa pressure range considered in our study. The major contribution to the PHDOS comes from the Te vibrations, but the spectrum lacks the phonon branches solely related to the Te vibrations present in the LP phase in the 14–18 meV range, since now the long and short Te–Te interlayer distances are very close to each other (Fig. 2(b)). In addition, the vibrational modes associated with the Au and Ag atoms harden under pressure as shown in the PHDOS (Fig. 8(b) and ESI,† Fig. S6(b)–(d)). The low- and high-frequency regions (region-1 and region-2) remain separated by a small gap centered around 15–16 meV at all pressure points. As in the LP phase, the optical phonon branches are hardening under compression, while the low-energy acoustic modes along the D–B direction and at the E-point soften. As for the electronic structure, the spin–orbit coupling has a minimal effect on the phonon spectrum, as shown in ESI,† Fig. S7(b).
4.3 Superconductivity
In order to investigate superconducting properties, we first evaluated the isotropic Eliashberg spectral function α2F(ω) and the cumulative e–ph coupling strength λ(ω). As depicted in Fig. 8(a), for the LP phase at 2 GPa, the low-frequency modes below 11 meV associated with vibrations from all atoms contribute 55% to the total e–ph coupling λ = 0.097. An almost four fold increase in the total e–ph coupling (λ = 0.386) is found at 4 GPa as the DOS at the Fermi level rises by a similar factor as shown in Fig. 7. From the division of the phonon spectrum, we found that region-1 below 11 meV supplies 59% of λ, slightly more than at 2 GPa since now the coupling due to the acoustic modes has strengthened with the phonon softening.In the HP phase at 6 GPa, the low-energy phonons below 15 meV (region-1) make up approximately 80% of the total λ = 0.429 (Fig. 8(b)). A comparative analysis of the α2F(ω) in the two phases shows that the HP phase lacks in region-2 (above 15 meV) the coupling coming solely from the Te-derived vibrations in the LP phase (14–18 meV range), leading to a factor of two reduction in λ in the upper frequency region. Under further compression, the ratio of the low- to high-frequency phonon contribution to the total e–ph remains nearly constant and λ decreases slowly following the same trend as the total DOS at the Fermi level.
Finally, to estimate the superconducting critical temperature (Tc), we solved the isotropic Migdal–Eliashberg equations implemented in the EPW code13,14,15 using a Coulomb pseudopotential 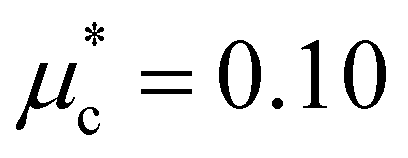 . Fig. 4 shows the calculated Tc and its comparison with the experiment. For the LP phase, we estimate Tc to be very close to zero at 2 GPa, consistent with the experiments at 1.8 GPa, with an increase to 0.6 K at 4 GPa. After the phase transition, we obtain a maximum Tc of 1.2 K at 6 GPa for the HP phase. In line with the resistivity measurements, the superconducting critical temperature decreases slowly in response to pressure, as shown in Fig. 4. Altogether, we find that the dome-shape behavior of the Tc mirrors the trends for the DOS at the Fermi level and the e–ph coupling strength under pressure (see Fig. 4 and 7). While our predicted Tc values are underestimated compared to the onset superconducting temperatures from the resistivity curves, they show a nice qualitative trend consistent with the experiments.
. Fig. 4 shows the calculated Tc and its comparison with the experiment. For the LP phase, we estimate Tc to be very close to zero at 2 GPa, consistent with the experiments at 1.8 GPa, with an increase to 0.6 K at 4 GPa. After the phase transition, we obtain a maximum Tc of 1.2 K at 6 GPa for the HP phase. In line with the resistivity measurements, the superconducting critical temperature decreases slowly in response to pressure, as shown in Fig. 4. Altogether, we find that the dome-shape behavior of the Tc mirrors the trends for the DOS at the Fermi level and the e–ph coupling strength under pressure (see Fig. 4 and 7). While our predicted Tc values are underestimated compared to the onset superconducting temperatures from the resistivity curves, they show a nice qualitative trend consistent with the experiments.
We also investigated whether the estimated critical temperature is affected when the non-local van der Waals (vdW) functional optB86b21–26 is included in the DFT calculations. In the case of the LP phase, the phonons remain almost unchanged and as a result the Tc is unaffected. For the HP phase, the lowest optical phonon branches along the D–B direction soften compared to the calculations without vdW. The softening varies from 3 meV at 6 GPa to 1.5 meV at 15 GPa, respectively, and it is due to a small compression along the out-of-plane direction. This led to a modest increase in the e–ph coupling, resulting in a rise in the Tc of about 25% on average, which still falls short of the experimental values. Similar underestimation of the computed Tc has been found in other layered compounds under pressure, such as MoTe227 and SnSe.28 The discrepancy between experiments and computations was attributed to the sensitivity of the electronic structure to the crystal parameters,27 the coexistence of multiple phases,28 and the substantial difference between the measured onset and zero resistance Tc27,28 under pressure.
