
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulation of hyperthermic and relaxometric responses of magnetic iron oxide nanoparticles through ligand exchange provides design criteria for dual-functionality†
Esther Rani
Aluri‡
a,
Sameer D.
Shingte‡
 a,
Eoin P.
McKiernan
a,
Steven
Ferguson
b and
Dermot F.
Brougham
*a
a,
Eoin P.
McKiernan
a,
Steven
Ferguson
b and
Dermot F.
Brougham
*a
aSchool of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: dermot.brougham@ucd.ie; Tel: +353 01 716 2077
bSchool of Chemical and Bioprocess Engineering, University College Dublin, Belfield, Dublin 4, Ireland
First published on 18th April 2023
Abstract
Magnetic iron oxide nanoparticles (MNPs) are the subject of intense study as theranostic tools that combine magnetic resonance imaging (MRI)-trackability with AC-field responsive heating. In this study, we report MNPs synthesised using a modification of an established thermal decomposition method which, following extensive parameter optimisation, provides strong hyperthermic heating efficacy that is reproducible batch to batch. The suspensions have an exceptional specific absorption rate of ∼2800 W g−1 (intrinsic loss power, ILP ∼20.4 W m2 g−1 kA−2 kHz−1) within a high concentration range (collective particle scenario) falling to ∼1000 W g−1 (∼7.9 W m2 g−1 kA−2 kHz−1) on full dispersion by dilution. The effect of stabilising ligand surface chemistry on the concentration-dependent hyperthermic and MRI efficacies was evaluated by ligand exchange/phase transfer from organic to aqueous and back to organic suspension, and on the formation of organogels. Fast field-cycling NMR relaxometry of the different suspensions reveals the role of moment dynamics and of subtle differences in particle and solvent diffusion in determining both the hyperthermic and relaxometric efficacies. These insights identify particle design compromises that are required to simultaneously optimise MNPs for the two applications.
1. Introduction
Magnetic iron oxide nanoparticles (MNPs) in the sub 20 nm size range are under intense scrutiny for biomedical applications as AC-field responsive nanovectors that can be guided magnetically and tracked by magnetic resonance imaging.1,2 After exposure to AC-fields, MNPs in suspension, in hydrogels, or in the body can generate localised targetable heat (hyperthermia) which can be used to ablate tumours, or for thermally triggered drug release/increasing local diffusivity.3,4 The hyperthermic efficacy is quantified by the suspensions specific absorption rate (SAR, units W g−1, of Fe),5 and there is an ongoing need for biocompatible magnetic materials with high SARs within biologically and physiologically safe AC-field ranges.6,7 Prominent studies describe efforts to maximise the SAR by optimising MNP size,8 phase,9 and shape10–13 and even by assembling MNPs (see below). Recent advances in combining magnetic particle imaging (MPI) approaches to rastering field-free volumes with AC-fields14 may enable safe localisation of thermal load from high-heating particles.Hyperthermic responses can be generated through Néel and Brownian relaxation processes. In the former MNPs moments re-orient in their magnetocrystalline field. The Néel correlation time, τN, can be increased (slower motion) by increasing the MNP size or through inter-MNP coupling.15 Hence the formation of MNP assemblies usually results in suppression of the SAR,16,17 although there are reports of SAR enhancement, for instance on forming short dipole-ordered chains.11,18,19 The Brownian contribution arises from diffusional motion of the particles, characterised by τD, which also enables energy transfer within the field. Irrespective of the mechanism, mismatch of the timescales of moment realignment with the AC-field rotation leads to heat dissipation.3 Typically calorimetric methods are used to measure hyperthermic efficacy, in which the initial temperature rise (quasi-adiabatic condition) is used to extract a SAR value.20 However complications arise due to differences in heat dissipation from the sample, arising due to concentration, sample size, and other effects,9,21–23 and these are not always addressed, even in some prominent papers. AC-magnetometry24 can address the non-adiabaticity, but is not generally available within the relevant range (νAC in the low 100's of kHz, HAC in the low 10's of kA m−1).
MNPs have also been used clinically as magnetic resonance imaging (MRI) contrast agents,1,15 as they can provide diagnostically valuable image contrast, and unusually for inorganic phases are relatively non-toxic and are tolerated in humans.25 MNPs also have the key advantages of (i) improving core crystallinity, which is possible through synthesis,26 providing strong saturation magnetisation, Ms, (although there are few approaches that are shown to provide particles with reproducible magnetisation at scale), and (ii) suitable surface chemistries can be used to retain dispersion in complex environments,27 and so extend blood circulation times.28 Full dispersion provides superparamagnetism, i.e. minimal room temperature magnetic remanence, Mr, and hence shelf-life. High Ms helps provide strong magnetic resonance imaging contrast, quantified by the suspension concentration-corrected 1H NMR relaxation rate enhancement, or relaxivity (r1 or r2, units s−1 mM−1 of Fe). High Ms and Mr are also associated with high magnetic susceptibility χ, which, along with the details of moment dynamics, gives rise to a strong SAR. However, the effect of the ligands on the magnetic properties of MNP suspensions is complex and is not fully understood. In addition, there are few syntheses that provide reproducible particle size, shape and magnetisation batch-to-batch.29 These aspects provide significant barriers to development.
There are a great many interesting studies in which suspensions of dispersed or clustered MNPs are evaluated for multiple functions, typically MRI, AC-field hyperthermia and MPI. For instance, Hayashi et al.30 prepared 80–100 nm PEG-stabilised clusters of MNPs and assessed their MRI and hyperthermic efficacy in vitro and in vivo. Piazza prepared 12 nm folate-functionalised MNPs and assessed their hyperthermic efficacy in suspension31 where, again, they formed large clusters of c.90 nm. Dadfar et al.32 described a centrifugation protocol to perform size-based separation for polydisperse dispersed MNP suspensions, with evaluation of the MRI, MPI and hyperthermic efficacy of the fractions. The authors used a coprecipitation technique to synthesize MNPs; however, it has been reported in the literature that the MNPs produced using coprecipitation may have poor crystallinity, colloidal stability and poor size control leading to poor magnetic properties.33 These are typical of the field in that they do not include combined MRI and hyperthermia (dependency on the applied frequency, concentration and temperature probe depth) analysis to evaluate the physical basis of the efficacies for both modes. Simultaneous optimisation of the SAR and relaxivity of MNP suspensions is critical for the development of multi-functional agents, and can provide insights into how moment dynamics determines both properties.33,34 However this approach has not been reported for spherical MNPs.
In particular the effect of the ligand anchor group on SAR and r1,2 is not well understood. It has been reported that the surface linkers can have direct effects,35 as binding strength can alter the thickness of outer oxide disordered, or canted, layers. The ligands (headgroups and tails) can also have indirect effects by altering particle dispersion/aggregation in different media. Furthermore, the direct and indirect effects depend on the MNP size in different ways. These complexities have resulted in conflicting literature reports,36,37 which have hampered the rational and systematic design of reproducible multifunction agents utilizing MNP suspensions.
These limitations motivated this study, in which we describe the optimisation of a thermal decomposition MNP synthesis protocol, to provide 16 nm Fe3O4 MNPs with strong reproducible SARs from batch-to-batch. We evaluate the sensitivity of the SAR measurement to particle concentration and other conditions, identifying a collective-particle scenario (reversible aggregation into short chains) as responsible for extraordinarily high SARs at high concentration. Evaluation of the materials following sequential phase transfer from organic to aqueous and back to organic suspension, and also of organogels, allowed evaluation of the effect of ligand headgroups and tails, of diffusion, and of aggregation on the concentration-dependence of SARs. Field-cycling NMR relaxometry measurements, of the 1H Larmor frequency dependence of r1, pinpointed the effects of the surface modifications on moment dynamics, and this provides new insights into the drivers of high SARs. Recommendations for the development of dual-functional MNPs and their correlation with the magnetic characteristics are presented.
