
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular engineering of naphthalene spacers in low-dimensional perovskites†
Andrei
Mitrofanov
 ab,
Yonder
Berencén
c,
Elaheh
Sadrollahi
d,
Regine
Boldt
a,
David
Bodesheim
e,
Hendrik
Weiske
fg,
Fabian
Paulus
h,
Jochen
Geck
d,
Gianaurelio
Cuniberti
ei,
Agnieszka
Kuc
*g and
Brigitte
Voit
*ab
ab,
Yonder
Berencén
c,
Elaheh
Sadrollahi
d,
Regine
Boldt
a,
David
Bodesheim
e,
Hendrik
Weiske
fg,
Fabian
Paulus
h,
Jochen
Geck
d,
Gianaurelio
Cuniberti
ei,
Agnieszka
Kuc
*g and
Brigitte
Voit
*ab
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: voit@ipfdd.de
bChair of Organic Chemistry of Polymers, Technische Universität Dresden, 01069 Dresden, Germany
cInstitute of Ion Beam Physics and Materials Research, Helmholtz-Zentrum Dresden-Rossendorf, Bautzner Landstr. 400, 01328 Dresden, Germany
dInstitute of Solid State and Materials Physics, Technische Universität Dresden, 01069 Dresden, Germany
eInstitute of Materials Science and Max Bergmann Center of Biomaterials, Technische Universität Dresden, 01062 Dresden, Germany
fWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstr. 2, 04103 Leipzig, Germany
gInstitute of Resource Ecology, Helmholtz-Zentrum Dresden-Rossendorf, Permoserstr. 15, 04318 Leipzig, Germany. E-mail: a.kuc@hzdr.de
hCenter for Advancing Electronics Dresden, Technische Universität Dresden, 01069 Dresden, Germany
iDresden Center for Computational Materials Science (DCMS), Technische Universität Dresden, 01062 Dresden, Germany
First published on 27th March 2023
Abstract
Hybrid organic–inorganic lead halide perovskites have drawn much interest due to their optical and electronic properties. The ability to fine-tune the structure by the organic component allows for obtaining a wide range of materials with various dimensionalities. Here, we combine experimental and theoretical work to investigate the structures and properties of a series of low-dimensional hybrid organic–inorganic perovskites, based on naphthalene ammonium cations, 2,6-diaminonaphthalene (2,6-DAN), 1-aminonaphthalene (1-AN) and 2-aminonaphthalene (2-AN). All materials exhibit edge- or face-sharing 1D chain structures. Compared to the 2D counterpart containing isomeric 1,5-diaminonaphthalene (1,5-DAN), 1D hybrid materials exhibit broadband light emission arising from the self-trapped excitons (STEs) owing to their highly distorted structure. This work expands the library of low-dimensional hybrid perovskites and opens new possibilities for obtaining broadband-light-emitting materials.
Introduction
Organic–inorganic hybrid low-dimensional perovskites are attracting significant attention for optoelectronic applications due to their higher stability, uncomplicated manufacturing process, and wide tunability of the optical and electronic properties. Dimensionality and properties are strongly dependent on the structure of the modulating cations used in the material synthesis.1,2 The use of various organic cations leads to the formation of two-dimensional (2D) layers, one-dimensional (1D) chains, or isolated zero-dimensional (0D) clusters depending on how the metal halide octahedra are connected.3,4 Even minimal differences in the structure of the organic cations can cause remarkable changes in the structural arrangement and, as a consequence, a modification of the optoelectronic properties of the perovskite system.5 Likewise, the incorporation of functionalized ammonium cations in perovskites allows the design of a variety of new organic–inorganic hybrid materials, in which functionalized and even semiconducting organic layers are assembled with a semiconducting inorganic layer at the molecular scale.6–11In recent works devoted to low-dimensional inorganic–organic halide perovskites much attention has been paid to the broadband emission properties primarily originating from the self-trapped excitons (STEs).12–14 High structural distortion of low-dimensional perovskite structures induces electron–phonon coupling and enhances the STE process, resulting in an enhanced broadband emission. In contrast to 2D counterparts, 1D perovskites offer more freedom for fine-tuning the structure and control of the photoluminescence (PL) properties. For instance, 1D structures based on corner-sharing chains show an unprecedented PL quantum yield of around 60%.15 While 2D perovskites have been extensively investigated for structural modification and property improvement over the past decade, there are only a few works on 1D structures to date.16–18 Expanding the library of 1D structures by designing new organic–inorganic perovskites with promising optoelectronic properties will open new opportunities for the application of this material class.
Among the variety of organic cations, which can be incorporated into hybrid perovskite, π-conjugated molecules, especially acene ammonium salts, have attracted particular attention.19–21 The molecules can be flexibly designed, changing the number and position of substituents as well as the size of the π-system. Moreover, owing to the smaller band gap compared to the aliphatic molecules, the use of π-conjugated molecules as cations in the perovskite structure can lead to band alignments between organic and inorganic parts, which increase the electronic interactions between the perovskite layers.22–24
Herein, we report a systematic structural and optoelectronic study of low-dimensional hybrid perovskite series, based on isomeric naphthalene ammonium salts and lead iodide. Examination of the influence of organic cations on the structural properties reveals that the number and position of amino groups at the naphthalene ring affect the dimensionality of the perovskite, resulting in 2D corner-sharing structure and 1D corner- and face-sharing structures. Compared to the 2D counterpart, the 1D perovskites exhibit broadband light emissions originating from STEs due to the highly distorted structures. Experimental and calculated results indicate, that the different optical behaviour observed for different types of structures points to variations in the light emission mechanisms.
Experimental
Chemicals
Lead iodide (PbI2, 99.999%), 1-naphthylamine (>99.0%), 2-naphthylamine (for synthesis), 1,5-diaminonaphthalene (97%), dimethylformamide (DMF, 99.8%, anhydrous), were purchased from Sigma-Aldrich. Hydroiodic acid (HI, 57% w/w aq. soln. stab with 1.5% H3PO2), naphthalene-2,6-dicarboxylic acid (98+%), magnesium sulfate (MgSO4, 99.5%) were purchased from Alfa Aesar. Thionyl chloride (SOCl2, ≥99.0%), and sodium hydroxide (NaOH, ≥98.0%) were purchased from Fluka. Isopropanol (≥99.8%), ethyl acetate (EtOAc, ≥99.8%), acetone (≥99.8%), and sulfuric acid (H2SO4, ≥95%) were purchased from Fisher Chemicals. Sodium azide (NaN3, 99%) was purchased from Acros Organics. Diethyl ether (Et2O, 99.8%) was purchased from Chemsolute. All reagents were used as received without further purification.Synthesis of 2,6-diaminonaphthalene
A protocol adapted from Meng et al.25 was used to synthesize 2,6-diaminonaphthalene. First, 0.4 g (1.85 mmol, 1 equiv.) of naphthalene-2,6-dicarboxylic acid was mixed with 4 mL of SOCl2, and five droplets of DMF were added. The atmosphere was purified by evacuation/argon refill three times. The reaction mixture was heated under reflux at 99 °C under Ar flow for 5 h. The solvent was removed under vacuum and the residue was dissolved in 85 mL of acetone. Then 1 g (15.4 mmol, 8.3 equiv.) of NaN3 was dissolved in 5 mL of H2O cooled to 0 °C. The acetone solution was added dropwise to the NaN3 solution, forming a white precipitate. The resulting mixture was stirred overnight. The white solid was collected using suction filtration, washed with cold water, and dried under vacuum for 1 h.The naphthalene-2,6-dicarbonylazide obtained was added portion-wise to 10 mL of concentrated H2SO4 at 0 °C. The solution turned immediately to bright yellow colour and gas evolution was observed. The ice bath was removed and the reaction mixture was stirred at room temperature for 2 h. After about 10 min, the bright yellow colour faded to light yellow. After stirring, the mixture was poured onto 20 mL of ice. The resulting white solution was made alkaline by adding a 50% aqueous solution of NaOH. The organic part was extracted with EtOAc, dried over MgSO4, and the solvent was evaporated to give the desired product naphthalene-2,6-diamine as a brownish-yellow solid (78 mg, 31% overall).