Finally, to gain insight into the anisotropy of the e–ph coupling, we evaluated the momentum-resolved e–ph coupling strength λk for the LP and the HP phase at various pressures. As shown in Fig. 9(a) and (b), λk displays a single peak with relatively weak anisotropy in the momentum space. For 6 GPa (HP phase), we also solved the anisotropic full-bandwidth Eliashberg equations29 where the sparse sampling approach with the intermediate representation30–32 was employed to perform the summation over the Matsubara frequencies. We found that the multiple-sheet Fermi surface gives rise to a single anisotropic gap with a distinguished peak at about 0.1 meV in the T = 0 K limit, as shown in Fig. 9(c). The energy distribution of the superconducting gap reflects closely the anisotropy in λk. We obtain an anisotropic Tc of 1.1 K for 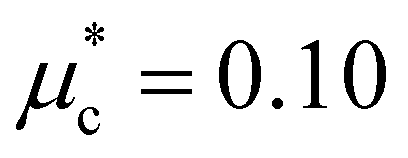 , a value identical to the one found for the isotropic gap calculation at 6 GPa.
, a value identical to the one found for the isotropic gap calculation at 6 GPa.
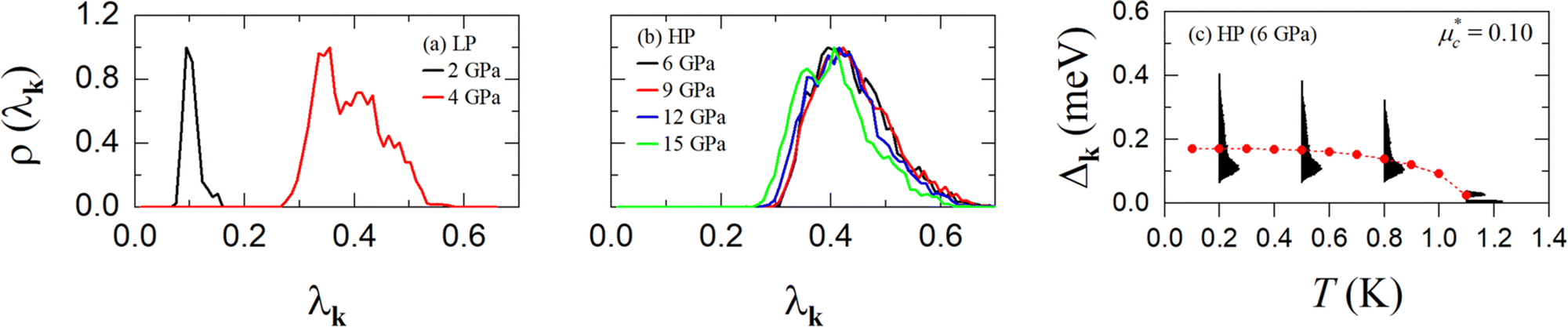 | ||
Fig. 9 Distribution of the e–ph coupling strength λk of (a) the LP and (b) the HP phase of AuAgTe4 at various pressures. (c) Energy distribution of the anisotropic superconducting gap Δk of the HP phase as a function of temperature at 6 GPa. The red symbols represent the isotropic superconducting gap at 6 GPa. The Coulomb parameter is set to 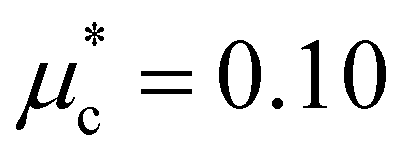 . . | ||
5 Discussion and conclusions
Summarizing the obtained experimental data we can conclude that AuAgTe4 undergoes a first order structural phase transition from the LP P2/c to the HP P2/m phase upon pressure at about 5 GPa. This phase transition is manifested by the observed changes in XRD patterns, Raman spectra, dramatic changes of the R(T) behavior and is in excellent agreement with first-principles calculations. We note that there is no sign of hysteresis: upon decompression the LP phase returns at ∼5 GPa. The transition to the HP phase is accompanied by a lattice volume contraction of ∼4%. The LP phase is characterized by a significant distortion of the AuTe6 and AgTe6 octahedra due to the linear and second-order Jahn–Teller effect: the six Te atoms form elongated (4 short and 2 long Au/Ag–Te bonds) and compressed (2 short and 4 long Au/Ag–Te bonds) octahedra around the Au and Ag atoms. These strong Jahn–Teller distortions also give rise to two rather different Te–Te interlayer distances,2 see Fig. 1(a) and 2(b).As usual, with increasing pressure one expects suppression of the Jahn–Teller distortions and indeed the difference between the two sets of Te–Te distances is reduced and then abruptly disappears at ∼5 GPa at the transition to the P2/m structure. A similar pressure dependence is also observed for the short and long Au–Te and Ag–Te intralayer bond lengths (Fig. 2(b) and (c)). These changes are due to the sliding of the atomic layers with respect to each other during compression, leading finally to the transition to the P2/m structure, which has regular Te octahedra around Au and Ag atoms (Fig. 1(b)).