2. Materials and methods
Materials
Iron(III) acetylacetonate (97% and 99%) from Alfa Aesar, and oleic acid, oleylamine, and dioctyl ether from Aldrich were purchased. Polyethylene glycol (2000 g mol−1), gallic acid, N,N′-dicyclohexylcarbodiimide, 4-dimethylaminopyridine and all solvents were purchased from Sigma Aldrich. Milli-Q water (18.2 MΩ, filtered with a filter pore size of 0.22 ΩM) was from Millipore.Synthesis of iron oxide nanoparticles
The experiment was carried out in 100 mL three-neck round bottom flasks connected to water-cooled condensers. Various factors were changed for the synthesis and Fe3O4 nanoparticles were prepared using modification of a procedure from Cheon.38 Fe(acac)3 (2 mmol, 0.71 g), oleic acid (6 mmol, 1.70 g), oleylamine (6 mmol, 1.61 g), and dioctyl ether (20 mL) were mixed first and heated to 200 °C under a N2 atmosphere and held at this temperature for an hour. The reaction mixture was refluxed to 300 °C with a heating ramp of 5 °C min−1 and held for an hour. The reaction mixture was cooled down to room temperature and MNPs were precipitated and washed with ethanol five times (5 × 10 mL). The wet nanoparticles were dispersed in a toluene solvent. The typical yield of the nanoparticles was 70 mg and the percentage yield was around 45%.Synthesis optimisation
The inclusion of a hold step at 200 °C for 1 h enables full conversion of the precursor to monomer below the nucleation temperature. Extensive study of the reaction conditions highlighted the necessity of this hold step, the correct precursor amount and surfactant ratio (L/P), and the use of dioctyl ether as the solvent in preparing monodisperse particles of good morphology (TEM), monodispersity, and consistently high SAR batch-to-batch.8,39,40 The complete set of factors investigated was an initial degassing step at 110 °C, the 1 h hold step at 200 °C, reflux time, precursor amount and source, precursor to surfactant ratio, octyl ether solvent ageing, and the use of an N2 blanket. Full details showing the effect on size, crystallinity, and heating efficiency of the particles are shown in Table S1B (ESI†). The hydrodynamic sizes of these particles are small (dhyd ∼ 18–20 nm) compared to the original report (dhyd ∼ 23 nm). We observed a lower heating efficiency (SAR) of the suspensions when the reaction was performed under an Ar blanket, as shown in Table S1A (ESI†), and it may be that changes at the surface interface affect nucleation, hence an N2 atmosphere without flow is recommended. The SAR is also found to be dependent on the precursor to surfactant ratio over the range 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 1:5, and 1:6 (Table S1C, ESI†), and the highest value measured for the suspension prepared with a ratio of 1:6. At a 1:6 precursor to surfactant ratio, there would be sufficient OA/OAm ligands covering the nanoparticle surface to avoid any dipole–dipole interaction and further aggregation, which could have lead to a high SAR for this ratio.
:4, 1:5, and 1:6 (Table S1C, ESI†), and the highest value measured for the suspension prepared with a ratio of 1:6. At a 1:6 precursor to surfactant ratio, there would be sufficient OA/OAm ligands covering the nanoparticle surface to avoid any dipole–dipole interaction and further aggregation, which could have lead to a high SAR for this ratio.
The optimised synthesis (with L/P ratio 6/1) involves mixing the precursor (0.71 g, 2 mmol), surfactants (1.70 and 1.61 g, 6 mmol) and solvent (20 mL) in a 100 mL round bottomed flask using a stirrer at 400 rpm for 5 mins, then heating to 200 °C under a N2 atmosphere and holding at that temperature for one hour. The temperature was then increased to 300 °C with a heating ramp of 5 °C min−1 and refluxed for one hour. During the reflux time, the stirring rate was reduced to 50 rpm and nitrogen flow was reduced to a minimum to avoid any disturbances that could trigger nucleation. The flask was then allowed to cool to room temperature and the solids were precipitated by the addition of 100 mL ethanol. The physical properties of the resulting oleate/oleylamine stabilised particles (OA/OAm-MNPs), prepared in typical yield of 45 ± 5% using the optimised synthesis, are shown below.
The reproducibility of the final properties (hyperthermic response, DLS and TEM) of the OA/OAm-MNP samples prepared with the optimised procedure and using different Fe(acac)3 sources are shown in Fig. S1 and Table S2 (ESI†). TEM analysis confirms the spherical shape and monodisperse size with a reproducible dTEM of ∼16 nm. The different batches show similar dhyd of ∼23 nm with narrow size distribution, PDI ∼ 0.15, demonstrating similar dispersibility. The heating efficiency of batches from the same Fe(acac)3 source are reproducible; with average SAR values measured (in the high concentration range, see below) to be 2700 ± 95 or 1955 ± 222 W g−1, for 97 and 99% pure Aesar, respectively. Hence Aesar 97% was used for the rest of the study. Finally, the hyperthermic response of OA/OAm-MNPs was significantly affected by the age of the octyl ether solvent after opening the bottle, Fig. S1 (ESI†). This is despite storing under N2 and/or in a glove box.
Dynamic light scattering (DLS) and zeta potential
DLS measurements were performed at 25 °C on a Malvern NanoZS (Malvern Instruments, Malvern UK) instrument, which uses a detection angle of 173°, and a 3 mW He–Ne laser operating at a wavelength of 633 nm. The Z-Average (mean hydrodynamic) diameter, dhyd, and the polydispersity index (PDI) values were obtained from analysis of the correlation functions, which was performed via cumulants analysis using the Dispersion Technology software (v. 4.10, Malvern Instruments, Worcestershire, U.K.)AC-field hyperthermia
Measurements on bulk homogeneous samples were carried out using a NanoTherics NAN201003 MagneTherm™ AC-field generator (NanoTherics Ltd; Newcastle-under-Lyme, United Kingdom). The system enables the temporal evolution of temperature upon the application of an AC-field to be measured. 1 mL of MNP suspension was transferred into a cylindrical shaped Eppendorf tube and placed in a thermally insulating polystyrene sample holder to perform measurements close-to-adiabatic conditions. The sample temperature was measured by using a non-metallic temperature sensor (Opsens Ltd, Canada) to avoid eddy currents. All samples were of 1 mL total volume, and hence were 20 mm in height in standard Eppendorf tubes. For most measurements the probe was placed 5 mm from the bottom of the sample. Measurements were also performed at different probe heights and at different particle concentrations, see below. The temperature of the sample was equilibrated in the instrument before the desired field was applied. All these measurements were carried out at a frequency νAC of 535 kHz and magnetic field strength HAC of 16 kA m−1. These will be referred to as ‘closed system’ measurements.The temperature rise upon the application of AMF as a function of time was measured to calculate the initial slope of the response, which was used to calculate SAR using the following equation:

Magnetic resonance characterisation
The spin-lattice relaxivity, r1, of the solvent is defined as the 1H spin-lattice relaxation rate enhancement per mM of Fe in the suspensions. The r1 value (units s−1 mM−1) is calculated using the following equation:where, R1,sus and R1,sol are the spin-lattice relaxation rates (units, s−1) of the MNP suspension and pure solvent, respectively, and [Fe] is the iron concentration in mM. r1 values were measured in the frequency range of 0.01–40 MHz using a Spinmaster FFC-2000 Fast Field Cycling NMR Relaxometer (Stelar SRL, Mede, Italy). The system operated at a measurement frequency of 16.3 MHz for 1H, with a 90° pulse of 7 µs. The temperature of the samples was maintained at 25 °C using a thermostated airflow system. T1 measurements were performed as a function of external field, B0, with standard pulse sequences incorporating field excursions. The dhyd values of all toluene and water suspensions were unchanged after the FFC-NMR measurements, suggesting no field-induced aggregation.