1H NMR (DMSO, 400 MHz): 7.25 (2H, d, J 8.80, 4-naphthalene), 6.78 (2H, dd, J 8.50, 1.90, 3-naphthalene), 6.68 (2H, d, J 1.90, 1-naphthalene), 4.86 (4H, br, NH2).
Synthesis of naphthalene ammonium salts
The organic salt precursors were prepared by the following procedure: 5 mmol of naphthalene amine was dissolved in 20 mL of EtOH in a round bottom flask and placed in the ice/H2O bath. The equimolar amount of HI was then added dropwise under continuous stirring. The bath was removed and the mixture was stirred for 2 h. The resulting solution was evaporated at 50 °C to get the precipitate. The product was purified by recrystallization from EtOH/Et2O mixture three times and finally dried at 60 °C in a vacuum oven for 24 h.Perovskite thin film fabrication
The perovskite precursor solutions were prepared by dissolving stoichiometric amounts of the naphthalene ammonium salt and PbI2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for divalent cation and 2:1 for monovalent cation) at 0.1 M concentration in DMF followed by stirring with heating at 70 °C for 30 min to obtain a clear solution. The substrates were cleaned by ultrasonication in acetone and isopropanol for 10 min each. Later the washed and dried substrates were treated in a UV-ozone cleaner for 20 min before depositing perovskite films. 50 μL of the perovskite precursor solution was spin-coated with a two-step program at 1000 and 4000 rpm for 10 and 28 s, respectively, followed by annealing at 100 °C for 10 min to complete the crystallization of perovskites. The perovskite films obtained were used for further characterizations. All solutions and thin films were prepared in a N2-filled glovebox.
:1 for divalent cation and 2:1 for monovalent cation) at 0.1 M concentration in DMF followed by stirring with heating at 70 °C for 30 min to obtain a clear solution. The substrates were cleaned by ultrasonication in acetone and isopropanol for 10 min each. Later the washed and dried substrates were treated in a UV-ozone cleaner for 20 min before depositing perovskite films. 50 μL of the perovskite precursor solution was spin-coated with a two-step program at 1000 and 4000 rpm for 10 and 28 s, respectively, followed by annealing at 100 °C for 10 min to complete the crystallization of perovskites. The perovskite films obtained were used for further characterizations. All solutions and thin films were prepared in a N2-filled glovebox.
Preparation of single crystals
For the growth of single crystals, a 57% w/w aqueous HI solution was diluted to 47% w/w. PbI2 was dissolved in freshly prepared HI (47%) to get a 0.3 M solution, which was kept at 120 °C. At the same time, the amine precursor solution was prepared by dissolving naphthalene ammonium salt at 0.03 M concentration in HI (47%) with stirring under heating at 130 °C to obtain a clear solution. Then 224 μL of amine precursor solution were rapidly added to the PbI2 solution. Single crystals were obtained by cooling the mixture to room temperature at a rate of 5 °C h−1. The crystals were isolated, gently washed with Et2O, and dried under reduced pressure.Characterization
Single-crystal X-ray diffraction (XRD) data were collected using a Bruker Kappa APEX II at 100 K. The crystal structures were solved and refined by Shelxl software. Diffraction measurements of perovskite films on glass were conducted under ambient conditions on a Bruker D8 Discover diffractometer equipped with a 1.6 kW copper anode (λ = 1.5406 Å). The measurements were performed in parallel beam geometry as coupled θ/2θ scan using a height-limiting slit of 0.2 mm and a LYNXEYE-1D detector. Additional measurements were also performed on a Seifert XRD 3003 Θ/Θ equipped with a copper anode. Optical absorption spectra were obtained using a commercial Cary 50 (Varian) spectrophotometer. The luminescent properties in the perovskite thin films were investigated using temperature-dependent photoluminescence (PL) and photoluminescence excitation (PLE) measurements. The PLE measurements were carried out using a Xenon lamp coupled to a monochromator for selective PL excitation. The emitted PL light from the sample was guided through a set of lenses to a Jobin Yvon Triax 320 monochromator and then recorded by a Hamamatsu Si photomultiplier. A lock-in amplifier synchronized to an optical chopper at 20 Hz was employed to enhance the signal-to-noise ratio. A closed-cycle helium gas cooling system was used to perform the temperature-dependent PL measurements in the temperature range of 20–300 K. Lasers with wavelengths of 325 nm and 405 nm were used for the power- and temperature-dependent measurements.Theoretical methods
All structures were fully relaxed (atomic positions and lattice parameters) using FHI-aims26 employing the PBE functional27 on tight tier 1 numeric atom-centered orbitals, including the Tkatschenko-Scheffler dispersion correction28 and scalar relativistic corrections (ZORA) on a 3 × 3 × 3, 4 × 2 × 6, 2 × 4 × 8, and 2 × 4 × 8 Γ-centered k-grid for 1,5-DAN, 2,6-DAN, 1-AN, and 2-AN based perovskite, respectively. The electronic band structure, the Mulliken projections, and the density of states (DOS) were calculated including spin–orbit coupling (SOC) and hybrid HSE06 functionals29,30 on a 6 × 6 × 6, 6 × 3 × 9, 3 × 6 × 12, and 3 × 6 × 12 Γ-centered k-grid for 1,5-DAN, 2,6-DAN, 1-AN, and 2-AN based perovskite, respectively. The cif files of all optimized models are given as ESI.†The structures of perovskites containing 2,6-DAN and 1-AN were based on the cif files obtained from experiments (CCDC ref. no.: 2210029 and 2210030†). For the (1,5-DAN)PbI4 the structure from ref. 31 (CCDC ref. no.: 853212†) was used. The structure of (2-AN)PbI3 was created based on that of (1-AN)PbI3. The organic spacer was correspondingly changed to 1-AN and the unit cell was adjusted such that the cell parameters corresponded to the experimental powder XRD (PXRD) pattern. A coarse relaxation was performed using The Vienna Ab initio Simulation Package (VASP),32 with the PBE functional,27 DFT-D3 dispersion correction.33 Furthermore, a k-spacing of 0.5 Å−1 and the precision “Normal” was used. The resulting structure was further reoptimized and electronic properties were calculated as specified above for all systems.
