
Rational design of hybridized local and charge transfer emitters towards high-performance fluorescent blue OLEDs†
Shuxin
Wang
ab,
Hanlin
Li
c,
Zhen
Song
 d,
He
Jiang
ab,
Xiandi
Zhang
a,
Chui-Shan
Tsang
a,
Quanlin
Liu
d,
Lawrence Yoon Suk
Lee
a,
Dongge
Ma
*c and
Wai-Yeung
Wong
*ab
d,
He
Jiang
ab,
Xiandi
Zhang
a,
Chui-Shan
Tsang
a,
Quanlin
Liu
d,
Lawrence Yoon Suk
Lee
a,
Dongge
Ma
*c and
Wai-Yeung
Wong
*ab
aDepartment of Applied Biology and Chemical Technology and Research Institute for Smart Energy, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, P. R. China
bThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen 518057, P. R. China. E-mail: wai-yeung.wong@polyu.edu.hk
cState Key Laboratory of Luminescent Materials and Devices, Center for Aggregation-Induced Emission, South China University of Technology, Guangzhou 510640, China. E-mail: msdgma@scut.edu.cn
dThe Beijing Municipal Key Laboratory of New Energy Materials and Technologies, School of Materials Sciences and Engineering, University of Science and Technology Beijing, Beijing 100083, China
First published on 3rd March 2023
Abstract
Hybridized local and charge-transfer (HLCT) emitters are promising for the realization of high-performance blue organic light-emitting diodes (OLEDs). However, the rational design of efficient HLCT emitters remains challenging. Here, we present two blue emitters (TAP1 and TAP2) with the HLCT state through the construction of D–π–A molecules. Theoretical calculations reveal the large overlap and partial separation of “hole” and “particle” orbitals for S1 excited states, demonstrating the HLCT nature of the emitters. Together with the clear solvatochromic phenomenon, we propose that introducing the π-conjugated anthracene unit into an appropriate donor–acceptor segment is an effective strategy to finely regulate the locally excited and charge transfer components and construct versatile HLCT emitters. Employing TAP2 and TAP1 as emitters, the blue OLEDs exhibit good color purity, high efficiency, and extremely low efficiency roll-offs with Commission internationale de l’éclairage coordinates of, respectively, (0.14, 0.10) and (0.15, 0.11), narrow full width at half maximum values of 45 nm and 52 nm, maximum external quantum efficiency values of 5.53% and 5.16%, and efficiency roll-off values of 0.5% and 1% at the practical brightness of 1000 cd m−2. By virtue of the transient photoluminescence decay curves, excited state energy levels and natural transition orbital analysis, we classify that the reverse intersystem crossing at high-lying excited states from T2 to S1 serves as an efficient approach to harvest triplet excitons and thus boost the exciton utilization efficiency. The superior properties of HLCT emitters in this work provide inspiration for the rational design of promising materials for high-performance blue OLEDs.
10th Anniversary StatementMy warmest congratulations to the 10th anniversary of the Journal of Materials Chemistry C. It has been my great honour to serve on the journal board as Associate Editor for nine years, from 2013 to 2022. Furthermore, I have published a very high number of research papers and review articles in the journal over the years. I have witnessed the tremendous growth and development of the journal to the current stage and I am sure that it is a flagship journal for research work on the electronic and optical properties of molecular and nano materials. I will definitely continue to make contributions to it in the years to come. Happy anniversary again! |
Introduction
Organic light emitting diodes (OLEDs) have emerged as promising candidates for the next generation of full-color flat-panel displays and solid-state lighting technologies.1–3 The development of efficient blue (especially deep-blue) emitters has sparked tremendous interest due to their critical role as either emitting or excitation sources.4–6 However, emitters matching the standard deep-blue Commission internationale de l’éclairage (CIE) coordinates of (0.14, 0.08) by the National Television System Committee (NTSC) generally have a lower efficiency and stability than their green and red counterparts because of their intrinsically large bandgaps, unbalanced carrier transport capability, and inferior energy level matching with other functional layers of OLEDs.7–10 To address this issue, several strategies have been proposed for utilizing triplet excitons and thus realizing high-efficiency blue emitters. Thermally activated delayed fluorescence (TADF) and triplet–triplet annihilation (TTA) emitters are regarded as promising candidates due to their high maximum internal quantum efficiency of electroluminescence values of 100% and 62.5%, respectively.11–13 Currently, numerous blue OLEDs with high external quantum efficiency values have been achieved based on TADF emitters. Unfortunately, TADF-type OLEDs suffer from severe efficiency roll-off due to slow up-conversion from the triplet charge-transfer state (3CT) to the singlet charge-transfer state (1CT) as the result of their first-order coupling character.14–18 By contrast, blue TTA-type OLEDs usually show mild efficiency roll-off, and their external quantum efficiency (EQE) is relatively low owing to their limited maximum exciton utilization efficiency (EUEmax; ≤62.5%).19–23More recently, the hybridized local and charge-transfer (HLCT) excited state with balanced locally excited (LE) and intramolecular charge transfer excited (CT) states has been developed as a feasible strategy for efficient blue OLEDs.24–28 It is proved to be an effective means of achieving 100% EUE through the harvesting of triplet excitons via efficient reverse intersystem crossing (RISC) at high-lying triplet and singlet excited states (hRISC), such as T2 to S1.29–31 The fast hRISC process can effectively suppress the accumulation of triplet excitons and thereby provide an HLCT emitter with a high EQE and a low efficiency roll-off simultaneously.32–35 In addition, the LE state with a large transition moment and orbital overlap contributes to the enhancement of the fluorescence quantum efficiency, and the relatively weak CT state is beneficial for the blue emission.36 The HLCT molecules are typically composed of donor–acceptor (D–A) segments and π-conjugated (π) spacers.33,37 By regulating the dihedral angles of the D–A segment, the character of the excited states can be modulated to form HLCT states and thus promote the hRISC process and radiative decay.38 So far, although several HLCT-type OLEDs with nearly 100% EUE and mild efficiency roll-off have been developed, the EQE of blue HLCT emitters remains limited and rarely reaches 10%, which hardly meets the practical requirements (Table S1, ESI†).39,40 Therefore, enormous efforts are required to enrich the HLCT molecules and further improve their performance for blue OLEDs.