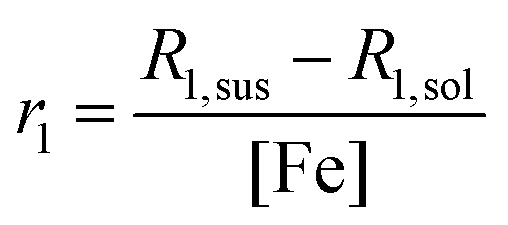
Iron determination using atomic absorption spectroscopy (AAS)
The elemental analysis of iron was performed by digesting 40 µL of a stock solution in 1.5 mL concentrated HCl (12 M) and kept for 2 h till all iron was dissolved in HCl. These samples were further diluted to 50 mL with 1 M HNO3 using a volumetric flask. AAS was performed using a Varian SpectrAA 55B atomic absorption spectrometer. The error in determining the concentration of Fe using AAS is estimated to also be ∼2.5%.Transfer of toluene based MNPs into water and back to toluene
Toluene based MNPs (5 mg mL−1) were dispersed in 5 mL of tetramethylammonium hydroxide (TMAOH) and 10 mL of ethanol.19,42 The ratio of the particles/ethanol/TMAOH was kept at 1:10:5 in all the cases. The above suspension was sonicated for 30 min and the slurry of bare MNPs in the form were separated using a strong magnet. After washing, the obtained MNPs dispersed well in water. Bare nanoparticles in water were transferred back to organic solvent (toluene) using oleic acid ligand. The first 10 mg of oleic acid was dissolved in 2 mL of toluene. 10 mg of the slurry of bare MNPs was dried using nitrogen gas and then added to the oleic acid solution. The MNP suspension was kept on a shaker overnight.
Synthesis of PEG2000-Gallol
The synthesis was adapted from Guardia et al.8 Poly(ethylene glycol) (1.0 g, 0.5 mmol, MW 2000 g mol−1) was dissolved in 70 mL tetrahydrofuran through sonication for 30 min and mild heating. 4-Dimethylaminopyridine (DMAP) (0.0089 g, 0.07 mmol) was dissolved in THF (1 mL) and N,N′-dicycyclohexylcabodiimide (DCC) (0.5256 g, 2.5 mmol) was separately dissolved in THF (5 mL). Gallic acid (0.086 g, 0.5 mmol) and the DMAP solution (1 mL) were added to the dissolved PEG solution. The solution was magnetically stirred at room temperature and the DCC solution (5 mL) was added dropwise. The reaction mixture was left to stir for ∼20 h. After the reaction, THF solvent was removed by a rotary evaporator and 200 mL de-ionised water was added to dissolve the solid. The pH of the solution was adjusted to pH 2 using 1 M HCl and allowed to sit for an hour. The aqueous solution was decanted using a centrifuge (5500 rpm for 10 min) and the solid precipitate was discarded. The product was extracted from the aqueous phase with chloroform (4 × 100 mL). CHCl3 was removed using a rotary evaporator and a pale, yellow-coloured PEG Gallol solid was dried under N2 and stored in a pre-weighed vial.In situ coating of PEG2000-Gallol on MNP surfaces
Toluene suspensions of OA/OAm-MNPs were transferred to aqueous media using a PEG2000-Gallol ligand8 without workup. Once the reaction duration was complete, the reaction temperature was reduced from 300 to 70 °C. 5 mL of PEG2000-Gallol solution (0.69 g of PEG2000-Gallol dissolved in 5 mL chloroform containing 0.5 mL triethylamine) was added to the 10 mL of reaction mixture and stirred overnight at 70 °C. The reaction mixture was cooled to room temperature and transferred to a separating funnel. 10 mL of de-ionised water was added to the separating funnel, which forms two phases. The aqueous phase containing PEG2000-Gallol MNPs was separated, and this was repeated until most of the MNPs had been visually transferred into water. Excess water was removed using a rotavapor and the volume was reduced to 10 mL. MNP suspensions were dialyzed against 2.5 L deionised water overnight using a 3.5 kDa pore sized cellulose membrane to remove excess PEG2000-Gallol. The dialysis step was repeated three times.Magnetic organogels
Poly(styrene-b-acrylic acid) was used to prepare the magnetic organogels. An appropriate amount of wet magnetic particles (0.5–8.0 mg) was dispersed in 1 mL chloroform, which forms a good suspension without aggregation. 0.10 g of poly (styrene-b-acrylic acid) was added to the chloroform based MNP. The suspension was sonicated for 15 min and refrigerated at 2 °C for two hours. The MNP organogel was kept at room temperature overnight before measuring the SAR.3. Results and discussion
3.1 MNP synthesis and primary characterisation
Spherical magnetic iron oxide nanoparticles were synthesised by thermal decomposition of iron(III) acetyl acetonate, Fe(acac)3, at 300 °C for 1 h with the surfactants oleic acid and oleylamine in high boiling point non-polar dioctyl ether. These will be referred to as OA/OAm-MNP suspensions. We followed the well-known procedure from Cheon and co-workers38 who describe ∼10 g scale synthesis of high SAR Zn- and Mn-doped ferrite MNPs.38 Following optimisation, the procedure provides Fe-only oxide MNPs with very high and reproducible SARs. Full details of the optimisation, the final protocol and a demonstration of reproducibility are provided in the Experimental section. In summary by using (i) an intermediate temperature hold step (200 °C for 1 h) to ensure full decomposition of the precursor; (ii) an appropriate precursor source; (iii) a surfactant to precursor mole ratio L/P of 1:6, and; (iv) dioctyl ether as the solvent, it was possible to prepare consistent batches with high, reproducible SARs. Rinaldi and co-workers have shown reproducibility for the Fe(oleate) system in docosane,43 a precursor that is difficult to provide reproducibly.34 To the best of our knowledge, ours is the first reproducibility study reported for the key OA/OAm/Fe(acac)3 system, a stable, readily available precursor. This advance was key for the analysis into the effects of MNP concentration, surface ligands and suspending medium that follows, as multiple batches were required.
Transmission electron microscopy, Fig. 1A, shows that OA/OAm-MNPs are spheres with a narrow size distribution; particles are well distributed on the TEM grids with a (sample) average size, dTEM, of ∼16.2 ± 1.8 nm. The size distribution conforms to a lognormal function, as shown. Dynamic light scattering, Fig. 1B, shows a z-average hydrodynamic size, dhyd, of 23.5 nm in toluene and low polydispersity index, PDI, of 0.11. The ∼3.5 nm thick organic layer inferred suggests full particle dispersion/absence of aggregation. The X-ray diffraction pattern of dried OA/OAm-MNPs, Fig. S2 (ESI†), is consistent with magnetite with peaks indexed to cubic spinel (Fe3O4-JCPDS index card number 49549-Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) mz).44 The average crystallite size of ∼11 nm obtained from Scherrer analysis is smaller than dTEM, which suggests the presence of a canted or disordered layer,45,46 see below. The presence of some γ-Fe2O3 cannot be ruled out, however, the suspensions remain very black (for >12 months) suggesting magnetite is predominant. More importantly we show below that the colloidal, hyperthermic and magnetic resonance properties of the suspensions are unchanged over many months, demonstrating stability toward further oxidation.
mz).44 The average crystallite size of ∼11 nm obtained from Scherrer analysis is smaller than dTEM, which suggests the presence of a canted or disordered layer,45,46 see below. The presence of some γ-Fe2O3 cannot be ruled out, however, the suspensions remain very black (for >12 months) suggesting magnetite is predominant. More importantly we show below that the colloidal, hyperthermic and magnetic resonance properties of the suspensions are unchanged over many months, demonstrating stability toward further oxidation.