Our resistivity measurements revealed superconductivity in both AuAgTe4 phases, the LP and HP. However, the LP phase becomes superconducting only above ∼1.5 GPa and shows an almost linear increase of the critical temperature with pressure up to ∼6 GPa (with a maximum Tc ≈ 2.5 K) followed by a slower decrease. The HP P2/m phase, once it occurs, has a higher critical temperature of ∼3.5 K (i.e. even higher than in pure AuTe2, with the maximum Tc ≈ 2.3 K33), and shows a trend to a sluggish Tc decrease under pressure. It is noteworthy that in the case of calaverite AuTe2 the superconductivity had been proposed to be induced by breaking of Te–Te dimers, which exist in the LP C2/m phase, but disappear in the superconducting HP P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 phase.3 Alternatively, it was also proposed that the breaking of the Te–Te dimers is not directly related to the onset of the superconductivity but that the tendency of charge disproportionation of Au2+ into, nominally, Au1+ and Au3+, could be crucial in the formation of Cooper pairs leading to superconductivity under pressure.2 Present results show that the situation is even more delicate here, since superconductivity appears already in the low-pressure “dimerized” phase.
m1 phase.3 Alternatively, it was also proposed that the breaking of the Te–Te dimers is not directly related to the onset of the superconductivity but that the tendency of charge disproportionation of Au2+ into, nominally, Au1+ and Au3+, could be crucial in the formation of Cooper pairs leading to superconductivity under pressure.2 Present results show that the situation is even more delicate here, since superconductivity appears already in the low-pressure “dimerized” phase.
Our first-principles computations demonstrate that most probably the superconductivity here is of a conventional type, with the low-energy phonon modes dominating the electron–phonon interactions. Although breaking of the Te–Te dimers is not directly responsible for the onset of superconductivity in AuAgTe4, it results in an appreciable increase in the critical temperature following the transition into the P2/m phase. This is due to an increase in the electronic density of states at the Fermi level related to closing of the pseudogap and to the phonon softening. Overall, despite some stoichiometric distinctions in the experimentally studied and calculated systems our theoretical estimates of the superconducting critical temperature are in good agreement with the experimental results, following a similar trend under applied pressure. Meanwhile some quantitative underestimate of the computed Tc compared to the experimental one may be caused by the appearance of Au in the Ag02 site. These findings can be important not only for silvanite, but also for other similar materials such as puzzling IrTe2. In the case of IrTe2, there is an anomalous structural transition at 270 K, the origin of which is debated, superconductivity induced by intercalation or doping, and complete reconstruction of the electronic structure and Ir–Ir dimerization in a monolayer.34–39
Author contributions
Yehezkel Amiel: experiment (electrical transport measurements), data analysis, and writing – review & editing; Gyanu P. Kafle: investigation (DFT calculations), formal analysis, writing – original draft, and visualization; Evgenia V. Komleva: investigation (DFT calculations) and visualization; Eran Greenberg: experiment (XRD), data analysis, visualization, and writing – review & editing; Yuri S. Ponosov: experiment (Raman), and writing – original draft; Stella Chariton: experiment (XRD) and data naalysis; Barbara Lavina: experiment (XRD) and data analysis; Dongzhou Zhang: experiment (XRD) and data analysis; Alexander Palevski: resources and writing – review & editing; Alexey V. Ushakov: investigation (DFT calculations); Hitoshi Mori: methodology and software (code development); Daniel I. Khomskii: idea and conceptualization; Igor I. Mazin, Sergey V. Streltsov, and Elena R. Margine: resources, conceptualization, writing – original draft & review and editing, and supervision; Gregory