Herein, we report two HLCT fluorescent emitters, 4-(10-(4-methylpyridin-2-yl)anthracen-9-yl)-N,N-diphenylaniline (TAP1) and 4-(10-(5-methylpyridin-2-yl)anthracen-9-yl)-N,N-diphenylaniline (TAP2), for blue OLEDs. It is demonstrated that grafting a conjugated anthracene unit in an appropriate D–A segment is an effective strategy to finely regulate the LE and CT components and construct versatile HLCT emitters. The properties of the physical and HLCT states are investigated in detail through UV-vis absorption and photoluminescence (PL) spectroscopy, thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), and theoretical calculations. As expected, the blue OLEDs with TAP2 and TAP1 as emitters exhibit good color purity, high efficiency, and extremely low efficiency roll-off characteristics with, respectively, CIE coordinates of (0.14, 0.10) and (0.15, 0.11), narrow full width at half maximum (FWHM) values of 45 nm and 52 nm, maximum external quantum efficiency (EQEmax) values of 5.53% and 5.16%, and efficiency roll-off values of 0.5% and 1% at the practical brightness of 1000 cd m−2. By virtue of the transient PL decay curves, excited state energy levels and natural transition orbital (NTO) analysis, hRISC from T2 to S1 is proposed to be an efficient approach for harvesting triplet excitons and thus boosting the exciton utilization efficiency. This work points to the superior properties of HLCT emitters, providing inspiration for the rational design of promising materials for high-performance blue OLEDs.
Results and discussion
Molecular design and synthesis
Based on the state-mixing equation, the most critical part for HLCT molecular design is manipulation of the energy gap between the LE and CT states, making it small enough to enable state mixing and HLCT state formation.38 Anthracene is selected as the π-conjugated spacer in consideration of its highly bulky and rigid skeleton and thus its outstanding performance for blue OLEDs.41,42 The further introduction of appropriate D–A segments helps manage the electron push–pull strength and effectively regulates the LE and CT components towards HLCT excited states (Fig. 1a).43 Moreover, the introduction of D–A segments can also enhance the carrier-transporting ability and thereby the exciton utilization efficiency of the as-prepared OLEDs.36 It has been proved that triphenylamine is a moderate electron-donating group with a unique space propeller conformation and thereby large steric hindrance, which has the potential to form CT transitions with an energy close to the LE state and thus facilitate the formation of the HLCT state. However, reported triphenylamine-anthracene-based blue HLCT emitters generally suffer from poor color purity with CIEy ≥ 0.12 due to the adoption of electron-accepting groups with large conjugation or strong electron-accepting ability such as phenanthroimidazole and triazine derivatives (Table S2, ESI†).44 Therefore, in this work, in order to achieve blue HLCT emitters with credible color purity, N-methylpyridine moieties with reduced conjugation and moderate electron-accepting ability are selected as electron-accepting groups to tune the LUMO energy and regulate the LE and CT components. The triphenylamine-anthracene moiety is linked to methylpyridine at the ortho-position to feature the potential CH⋯N hydrogen bond, which can benefit the construction of rigid structures, endowing the emitters with a high radiative transition rate.45 It has been reported that manipulation of the alkyl groups is a powerful tool to regulate the crystal structure and thus optimize the photophysical properties of organic emitters for OLEDs.46 Therefore, in this work, in order to explore the impact of the methyl group at different positions on the crystal structure and photophysical properties, methylpyridine moieties with the methyl group at the para- and meta-positions are selected as the electron-accepting groups for TAP1 and TAP2, respectively.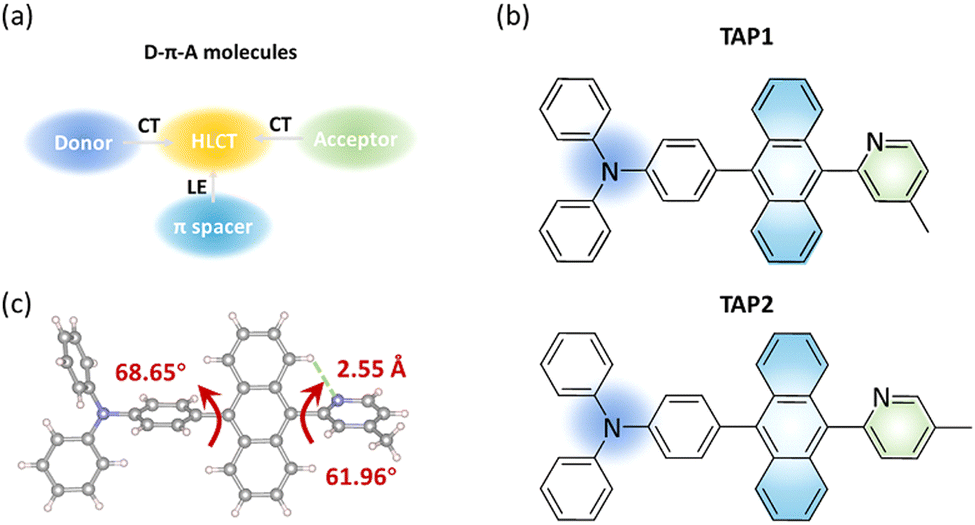 | ||
| Fig. 1 (a) Molecular design of D–π–A molecules. (b) Molecular structures of TAP1 and TAP2. (c) Single crystal structure of TAP1. The grey, blue, and white spheres denote carbon, nitrogen, and hydrogen atoms, respectively. | ||