Results and discussion
Isomeric naphthalene cations, used in the current work are shown in Fig. 1. The mixing of the cation and PbI2 in an aqueous solution of HI at high temperature with subsequent slow cooling allows to grow a single crystal for structural analysis.31 The determination of the exact crystal structure is important to understand the behaviour of charge carriers and correctly describe the optoelectronic properties of the perovskites. For the single crystal growth, we modified the aforementioned procedure dividing it into two steps: (1) preparation of separate PbI2 and naphthalene ammonium solutions; (2) mixing the two solutions and slow cooling from 120 °C to room temperature. We noticed that the simultaneous dissolution of both precursors results in the formation of a pure amine phase during the synthesis. This feature can be associated with the different solubility of individual precursors.34 For the same reason, the amines were used as the reaction-limiting reagent in each particular case. The crystal structures of the perovskite with naphthalene amines were determined using single-crystal XRD. The selected crystallographic information of the synthesized compounds is shown in Table 1.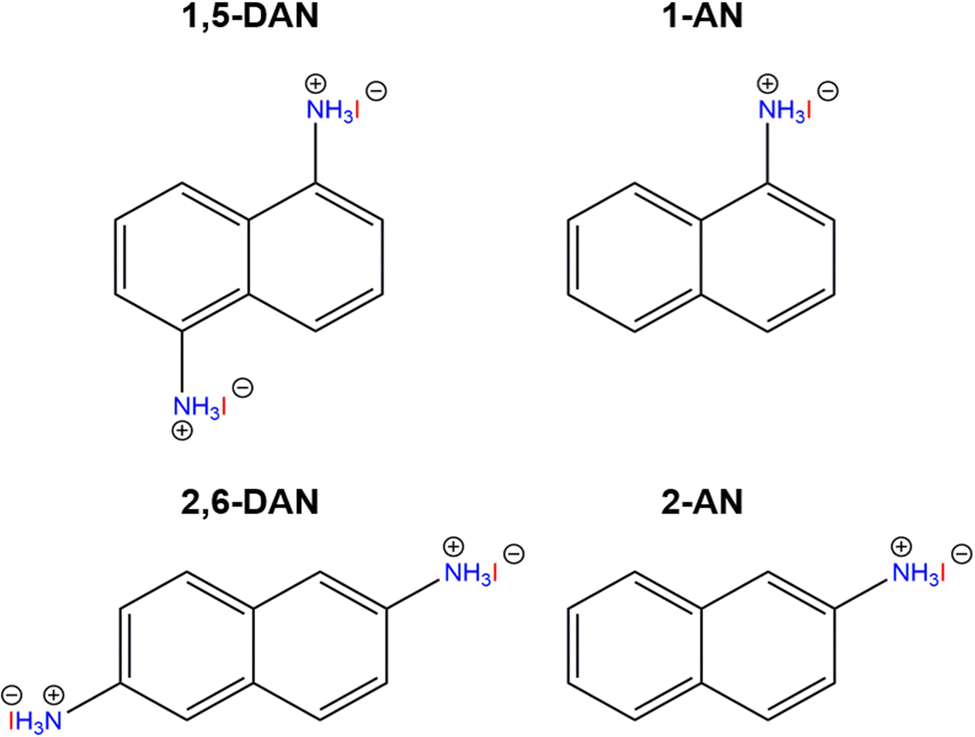 | ||
| Fig. 1 Organic cations used in this work: 1,5-DAN = 1,5-diaminonaphthalene; 1-AN = 1-aminonaphthalene; 2,6-DAN = 2,6-diaminonaphthalene; 2-AN = 2-aminonaphthalene. | ||
| Amine | 1,5-DAN31 | 2,6-DAN | 1-AN | 2-ANa |
|---|---|---|---|---|
| a Obtained from DFT. | ||||
| Formula | C10H12I4N2Pb | C10H12I4N2Pb | C10H10I3NPb | C10H10I3NPb |
| T/K | 173 | 100 | 100 | — |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Triclinic |
| Space group | P21/c | P21/c | Pbca | P1 |
| a/Å | 11.163(3) | 12.000(4) | 15.387(3) | 28.655 |
| b/Å | 8.960(2) | 18.768(7) | 7.8556(14) | 13.871 |
| c/Å | 8.737(2) | 8.072(3) | 25.135(3) | 7.7965 |
| α (°) | 90 | 90 | 90 | 89.972 |
| β (°) | 90.314(4) | 102.396(7) | 90 | 90.008 |
| γ (°) | 90 | 90 | 90 | 90.007 |
| V/Å3 | 873.8(4) | 1775.56 | 3038.17 | 3098.98 |
| Z | 2 | 4 | 1 | 1 |
The structure of (1,5-DAN)PbI4 was reported previously31 and exhibited a two-dimensional arrangement, in which the diammonium cations are incorporated between two inorganic layers (see Fig. 2a). This system was found to crystallize monoclinic with the P21/c space group. Although the formation of layered structures is not typical using cations with the amino group directly bonded to the aromatic ring, there are several examples of 2D perovskites with such cations.35–37 For example, Gao et al.37 reported a new series of Dion–Jacobson perovskite phases containing m-phenylene diammonium cation. They were grown in a single-crystal form as well as the thin films with considerable environmental stability. Whereas 1,5-DAN is forming an alternating inorganic–organic layered structure, its structural isomer 2,6-DAN leads to the formation of a 1D framework. The (2,6-DAN)PbI4 crystal has a monoclinic symmetry with the P21/c space group. The inorganic part of the (2,6-DAN)PbI4 consists of lead iodide chains, assembled from edge-sharing [PbI6]4− octahedra extending along the c direction forming zigzag-like chains (see Fig. 2b), similar to previously observed perovskite with 1,4-benzene diammonium,38 a cation belonging to the acene diamine homological series. The connection of the chains is provided by ionic and hydrogen bonding interactions between the 2,6-DAN cations and iodide anions, which leads to the formation of a 1D perovskite structure. Despite the structural similarity, 1,5- and 2,6-DAN cations form different perovskite phases (2D and 1D, respectively). Similarly, it was reported for lead iodide perovskites with other pair of cations, where amino group were directly connected to the aromatic core. The m-phenylene diammonium forms the 2D structure37 whereas its linear isomer p-phenylene diammonium forms a 1D structure with edge-sharing octahedra.38