 | ||
| Fig. 1 Characterisation of the OA/OAm-MNP toluene suspensions from the optimised procedure. (A) Representative TEM image of the as-synthesised OA/OAm-MNPs (n = 185), a lognormal fit is included for illustration. (B) Hydrodynamic size and (cumulants) fitted DLS correlograms, confirming the appropriateness of the z average as a measure of dhyd. (C) Temperature as a function of AC-field irradiation time keeping the thermal probe at 0.25 cm from the bottom of the Eppendorf. (D) Temporal evolution of SAR (νAC 535 kHz, HAC 16 kA m−1) and OA/OAm-MNP size during the reaction; the scale bars are 10 nm. | ||
3.2 Measurement of SAR in strongly heating MNP suspensions
The AC-field heating efficiency, SAR, of the optimised OA/OAm-MNP toluene suspensions was measured, at νAC 535 kHz and HAC 16 kA m−1, using a thermal probe at 0.5 cm from the bottom of an insulated Eppendorf (see the Experimental section). A typical experiment is shown in Fig. 1C for which the initial slope was 4.9 °C s−1, corresponding to a SAR value of 2797 ± 140 W g−1 (a 5% error estimate, see the Experimental section) at a relatively high Fe concentration of 2.6 mg mL−1. This is equivalent to an ILP of 20.4 W m2 g−1 kA−2 kHz−1. The response is in the range of the highest reported values,47,48 hence it is further investigated below. There are two points worth noting.Firstly, Cheon et al.38 reported a SAR of 432 W g−1 for Zn0.4Mn0.6Fe2O4, equivalent to an ILP of 0.5 W m2 g−1 kA−2 kHz−1 using the procedure on which this study is based. For comparison we reproduced the reported procedure, in so far as is possible, to prepare Fe-only MNPs and measured a lower value of 317 W g−1 at 2.8 mg mL−1 Fe, under our AC-field conditions, which is equivalent to a much lower ILP of 2.3 W m2 g−1 kA−2 kHz−1 This further demonstrates the value of the optimisation and the critical role of the hold step. Secondly, the temporal evolution of OA/OAm-MNP properties was evaluated by taking aliquots during a reaction, Fig. 1D. The SAR recorded at the end of this experiment was slightly lower than usual for the optimised reaction, presumably due to the repeated disturbance. The highest SAR, 1698 W g−1, at 105 mM Fe, was measured for the 60 min reflux suspension. The 45 min reaction time produced a far weaker response even though the final size was almost achieved. Note that there was no evidence of sub-populations of smaller MNPs in the images recorded for the aliquots. Interestingly, the SAR decreased to 145 W g−1 on extending the reflux to 2 hr, suggesting reduced crystallinity. This further illustrates the sharpness of the ‘sweet spot’ in the parameter space for the optimised reproducible synthesis.
The robustness of the high heating response of the optimised OA/OAm-MNP toluene suspensions was evaluated. First the thermal probe was placed at different locations within the insulated Eppendorf tube, see Fig. 2A. It is clear that, at the concentration used (70 mM, or 3.9 mg mL−1 Fe), the response is depth-dependent; the values obtained increase moving down from the upper surface and stabilise with a variation of c.4% at 0.5 and 0.0 cm from the bottom. This suggests that the sample is not homogeneous or heat dissipation is greater at the top of the tube. Temperature differences of up to 7.5 °C between the top and bottom of the sample holder have been described previously20,22 and ascribed to steady state diffusion and convection effects. In most reports of high SAR materials8,10,38,42,48–50 the probe location is not specified.
 | ||
| Fig. 2 Characterisation of the optimised OA/OAm-MNP toluene suspensions: (A) SAR (535 kHz, 16 kA m−1) values recorded with the thermal probe at different depths (0.0 cm is closest to the bottom of the 2 cm tall, 1 mL samples) at Fe 3.9 mg mL−1. Inset corresponding temperature changes as a function of irradiation time. (B) SAR recorded as a function of Fe concentration for three different optimised batches, with the probe placed 0.50 cm from the bottom. Characterisation of OA/OAm-MNP-organogels: (C) SAR values recorded with the probe at different depths (Fe 1.38 mg mL−1). (D) SAR (green circles) and heating ramps (blue squares) as a function of Fe concentration, with the probe at 0.50 cm. | ||
Secondly, the dependence of SAR on Fe concentration was evaluated for three independently prepared optimised suspensions, Fig. 2B, with the probe placed at 0.50 cm from the bottom. A ∼63% decrease in the average measured SAR, from ∼2700 to ∼1000 W g−1, was measured on decreasing the Fe concentration from ≥3 to ≤1 mg mL−1. The dependence of SAR on concentration is not always evaluated, despite the fact that significant changes with concentration have been described.21,22,51 For instance Ivan et al.21 reported increased efficiency on increasing Fe concentration over a similar range to that studied here. The effect was ascribed to local dipolar interactions, i.e. a collective particle scenario, although no measurement of depth dependence was reported. Hence higher SAR values at higher concentrations for the optimised OA/OAm-MNP suspensions probably arise from the reversible formation of ordered aggregates, e.g. short chains11 which have been shown to open up the AC-field hysteresis loop.
For OA/OAm-MNP when the probe was placed close to the upper surface (at 1.5 cm) the measured SAR values were lower, presumably due to heat dissipation; on average a reduction to ∼55% of the original was measured over the same concentration range between 0.3 and 8 mg mL−1, as shown in Fig. S3 (ESI†). Nevertheless, a significant increase in SAR on increasing Fe concentration into the higher range was maintained. These findings confirm that OA/OAm-MNP suspensions are homogeneous at all concentrations and that a concentration-dependent SAR enhancement (presumably arising from the formation of short-chains) is observed irrespective of the probe position.
Finally, the heating efficiency of organogels, formed using p(styrene-b-acrylic acid) and OA/OAm-MNPs, see the Experimental section, was measured at different probe locations, Fig. 2C, and different Fe concentrations, Fig. 2D. It was found that the SAR of the gel (calculated using a heat capacity of 1.56 J g−1 °C−1) was lower than in suspension with an average of 215 ± 4% W g−1. The reduced response arises from the loss of a significant Brownian (MNP diffusion) hyperthermic contribution due to the matrix. The value was independent of probe position, confirming that the gels are homogeneous (as they appear to the eye) and that heat dissipation at the upper surface is not a significant issue, presumably due to the reduced temperature increase. The absence of a transition to a higher SAR at a higher concentration demonstrates that, as expected, gelation prevents the collective particle scenario, i.e. there is full MNP dispersion in the gels across the concentration range studied, and the enhancement is not due to dipolar interactions between dispersed particles.
The data shown in Fig. 2 re-emphasises7 the value of evaluating concentration- and probe depth-dependence for high SAR suspensions. Low concentration (dispersed) SAR values should be reported both to enable comparisons and because the upper range is not normally achievable for applications. The key result is that for fully dispersed OA/OAM-MNPs in toluene the (dispersed particle) SAR is 1000 ± 50 W g−1, an ILP of 7.9 W m2 g−1 kA−2 kHz−1. This is achievable reproducibly and is sufficiently high for hyperthermic applications.