The molecular structures of TAP1 and TAP2 are shown in Fig. 1b. The synthetic routes are outlined in Scheme S1 (ESI†), and the detailed synthetic procedures are described in the ESI.† The intermediates BrAP1 and BrAP2 were first synthesized, and then reacted with ((4-diphenylamino)phenyl)boronic acid to synthesize the targeted products TAP1 and TAP2 through the Suzuki cross-coupling reaction. The two emitters were characterized using 1H/13C NMR spectroscopy and mass spectrometry, as shown in the ESI.† TAP1 and TAP2 exhibit good solubility in common solvents such as toluene, dichloromethane, and chloroform. Single crystals of TAP1 were grown in the dichloromethane solvent covered with hexane as the top layer through slow solvent evaporation. The crystal structure has a highly twisted geometry with dihedral angles between anthracene and the neighboring triphenylamine and N-methylpyridine units of 68.65° and 61.96°, respectively (Fig. 1c), which suppresses intramolecular π-conjugation to realize the blue emission. The H⋯N distance is 2.55 Å, which falls in the range of typical H⋯N distances for the CH···N hydrogen bond, indicating the formation of the CH⋯N hydrogen bond and thus leading to more rigid structure.47
Thermal and electrochemical properties
The thermal properties were investigated via thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements. TAP1 and TAP2 demonstrate an outstanding thermal stability with high decomposition temperatures (Td, corresponding to 5% weight loss) at 466 °C and 464 °C, respectively (Fig. S1, ESI† and Table 1). The DSC results show the high melting temperature (Tm) values of TAP1 and TAP2 at 299 °C and 286 °C, respectively (Fig. S2, ESI† and Table 1), which generally benefit the formation of a uniform evaporated film for the fabrication of OLEDs. Cyclic voltammetry (CV) measurements were conducted to investigate the electrochemical properties of the emitters. As shown in Fig. S3 (ESI†), the oxidation onset against the ferrocenium/ferrocene (Fc+/Fc) redox couple for both TAP1 and TAP2 is 0.89 eV, and the corresponding HOMO energy is calculated to be −5.28 eV. Combined with the band gap (Eg) energy of 2.94 eV for both TAP1 and TAP2, estimated from the onset absorption (Fig. 2a), the LUMO energy of both TAP1 and TAP2 is calculated to be −2.34 eV (Table 1).| Emitter | λ abs (nm) | λ em (nm) | FWHMc (nm) | PLQYd (%) | Lifetime (ns) | E g (eV) | HOMO/LUMOf (eV) | T d/Tmg (°C) |
|---|---|---|---|---|---|---|---|---|
| a Absorption maximum. b Emission peak. c Full width at half maximum. d Absolute PLQY (left) and relative PLQY using 9,10-diphenylanthracene as a standard (right). e Optical energy gap measured from the absorption onset in 10−5 mol L−1 toluene. f HOMO calculated via cyclic voltammetry; LUMO derived from HOMO and Eg. g Decomposition temperature Td and melting temperature Tm. | ||||||||
| TAP1 | 355, 375, 395 | 457 | 64 | 41.7/44.3 | 1.46 | 2.94 | −5.28/−2.34 | 466/299 |
| TAP2 | 355, 375, 395 | 457 | 64 | 36.0/65.4 | 1.48 | 2.94 | −5.28/−2.34 | 464/296 |
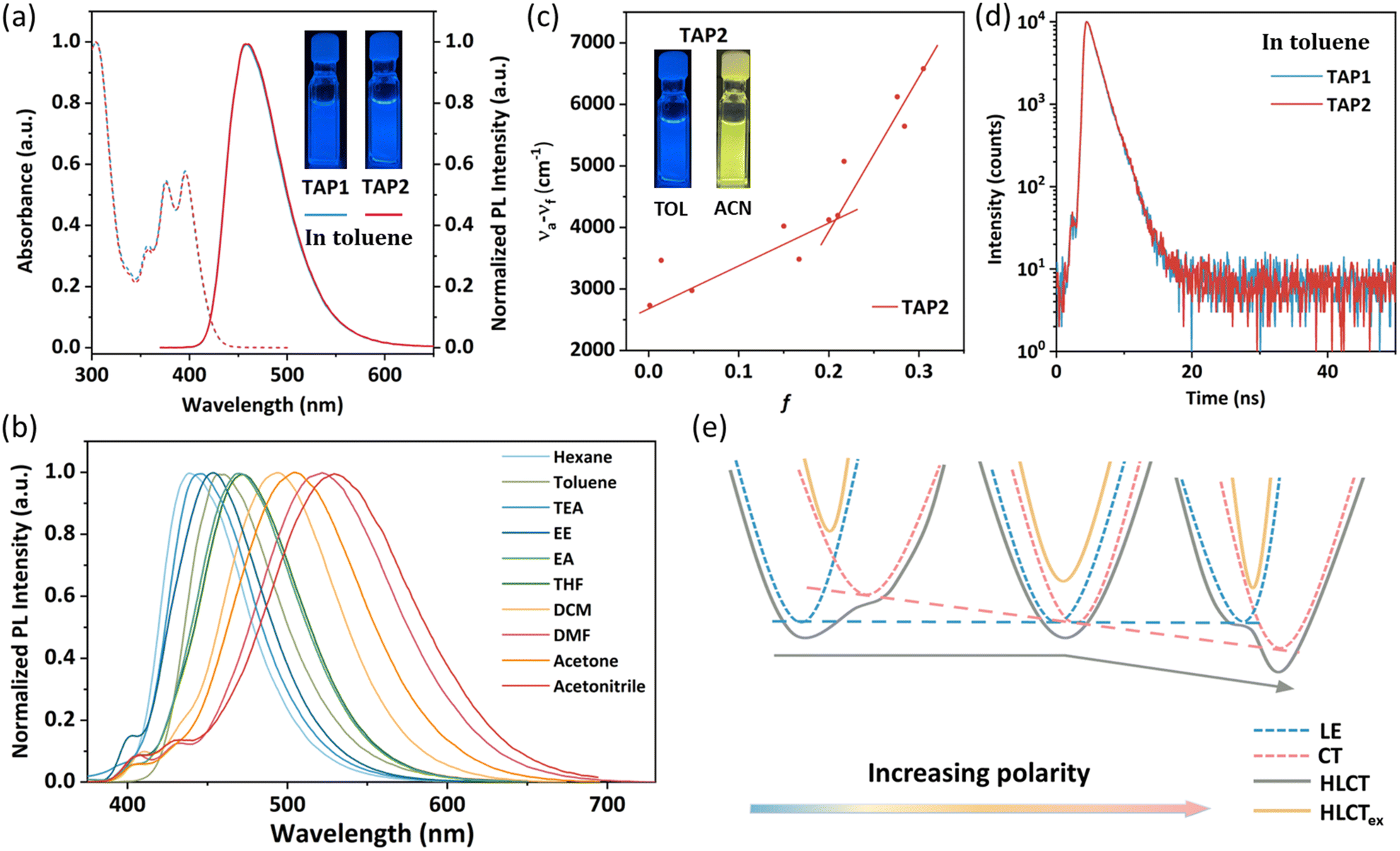 | ||
| Fig. 2 (a) UV-vis absorption and PL spectra of TAP1 and TAP2 in toluene (10−5 mol L−1). Insets show photographs of TAP1 and TAP2 in dilute toluene under 365 nm excitation. (b) PL spectrum of TAP2 in different solvents under 355 nm excitation. (c) Solvatochromic Lippert–Mataga models of TAP2 under 355 nm excitation. Insets show photographs of TAP2 in dilute toluene (left) and acetonitrile (right) under 365 nm excitation. (d) Lifetime decay curves of TAP1 and TAP2 in toluene (10−5 mol L−1). (e) Illustration of HLCT state formation in solvents with different polarities. | ||
Photophysical properties
The ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra of TAP1 and TAP2 in toluene (10−5 mol L−1) are shown in Fig. 2a. Both emitters exhibit similar absorption and emission properties. The vibronically structured absorption band in the range of 350–430 nm can be assigned to the intramolecular charge transfer transition of the anthracene unit, while the absorption band within 300–340 nm can be attributed to the π→π* transitions of triphenylamine as well as N-methylpyridine.48–51 As shown in Fig. 2a, both emitters show blue emission peaking at 457 nm with an FWHM of 64 nm. The absolute photoluminescence quantum yield (PLQY) of TAP1 and TAP2 in toluene was measured to be 41.7% and 36%, respectively. Using 9,10-diphenylanthracene as the reference, the relative PLQY was also tested and calculated to be 44.3% for TAP1 and 65.4% for TAP2.52The PL spectra of TAP2 and TAP1 in solvents with different polarities are displayed in Fig. 2b for TAP2 and Fig. S4 (ESI†) for TAP1. Both emitters exhibit a clear solvatochromic effect with a large redshift of 91 nm in different solvents from low-polarity hexane to high-polarity acetonitrile, which indicates that the emitters possess intramolecular CT character in the excited state. The correlation between the Stokes shift and the solvent polarity is plotted in Fig. 2c for TAP2 and in Fig. S5 (ESI†) for TAP1. According to the Lippert–Mataga equation,53 the non-linear relationship with two independent slopes of the fitted lines in low- and high-polarity solvents suggests the co-existence of two different excited state characters as well as the formation of HLCT states. The small slope in low-polarity solvents corresponds to the small dipole moment attributed to the LE state, while the large slope in high-polarity solvents corresponds to the large dipole moment attributed to the CT states.