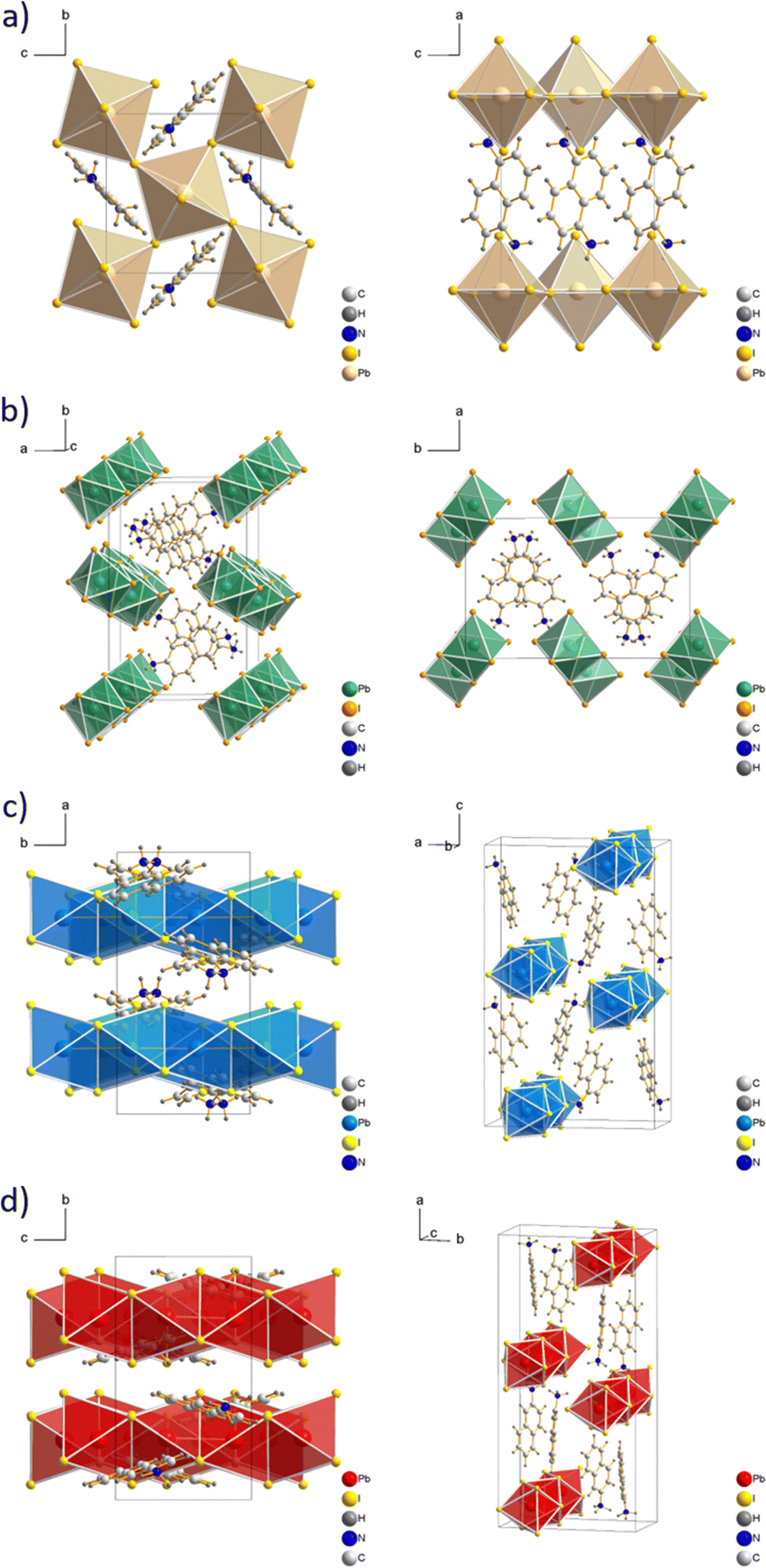 | ||
| Fig. 2 Crystal structures of (a) (1,5-DAN)PbI4, (b) (2,6-DAN)PbI4, (c) (1-AN)PbI3, and (d) (2-AN)PbI3; top and side views are included to show explicitly the 2D nature of 1,5-DAN and the 1D character of the 2,6-DAN, 1-AN, and 2-AN perovskites. | ||
We confirmed the crystal structure of (1-AN)PbI3, first reported in ref. 39 with slightly different lattice parameters. It has a similar atomic arrangement as in (2,6-DAN)PbI4. Still, the inorganic motif is built up from face-sharing [PbI6]4− octahedra along the b direction (see Fig. 2c). Each chain is surrounded by six 1-AN molecules, stacked into the columns. The crystal has an orthorhombic symmetry with a Pbca space group. A single crystal with 2-AN could not be grown using conventional solution methods. The structure was obtained by interchanging the 1-AN cations with 2-AN and adjusting cell parameters to align them with the experimental PXRD. Subsequent relaxation was performed using DFT calculation (see Theoretical methods). The resulting calculated 1D structure (see Fig. 2d) has a triclinic symmetry with the P1 space group. The inorganic part is presented as chains of face-shared octahedra extended along the c direction.
A critical factor, which affects the perovskite's optical properties is the structural distortion of the lead halide octahedra. It can be quantified by the bond length distortion (Δd) as well as the octahedral angle variance (σoct2) using the following formulas
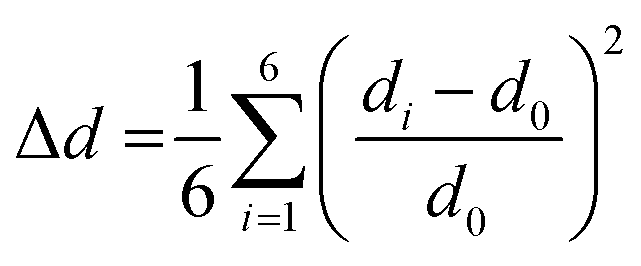 | (1) |
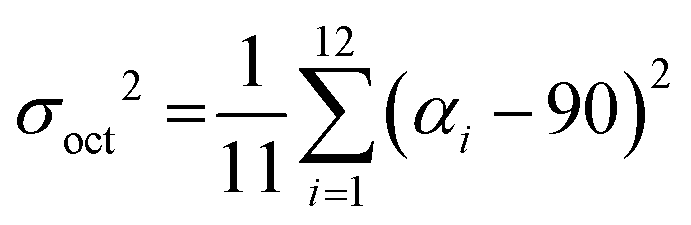 | (2) |
Thin films were spin-coated onto glass substrates from a DMF solution containing ammonium salts and PbI2 in a specific stoichiometric ratio followed by thermal annealing (see Experimental section), and the resulting perovskite materials were characterized with PXRD. Comparison of experimentally obtained PXRD patterns with the simulated patterns (Fig. S1, ESI†), derived from single-crystal diffraction, allows the establishment of the structure of the resulting thin films in a reliable manner. The film structures of (1,5-DAN)PbI4, (2,6-DAN)PbI4, and (1-AN)PbI3 are in full agreement with the single crystal structure. The predicted XRD pattern for (2-AN)PbI3 (Fig. S1d, ESI†) matches the majority of the reflexes in the observed diffraction pattern and simultaneously reveal some differences between calculated and experimental results. There is no peak at 12.7° from PbI2 (001) planes, which indicates the absence of a lead iodide phase in the perovskite. The (2,6-DAN)PbI4 film firstly annealed at 100 °C did not exhibit any reflections in the diffractogram, which indicates an amorphous nature of the film. Only after increasing the annealing temperature to 150 °C (Fig. S1b, ESI†), several reflections appear. We believe that because of the rigid structure of the 2,6-diaminonaphthalene cation, higher energy might be needed to form a good crystalline thin film.
The optical absorption spectra of the investigated perovskites also support their dimensionality. As a typical layered perovskite, (1,5-DAN)PbI4 exhibits a characteristic excitonic transition centered at 508 nm (Fig. 3a). We also observed the second peak at 462 nm, which vanishes after additional annealing of the (1,5-DAN)PbI4 film at 150 °C (Fig. S2, ESI†). In ref. 19 where this absorption feature was not observed, the films of 1,5-DAN hybrid perovskites were annealed at 140 °C. To determine the optical band gap energy Tauc plot was used assuming both a direct and an indirect band gap. Obtained values correspond to the onset of the excitonic absorption. The optical band gap energy obtained from the Tauc plot (Fig. S3, ESI†) is 2.39 eV for the direct band gap and 2.34 eV for the indirect band gap. The tail at longer wavelengths in the absorption spectrum became clear upon normalization. The overall absorbance of (1,5-DAN)PbI4 was lower than the one from the other compounds. The (2,6-DAN)PbI4 perovskite has a higher absorption energy, with an absorption edge peaked at 410 nm, which is much more blue-shifted than for (1,5-DAN)PbI4. This absorption is characteristic of the electronic transitions in 1D lead-iodide chains.40,41 The corresponding Tauc plot yields a direct band gap of 2.88 eV and an indirect one of 2.60 eV. A similar absorption, with a peak centered at 379 nm, is also observed in the case of (1-AN)PbI3, whose structure also consists of inorganic [PbI6]4− chains. The corresponding optical band gap values obtained from the Tauc plot are 3.08 eV and 2.89 eV for the direct and indirect transitions, respectively. (2-AN)PbI3 exhibits the first absorption peak at 415 nm, equivalent to an optical band gap energy for the direct transition of 2.86 eV and 2.71 eV for the indirect one. The optical properties of the investigated materials depend on the connectivity of the [PbI6]4− octahedra. The band gap increases following the general trend of “corner-sharing < edge-sharing < face-sharing”.42
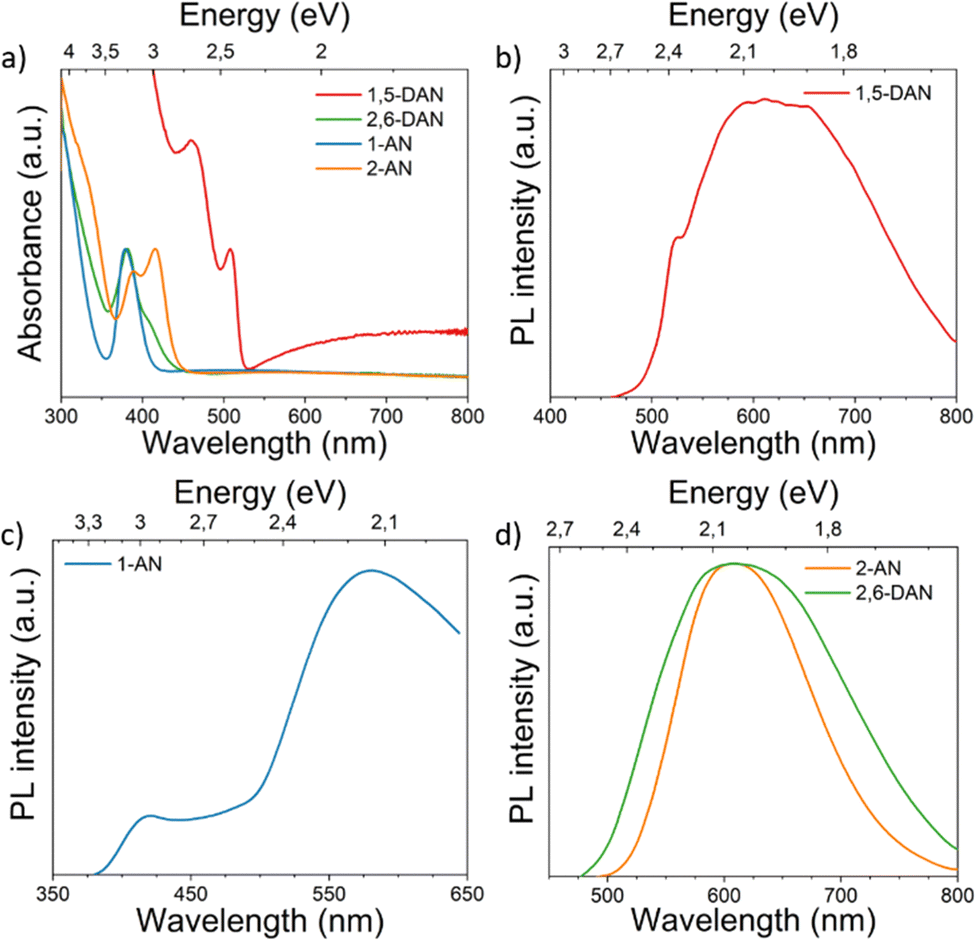 | ||
| Fig. 3 (a) UV-vis absorption at room temperature and (b)–(d) PL spectra at 20 K of all investigated perovskite systems. | ||
The 20K-PL spectrum of (1,5-DAN)PbI4 (Fig. 3b) under a 405 nm-laser excitation shows a shoulder located at 523 nm close to the expected direct band gap and a broad emission band centered at around 620 nm. The high-energy shoulder is attributed to free excitons (FE)/carriers,19 whereas the broadband PL can be originated from defect-related processes.43 Previous investigation of (1,5-DAN)PbI4 also revealed low intense broadband emission, which was attributed to the substrate or material impurity.19 To get more information about the luminescence origin, we recorded PLE spectra. Fig. S4a (ESI†) shows a PLE spectrum monitored at 520 nm of (1,5-DAN)PbI4 with a peak centered at 416 nm. After exciting the sample using a resonance wavelength to the emission peak, the broadband emission is quenched, giving rise to a clear excitonic peak centered at 530 nm (Fig. S5, ESI†). It allows us to conclude that the broad emission band is a result of extrinsic effects like defects. We did not observe such changes in other samples. In the PL spectrum of (1-AN)PbI3 (Fig. 3c) along with the low-energy broadband centered at around 577 nm, which can be attributed to the self-trapped excitons (STE) state, a small peak around 420 nm was also identified. The high energy band correlates with the indirect band gap calculated from the Tauc plot (Fig. S3, ESI†). Similar optical properties for 1D perovskites were previously demonstrated in ref. 14 (2,6-DAN)PbI4 (Fig. 3d) shows, in turn, broad PL centered at 610 nm with a Stokes shift of 190 nm, indicating the STE nature of the emission. A similar optical behavior was observed for the (2-AN)PbI3 perovskite, which exhibits broadband light emission centered at 610 nm, far from the absorption edge, showing a significant Stokes shift of 195 nm. This suggests an excitonic self-trapped emission, which is common for low-dimensional perovskites.44 The excitation power of the laser was selected in a way, that the structure of the samples was not modified by the laser power.
To investigate the nature of these emissions, we have inspected variable-temperature PL spectra (Fig. 4). The spectra acquired at 300 K are additionally shown on Fig. S6 (see ESI†). The temperature-dependent PL measurement of (1,5-DAN)PbI4 (Fig. 4a) shows that FE emission peak becomes narrower with decreasing temperature. At low temperatures the FE emission should be dominant over the broadband emission, however, the high broadband intensity still remains, coming probably from defect-related states. For (1-AN)PbI3 the peak position almost remains unchanged whereas the intensity increases with decreasing temperature. Similar behavior was observed in the case of (2,6-DAN)PbI4. For (2-AN)PbI3 the peak intensity increases monotonously and narrows as the temperature decreases. We suggest for 1-AN, 2-AN, and 2,6-DAN an STE origin of the broadband emission. The carriers in the STE state cannot tunnel back to the FE state, which suggests highly distorted structures.3,45 Neither a splitting of the peak nor a shift of the peak position was observed when cooling the samples from 300 K down to 20 K, which indicates that the emission does not arise from multiple radiative transitions.1
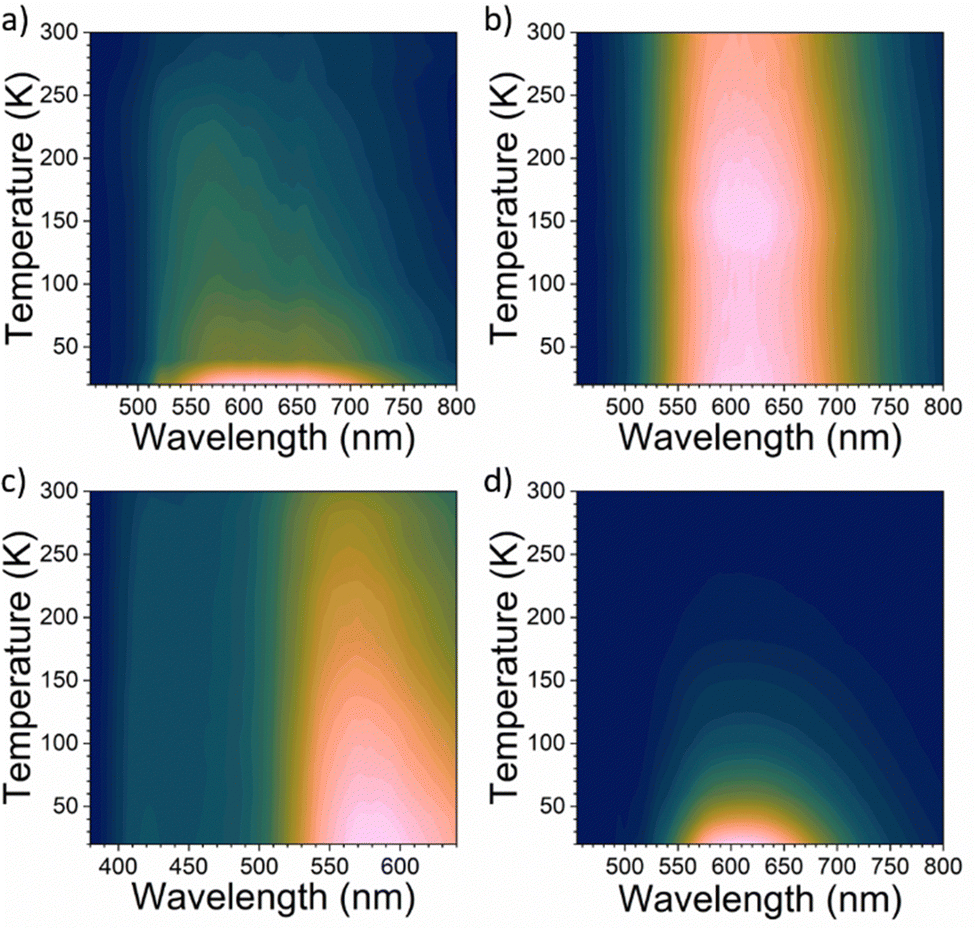 | ||
| Fig. 4 Temperature-dependent steady-state PL spectra of (a) (1,5-DAN)PbI4, (b) (2,6-DAN)PbI4, (c) (1-AN)PbI3 and (d) (2-AN)PbI3. | ||
The relationship between PL intensity and temperature can be described using the Arrhenius equation:
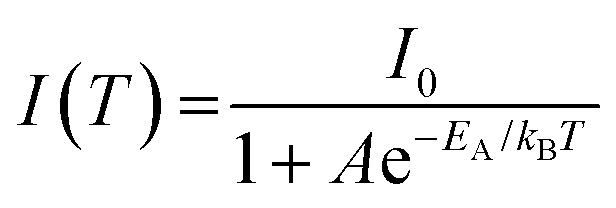 | (3) |
To support the experiment and estimate the electronic properties of the obtained structures, DFT calculations were performed as discussed in the Experimental Section. The resulting band structures and corresponding DOS, both projected on atomic contributions, are shown in Fig. 5. All systems are semiconductors with direct band gaps at the X point in the case of both DAN-based systems (2.08 eV and 2.91 eV for 1,5- and 2,6-DAN, respectively) and indirect gaps for both AN-based systems (3.26 eV and 3.12 eV for 1- and 2-AN, respectively) between the Γ point and the low-symmetry point. The results are in reasonable agreement with the experimental values of 2.39, 2.88, (both from direct band gap approximation) 2.89, and 2.71 (both from indirect band gap approximation) eV for 1,5-DAN-, 2,6-DAN-, 1-AN-, and 2-AN-based perovskites, respectively, and show an increase in the band gap arising from the dimensionality reduction of these hybrid systems.