3.3 Colloidal dispersion in different media and magnetic resonance characterisation
OA/OAm-MNPs in toluene were transferred into aqueous suspension by stripping off the organic ligands (oleic acid and oleylamine) using the base TMAOH, to form aqueous suspensions, as shown schematically in Fig. 3A. For the bare particle, or B-MNP, suspensions the SAR, in water, was lower with indistinguishable values of 211 and 205 W g−1 recorded in the higher and lower concentration ranges (down from 2211 and 1000 W g−1, in toluene). The aqueous B-MNP suspension had dhyd ∼ 34 nm, and slightly higher PDI of 0.14, Fig. 3C. It is clear that there is some loss of dispersion for B-MNPs in water. Presumably the clusters formed are random aggregates which, unlike ordered chains, are known to suppress SAR.23 Although the hyperthermia measurements are made at a higher concentration than is used for DLS, it is clear that the B-MNP size is unchanged across the concentration range studied.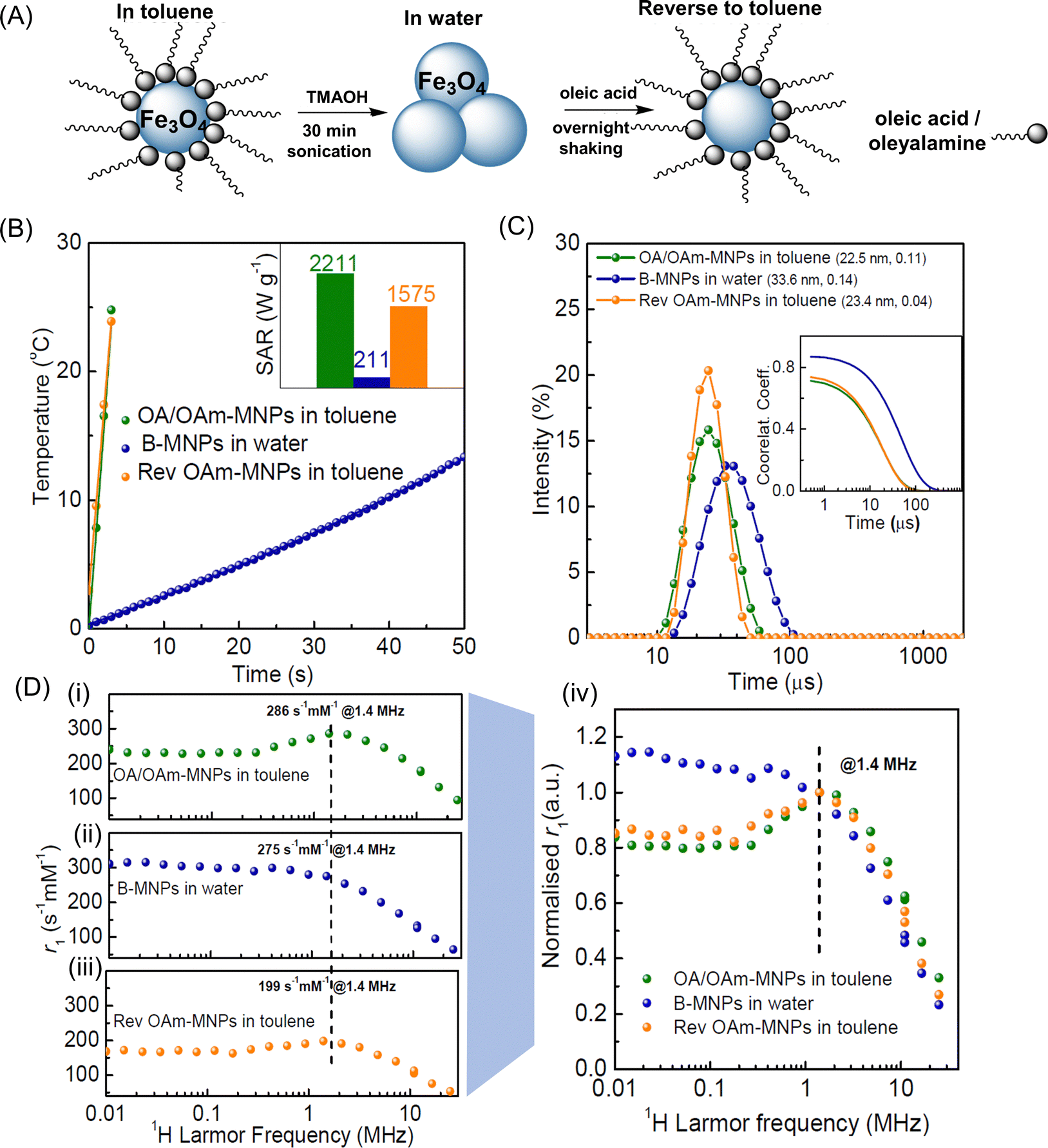 | ||
| Fig. 3 Colloidal characterisation of MNPs in different suspending media. (A) Schematic representation of MNPs in toluene suspension transferred to water (clusters are formed) and then redispersed back into toluene. (B) AC-field heating ramps of nanoparticles in toluene, water, and re-dispersed in toluene (inset are the respective SAR values) measured within the high concentration range (at 3.88, 4.75, and 4.75 mg mL−1, for OA/OAm-, B-, and OA-MNPs respectively) with the probe at 0.5 cm. (C) Hydrodynamic size (intensity) distributions of the same MNP suspensions with, inset, DLS correlograms. (D) FFC-NMR profiles (spin-lattice relaxivity, r1, as a function of 1H Larmor frequency) for dispersed MNP suspensions in; (i) toluene (OA/OAm-MNPs); (ii) water (B-MNPs), and; (iii) back in toluene (OA-MNPs); (iv) normalised relaxivity (arbitrary units) of the suspensions scaled to a common value at 1.4 MHz. | ||
B-MNPs were subsequently transferred back to toluene suspension using oleic acid (only) as the stabilising ligand to form OA-MNPs, as shown in Fig. 3A. The Fe yield for the first step was found to be 88% and for the second 93%, returning to 82% of the original toluene suspension. The two toluene suspensions, OA/OAm-MNP and OA-MNP (before and after transfer) are fully dispersed within the DLS concentration range (<0.5 mM) showing very similar dhyd ∼ 23–24 nm and low PDI (≤0.10). The correlograms are superimposable, Fig. 3C, so the aggregates observed in H2O are removed, and the original particle diffusion correlation time, τD, is recovered (within error). This view is supported by the fact that the dTEM value is unchanged for OA-MNPs at 16.2 ± 1.9 nm. Hence the 18% loss of material is from across the size distribution, i.e. not preferentially from larger (more magnetic) or smaller (less magnetic) particles. AC-field heating ramps of MNPs suspended in toluene, water, and on re-suspending back to toluene are shown in Fig. 3B. On returning to toluene the OA-MNPs regained most of their hyperthermic response with a value of 1575 W g−1 measured in the high concentration range, at 4.75 mg mL−1, which is ∼71% of the original OA/OAm-MNP value. In the dispersed range the recovery was 91%, with a value of 910 W g−1 recorded, down from 1000 W g−1. As there is no size dependence to the MNP loss SARBrown (the Brownian contribution) is expected to remain unchanged. This points to changes in SARNéel, arising from the effect of the different ligand binding groups, as the cause. These issues are discussed in Section 3.5.
The 1H spin-lattice relaxation of the aqueous and organic MNP suspensions was evaluated, in the dispersed concentration range (0.5–1.0 mM), using fast field-cycling NMR relaxometry (FFC-NMR), Fig. 3D. In this approach r1 is measured as a function of field strength and hence 1H Larmor frequency, νL. The shape of the FFC-NMR profile is highly sensitive; (i) to MNP size (which determines, τB, the solvent diffusion time past the quasi-static MNP); (ii) to crystallinity (which determine magnetisation, Ms) and; (iii) to the inherent magneto-crystalline anisotropy of the particles and to inter-MNP interactions, which together determine the Néel correlation time, τN, which characterises moment re-orientation.14 The profile of OA/OAM-MNPs in toluene Fig. 3D-i, is the characteristic superparamagnetic response well-documented for water suspensions.14 The low frequency r1 plateau, prominent r1 maximum in the low MHz range, and strong decrease in r1 at higher frequencies further demonstrate that the particles are fully dispersed. The r1 values at low frequency are high, as compared to suspensions of smaller MNPs,46 which is consistent with longer τN (higher magneto-crystalline anisotropy) as expected given the higher magnetic volume of the particles studied here. An accepted model, SPM-theory, was developed15 for aqueous suspensions of dispersed superparamagnetic nanoparticles, which enables semi-quantitative evaluation of the profiles, see below. However detailed interpretation of the toluene profiles will not be attempted, as they may be complicated by the different 1H environments present in the solvent molecules.