Kh. Rozenberg: conceptualization, project supervision, and writing – original draft & review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. L. Bohatý and Prof. P. Becker-Bohatý for providing us with natural single-crystals. We thank I. Silber and G. Tuvia for assisting with the resistance measurements. This work was supported by the National Science Foundation under grant no. DMR-2035518 (for superconductivity analysis) and grant no. OAC-2103991 (for code development). This research was supported by the Israel Science Foundation (grants no. 1552/18 and 1748/20). This work used the Expanse system at the San Diego Supercomputer Center via allocation TG-DMR180071 and the Frontera supercomputer at the Texas Advanced Computing Center via the Leadership Resource Allocation (LRAC) award DMR22004. Expanse is supported by the Extreme Science and Engineering Discovery Environment (XSEDE) program40 through NSF award no. ACI-1548562, and Frontera is supported by NSF award no. OAC-1818253.41 Portions of this work were performed at GeoSoilEnviroCARS (The University of Chicago, Sector 13), Advanced Photon Source (APS), Argonne National Laboratory. GeoSoilEnviroCARS is supported by the National Science Foundation – Earth Sciences (EAR – 1634415). Use of the COMPRES-GSECARS gas loading system was supported by COMPRES under NSF Cooperative Agreement EAR – 1606856 and by GSECARS through NSF grant EAR – 1634415 and DOE grant DE-FG02-94ER14466. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. E. V. K., Yu. S. P., A. V. U., and S. V. S., thank the Russian Ministry of Science and High Education (project “Quantum” no. 122021000038-7).Notes and references
- B. Dam, A. Janner and J. D. H. Donnay, Phys. Rev. Lett., 1985, 55, 2301 CrossRef CAS PubMed.
- S. V. Streltsov, V. V. Roizen, A. V. Ushakov, A. R. Oganov and D. I. Khomskii, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9945–9950 CrossRef CAS PubMed.
- K. Kudo, H. Ishii, M. Takasuga, K. Iba, S. Nakano, J. Kim, A. Fujiwara and M. Nohara, J. Phys. Soc. Jpn., 2013, 82, 063704 CrossRef.
- W. J. de Haas and F. Jurriaanse, Naturwissenschaften, 1931, 19, 706 CrossRef CAS.
- E. A. Wood and B. T. Matthias, Acta Crystallogr., 1956, 9, 534 CrossRef CAS.
- M. Rahm, R. Hoffmann and N. W. Ashcroft, J. Am. Chem. Soc., 2017, 139, 8740–8751 CrossRef CAS PubMed.
- M. Isobe, H. Yoshida, K. Kimoto, M. Arai and E. Takayama-Muromachi, Chem. Mater., 2014, 26, 2155–2165 CrossRef.
- L. Orgel, J. Chem. Soc., 1958, 4186 RSC.
- J. K. Burdett and O. Eisenstein, Inorg. Chem., 1992, 31, 1758–1762 CrossRef CAS.
- A. V. Ushakov, S. V. Streltsov and D. I. Khomskii, J. Phys.: Condens. Matter, 2019, 31, 235601 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli and M. Cococcioni, et al. , J. Phys: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- F. Giustino, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165108 CrossRef.
- S. Poncé, E. R. Margine, C. Verdi and F. Giustino, Comput. Phys. Commun., 2016, 209, 116 CrossRef.
- E. R. Margine and F. Giustino, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 024505 CrossRef.
- G. Tunnell and L. Pauling, Acta Crystallogr., 1952, 5, 375–381 CrossRef.
- G. Tunell, Am. Mineral., 1941, 26, 457–477 CAS.
- F. Wagner, J. Sawicki, J. Friedl, J. Mandarino, D. Harris and L. Cabri, Can. Mineral., 1994, 32, 189–201 Search PubMed.
- G. Kitahara, A. Yoshiasa, M. Tokuda, M. Nespolo, H. Hongu, K. Momma, R. Miyawaki and K. Sugiyama, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2022, 78, 117–132 CrossRef CAS PubMed.
- O. L. Anderson, Equations of state of solids for geophysics and ceramic science, Oxford University Press on Demand, 1995 Search PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- T. Thonhauser, V. R. Cooper, S. Li, A. Puzder, P. Hyldgaard and D. C. Langreth, Phys. Rev. B, 2007, 76, 125112 CrossRef.
- T. Thonhauser, S. Zuluaga, C. A. Arter, K. Berland, E. Schröder and P. Hyldgaard, Phys. Rev. Lett., 2015, 115, 136402 CrossRef CAS PubMed.
- K. Berland, V. R. Cooper, K. Lee, E. Schröder, T. Thonhauser, P. Hyldgaard and B. I. Lundqvist, Rep. Prog. Phys., 2015, 78, 066501 CrossRef PubMed.