The formation of HLCT states can also be verified by the lifetime measurements. The transient PL decay curves show mono-exponential profiles with the lifetime of 1.46 ns and 1.48 ns for TAP1 and TAP2, respectively (Fig. 2d and Table 1), which indicate that the excited state responsible for the fluorescence emission stems from one single excited state instead of a mixture of two separate LE and CT states.38 Based on the solvatochromic effect analysis above, it is reasonable to infer that the LE and CT states are integrated into a HLCT state rather than two separated components. The HLCT feature of the two emitters can also be verified through NTO analysis, which will be discussed in detail in the next section. In order to visualize the formation of the HLCT state and further explain the non-linear relationship between the Stokes shift and the solvent polarity, the schematic diagram is depicted in Fig. 2e. As the solvent polarity increases, the LE energy level barely changes, while the CT energy level decreases, driven by its strong interaction with the solvent field. Therefore, for the case that the LE component dominates the HLCT state in low-polarity solvents, the CT energy level decreases as the polarity increases and gradually dominates in the HLCT states, resulting in an intercrossed character of LE and CT states and thereby a non-linear relationship between the Stokes shift and the solvent polarity.38
Theoretical calculations
To gain insight into the excited state features and explore the electronic properties of the two emitters, density functional theory (DFT) calculations were conducted using ORCA software at the def2-TZVP basis set54 with def2/J55 or def2/JK56 as the auxiliary basis set. The optimized molecular configurations are shown in Fig. S6 (ESI†). TAP1 and TAP2 exhibit a twisted geometry with dihedral angles of 69–73° between anthracene and the neighboring triphenylamine and N-methylpyridine moieties. Such twisted configurations not only suppress the intramolecular π-conjugation to guarantee the pure blue emission but also limit the intermolecular π–π stacking, restraining the energy dissipation via the non-radiative channel. As shown in Fig. S7 (ESI†), the LUMO of TAP1 and TAP2 is localized on the anthracene unit, while the HOMO is mainly distributed over the triphenylamine moiety with a fraction extended to the anthracene unit.To further explore the excited state features of TAP1 and TAP2, the NTOs were investigated using time-dependent density functional theory (TD-DFT) at the def2-TZVPP basis set54 with def2/J55 as the auxiliary basis set. The distributions of the “hole” and “particle” for S0→S1 transition are intuitively displayed in the bottom and top images, respectively, of Fig. 3a. The large overlap and the partial separation of the “hole” and “particle” orbitals reveal the HLCT features of the S0→S1 transition. Combining analyses of the solvatochromic effect, transient PL decay curves, and NTO calculation results, it is reasonable to come to the conclusion that the two emitters are HLCT-type materials. For the S0→T1 transition, the “hole” and “particle” are both localized on the anthracene unit (Fig. S8, ESI†), exhibiting a clear LE feature. Interestingly, the S0→T2 transition is verified to behave as an HLCT transition, since its “hole” and “particle” present a partial overlap and partial separation character (Fig. S9, ESI†). To gain insight into the RISC process, the energy level arrangements and spin–orbit coupling (SOC) matrix elements were analysed. As shown in Fig. 3b, the two emitters exhibit a large energy gap between T1 and T2 (1.29 eV) but a small energy gap between S1 and T2 (0.01 eV), which can suppress the interconversion transition from T2 to T1 and trigger the spin-flip at high-lying excited states, enabling a fast hRISC process from T2 to S1 and thus realizing the efficient triplet exciton utilization. The calculated high SOC constant (0.36 cm−1) further verifies the efficient spin-flip between S1 and T2. On the basis of the structure, properties, and theoretical calculation analyses, the two emitters show similar optimized molecular structures (Fig. S6, ESI†) as well as nearly identical photophysical properties (Fig. 2 and Fig. S4 and S5, ESI†) and energy level distributions (Fig. 3), which indicate that the methyl group at the para- or meta-position in methylpyridine has a negligible effect on the crystal structure and photophysical properties of TAP1 and TAP2. All of these results demonstrate that our designed strategy of introducing the π-conjugated anthracene unit into an appropriate D–A segment is a practical approach to finely tune the excited state features and thus construct versatile HLCT emitters for high-performance blue OLEDs.
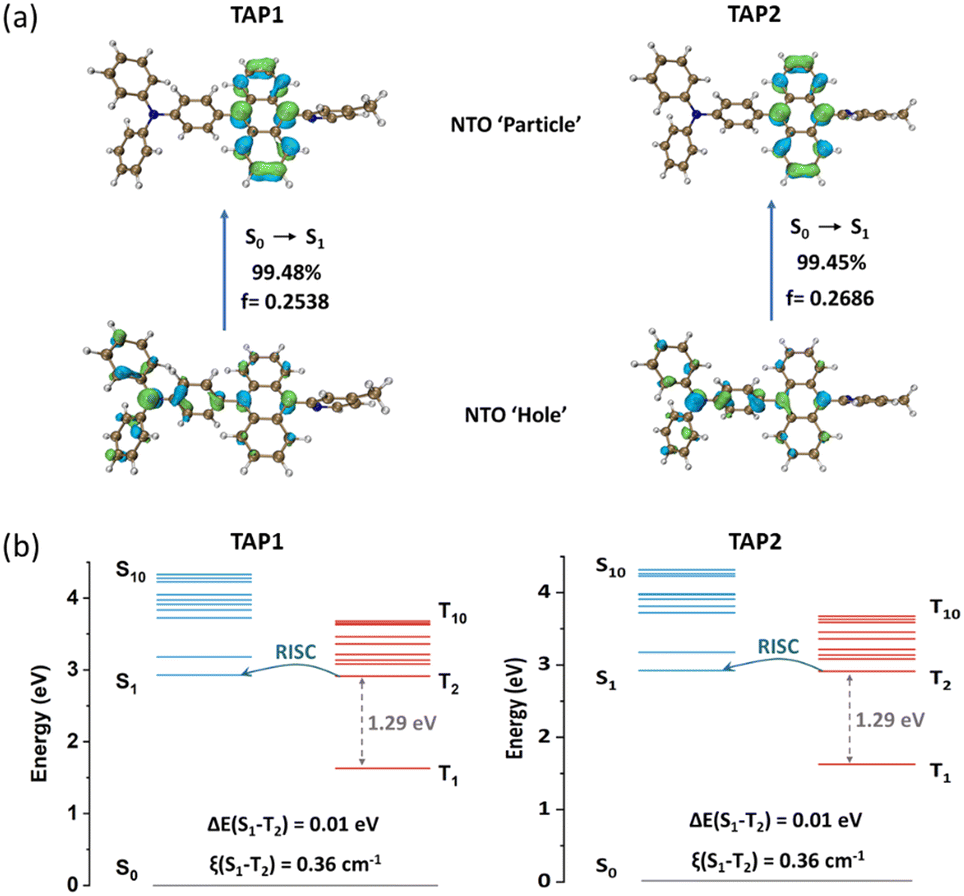 | ||
| Fig. 3 (a) NTO distributions for the S0→S1 transition and (b) energy level diagrams including SOC constants (ξ) of TAP1 (left) and TAP2 (right). | ||
Electroluminescence properties
To explore the potential application of TAP1 and TAP2 for blue OLEDs, their electroluminescence (EL) performance was investigated. Through the thermal evaporation method, the doped OLEDs were fabricated with the device structure of ITO/HATCN(15 nm)/TAPC(60 nm)/TCTA(5 nm)/EML(20 nm)/TmPyPB(45 nm)/LiF/Al (Fig. 4a), in which 1,4,5,8,9,11-hexacarbonitrile (HATCN), di-(4-(N,N-ditolylamino)phenyl)cyclohexane (TAPC), tris(4-carbazoyl-9-ylphenyl)amine (TCTA), and 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPB) act as the hole-injecting, hole-transporting, exciton-blocking, and exciton-transporting layers, respectively (Fig. S10, ESI†). As for the emitting layer, 9-[4-(10-phenyl-9-anthryl)phenyl]-9H-carbazole (CZPA) was selected as the host matrix due to its appropriate energy level and effective energy transfer for the blue fluorescent emitters.57,58 The doping concentration of the TAP1 and TAP2 devices is 7% and 10%, respectively. The PL spectra of the pure CZPA film, the CZPA:7% TAP1 film, and the CZPA:10% TAP2 film are displayed in Fig. S11 (ESI†). The different emission profiles of the host and doped films show that the EL of the OLEDs is from the emitters instead of the host materials, which indicates efficient energy transfer from the CZPA host to the TAP1 and TAP2 emitters.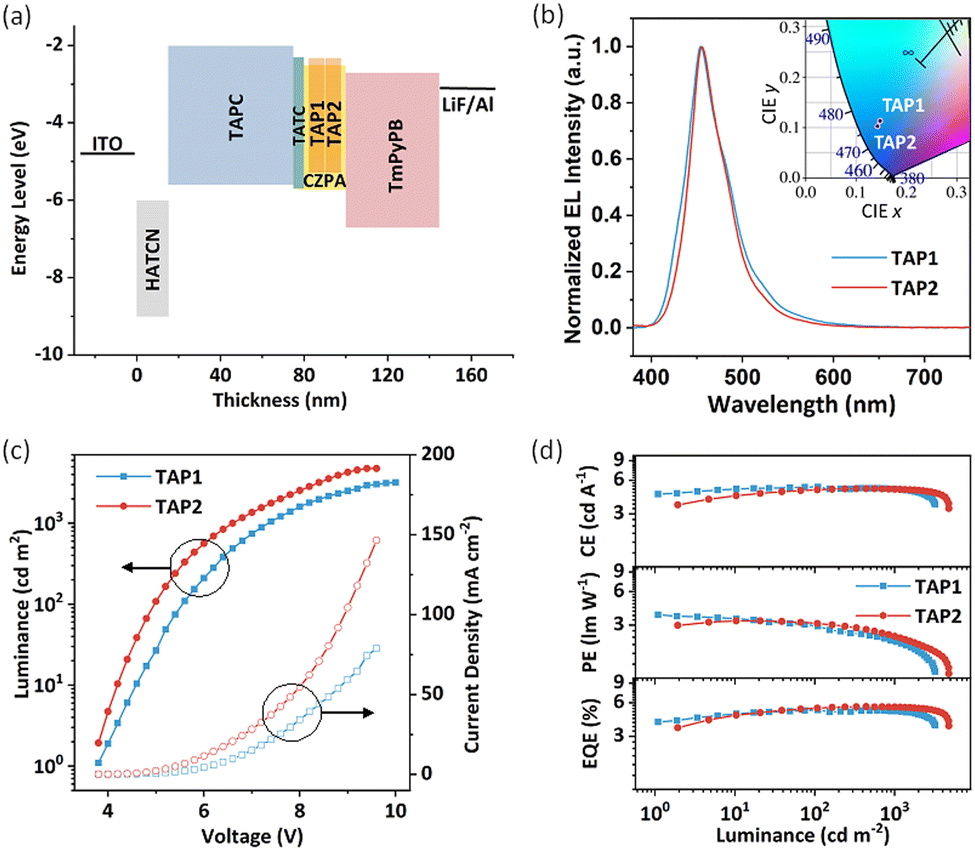 | ||
| Fig. 4 (a) Energy level diagram; (b) electroluminescent spectra (inset shows the corresponding CIE coordinates); (c) luminance–voltage–current density plots and (d) efficiency–luminance characteristics of the blue OLEDs. | ||
The EL performance features of the fabricated OLEDs are illustrated in Fig. 4, and some critical parameters are summarized in Table 2. Both devices exhibit a low turn-on voltage (3.8 V), indicating the high carrier mobilities of the emitters and the small injection barriers in the devices. As shown in Fig. 4b, the TAP1 and TAP2 devices show similar blue emissions peaking at 455 nm, while the TAP2 device possesses a narrower emission band with an FWHM of 45 nm and a better corresponding color purity with CIE coordinates of (0.14, 0.10) compared with the TAP1 device (FWHM of 52 nm, CIE coordinates of (0.15, 0.11)). Moreover, the TAP2 device also shows a higher EQEmax of 5.53% and a higher maximum luminance (Lmax) of 4751 cd m−2 compared with that of the TAP1 device (EQEmax = 5.16%, Lmax = 3198 cd m−2), while the maximum power efficiency and the maximum current efficiency of the two devices are comparable, as shown in Table 2. Surprisingly, both devices exhibit extremely low efficiency roll-off values of 1% and 0.5%, which are maintained at 5.11% for TAP1 and 5.50% for TAP2 at the practical brightness of 1000 cd m−2, which are among the best for blue HLCT emitters.23,37,40 This can be attributed to the large energy gap between T1 and T2 (1.29 eV) and the small energy gap between S1 and T2 (0.01 eV), which enable a fast hRISC process from T2 to S1 and thereby a suppressed efficiency roll-off.
| Device | λELa (nm) | EQEb (%) | CIE (x, y) | FWHMc (nm) | V on (V) | L max (cd m−2) | CEf (cd A−1) | PEg (lm W−1) | η r (%) |
|---|---|---|---|---|---|---|---|---|---|
| a EL emission peak. b Maximum external quantum efficiency. c Full width at half maximum of EL spectra. d Turn-on voltage at the luminance of 1 cd m−2. e Maximum luminance. f Maximum current efficiency. g Maximum power efficiency. h Exciton utilization efficiency estimated from the absolute PLQY. | |||||||||
| TAP1 | 455 | 5.16 | (0.15, 0.11) | 52 | 3.8 | 3198 | 5.228 | 3.719 | 41.25–61.87 |
| TAP2 | 455 | 5.53 | (0.14, 0.10) | 45 | 3.8 | 4751 | 5.041 | 3.264 | 51.20–76.81 |