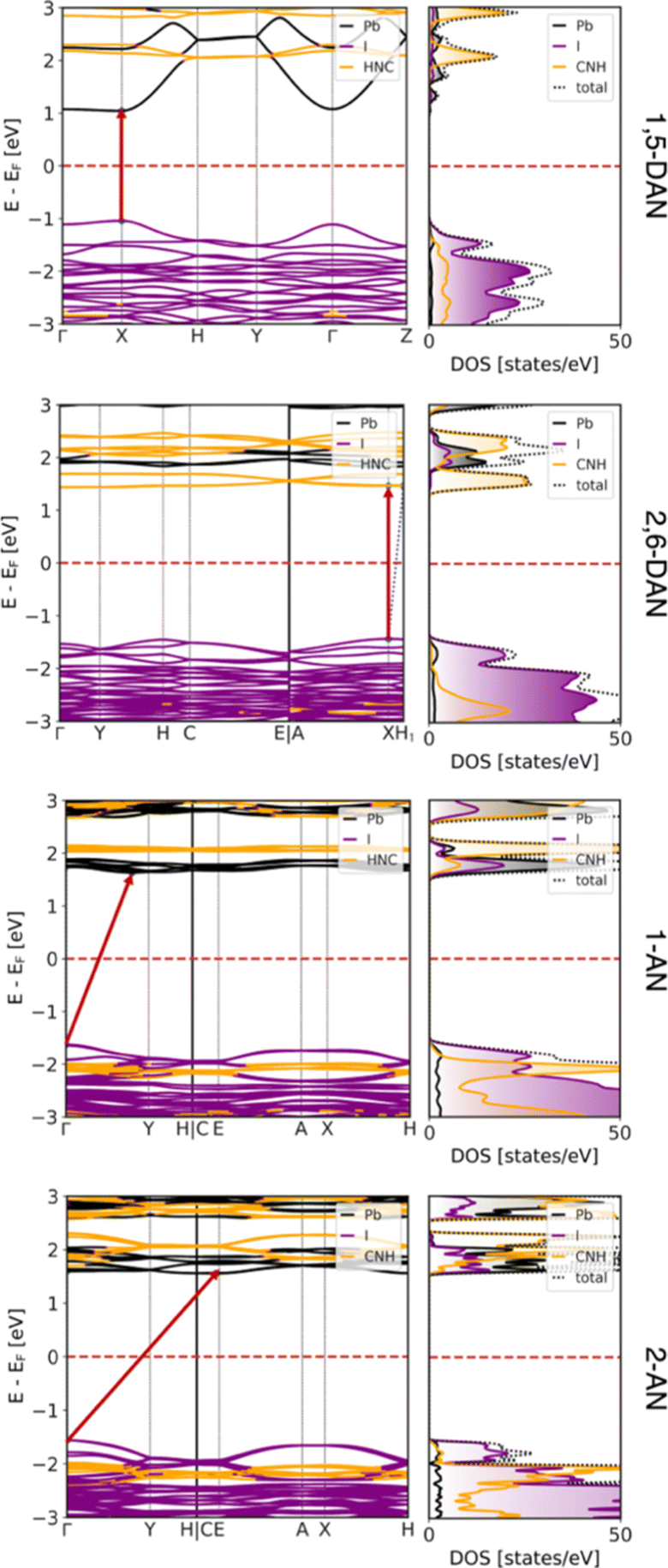 | ||
| Fig. 5 Electronic band structure and density of states projected on atomic contributions of (from top to bottom) (1,5-DAN)PbI4, (2,6-DAN)PbI4, (1-AN)PbI3, and (2-AN)PbI3 perovskites, calculated at the DFT/HSE06 level of theory. Fermi level was shifted to zero and marked as red dashed lines. | ||
The majority contributions to the band structure and the corresponding DOS show a very typical behavior of a 2D perovskite in the case of (1,5-DAN)PbI4. The valence band maximum (VBM) is composed mostly of I 5p states, while the conduction band minimum (CBM) is formed from Pb 6p states. The states corresponding to the organic π-conjugated molecules are at least 1 eV away from the band edges and do not strongly hybridize with the [PbI6]4− layers. This means that any electron–hole pair (or excitons) will stay in the inorganic network of the perovskite.
The situation is, however, different for the 1D perovskites: In the case of (2,6-DAN)PbI4, the VBM is still dominated by the I 5p states, while the CBM is formed from states of the organic π-conjugated molecules. This suggests that even if electrons are excited within the [PbI6]4− chains, relaxation will relocate the electrons to the organic cations. In this case, spatially separated electrons and holes with longer lifetimes should appear. In the case of (1-AN)PbI3 and (2-AN)PbI3, we observe a slightly stronger hybridization between states from organic and inorganic parts. The VBM is dominated by I 5p states, however, states from the organic molecules are only 400 meV below them. The CBM is formed by Pb 6p states with organic states lying about 400 meV or less above them. The DOS, however, shows contribution to the CBM from all species to some extent, supporting a good hybridization. It can result in carrier transport between the organic and inorganic part, which would be interesting for optoelectronic applications. Moreover, strong interaction of lead iodide chains with the organic cation can additionally contribute to the low-energy bands,46 but more complex spectroscopy experiments are required to draw strong conclusion. The overall dispersion of the VBM and CBM reduces when going from 2D to 1D systems, indicating heavier charge carriers.
Conclusions
The configurations of naphthalene derivatives significantly influence the structures of the inorganic framework in metal halide perovskites, causing the transformation of the inorganic network from 2D to 1D corner- or face-sharing structures. Theoretical results reveal, that the organic cations in the 1D compounds strongly contribute to the band structure resulting in orbitals hybridization. Unlike the 2D compound containing 1,5-DAN, highly distorted 1D perovskites show broadband emissions originating from STEs. This work expands the library of underexplored low-dimensional hybrid perovskites and serves as a fundamental tool to explore the structure–property relationships of organic–inorganic hybrid lead halides.Author contributions
B. V., A. M., and A. K. conceived and planned the experiments. B. V. supervised the work. A. M. synthesized and characterized samples. F. P. provided support by thin film preparation. E. S. performed SCXRD measurements and analysis with the supervision of J. G. R. B. and F. P. performed thin film XRD measurements. Y. B. designed the spectroscopy experiments. A. M. and Y. B. performed the spectroscopy measurements along with the data analysis and the interpretation of the results. H. W. and A. K. performed DFT calculations and analyzed the theory results. D. B. performed the prerelaxation study of (2-AN)PbI3 with the supervision of G. C. A. M. wrote the manuscript with contributions from A. K. All authors have discussed the results and commented on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M., D. B., H. W., J. G., G. C., A. K., E. S., and B. V. acknowledge support by the DFG CRC 1415 project. A. K. acknowledges association with SPP 2244 project. D. B., H. W., G. C., and A. K. acknowledge the Center for Information Services and HPC (ZIH) at TU Dresden for computer time. We gratefully thank Dr Olga Kataeva for the helpful discussion of the SCXRD results and Prof. Alexey Chernikov for the discussion about the photophysical properties. The Scientific color map batlow (Crameri 2018) is used in this study to prevent visual distortion of the data and exclusion of readers with color vision deficiencies (Crameri et al., 2020).References
- J. Q. Zhao, C. Q. Jing, J. H. Wu, W. F. Zhang, L. J. Feng, C. Y. Yue and X. W. Lei, J. Phys. Chem. C, 2021, 125, 10850–10859 CrossRef CAS.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed.
- L. Mao, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS PubMed.
- D. Cortecchia, S. Neutzner, A. R. S. Kandada, E. Mosconi, D. Meggiolaro, F. de Angelis, C. Soci and A. Petrozza, J. Am. Chem. Soc., 2017, 139, 39–42 CrossRef CAS PubMed.
- K. Z. Du, Q. Tu, X. Zhang, Q. Han, J. Liu, S. Zauscher and D. B. Mitzi, Inorg. Chem., 2017, 56, 9291–9302 CrossRef CAS PubMed.
- X. Li, J. Yang, Z. Song, R. Chen, L. Ma, H. Li, J. Jia, J. Meng, X. Li, M. Yi and X. Sun, ACS Appl. Energy Mater., 2018, 1, 4467–4472 CrossRef CAS.
- Y. Gao, Z. Wei, S. N. Hsu, B. W. Boudouris and L. Dou, Mater. Chem. Front., 2020, 4, 3400–3418 RSC.
- D. B. Mitzi, K. Chondroudis and C. R. Kagan, Inorg. Chem., 1999, 38, 6246–6256 CrossRef CAS PubMed.
- A. H. Proppe, M. H. Tremblay, Y. Zhang, Z. Yang, R. Quintero-Bermudez, S. O. Kelley, S. Barlow, S. R. Marder and E. H. Sargent, J. Phys. Chem. C, 2020, 124, 24379–24390 CrossRef CAS.
- A. Mishra, P. Ahlawat, G. C. Fish, F. Jahanbakhshi, M. Mladenović, M. Almalki, M. A. Ruiz-Preciado, M. C. Gelvéz-Rueda, D. J. Kubicki, P. A. Schouwink, V. Dufoulon, T. Schneeberger, A. Aslanzadeh, F. C. Grozema, S. M. Zakeeruddin, J. E. Moser, U. Rothlisberger, L. Emsley, J. V. Milić and M. Grätzel, Chem. Mater., 2021, 33, 6412–6420 CrossRef CAS.
- J. v Milić, J. Mater. Chem. C, 2021, 9, 11428–11443 RSC.
- W. F. Zhang, W. J. Pan, T. Xu, R. Y. Song, Y. Y. Zhao, C. Y. Yue and X. W. Lei, Inorg. Chem., 2020, 59, 14085–14092 CrossRef CAS PubMed.
- Z. Qi, H. Gao, X. Yang, Y. Chen, F. Q. Zhang, M. Qu, S. L. Li and X. M. Zhang, Inorg. Chem., 2021, 60, 15136–15140 CrossRef CAS PubMed.
- G. Wu, C. Zhou, W. Ming, D. Han, S. Chen, D. Yang, T. Besara, J. Neu, T. Siegrist, M. H. Du, B. Ma and A. Dong, ACS Energy Lett., 2018, 3, 1443–1449 CrossRef CAS.
- G. Yu, F. Lin, K. Zhou, S. Fang, Y. Shi, W. Liu, H. Hu, B. Ma and H. Lin, Chem. Mater., 2021, 33, 5668–5674 CrossRef CAS.
- X. Y. Sun, M. Yue, Y. X. Jiang, C. H. Zhao, Y. Y. Liao, X. W. Lei and C. Y. Yue, Inorg. Chem., 2021, 60, 1491–1498 CrossRef CAS PubMed.
- D. Li, W. Wu, S. Wang, X. Zhang, L. Li, Y. Yao, Y. Peng and J. Luo, J. Mater. Chem. C, 2020, 8, 6710–6714 RSC.
- Q. Hu, H. Yu, S. Gong, Q. Han and W. Wu, J. Mater. Chem. C, 2022, 10, 6002–6008 RSC.
- Y. H. L. Lin and J. C. Johnson, J. Phys. Chem. Lett., 2021, 12, 4793–4798 CrossRef CAS PubMed.
- W. Zhao, R. Zhang, S. Wang, D. Zheng, Q. Kong, J. Liu, J. Li, J. Guo, S. Yang and K. Han, J. Phys. Chem. C, 2021, 125, 23889–23894 CrossRef CAS.
- Yukta, R. D. Chavan, D. Prochowicz, P. Yadav, M. M. Tavakoli and S. Satapathi, ACS Appl. Mater. Interfaces, 2022, 14, 850–860 CrossRef CAS PubMed.
- Q. Dai, H. Li, G. Sini and J. Bredas, Adv. Funct. Mater., 2022, 32, 2108662 CrossRef CAS.
- Y. Gao, E. Shi, S. Deng, S. B. Shiring, J. M. Snaider, C. Liang, B. Yuan, R. Song, S. M. Janke, A. Liebman-Peláez, P. Yoo, M. Zeller, B. W. Boudouris, P. Liao, C. Zhu, V. Blum, Y. Yu, B. M. Savoie, L. Huang and L. Dou, Nat. Chem., 2019, 11, 1151–1157 CrossRef CAS PubMed.
- W. A. Dunlap-Shohl, E. T. Barraza, A. Barrett, S. Dovletgeldi, G. Findik, D. J. Dirkes, C. Liu, M. K. Jana, V. Blum, W. You, K. Gundogdu, A. D. Stiff-Roberts and D. B. Mitzi, Mater. Horiz., 2019, 6, 1707–1716 RSC.
- W. Meng, T. K. Ronson and J. R. Nitschke, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10531–10535 CrossRef CAS PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- A. Lemmerer and D. G. Billing, CrystEngComm, 2012, 14, 1954–1966 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. C. Stoumpos, D. H. Cao, D. J. Clark, J. Young, J. M. Rondinelli, J. I. Jang, J. T. Hupp and M. G. Kanatzidis, Chem. Mater., 2016, 28, 2852–2867 CrossRef CAS.
- L. Dobrzycki and K. Woźniak, CrystEngComm, 2008, 10, 577 RSC.
- R. Chen, H. Gu, Y. Han, J. Yin, G. Xing and B.-B. Cui, J. Mater. Chem. C, 2022, 10, 9465–9470 RSC.
- L. Gao, X. Li, B. Traoré, Y. Zhang, J. Fang, Y. Han, J. Even, C. Katan, K. Zhao, S. Liu and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 12063–12073 CrossRef CAS PubMed.
- C. Ma, D. Shen, B. Huang, X. Li, W. C. Chen, M. F. Lo, P. Wang, M. Hon-Wah Lam, Y. Lu, B. Ma and C. S. Lee, J. Mater. Chem. A, 2019, 7, 8811–8817 RSC.
- A. Lemmerer and D. G. Billing, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m904–m906 CrossRef CAS.
- B. Febriansyah, T. M. Koh, R. A. John, R. Ganguly, Y. Li, A. Bruno, S. G. Mhaisalkar and J. England, Chem. Mater., 2018, 30, 5827–5830 CrossRef CAS.
- J. I. Fujisawa and T. Ishihara, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 113203 CrossRef.
- M. E. Kamminga, G. A. de Wijs, R. W. A. Havenith, G. R. Blake and T. T. M. Palstra, Inorg. Chem., 2017, 56, 8408–8414 CrossRef CAS PubMed.
- H. Hu, Y. Liu, Z. Xie, Z. Xiao, G. Niu and J. Tang, Adv. Opt. Mater., 2021, 9, 2101423 CrossRef CAS.
- J. Li, H. Wang and D. Li, Front. OptoElectron., 2020, 13, 225–234 CrossRef PubMed.
- X. Li, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, C. C. Stoumpos, R. D. Schaller and M. G. Kanatzidis, Chem. Mater., 2019, 31, 3582–3590 CrossRef CAS.
- A. Samet, S. Triki and Y. Abid, J. Phys. Chem. C, 2019, 123, 6213–6219 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2210029 and 2210030. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc00132f |
| This journal is © The Royal Society of Chemistry 2023 |