- D. C. Langreth, B. I. Lundqvist, S. D. Chakarova-Käck, V. R. Cooper, M. Dion, P. Hyldgaard, A. Kelkkanen, J. Kleis, L. Kong and S. Li, et al. , J. Phys.: Condens. Matter, 2009, 21, 084203 CrossRef CAS PubMed.
- R. Sabatini, E. Küçükbenli, B. Kolb, T. Thonhauser and S. De Gironcoli, J. Phys.: Condens. Matter, 2012, 24, 424209 CrossRef PubMed.
- H. Paudyal, S. Poncé, F. Giustino and E. R. Margine, Phys. Rev. B, 2020, 101, 214515 CrossRef CAS.
- G. Marini, P. Barone, A. Sanna, C. Tresca, L. Benfatto and G. Profeta, Phys. Rev. Mater., 2019, 3, 114803 CrossRef CAS.
- H. Lee, S. Poncé, K. Bushick, S. Hajinazar, J. Lafuente-Bartolome, J. Leveillee, C. Lian, F. Macheda, H. Paudyal and W. H. Sio, et al., arXiv, 2023, preprint, arXiv:2302.08085, DOI: 10.48550/arXiv.2302.08085.
- H. Shinaoka, J. Otsuki, M. Ohzeki and K. Yoshimi, Phys. Rev. B, 2017, 96, 035147 CrossRef.
- J. Li, M. Wallerberger, N. Chikano, C.-N. Yeh, E. Gull and H. Shinaoka, Phys. Rev. B, 2020, 101, 035144 CrossRef CAS.
- M. Wallerberger, S. Badr, S. Hoshino, S. Huber, F. Kakizawa, T. Koretsune, Y. Nagai, K. Nogaki, T. Nomoto and H. Mori, et al. , SoftwareX, 2023, 21, 101266 CrossRef.
- S. Kitagawa, H. Kotegawa, H. Tou, H. Ishii, K. Kudo, M. Nohara and H. Harima, J. Phys. Soc. Jpn., 2013, 82, 113704 CrossRef.
- J. J. Yang, Y. J. Choi, Y. S. Oh, A. Hogan, Y. Horibe, K. Kim, B. I. Min and S. W. Cheong, Phys. Rev. Lett., 2012, 108, 116402 CrossRef CAS PubMed.
- Q. Li, W. Lin, J. Yan, X. Chen, A. G. Gianfrancesco, D. J. Singh, D. Mandrus, S. V. Kalinin and M. Pan, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- S.-I. Ideta, D. Zhang, A. G. Dijkstra, S. Artyukhin, S. Keskin, R. Cingolani, T. Shimojima, K. Ishizaka, H. Ishii and K. Kudo, et al. , Sci. Adv., 2018, 4, 3867 CrossRef PubMed.
- S. Pyon, K. Kudo and M. Nohara, J. Phys. Soc. Jpn., 2012, 81, 053701 CrossRef.
- Y. Song, F. Meng, T. Ying, J. Deng, J. Wang, X. Han, Q. Zhang, Y. Huang, J.-G. Guo and X. Chen, J. Phys. Chem. Lett., 2021, 12, 12180–12186 CrossRef CAS PubMed.
- S. Park, S. Y. Kim, H. K. Kim, M. J. Kim, T. Kim, H. Kim, G. S. Choi, C. J. Won, S. Kim and K. Kim, et al. , Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither, A. Grimshaw, V. Hazlewood, S. Lathrop, D. Lifka, G. D. Peterson, R. Roskies, J. R. Scott and N. Wilkins-Diehr, Comput. Sci. Eng., 2014, 16, 62–74 Search PubMed.
- D. Stanzione, J. West, R. T. Evans, T. Minyard, O. Ghattas and D. K. Panda, Frontera: The Evolution of Leadership Computing at the National Science Foundation, ACM, New York, NY, USA, 2020, pp. 106–111.
Footnotes |
| † Electronic supplementary information (ESI) available: Technical details about the experimental and calculation methods used; discussion of the Raman spectroscopy and DFT results; additional figures and tables. CCDC 2218994–2219002 and 2215501. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc00787a |
| ‡ These authors contributed equally to this work. |
| § The XRD and resistance R(P,T) experiments were performed with a natural single-crystal of silvanite from the classical Transylvania locality from the private collection of Ladislav Bohatý and Petra Becker–Bohatý, University of Cologne. The Raman experiment was carried out using natural single-crystals of silvanite from the Kochbulak deposit, Kuraminsky range, Uzbekistan. XRD results for both samples are in agreement with each other. |
| This journal is © The Royal Society of Chemistry 2023 |