For B-MNP suspensions in water the shape of the profile is quite different, Fig. 3D-ii, with clear suppression of the r1 maximum and significantly increased low frequency r1 evident. This change can be unambiguously ascribed36 to increased τN, due to inter-MNP dipolar interactions within clusters. This loss of superparamagnetism also apparently suppresses SAR. Hence DLS (which is weighted by the 6th power of scatterer size) suggests the presence of clusters, and the SAR and r1 measurements (which provide number averages for the suspensions) confirm this view.
On re-dispersion back in toluene, Fig. 3D-iii, the ‘SPM-like’ profile re-emerges for the OA-MNP suspension with a 70% recovery (at 1.4 MHz) of the original OA/OAm-MNP r1 value. Scaling the profiles recorded in toluene by this %, Fig. 3D-iv, shows that the original OA/OAm-MNP profile shape is almost fully restored. The almost exact agreement at high frequency is expected, given DLS shows the original dhyd/τD values are recovered. The scaled relaxivity at low frequency for OA-MNPs is slightly higher, which suggests slightly increased τN.
As noted above, the retention of the original τD on redispersion in toluene (from DLS) shows that the SAR recovery of only 91% is associated with changes to SARNéel. FFC-NMR shows that the τN value increases slightly, despite full dispersion. This may arise due to stronger binding of carboxylate (only) groups, altering the outer disordered oxide layer.37 In any case the r1 suppression is a stronger effect, and it is observed across the frequency range. As noted, Néel dynamics dominate the shape at low frequency, and Brownian dynamics at high frequency, giving rise to the dispersive features. The absolute value of r1, is determined by the strength of the solvent 1H to particle moment coupling, which in effect scales the profiles up and down. We suggest that a thicker conformal oleic layer with densely packed fatty acid tails reduces r1 (at all frequencies) by increasing the distance of closest approach of the diffusing solvent molecules. Differences in surface packing for oleic acid and oleylamine are well-documented.52 The fact that re-dispersion with OA only results in 70% recovery of r1 and 91% of SAR shows that hyperthermia is less sensitive to solvent-particle interactions, as might be expected.
3.4 Aqueous dispersions of PEGylated MNPs
PEG surface grafting was undertaken before work-up for an optimised MNP batch to assess the properties of fully dispersed aqueous suspensions. An in situ method for PEG-gallol ligand exchange on magnetic nanocubes has been reported8 in which catechol-like trihydroxy gallol groups strongly bind to FeO, and the PEG chains provide good steric stabilisation. Hence in situ PEG2000-gallol surface grafting was undertaken before work-up of an OA/OAm-MNP batch. This functionalised PEG has been recently reported,53 and full details of the procedure are provided in the Experimental section. The typical Fe yield for the in situ reaction was ∼40% (∼45% for OA/OAm-MNPs). The particle characterisation and hyperthermic and relaxivity data for aqueous P-MNP suspensions are shown in Fig. 4. The dTEM value obtained for OA/OAm-MNPs of ∼16.9 ± 2.1 nm (sample average) shows that P-MNP morphology and size were unchanged, within error, as compared to OA-MNPs. The dhyd in H2O was ∼33 nm, which is greater than the value of 23 nm measured for OA/OAm-MNPs in toluene, due the grafted PEG2000 chains.49,53 The relatively small size and low PDI of 0.09 suggest full particle dispersion. | ||
| Fig. 4 Characterisation of the PEG2000 grafted MNPs (P-MNPs). (A) Hydrodynamic size and (cumulants) fitted DLS correlograms for PEGylated MNPs in aqueous suspension. Inset representative TEM image of the PEGylated MNPs. (B) Temperature as a function of AC-field irradiation time (probe at 0.5 cm) for the same aqueous suspension, at Fe 5.70 mg mL−1. Inset, the SAR values of these MNPs recorded over a 12 month period for Fe 2.79 mg mL−1. (C) SAR in suspension as a function of iron concentration. For (B and C) the thermal probe was placed at 0.5 cm from the bottom. (D) FFC-NMR profile for an aqueous P-MNP suspension at Fe 0.94 mg mL−1 and for an OA-MNP suspension in toluene at Fe 0.76 mg mL−1. The solid line is a simulation of the P-MNP profile using SPM theory (dcore 16.9 nm, Ms 69 emu g−1, τN 65 ns, ΔEanis 3.0 GHz). | ||
The hyperthermic response of the aqueous P-MNP suspensions is significantly reduced to 245 W g−1, Fig. 4B, far lower than 1000 and 910 W g−1 measured for dispersed OA/OAm- and OA-MNPs, respectively, in toluene. The value for P-MNPs remains stable over a 12 month period, and is independent of Fe concentration, Fig. 4C, even in the high SAR range. This confirms full dispersion of P-MNPs in water at all concentrations studied, due to strong steric repulsion, and the absence of a collective particle scenario. The lower SAR may arise from the effect of grafted PEG on the Brownian contribution,45 and/or, as noted above, from the effects of the binding groups.26
The FFC-NMR profile of aqueous P-MNP suspensions, Fig. 4D (note the r1 values in this figure are not scaled), is revealing. The r1 maximum at 0.9–1.0 MHz is in the expected range for superparamagnetic particles of this size, confirming that the suspension comprises fully dispersed single cores. Simulation of the aqueous P-MNP profile was undertaken using SPM-theory.15 The agreement is very good with all the features clearly captured despite the relatively large MNP size, which places these particles in the upper part of the superparamagnetic range for which the model was developed. Good agreement was achieved using the TEM core size and a saturation magnetisation, Ms, value of 69 emu g−1, which is in the expected range for Fe3O4 MNPs of this size,45 but only when a long τN of 65 ns was specified, Fig. 4D.
It is very interesting that the P-MNP profile is almost superimposable with that of the re-dispersed OA-MNPs in toluene. As noted above, the different 1H environments in toluene complicate quantification of the profiles, and so simulations are not appropriate and we do not interpret the absolute r1 values for the toluene suspensions. Nevertheless, it is clear that the profiles of the three suspensions of fully dispersed particles (OA/OAM-, OA-, and P-MNPs) are superimposable on scaling. This demonstrates that the key correlation times are largely unchanged and differences in the hyperthermic and relaxometric responses arise from ligand-induced changes in the particle diffusion and the surface solvent access, respectively.