The exciton utilization efficiency for the two devices is estimated to be 41–77% by employing the absolute PLQY and 28–58% by employing the relative PLQY,40,59,60 which far exceed the upper limit of 25% spin statistics for conventional fluorescent OLEDs, indicating the efficient triplet–singlet conversion process in the EL process. The calculation process is described in the ESI.† Based on the excited state energy levels and NTO distribution analysis above, the similar HLCT feature for the NTOs of S1 and T2 excited states together with the lifetime at the nanosecond scale as well as the small energy gap and large SOC constant between the S1 and T2 energy levels suggest that the hRISC from T2 to S1 could serve as an efficient approach to harvest the triplet exciton and thus boost the exciton utilization efficiency.30,32 Our results demonstrate that the OLEDs based on HLCT emitters with the advantages of a high EQE, a low efficiency roll-off, and good color purity are promising candidates for high-performance blue OLEDs.
Conclusions
In summary, we have successfully designed and synthesized two blue HLCT fluorescent emitters TAP1 and TAP2 by grafting a conjugated anthracene unit in the triphenylamine and N-methylpyridine (D–A) segment. Theoretical calculations have revealed the large overlap and partial separation of “hole” and “particle” orbitals for the S1 excited states, demonstrating the HLCT nature of the emitters. Together with a clear solvatochromic phenomenon, we have shown that introducing the π-conjugated anthracene unit into an appropriate donor–acceptor segment is an effective strategy to finely regulate the LE and CT components and construct versatile HLCT emitters. Benefiting from the advantages of the large transition moment and high exciton utilization efficiency for HLCT emitters, blue OLEDs with TAP2 or TAP1 as emitters exhibit good color purity and high efficiency with CIE coordinates of (0.14, 0.10) and (0.15, 0.11), narrow FWHM values of 45 nm and 52 nm, and EQEmax values of 5.53% and 5.16%, respectively. What is more, both devices exhibit extremely low efficiency roll-off values of 1% and 0.5%, retained at 5.50% for TAP2 and 5.11% for TAP1 at the practical brightness of 1000 cd m−2. Finally, based on their transient PL decay curves, excited state energy levels and NTO analysis, we have proposed that hRISC from T2 to S1 can serve as an efficient approach to harvest the triplet excitons and thus boost the exciton utilization efficiency. This work demonstrates that the rational design of HLCT emitters is a promising strategy towards high-performance blue OLEDs.Author contributions
S. X. W. designed and synthesized the compounds, measured characteristics, analysed the photophysical, electrochemical and electroluminescence properties, and wrote the manuscript with the help from J. H. H. L. L. and D. G. M. fabricated the OLED devices and measured the characteristics. Z. S. and Q. L. L. performed the theoretical calculations. X. D. Z., C. S. T., and L. Y. S. L. measured the transient photoluminescence decay curves and the photoluminescence quantum yields. W. Y. W. revised the manuscript and supervised the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Key R&D Program of China (2022YFE0104100), ITC Guangdong-Hong Kong Technology Cooperation Funding Scheme (TCFS) (GHP/038/19GD), CAS-Croucher Funding Scheme for Joint Laboratories (ZH4A), the Hong Kong Research Grants Council (PolyU 15305320), Fundamental Research Funds for the Central Universities (QNXM20210022), the Hong Kong Polytechnic University, Miss Clarea Au for the Endowed Professorship in Energy (847S) and Research Institute for Smart Energy (CDAQ) for financial support. We thank Limin Zhang for help with the X-ray crystal structure analysis.References
- J. R. Sheats, H. Antoniadis, M. Hueschen, W. Leonard, J. Miller, R. Moon, D. Roitman and A. Stocking, Science, 1996, 273, 884–888 CrossRef CAS PubMed.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- X. Peng, W. Qiu, W. Li, M. Li, W. Xie, W. Li, J. Lin, J. Yang, X. Li and S. Su, Adv. Funct. Mater., 2022, 32, 2203022 CrossRef CAS.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- H. Zhang, B. Zhang, Y. Zhang, Z. Xu, H. Wu, P. Yin, Z. Wang, Z. Zhao, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 2002323 CrossRef CAS.
- J. Wei, C. Zhang, D. Zhang, Y. Zhang, Z. Liu, Z. Li, G. Yu and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 12269–12273 CrossRef CAS PubMed.
- K. Klimes, Z. Zhu and J. Li, Adv. Funct. Mater., 2019, 29, 1903068 CrossRef.
- M. Mamada, H. Katagiri, C. Chan, Y. Lee, K. Goushi, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Funct. Mater., 2022, 32, 2204352 CrossRef CAS.
- L. Xing, Z.-L. Zhu, J. He, Z. Qiu, Z. Yang, D. Lin, W.-C. Chen, Q. Yang, S. Ji, Y. Huo and C.-S. Lee, Chem. Eng. J., 2021, 421, 127748 CrossRef CAS.
- M. I. Alam, M. R. Nagar, S. R. Nayak, A. Choudhury, J. Jou and S. Vaidyanathan, Adv. Opt. Mater., 2022, 10, 2200376 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- H. Lim, S. Woo, Y. H. Ha, Y. Kim and J. Kim, Adv. Mater., 2022, 34, 2100161 CrossRef CAS PubMed.
- P. Nalaoh, N. Sungworawongpana, P. Chasing, W. Waengdongbung, P. Funchien, C. Kaiyasuan, T. Sudyoadsuk and V. Promarak, Adv. Opt. Mater., 2021, 9, 2100500 CrossRef CAS.
- A. Khan, X. Tang, C. Zhong, Q. Wang, S. Yang, F. Kong, S. Yuan, A. S. D. Sandanayaka, C. Adachi, Z. Jiang and L. Liao, Adv. Funct. Mater., 2021, 31, 2009488 CrossRef CAS.
- Y. Luo, S. Li, Y. Zhao, C. Li, Z. Pang, Y. Huang, M. Yang, L. Zhou, X. Zheng, X. Pu and Z. Lu, Adv. Mater., 2020, 32, 2001248 CrossRef CAS PubMed.
- H. Liu, J. Li, W.-C. Chen, Z. Chen, Z. Liu, Q. Zhan, X. Cao, C.-S. Lee and C. Yang, Chem. Eng. J., 2020, 401, 126107 CrossRef CAS.
- D. Zhang, X. Song, M. Cai, H. Kaji and L. Duan, Adv. Mater., 2018, 30, 1705406 CrossRef PubMed.
- H. Yersin, R. Czerwieniec, L. Mataranga-Popa, J. Mewes, G. Cheng, C. Che, M. Saigo, S. Kimura, K. Miyata and K. Onda, Adv. Funct. Mater., 2022, 32, 2201772 CrossRef CAS.
- X. Tang, Q. Bai, T. Shan, J. Li, Y. Gao, F. Liu, H. Liu, Q. Peng, B. Yang, F. Li and P. Lu, Adv. Funct. Mater., 2018, 28, 1705813 CrossRef.
- W. Li, P. Chasing, P. Nalaoh, T. Chawanpunyawat, N. Chantanop, C. Sukpattanacharoen, N. Kungwan, P. Wongkaew, T. Sudyoadsuk and V. Promarak, J. Mater. Chem. C, 2022, 10, 9968–9979 RSC.
- Z. Wu, S. Song, X. Zhu, H. Chen, J. Chi, D. Ma, Z. Zhao and B. Z. Tang, Mater. Chem. Front., 2021, 5, 6978–6986 RSC.
- W. Liu, S. Ying, R. Guo, X. Qiao, P. Leng, Q. Zhang, Y. Wang, D. Ma and L. Wang, J. Mater. Chem. C, 2019, 7, 1014–1021 RSC.
- C. Fu, S. Luo, Z. Li, X. Ai, Z. Pang, C. Li, K. Chen, L. Zhou, F. Li, Y. Huang and Z. Lu, Chem. Commun., 2019, 55, 6317–6320 RSC.
- S. Zhang, W. Li, L. Yao, Y. Pan, F. Shen, R. Xiao, B. Yang and Y. Ma, Chem. Commun., 2013, 49, 11302 RSC.
- W.-C. Chen, Y. Yuan, S.-F. Ni, Q.-X. Tong, F.-L. Wong and C.-S. Lee, Chem. Sci., 2017, 8, 3599–3608 RSC.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- X. Tang, Q. Bai, Q. Peng, Y. Gao, J. Li, Y. Liu, L. Yao, P. Lu, B. Yang and Y. Ma, Chem. Mater., 2015, 27, 7050–7057 CrossRef CAS.
- Y. Pan, J. Huang, W. Li, Y. Gao, Z. Wang, D. Yu, B. Yang and Y. Ma, RSC Adv., 2017, 7, 19576–19583 RSC.
- H. Zhang, J. Xue, C. Li, S. Zhang, B. Yang, Y. Liu and Y. Wang, Adv. Funct. Mater., 2021, 31, 2100704 CrossRef CAS.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- J. Yang, Q. Guo, J. Wang, Z. Ren, J. Chen, Q. Peng, D. Ma and Z. Li, Adv. Opt. Mater., 2018, 6, 1800342 CrossRef.
- Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 510–515 CrossRef CAS.
- T. Liu, X. Chen, J. Zhao, W. Wei, Z. Mao, W. Wu, S. Jiao, Y. Liu, Z. Yang and Z. Chi, Chem. Sci., 2021, 12, 5171–5176 RSC.
- M. Chen, Y. Yuan, J. Zheng, W.-C. Chen, L.-J. Shi, Z.-L. Zhu, F. Lu, Q.-X. Tong, Q.-D. Yang, J. Ye, M.-Y. Chan and C.-S. Lee, Adv. Opt. Mater., 2015, 3, 1215–1219 CrossRef CAS.
- X. Lv, M. Sun, L. Xu, R. Wang, H. Zhou, Y. Pan, S. Zhang, Q. Sun, S. Xue and W. Yang, Chem. Sci., 2020, 11, 5058–5065 RSC.
- B. Liu, Z.-W. Yu, D. He, Z.-L. Zhu, J. Zheng, Y.-D. Yu, W.-F. Xie, Q.-X. Tong and C.-S. Lee, J. Mater. Chem. C, 2017, 5, 5402–5410 RSC.
- X. Qiu, S. Ying, C. Wang, M. Hanif, Y. Xu, Y. Li, R. Zhao, D. Hu, D. Ma and Y. Ma, J. Mater. Chem. C, 2019, 7, 592–600 RSC.
- W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- S.-Y. Yang, Y.-L. Zhang, A. Khan, Y.-J. Yu, S. Kumar, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2020, 8, 3079–3087 RSC.
- X. Lv, L. Xu, Y. Yu, W. Cui, H. Zhou, M. Cang, Q. Sun, Y. Pan, S. Xue and W. Yang, Chem. Eng. J., 2021, 408, 127333 CrossRef CAS.
- J.-Y. Hu, Y.-J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064–2071 CrossRef CAS.
- X. Zheng, T. Huang, G. Yang, A. Lin, K. Chen, X. Chen, J. Li and Q. Tong, Chem. – Eur. J., 2021, 27, 16181–16188 CrossRef CAS PubMed.
- L. Chen, Y. Jiang, H. Nie, R. Hu, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17215–17225 CrossRef CAS PubMed.
- (a) X. Lv, M. Sun, L. Xu, R. Wang, H. Zhou, Y. Pan, S. Zhang, Q. Sun, S. Xue and W. Yang, Chem. Sci., 2020, 11, 5058–5065 RSC; (b) F. Liu, X. Man, H. Liu, J. Min, S. Zhao, W. Min, L. Gao, H. Jin and P. Lu, J. Mater. Chem. C, 2019, 7, 14881–14888 RSC; (c) J. Zhang, Y. Zhao, H. Xu, D. Zhang, Y. Miao, R. Shinar, J. Shinar, H. Wang, B. Xu and Y. Wu, J. Mater. Chem. C, 2019, 7, 10810–10817 RSC; (d) W. Li, P. Chasing, W. Benchaphanthawee, P. Nalaoh, T. Chawanpunyawat, C. Kaiyasuan, N. Kungwan, S. Namuangruk, T. Sudyoadsuk and V. Promarak, J. Mater. Chem. C, 2021, 9, 497–507 RSC; (e) J. Jayabharathi, V. Thanikachalam, R. Ramya and S. Panimozhi, RSC Adv., 2019, 9, 33693–33709 RSC.
- V. Thangaraji, P. Rajamalli, J. Jayakumar, M.-J. Huang, Y.-W. Chen and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 17128–17133 CrossRef CAS PubMed.
- (a) S. K. Pathak, H. Liu, C. J. Zhou, G. H. Xie and C. L. Yang, J. Mater. Chem. C, 2021, 9, 7363 RSC; (b) M. M. Han, Y. W. Chen, Y. J. Xie, F. Y. Zhang, X. N. Li, A. R. Huang, Y. H. Fan, Y. Y. Fan, Y. B. Gong, Q. Peng, Q. Q. Li, D. G. Ma and Z. Li, Cell Rep. Phys. Sci., 2020, 1, 100252 CrossRef; (c) H.-Y. Chih, Y.-W. Chen, Y.-C. Hsieh, W.-C. Li, C.-W. Liao, C.-H. Lin, T.-Y. Chiu, W.-W. Tsai, C.-W. Lu and C.-H. Chang, Chem. – Eur. J., 2019, 25, 16699–16711 CrossRef CAS PubMed.
- M. Mazik, D. Blaser and R. Boese, Tetrahedron, 2001, 57, 5791–5797 CrossRef CAS.
- I. Cho, S. H. Kim, J. H. Kim, S. Park and S. Y. Park, J. Mater. Chem., 2012, 22, 123–129 RSC.
- R. Norman. Jones, Chem. Rev., 1947, 41, 353–371 CrossRef PubMed.
- S. Tang and J. Zhang, J. Phys. Chem. A, 2011, 115, 5184–5191 CrossRef CAS PubMed.
- M. L. Dell’Arciprete, C. J. Cobos, J. P. Furlong, D. O. Mártire and M. C. Gonzalez, ChemPhysChem, 2007, 8, 2498–2505 CrossRef PubMed.
- John V. Morris, Mary A. Mahaney and J. Robert Huber, J. Phys. Chem., 80, 969–974 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Weigend, J. Comput. Chem., 2008, 29, 167–175 CrossRef CAS PubMed.
- S. Xiao, X. Qiao, C. Lin, L. Chen, R. Guo, P. Lu, L. Wang and D. Ma, Adv. Opt. Mater., 2022, 10, 2102333 CrossRef CAS.
- A. Salehi, C. Dong, D.-H. Shin, L. Zhu, C. Papa, A. Thy Bui, F. N. Castellano and F. So, Nat. Commun., 2019, 10, 2305 CrossRef PubMed.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. Furno, R. Meerheim, S. Hofmann, B. Lüssem and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 115205 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2233673. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc05554f |
| This journal is © The Royal Society of Chemistry 2023 |