3.5 Comparing the hyperthermic and relaxometric Properties
1H NMR relaxation in MNP suspensions is driven by dynamic processes that modulate the angle that the vector which joins the 1H to the particle moment makes with the external field. At low frequency the Néel correlation time, τN, (for moment re-orientation) dominates r1. At high frequency the moments are locked to the external field and the Brownian correlation time, τB, (solvent diffusion) is determined. The key processes that contribute to hyperthermia are τN and τD (MNP diffusion), both of which generate motion of the particle moment. As the hyperthermic and relaxometric properties are sensitive to different processes some interpretation can be cautiously attempted by comparing the values for the different toluene suspensions/organogels in the dispersed-particle concentration range, Table 1.| [Fe] range (mM)a (mg mL−1) | 0.50 | >0.50 | 27–45 | 45–90 | Comment |
|---|---|---|---|---|---|
| 0.028 | 0.028 | 1.5–2.5 | 2.5–5.0 | ||
| d hyd (nm) | Character at 295 K from FFC-NMR | SAR low [Fe] (W g−1) | SAR high [Fe] (W g−1) | ||
| Measurement | |||||
| a 17.9 mM Fe ≡ 1 mg mL−1. | |||||
| OA/OAm-MNP in toluene | 24 | Superpara-magnetic | 1000 ± 50 | 2797 ± 90 | MNP chains at high [Fe] |
| OA-MNP in toluene | 24 | Superpara-magnetic | 910 ± 46 | 1575 ± 79 | Shorter chains at high [Fe] |
| B-MNP in H2O | 34 | Ferromagnetic | 219 ± 11 | 211 ± 11 | Clusters at all [Fe] |
| P-MNP in H2O | 33 | Superpara-magnetic | 245 ± 12 | 245 ± 12 | Dispersed at all [Fe] |
(i) For dispersed OA/OAm-MNPs, assuming that gelation suppresses all contributions except SARNéel which is unchanged, then; SARGel = SARNéel = 215 W g−1.
(ii) For dispersed OA/OAm-MNP suspensions, assuming only Brown and Néel processes contribute to SAR, then; SARBrown = 785 W g−1 (= SARSusp–SARNéel = 1000–215 W g−1), which is c.4 times SARNéel.
(iii) For dispersed OA-MNP suspensions, assuming SARBrown is unchanged on re-dispersion (which is reasonable as τD is unchanged, Fig. 3B) at 785 W g−1; then SARNéel = 125 W g−1 (= SARSusp–SARBrown = 910–785 W g−1) which is a suppression to 58% of the original OA/OAm-MNP value (215 W g−1). This is due, primarily, to the effect of carboxylate (only) binding groups on τN, an effect that also increases the low frequency relaxivity.
It is interesting that the SARNéel recovery on re-dispersing (58%) is in the same range as the r1 recovery (70%). The analysis suggests that the effects on SAR for OA/OAm- and OA-MNPs arise from differences in τN due to the carboxylate (only) binding, and an effect on r1 arising primarily from the thick conformal surface C18:1 alkyl layer.
Finally, comparing the P-MNP and OA-MNP relaxation profiles; the close agreement shows again that the τN and τB values are similar for these two very different suspensions. Therefore, differences in SARNéel are unlikely to be the cause of the reduced SAR for P-MNPs. The strong inference is that reduced diffusional correlation time, τD, of the P-MNPs arising from the PEG chains is determining. In the case of P-MNPs, the SAR suppression arises largely from the greater hydrodynamic size, but this does not significantly affect the 1H relaxation.
4. Conclusions
The optimised method described here, modified from the approach of Cheon and co-workers, reproducibly provides toluene suspensions of MNPs with SAR of ∼1000 W g−1 (ILP 7.9 W m2 g−1 kA−2 kHz−1) when dispersed, and ∼2800 W g−1 (ILP 20.4 W m2 g−1 kA−2 kHz−1) in the collective particle range. An in situ PEG grafting approach is described which provides PEGylated MNPs with exceptional long-term aqueous colloidal stability and SAR of ∼245 W g−1 (ILP 1.9 W m2 g−1 kA−2 kHz−1).The suspension relaxivity and SAR are sensitive to the magnetic properties and the dynamics of the system in subtly different ways. For particles in this size range, processes that contribute to hyperthermia are modulated by ligand-related effects that alter τD, and to a lesser extent τN, either directly or by inducing aggregation. Processes that contribute to relaxivity in the clinical field range are determined primarily by τB and can be influenced by solvation of the ligand shell. The findings illustrate how design of multifunctional MRI-trackable hyperthermic nanovectors may depend on a trade-off between high PEG brush thickness, necessary for colloidal stability both in complex media and in vivo, and minimizing dhyd to maintain high SARs. Comparison of the profiles of MNP dispersions, that differ only in the headgroups used, suggests a second trade-off (for smaller MNPs in particular) in ligand binding strength, which should be sufficiently strong to ensure colloidal stability, e.g. in buffers/media containing phosphate, while minimising the thickness of the non-magnetic surface oxide layer. The analysis presented in this study highlights the importance of ligand modulation for the development of dual-functional MNP suspensions with improved magnetic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from Science Foundation Ireland (16/IA/4584).References
- N. Lee, D. Yoo, D. Ling, M. H. Cho, T. Hyeon and J. Cheon, Chem. Rev., 2015, 115, 10637–10689 CrossRef CAS PubMed.
- J. Zeng, M. Gong, D. Wang, M. Li, W. Xu, Z. Li, S. Li, D. Zhang, Z. Yan and Y. Yin, Nano Lett., 2019, 19, 3011–3018 CrossRef CAS PubMed.
- S. Laurent, S. Dutz, U. O. Häfeli and M. Mahmoudi, Adv. Colloid Interface Sci., 2011, 166, 8–23 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, Y. Wang, W. Zhu, G. Li, X. Ma, Y. Zhang, S. Chen, S. Tiwari and K. Shi, Theranostics, 2020, 10, 3793 CrossRef CAS PubMed.
- S. Dutz and R. Hergt, Nanotechnology, 2014, 25, 452001 CrossRef PubMed.
- I. A. Brezovich, Med. Phys. Monogr, 1988, 16, 82–111 Search PubMed.
- D. Ortega and Q. A. Pankhurst, Magnetic hyperthermia, in Nanoscience: Nanostructures through Chemistry, ed. P. O'Brien, Royal Society of Chemistry, Cambridge, 2013, vol. 1, pp. 60–88 Search PubMed.
- P. Guardia, A. Riedinger, S. Nitti, G. Pugliese, S. Marras, A. Genovese, M. E. Materia, C. Lefevre, L. Manna and T. Pellegrino, J. Mater. Chem. B, 2014, 2, 4426–4434 RSC.
- S.-h Noh, S. H. Moon, T.-H. Shin, Y. Lim and J. Cheon, Nano Today, 2017, 13, 61–76 CrossRef CAS.
- J.-H. Lee, J.-t Jang, J.-s Choi, S. H. Moon, S.-h Noh, J.-w Kim, J.-G. Kim, I.-S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418–422 CrossRef CAS PubMed.
- C. Martinez-Boubeta, K. Simeonidis, A. Makridis, M. Angelakeris, O. Iglesias, P. Guardia, A. Cabot, L. Yedra, S. Estradé and F. Peiró, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- J. Muro-Cruces, A. G. Roca, A. López-Ortega, E. Fantechi, D. del-Pozo-Bueno, S. Estradé, F. Peiró, B. Sepúlveda, F. Pineider and C. Sangregorio, ACS Nano, 2019, 13, 7716–7728 CrossRef CAS PubMed.
- Z. Nemati, R. Das, J. Alonso, E. Clements, M. Phan and H. Srikanth, J. Electron. Mater., 2017, 46, 3764–3769 CrossRef CAS.
- Z. W. Tay, P. Chandrasekharan, A. Chiu-Lam, D. W. Hensley, R. Dhavalikar, X. Y. Zhou, E. Y. Yu, P. W. Goodwill, B. Zheng and C. Rinaldi, ACS Nano, 2018, 12, 3699–3713 CrossRef CAS PubMed.
- A. Roch, R. N. Muller and P. Gillis, J. Chem. Phys., 1999, 110, 5403–5411 CrossRef CAS.
- L. C. Branquinho, M. S. Carrião, A. S. Costa, N. Zufelato, M. H. Sousa, R. Miotto, R. Ivkov and A. F. Bakuzis, Sci. Rep., 2013, 3, 1–11 Search PubMed.
- A. E. Deatsch and B. A. Evans, J. Magn. Magn. Mater., 2014, 354, 163–172 CrossRef CAS.
- E. Alphandery, S. Faure, O. Seksek, F. Guyot and I. Chebbi, ACS Nano, 2011, 5, 6279–6296 CrossRef CAS PubMed.
- D. Serantes, K. Simeonidis, M. Angelakeris, O. Chubykalo-Fesenko, M. Marciello, M. D. P. Morales, D. Baldomir and C. Martinez-Boubeta, J. Phys. Chem. C, 2014, 118, 5927–5934 CrossRef CAS.
- R. Wildeboer, P. Southern and Q. Pankhurst, J. Phys. D: Appl. Phys., 2014, 47, 495003 CrossRef.
- I. Conde-Leboran, D. Baldomir, C. Martinez-Boubeta, O. Chubykalo-Fesenko, M. del Puerto Morales, G. Salas, D. Cabrera, J. Camarero, F. J. Teran and D. Serantes, J. Phys. Chem. C, 2015, 119, 15698–15706 CrossRef CAS.
- B. Lahiri, S. Ranoo and J. Philip, J. Phys. D: Appl. Phys., 2017, 50, 455005 CrossRef.
- J. G. Ovejero, D. Cabrera, J. Carrey, T. Valdivielso, G. Salas and F. J. Teran, Phys. Chem. Chem. Phys., 2016, 18, 10954–10963 RSC.
- I. Rodrigo, I. Castellanos-Rubio, E. Garaio, O. K. Arriortua, M. Insausti, I. Orue, J. Á. García and F. Plazaola, Int. J. Hyperthermia, 2020, 37, 976–991 CrossRef CAS PubMed.
- S. Dutz and R. Hergt, Int. J. Hyperthermia, 2013, 29, 790–800 CrossRef PubMed.
- D. A. Herman, P. Ferguson, S. Cheong, I. F. Hermans, B. J. Ruck, K. M. Allan, S. Prabakar, J. L. Spencer, C. D. Lendrum and R. D. Tilley, Chem. Commun., 2011, 47, 9221–9223 RSC.
- B. Pelaz, P. Del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernandez, J. De La Fuente, G. Nienhaus and W. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- W. Xue, Y. Liu, N. Zhang, Y. Yao, P. Ma, H. Wen, S. Huang, Y. Luo and H. Fan, Int. J. Nanomed., 2018, 13, 5719 Search PubMed.
- L. M. Liz-Marzán, C. R. Kagan and J. E. Millstone, Journal, 2020, 14, 6359–6361 CrossRef PubMed.
- K. Hayashi, M. Nakamura, W. Sakamoto, T. Yogo, H. Miki, S. Ozaki, M. Abe, T. Matsumoto and K. Ishimura, Theranostics, 2013, 3, 366 CrossRef CAS PubMed.
- R. D. Piazza, W. R. Viali, C. C. Dos Santos, E. S. Nunes, R. F. C. Marques, P. C. Morais, S. W. Da Silva, J. A. H. Coaquira and M. Jafelicci, Mater. Res. Express, 2020, 7, 015078 CrossRef CAS.
- S. M. Dadfar, D. Camozzi, M. Darguzyte, K. Roemhild, P. Varvarà, J. Metselaar, S. Banala, M. Straub, N. Güvener and U. Engelmann, J. Nanobiotechnol., 2020, 18, 1–13 CrossRef PubMed.
- M. Levy, A. Quarta, A. Espinosa, A. Figuerola, C. Wilhelm, M. García-Hernández, A. Genovese, A. Falqui, D. Alloyeau and R. Buonsanti, Chem. Mater., 2011, 23, 4170–4180 CrossRef CAS.
- S. D. Shingte, A. H. Phakatkar, E. McKiernan, K. Nigoghossian, S. Ferguson, R. Shahbazian-Yassar and D. F. Brougham, Chem. Mater., 2022, 34, 10801–10810 CrossRef CAS PubMed.
- Q. Zhang, I. Castellanos-Rubio, R. Munshi, I. Orue, B. Pelaz, K. I. Gries, W. J. Parak, P. Del Pino and A. Pralle, Chem. Mater., 2015, 27, 7380–7387 CrossRef CAS PubMed.
- J. K. Stolarczyk, A. Deak and D. F. Brougham, Adv. Mater., 2016, 28, 5400–5424 CrossRef CAS PubMed.
- J. Zeng, L. Jing, Y. Hou, M. Jiao, R. Qiao, Q. Jia, C. Liu, F. Fang, H. Lei and M. Gao, Adv. Mater., 2014, 26, 2694–2698 CrossRef CAS PubMed.
- J. t Jang, H. Nah, J. H. Lee, S. H. Moon, M. G. Kim and J. Cheon, Angew. Chem., Int. Ed., 2009, 48, 1234–1238 CrossRef CAS PubMed.
- H. S. Dehsari, A. H. Ribeiro, B. Ersöz, W. Tremel, G. Jakob and K. Asadi, CrystEngComm, 2017, 19, 6694–6702 RSC.
- P. Guardia, A. Labarta and X. Batlle, J. Phys. Chem. C, 2011, 115, 390–396 CrossRef CAS.
- E. R. Aluri, E. Gannon, K. Singh, S. Kolagatla, K. Kowiorski, S. Shingte, E. McKiernan, C. Moloney, K. McGarry and L. Jowett, J. Colloid Interface Sci., 2022, 611, 533–544 CrossRef PubMed.
- R. Das, J. Alonso, Z. Nemati Porshokouh, V. Kalappattil, D. Torres, M.-H. Phan, E. Garaio, J. Á. García, J. L. Sanchez Llamazares and H. Srikanth, J. Phys. Chem. C, 2016, 120, 10086–10093 CrossRef CAS.
- E. C. Vreeland, J. Watt, G. B. Schober, B. G. Hance, M. J. Austin, A. D. Price, B. D. Fellows, T. C. Monson, N. S. Hudak, L. Maldonado-Camargo, A. C. Bohorquez, C. Rinaldi and D. L. Huber, Chem. Mater., 2015, 27, 6059–6066 CrossRef CAS.
- M. Fleet, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1491–1493 CrossRef.
- X. L. Liu, H. M. Fan, J. B. Yi, Y. Yang, E. S. G. Choo, J. M. Xue and J. Ding, J. Mater. Chem., 2012, 22, 8235–8244 RSC.
- C. J. Meledandri, J. K. Stolarczyk, S. Ghosh and D. F. Brougham, Langmuir, 2008, 24, 14159–14165 CrossRef CAS PubMed.
- B. Mehdaoui, A. Meffre, J. Carrey, S. Lachaize, L. M. Lacroix, M. Gougeon, B. Chaudret and M. Respaud, Adv. Funct. Mater., 2011, 21, 4573–4581 CrossRef CAS.
- S. Tong, C. A. Quinto, L. Zhang, P. Mohindra and G. Bao, ACS Nano, 2017, 11, 6808–6816 CrossRef CAS PubMed.
- P. Guardia, R. Di Corato, L. Lartigue, C. Wilhelm, A. Espinosa, M. Garcia-Hernandez, F. Gazeau, L. Manna and T. Pellegrino, ACS Nano, 2012, 6, 3080–3091 CrossRef CAS PubMed.
- S. He, H. Zhang, Y. Liu, F. Sun, X. Yu, X. Li, L. Zhang, L. Wang, K. Mao and G. Wang, Small, 2018, 14, 1800135 CrossRef PubMed.
- S.-Y. Wang, S. Huang and D.-A. Borca-Tasciuc, IEEE Trans. Magn., 2012, 49, 255–262 Search PubMed.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- E. P. McKiernan, C. Moloney, T. R. Chaudhuri, S. Clerkin, K. Behan, R. M. Straubinger, J. Crean and D. F. Brougham, Acta Biomater., 2022, 152, 393–405 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc00489a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |