
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-mode enhanced Raman scattering spectroscopy using aggregation-free hybrid metal/metal-oxide nanoparticles with intrinsic oxygen vacancies†
Gemma
Davison
ab,
Yidan
Yin
 ab,
Tabitha
Jones
ab,
Ivan P.
Parkin
b,
William J.
Peveler
*c and
Tung-Chun
Lee
*ab
ab,
Tabitha
Jones
ab,
Ivan P.
Parkin
b,
William J.
Peveler
*c and
Tung-Chun
Lee
*ab
aInstitute for Materials Discovery, University College London, London, WC1H 0AJ, UK. E-mail: tungchun.lee@ucl.ac.uk
bDepartment of Chemistry, University College London, London, WC1H 0AJ, UK
cSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: william.peveler@glagsow.ac.uk
First published on 15th February 2023
Abstract
Surface-enhanced Raman scattering (SERS) spectroscopy, with strong and stable signals, was achieved in aqueous solution using colloidal hybrid nanoparticles, consisting of gold nanotriangles (Au NTs) with a nanoscale coating of tin dioxide (SnO2). The observed strong SERS signals can be attributed to an electromagnetic enhancement from the anisotropic Au NTs, and chemical enhancement resulted from the photo-induced charge transfer from SnO2 to Au NTs. The latter was enabled by the presence of persistent inter-band levels introduced by the intrinsic oxygen vacancies in SnO2, as well as by the nanoscale mixing of the two components. Moreover, the intrinsic oxygen vacancies in highly defected SnO2 nanostructures and the aggregation-free approach underpin the high stability of SERS signals. The multi-mode enhanced Raman signal could be further boosted by in situ UV-irradiation and, as a proof-of-concept application, detection of an explosive marker 2,4-dinitrotoluene (DNT) was demonstrated in aqueous solution, achieving a detection limit down to 6 nM (1 μg mL−1) with a significant signal enhancement of 22 times over and above the SERS signals of bare Au NTs.
Introduction
Surface-enhanced Raman scattering (SERS) spectroscopy is arguably the most studied Raman approach in analytical chemical applications to date. For typical SERS, intrinsically weak Raman scattering signals from molecules are significantly enhanced when they are in close proximity to nanostructured surface of plasmonic metals, e.g., Au, Ag, Cu and Al. The predominant contribution to SERS effects arises from the so-called electromagnetic (EM) enhancement which involves the interplay of local electric field of the plasmonic nanostructures with incoming and outgoing photons.1,2 In particular, gigantic field enhancements have been achieved at tips of anisotropic nanostructures or plasmonic nanojunctions and have become popular approaches for boosting sensitivity.3–5 EM-enhanced Raman has found applications in analysis of small molecules,6 environmental monitoring,7,8 and homeland security with abundant examples of sensitive detection of explosives in the literature,9–11 thanks to the ability to ‘fingerprint’ target compounds in a range of phases (e.g. solid, solution, condensed vapour, and within complex matrices) in a label-free manner. The method can be performed at stand-off distance or in a sealed volume, which further underpins versatile detection schemes for on-site applications.12Alongside the EM enhancement, there is the possibility of chemical enhancement (CE), whereby a molecule adsorbed on a substrate undergoes changes to its polarizability upon irradiation by the laser used for Raman excitation, which subsequently enhances its Raman cross-section and thus improves signal output by a smaller but still significant amount.13 To make the most of this additional enhancement mode, recent research has refocused on investigating SERS behaviour of dielectric and semiconducting materials, which are capable of displaying both EM enhancement, via improved light trapping and scattering, and plasmonics in certain specific scenarios; and CE of surface bound analytes.14–17 Whilst the light–matter interactions in dielectric SERS substrates are more complex than those of plasmonic metals, they offer advantages of versatile and robust surface chemistry, highly tuneable shape and size, low-cost, as well as switchable enhancement using external physical triggers.18–21
In this context, we have been refining the photo-induced enhanced Raman scattering (PIERS) technique,22 whereby irradiation of metal oxide surfaces with UV light creates oxygen defects and turns on local CE and EM enhancement in the presence of Au or Ag nanoparticles, to boost signals by ca. 1 order of magnitude over conventional SERS.23,24 Suitable metal oxides have been demonstrated to be defected TiO2,22,25 WO3,18,21 SnO2,20,26 and ZnO,25,27 amongst others, that are capable of mediating intrinsic or induced defects. In particular, titanium dioxide, the archetypal PIERS substrate, shows little or no Raman enhancement without the creation of metastable oxygen defects via UV-irradiation,28 whilst metal oxides with intrinsic defects (WO3, SnO2) can exhibit inherent SERS activity without prior photo-activation.29 However, all precedent examples of PIERS are based on solid substrates, limiting their application as in situ biological probes in aqueous media, and reducing the signal consistency across the substrate due to localized clustering of the Raman probe on the surface.
Herein we demonstrate that a combination of nanostructured, intrinsically defected, SnO2 coated on colloidal anisotropic gold nanotriangles (Au NTs, Fig. 1(a)) can enable significant Raman signal enhancement, of up to 16 times over that of bare Au NTs, without any UV-activation or externally triggered aggregation. Moreover, the signal enhancement can be further boosted with a UV light-triggered PIERS effect to achieve up to 22 times enhancement over the signal from bare Au NTs. The observed strong SERS signals can be attributed to a multi-mode enhancement mechanism, consisting of EM enhancement from the anisotropic Au NTs and CE from the photo-induced charge transfer from SnO2 to Au NTs. The latter was enabled by the presence of persistent inter-band levels introduced by the intrinsic oxygen vacancies in SnO2 as characterized using UV-vis and X-ray photoelectron spectroscopy (Fig. 1(b)), as well as by nanoscale mixing of the two components as evidenced by transmission electron microscopy (Fig. 1(c)). Meanwhile, the intrinsic oxygen vacancies in highly defected SnO2 nanostructures and the aggregation-free approach underpin the high stability of SERS signals. We envisage our aggregation-free hybrid SERS approach being applied in detection of trace chemicals in situ, with nanomolar detection limits for bio-sensing, and in detecting pollutants and chemical threats in wastewaters – the latter application we trialled here as a proof-of-concept, achieving a detection limit of 6 nM (1 μg mL−1) for an explosive marker 2,4-dinitrotoluene.
 | ||
| Fig. 1 Schematic showing (a) illustration of a Au NT–SnO2 hybrid nanoparticle, with intrinsic oxygen vacancies in the SnO2, formed by overcoating pre-formed Au NTs with SnO2 from Na2[Sn(OH)6]; and (b) the plausible hybrid SERS charge-transfer (CT) mechanism, where the incident Raman laser excites electrons from inter-band oxygen vacancy (VO) levels to the conduction band (CB) in the SnO2, and charge transfer to the Au NTs and then to the analyte occurs. Energy level values were extrapolated from experimental data in this work, and correlate with literature values. (c) TEM images of as-synthesized Au NTs and samples Au NT–SnO2-1, Au NT–SnO2-2 and Au NT–SnO2-3, with increasing numbers of SnO2 coating cycles. See Fig. S1 (ESI†) for photographs of the sample solutions. | ||
Experimental
Au NTs were synthesized using modified literature procedures as described in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 To coat Au NTs with SnO2, the pH of 10 mL of Au NT solution was adjusted to 10.5 by adding approximately 20 μL of 1 M sodium hydroxide, monitored with a Mettler Toledo FiveEasy pH meter. The solution was mixed with 2 mL of 0.4 mM sodium stannate for 5 minutes in a thermomixer at 600 rpm, followed by heating to 60 °C and maintaining the solution at this temperature for 1 hour under rapid mixing at 800 rpm.31 The Au NT–SnO2 solution was then centrifuged and re-dispersed in water (sample Au NT–SnO2-1). The coating cycle was repeated up to twice more to form successively thicker coatings of SnO2 (samples Au NT–SnO2-2 and -3 respectively).
30 To coat Au NTs with SnO2, the pH of 10 mL of Au NT solution was adjusted to 10.5 by adding approximately 20 μL of 1 M sodium hydroxide, monitored with a Mettler Toledo FiveEasy pH meter. The solution was mixed with 2 mL of 0.4 mM sodium stannate for 5 minutes in a thermomixer at 600 rpm, followed by heating to 60 °C and maintaining the solution at this temperature for 1 hour under rapid mixing at 800 rpm.31 The Au NT–SnO2 solution was then centrifuged and re-dispersed in water (sample Au NT–SnO2-1). The coating cycle was repeated up to twice more to form successively thicker coatings of SnO2 (samples Au NT–SnO2-2 and -3 respectively).
An ocean optics Raman spectrometer with a 632.8 nm helium–neon laser and a laser power of 22 mW was used to take the Raman measurements. For PIERS measurements, a UV LED with 12.5 mW power was used to irradiate the substrate with a light path perpendicular to that of the Raman laser. Stock solutions of 4-MBA and 2,4-DNT were added to Au NT–SnO2 solutions at various concentrations. Ten accumulations of 7 s scans were acquired for each measurement. Five measurements were taken per sample. The spectra were averaged, and baseline corrected using an asymmetric least-squares (ALS) plugin in Origin.
Full experimental and instrumentation details are given in the ESI.†
Results and discussion
Synthesis and characterization of hybrid nanostructures
Hybrid nanoparticles of Au NT–SnO2 were produced by first synthesizing gold nanotriangles (Au NTs) in the presence of cetyltrimethylammonium chloride (CTAC) in aqueous media using a modified protocol from the literature.30 The formation of the Au NTs was verified by UV-Visible (UV-Vis) spectroscopy showing the expected localized surface plasmon resonance (LSPR) bands at ca. 548 nm and ca. 624 nm (Fig. 2(a) and Fig. S4, ESI†), which correspond to the out-of-plane and in-plane dipolar modes respectively, consistent with the method report.30 Transmission electron microscopy (TEM) further confirmed the triangular planar shape of the Au NT (Fig. 1(c) and Fig. S2, ESI†), where the averaged side length is 67 ± 10 nm (Fig. S3, ESI†). | ||
| Fig. 2 (a) UV-vis spectra of Au NTs and Au NT–SnO2 with increasing numbers of SnO2 coating cycles. Data normalized at 580 nm for ease of comparison. The solvent was 17 mM aqueous CTAC for as-synthesized Au NTs and deionized water for Au NT–SnO2. (b) Bright-field STEM image and STEM-EDS elemental maps of a representative hybrid nanoparticle from Au NT–SnO2-2, verifying the presence of a SnO2 coating around a Au NT core and the existence of voids (marked by grey arrows) within the coating. | ||
As-synthesized CTAC-protected Au NTs were then coated with a nanoscale, defected layer of SnO2via condensation of sodium stannate (sample Au NT–SnO2-1). The coating cycle was repeated up to twice more to form successively thicker coatings of SnO2 (samples Au NT–SnO2-2 and -3 respectively, Fig. S1, ESI†) for subsequent investigation into the effects of SnO2 thickness on SERS enhancement. The shape of the Au NT cores remained the same after the coating process, as evidenced by the same overall shape of the LSPR band in their UV-vis spectra (Fig. 2(a)).32
Nanoscale morphology of the hybrid NPs was characterized by TEM (Fig. 1(c) and Fig. S2, S5–S7, ESI†), and no change was observed in size and shape of the Au NTs across the coating cycles, consistent with the UV-vis spectra. Interestingly, the morphology of the SnO2 evolved significantly over successive coating cycles. After the first coating cycle (Au NT–SnO2-1), a thin (<5 nm), discontinuous layer of SnO2 was grown on the surface of Au NTs. The surface coverage and thickness of the patchy SnO2 layer increased after the second and third coating cycles. For the latter, extended quasi-fractal networks of SnO2 NPs with embedded Au NTs can be observed. The resultant morphological diversity of the SnO2 coating gave us an opportunity to explore the effect on SERS, and potential insight into the design of nanoscale oxide coatings with morphology readily accessible via sol–gel processes. The voids within the SnO2 coating were further visualized using elemental mapping by scanning transmission electron microscopy-energy dispersive X-ray spectroscopy (STEM-EDS), as shown in Fig. 2(b). These voids were later shown to have a major impact on the accessibility of analyte molecules to the surface of Au NTs, which subsequently translate to the hybrid SERS enhancement.
Phase identification of the materials in the hybrid NPs was carried out using powder X-ray diffraction (XRD), with expected major peaks corresponding to cubic Au (ICSD collection code 64701) and tetragonal SnO2 (ICSD collection code 9163) confirmed (Fig. S8, ESI†). The broad SnO2 peaks were indicative of small and highly defected SnO2 crystallites.32
When Au NTs are coated with SnO2, the increase in refractive index of the dielectric environment around the Au NTs should cause a red shift in the wavelength of the LSPR. Interestingly however, in our case a subtle but consistent blue shift in both out-of-plane and in-plane LSPR modes was observed (Fig. 2(a) and Fig. S4, ESI†), which cannot be explained by any change in local surfactant concentration, since the refractive index of SnO2 (nD = 2.006) is significantly larger than that of CTAC solid (nD = 1.378). Herein we attribute the observed blue shift to the counteracting effects of electron injection from SnO2 to Au NTs. As per the work of Mulvaney, and previous work by Parkin et al., we quantified the electron injection density using eqn (1) for the observed blue shifts of −9 to −14 nm (for the 624 nm plasmon mode), and estimated change in electron densities (ΔN/N) to be 2.9% to 4.5% with increasing SnO2 coatings, comparable to previously found values.22,33 We note that the actual values of (ΔN/N) could be larger than our present estimation because we did not account for any expected red-shifting caused by the increase in local refractive index by the SnO2 coating. This finding provides an initial hint of possible electron transfer effects from SnO2 to Au NT that underpins the enhancement mechanism.
 | (1) |
The optical band gaps of the SnO2 within the Au NT–SnO2 were extracted from the Tauc plots calculated based on their UV-vis spectra (Fig. S9–S13, ESI†). The optical band gap of SnO2 NPs alone is estimated to be 3.9 eV (Fig. S10, ESI†), consistent with literature values;34 whereas for Au NT–SnO2 the values at ∼3 eV correspond to the band gap of SnO2 within the hybrid nanostructures,35,36 which widens with increasing SnO2 thickness (2.83 to 3.05 eV), consistent with the expected decrease in oxygen vacancy concentration in thicker coatings. We note that whilst the trend observed is consistent with expectations (decreasing oxygen vacancies increase the observed band gap), the absolute values cannot be entirely relied upon due to influence of variable crystallite size and background influence of the Au NTs in the Tauc plots.34
X-ray photoelectron spectroscopy (XPS) was used to further investigate the electronic structure of the defected SnO2. In particular, the oxygen 1s region shows two peaks, O 1s which relates to oxygen bound to Sn atoms, and O 1sv which results from oxygen vacancies, i.e. the signal arising from O atoms bound to non-saturated Sn atoms (Fig. S14, ESI†).37 For Au NT–SnO2, the higher intensity of the O 1sv peak compared to the O 1s peak indicates that the SnO2 was defected with more oxygen vacancies than a reference SnO2 powder (Acros Organics), which exhibits an O 1s peak with higher intensity than that of the O 1sv peak (Fig. S16a, ESI†). This finding was verified by a sample of core@shell Au NP@SnO2, accessible via the same but scaled-up protocol. The larger sample size allowed XPS spectra with better resolved peaks, showing energies corresponding to Au 4f7/2, Au 4f5/2, Sn 3d5/2 and Sn 3d3/2 (Fig. S15, ESI†), consistent with that of the Au NT–SnO2. Due to the oxygen vacancies in the defected SnO2, the valence band edge was measured at 1.89 eV above that found in normal SnO2 (1.45 eV vs. 3.34 eV), which indicates the abundance of electron-rich oxygen vacancies that reduce the effective band gap due to their corresponding inter-band levels (Fig. 1(b) and Fig. S16, ESI†).32,38–42
Hybrid SERS with stable enhancement in aqueous media
We demonstrate intense signals from our hybrid SERS approach for trace chemical detection in aqueous media, where the charge transfer (CT) effects enable additional CE over and above the EM enhancement from the Au NTs alone. We note that the observed additional boost in signal intensity can also be partially attributed to the increased EM enhancement resulting from the injection of electron density from the SnO2 to the Au NTs via band alignment.43 Moreover, the hybrid SERS signals were shown to be highly stable over long timeframes, due to the intrinsic nature of the oxygen vacancies and the aggregation-free approach of the sensing scheme.As an initial test of the synergistic enhancement, we employed 4-mercaptobenzoic acid (4-MBA) as the model analyte, which is known to be highly Raman active and can bind strongly to the Au NT/SnO2 surface via its thiol moiety. Its Raman activity allows us to observe strong and distinctive signals for quantitative comparison between the Au NT–SnO2 hybrid NPs and the control, bare Au NTs. The strong preferential affinity for Au, accessed via gaps in the SnO2 coating, is expected to help minimize differences introduced by surface binding effects from the SnO2 coating (e.g. change in surface charge) to allow fair comparison.
Upon addition of 4-MBA to a solution of bare Au NTs, characteristic Raman peaks of 4-MBA can be observed at 1080 cm−1 and 1588 cm−1, which are characteristic of the ν8 aromatic ring vibration with the C–S stretching mode, and the ν12 aromatic ring breathing mode, respectively (Fig. 3(a)).44 A titration was then repeated by using the same concentration of Au NT–SnO2 hybrid NPs (here Au NT–SnO2-2). The intensities of the main Raman peaks were higher across all concentrations of 4-MBA with the hybrid system, compared to the SERS of bare Au NTs that exhibited plasmonic (EM) enhancement alone, with the difference in intensity increasing with the concentration of 4-MBA (Fig. 3(b)). The hybrid-SERS enhancement (Ehybrid) is defined as:
 | (2) |
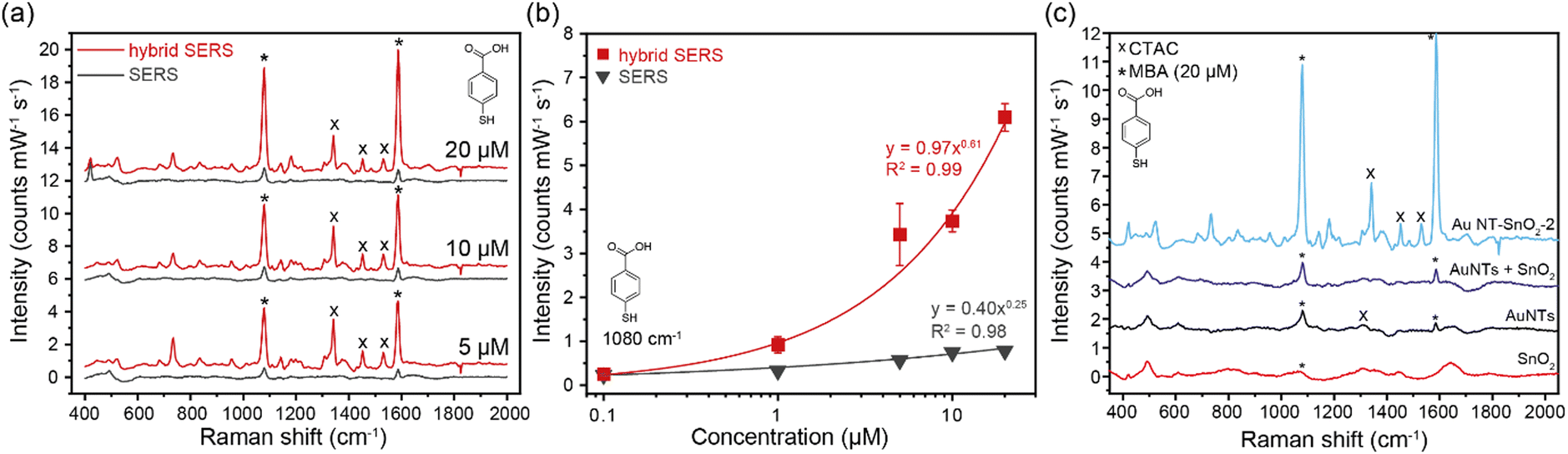 | ||
| Fig. 3 (a) Spectra of SERS using Au NTs and hybrid SERS using Au NT–SnO2-2 in the presence of 5, 10, and 20 μM 4-MBA. Note that the CTAC signals gradually decrease upon increasing concentration of 4-MBA that displaces CTAC from the Au NT surface via the formation of strong Au–S bonds. (b) Plot of Raman intensity of the 1080 cm−1 peak intensity against 4-MBA concentration in log scale. Strong correlation was observed using power law fitting (R2 > 0.98). (c) Spectra of 20 μM 4-MBA in SnO2 NPs, Au NTs, a physical mixture of Au NTs and SnO2 NPs, and the hybrid Au NT–SnO2-2. Raman peaks of 4-MBA marked with * and CTAC with ×. Solvent was deionized water for Au NT–SnO2, and 17 mM aqueous CTAC for Au NTs alone. Baseline offset for clarity. | ||
Control measurements were performed using SnO2 NPs synthesized in an identical fashion but without Au NTs. As expected, no Raman signal from 4-MBA can be observed (red spectrum in Fig. 3(c)), owing to the lack of EM enhancement from Au NTs and to the broken CT pathway via the Au NTs. Another control spectrum was obtained using a physical mixture of Au NTs and SnO2 NPs, where no addition enhancement can be observed, i.e., Ehybrid ∼ 1 (purple spectrum in Fig. 3(c)). This finding highlights the importance of close physical contact between Au NTs and SnO2 NPs which is essential for the hypothesized CT mechanism and the electron injection effects, as opposed to other long-range radiative energy transfer pathways.
We note that the SERS enhancement of 4-MBA increased with a thicker coating of SnO2 on the Au NTs (Fig. S18, ESI†), but with the largest improvement seen after the second coating cycle. This is likely because Au NT–SnO2-2 has the best balance between the accessible surface area on plasmonic Au NTs via the voids of the SnO2 coating (Fig. 2(b)) and the mass of chemically-enhancing SnO2 which are all important considerations in such systems.45 The sample Au NT–SnO2-2 was therefore selected for all subsequent measurements, unless otherwise stated.
We attribute the increase in intensity of the Raman signal to the photo-induced charge transfer from SnO2 to Au NTs in our hybrid system. As evidenced by the UV-vis (Fig. 2(a)) and XPS data (Fig. S14–S16, ESI†), the SnO2 contains intrinsic oxygen vacancies (VO), which create donor states approximately 2 eV below the conduction band edge of the SnO2.46–48 When the substrate was illuminated by a 633 nm Raman laser with energy of ca. 2 eV, electrons in the VO levels were excited to the conduction band of the SnO2, and then transferred to the Au NTs (Fig. 1(b)).22,49 The photo-induced charge injection resulted in a negative shift in the Fermi level of the Au NTs, where the increased charge density improved the probability of charge transfer to a range of molecules and resulted in chemical enhancement in the Raman signal. Furthermore, the signal enhancement was shown to be stable over an hour, and in theory should remain stable over an extended period of time given occasional agitation to keep the hybrid NPs well-dispersed against gravity, due to the intrinsic and persistent nature of the defects in the SnO2 coating (Fig. S19, ESI†).
PIERS enhancement of the hybrid system for explosive detection
We could further boost the Raman signal enhancement of our hybrid system by undertaking a photo-induced enhancement (PIERS) approach. UV irradiation of the SnO2 coating in situ generated additional oxygen vacancies that could further enhance the signal,22,25 enabling nM detection levels.These additional oxygen vacancies were evidenced by further blue-shifting of the LSPR band upon UV irradiation (Fig. S20, ESI†).22
Following success with 4-MBA, a more challenging analyte was targeted in solution, 2,4-dinitrotoluene (DNT), which adsorbs less readily to the hybrid nanomaterial surface, lacking a thiol, amine, or carboxylic acid. DNT was chosen as a model compound for many toxic aromatic environmental pollutants and explosive threats.
Raman spectra of DNT with concentrations ranging from 0 to 50 μM were collected with Au NTs (SERS), Au NT–SnO2-2 (hybrid SERS), and Au NT–SnO2-2 with UV illumination from a 12.5 mW, 365 nm LED (PIERS), as shown in Fig. 4, where a clear further enhancement in the DNT signals is observed after UV irradiation to prime the system. Sample Au NT–SnO2-2 was found to provide the best hybrid enhancement over thinner and thicker coatings (Fig. S21, ESI†). In particular, a significant decrease in signal intensity was observed for the sample with thick SnO2 coating (Au NT–SnO2-3), highlighting the importance of accessible surface on the Au NTs via the voids of the SnO2 coating, and metal–analyte distance especially for analytes with low binding affinity to metal surfaces, such as DNT.45 Meanwhile with this relatively low intensity UV source, it took approximately 1.5 hours of UV illumination for a maximum PIERS intensity to be reached (Fig. S22, ESI†). Notably, the observed trend in PIERS intensity is consistent with that of the blue-shift in LSPR upon UV irradiation (Fig. S20, ESI†), illustrating the correlation between the VO concentration and the PIERS activity, and further confirming the underpinning role of oxygen vacancy in our proposed CE mechanism.
 | ||
| Fig. 4 (a) SERS, hybrid SERS, and PIERS spectra of 50 μM 2,4-DNT. Baseline offset for clarity. (b) Plots showing Raman intensity against concentration of DNT for the 1359 cm−1 peak. Solutions were irradiated for 2 hours with 365 nm UV LED prior to PIERS measurements. Raman peaks of 2,4-DNT marked with * and CTAC with ×. Solvent was deionized water for PIERS and hybrid SERS using Au NT–SnO2-2, and 17 mM aqueous CTAC for SERS using Au NTs alone. | ||
Characteristic Raman peaks of DNT were observed at 889 cm−1 (NO2 out-of-plane bend), 1289 cm−1, and 1359 cm−1 (NO2 stretching mode) (Fig. 4 and Fig. S23, S27, ESI†). The peak at 1640 cm−1 is characteristic of the H–O–H bending mode of water, and the peaks at 1342, 1453, and 1531 cm−1 were due to residual CTAC (Fig. S24, ESI†). There was a large enhancement from Au NT–SnO2, and an additional boost from illumination with a UV LED. The PIERS enhancement can be attributed to the UV irradiation creating additional oxygen vacancies, as previously reported.22,25
The hybrid SERS and PIERS spectra show strong correlation, following approximately a power law between intensity and concentration range for all DNT peaks (R2 between 0.9 and 0.99, Fig. 4(b) and Fig. S23, ESI†).50 The DNT peaks at 889 cm−1 and 1289 cm−1 were not easily visible in the plain SERS spectra, however, with an FFT filter applied to filter out background noise (Fig. S25, ESI†), the 1359 cm−1 peak shows a good correlation with a power law between the intensity and the concentration (R2 = 0.94) (Fig. 4(b)). For 50 μM DNT, using eqn (2), the 1359 cm−1 peaks exhibit a 16 times hybrid SERS enhancement, and a 22 times PIERS enhancement.
The hybrid SERS signals of DNT can be observed at 1 μM, with a signal-to-noise ratio (SNR) of 11.4, implying that the theoretical detection limit was 88 nM. The SNR was calculated by dividing the intensity of the main DNT peak at 1359 cm−1 by the averaged amplitude of the background signal from 1650 to 1850 cm−1. The PIERS signal was visible down to 100 nM, with a signal to noise ratio of 16.7 and thus a theoretical detection limit of 6 nM (1 μg mL−1). According to the Environmental Protection Agency, soil or waste which produce leachate with DNT concentrations of more than 0.13 mg L−1 (714 nM) are classified as hazard waste and require treatment,51 therefore our hybrid SERS system offers a promising tool for environmental detection and monitoring.9,11
Conclusions
In summary, we have achieved aggregation-free multi-mode enhanced Raman scattering via combining colloidal, anisotropic Au NTs and VO-rich SnO2 coating within the same hybrid nanostructure, simultaneously enabling strong electromagnetic enhancement and effective chemical enhancement, respectively. The chemical enhancement has been attributed to the photo-induced charge transfer from the VO inter-band levels to the conduction band within the highly defected SnO2 and subsequently to the Au NTs and finally to the analyte molecules, as evidenced by our UV-vis, XPS and SERS data combined. The resultant SERS signals exhibit both strong intensity and high stability, which have been previously challenging to achieve using colloidal NPs, as aggregation of NPs is commonly required to turn on the SERS activity. Without UV activation, our aggregation-free hybrid SERS system achieved a theoretical detection limit of 88 nM for an explosive marker DNT in water, exhibiting signals 16 times stronger than those of bare Au NTs. Furthermore, the Raman signal can be further boosted by exploiting the PIERS approach which generates additional oxygen vacancies in SnO2 using UV irradiation, to yield a detection limit of 6 nM for DNT, showing promise in wastewater epidemiology and the detection of hazardous substances. Our hybrid SERS approach is generic and should be readily extendable to detect a range of molecular targets and to enhance other SERS systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TCL is grateful to the Research Project Grant (RPG-2016-393) funded by the Leverhulme Trust. WJP thanks the University of Glasgow for a Lord Kelvin Adam Smith Fellowship and the Royal Society for a Research Grant (RGS\R2\192190). GD and TJ would like to thank the EPSRC M3S CDT (EP/L015862/1) for sponsoring their studentship. TJ and TCL acknowledge Camtech Innovations for contribution to TJ's studentship. The authors acknowledge Dr Steve Firth for assistance with TEM and Mr Martin Vickers for XRD support.Notes and references
- J. Langer, D. de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J.-F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J.-M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L.-L. Tay, K. G. Thomas, Z.-Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS.
- E. L. Ru and P. Etchegoin, Principles of Surface-Enhanced Raman Spectroscopy: and Related Plasmonic Effects, Elsevier Science, 2008 Search PubMed.
- Y. Qin, Y. Wu, B. Wang, J. Wang, X. Zong and W. Yao, RSC Adv., 2021, 11, 19813–19818 RSC.
- F. Tian, F. Bonnier, A. Casey, A. E. Shanahan and H. J. Byrne, Anal. Methods, 2014, 6, 9116–9123 RSC.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef CAS.
- A. I. Pérez-Jiménez, D. Lyu, Z. Lu, G. Liu and B. Ren, Chem. Sci., 2020, 11, 4563–4577 RSC.
- W.-I. K. Chio, H. Xie, Y. Zhang, Y. Lan and T.-C. Lee, TrAC, Trends Anal. Chem., 2022, 146, 116485 CrossRef CAS.
- R. A. Halvorson and P. J. Vikesland, Environ. Sci. Technol., 2010, 44, 7749–7755 CrossRef CAS.
- W.-I. K. Chio, W. J. Peveler, K. I. Assaf, S. Moorthy, W. M. Nau, I. P. Parkin, M. Olivo and T.-C. Lee, J. Phys. Chem. C, 2019, 123, 15769–15776 CrossRef CAS.
- S. Ben-Jaber, W. J. Peveler, R. Quesada-Cabrera, C. W. O. Sol, I. Papakonstantinou and I. P. Parkin, Nanoscale, 2017, 9, 16459–16466 RSC.
- W. J. Peveler, S. Ben Jaber, I. P. Parkin, S. Ben Jaber and I. P. Parkin, Forensic Sci., Med., Pathol., 2017, 13, 490–494 CrossRef.
- E. Smith and G. Dent, Modern Raman Spectroscopy: A Practical Approach, 2005 Search PubMed.
- I. Alessandri and J. R. Lombardi, Chem. Rev., 2016, 116, 14921–14981 CrossRef CAS.
- R. Livingstone, L. G. Quagliano, N. Perez-Paz, M. Munoz, M. C. Tamargo, F. Jean-Mary and J. R. Lombardi, in Nanosensing: Materials and Devices II, ed. M. S. Islam and A. K. Dutta, 2005, vol. 6008, p. 60080A Search PubMed.
- Y. Wang, J. Zhang, H. Jia, M. Li, J. Zeng, B. Yang, B. Zhao, W. Xu and J. R. Lombardi, J. Phys. Chem. C, 2008, 112, 996–1000 CrossRef CAS.
- G. Demirel, R. L. M. Gieseking, R. Ozdemir, S. Kahmann, M. A. Loi, G. C. Schatz, A. Facchetti and H. Usta, Nat. Commun., 2019, 10, 5502 CrossRef CAS.
- M. Yilmaz, E. Babur, M. Ozdemir, R. L. Gieseking, Y. Dede, U. Tamer, G. C. Schatz, A. Facchetti, H. Usta and G. Demirel, Nat. Mater., 2017, 16, 918–924 CrossRef CAS.
- X. Hou, X. Luo, X. Fan, Z. Peng and T. Qiu, Phys. Chem. Chem. Phys., 2019, 21, 2611–2618 RSC.
- P. Tarakeshwar, D. Finkelstein-Shapiro, S. J. Hurst, T. Rajh and V. Mujica, J. Phys. Chem. C, 2011, 115, 8994–9004 CrossRef CAS.
- X. Jiang, L. Zhang, T. Wang and Q. Wan, J. Appl. Phys., 2009, 106, 104316 CrossRef.
- S. Cong, Y. Yuan, Z. Chen, J. Hou, M. Yang, Y. Su, Y. Zhang, L. Li, Q. Li, F. Geng and Z. Zhao, Nat. Commun., 2015, 6, 7800 CrossRef CAS.
- S. Ben-Jaber, W. J. Peveler, R. Quesada-Cabrera, E. Cortés, C. Sotelo-Vazquez, N. Abdul-Karim, S. A. Maier and I. P. Parkin, Nat. Commun., 2016, 7, 12189 CrossRef CAS.
- O. E. Dagdeviren, D. Glass, R. Sapienza, E. Cortés, S. A. Maier, I. P. Parkin, P. Grütter and R. Quesada-Cabrera, Nano Lett., 2021, 21, 8348–8354 CrossRef CAS.
- J. Ye, R. Arul, M. K. Nieuwoudt, J. Dong, W. Gao and M. C. Simpson, 2021, preprint, arXiv:2105.14468, https://arxiv.org/abs/2105.14468v1.
- D. Glass, E. Cortés, S. Ben-Jaber, T. Brick, W. J. Peveler, C. S. Blackman, C. R. Howle, R. Quesada-Cabrera, I. P. Parkin and S. A. Maier, Adv. Sci., 2019, 1901841 CrossRef CAS.
- K. Vijayarangamuthu and S. Rath, Int. J. Appl. Ceram. Technol., 2015, 12, 790–794 CrossRef CAS.
- D. Glass, R. Quesada-Cabrera, S. Bardey, P. Promdet, R. Sapienza, V. Keller, S. A. Maier, V. Caps, I. P. Parkin and E. Cortés, ACS Energy Lett., 2021, 6, 4273–4281 CrossRef CAS.
- A. Brognara, B. R. Bricchi, L. William, O. Brinza, M. Konstantakopoulou, A. L. Bassi, M. Ghidelli and N. Lidgi-Guigui, Small, 2022, 2201088 CrossRef CAS PubMed.
- L. Jiang, P. Yin, T. You, H. Wang, X. Lang, L. Guo and S. Yang, ChemPhysChem, 2012, 13, 3932–3936 CrossRef CAS.
- L. Chen, F. Ji, Y. Xu, L. He, Y. Mi, F. Bao, B. Sun, X. Zhang and Q. Zhang, Nano Lett., 2014, 14, 7201–7206 CrossRef CAS.
- G. Oldfield, T. Ung and P. Mulvaney, Adv. Mater., 2000, 12, 1520–1522 CrossRef.
- Y. Yang, Y. Wang and S. Yin, Appl. Surf. Sci., 2017, 420, 399–406 CrossRef CAS.
- P. Mulvaney, J. Pérez-Juste, G. Michael, L. Liz-Marzán and C. Pecharromán, Plasmonics, 2006, 1, 61–66 CrossRef CAS.
- N. Kamarulzaman, N. D. Abdul Aziz, M. F. Kasim, N. F. Chayed, R. H. Yahaya Subban and N. Badar, J. Solid State Chem., 2019, 277, 271–280 CrossRef CAS.
- J. C. Selvakumari, M. Ahila, M. Malligavathy and P. Padiyan, Int. J. Miner., Metall. Mater., 2017, 24, 1043–1051 CrossRef CAS.
- M. Karmaoui, A. B. Jorge, P. F. McMillan, A. E. Aliev, R. C. Pullar, J. A. Labrincha and D. M. Tobaldi, ACS Omega, 2018, 3, 13227–13238 CrossRef CAS.
- V. Kumar, V. Kumar, S. Som, J. Neethling, J. Olivier, M. Lee, O. Ntwaeaborwa and H. Swart, Nanotechnology, 2014, 25, 135701 CrossRef.
- B. Liu, Y. Jiang, Y. Wang, S. Shang, Y. Ni, N. Zhang, M. Cao and C. Hu, Catal. Sci. Technol., 2018, 8, 1094–1103 RSC.
- M. Ye, H. Zhou, T. Zhang, Y. Zhang and Y. Shao, Chem. Eng. J., 2013, 226, 209–216 CrossRef CAS.
- M. Liu, W. Tang, Z. Xie, H. Yu, H. Yin, Y. Xu, S. Zhao and S. Zhou, ACS Catal., 2017, 7, 1583–1591 CrossRef CAS.
- P. De Padova, M. Fanfoni, R. Larciprete, M. Mangiantini, S. Priori and P. Perfetti, Surf. Sci., 1994, 313, 379–391 CrossRef CAS.
- Y. Zhou and S. M. Davis, Catal. Lett., 1992, 15, 51–55 CrossRef CAS.
- X. Tang, X. Fan, L. Yao, G. Li, M. Li, X. Zhao, Q. Hao and T. Qiu, J. Phys. Chem. Lett., 2022, 13, 7816–7823 CrossRef CAS.
- L. Jiang, T. You, P.-G. Yin, Y. Shang, D. Zhang, L. Guo and S. Yang, Nanoscale, 2013, 5, 2784 RSC.
- J. Fernández-Vidal, A. M. Gómez-Marín, L. A. H. Jones, C.-H. Yen, T. D. Veal, V. R. Dhanak, C.-C. Hu and L. J. Hardwick, J. Phys. Chem. C, 2022, 126, 12074–12081 CrossRef.
- Y. Porte, R. Maller, H. Faber, H. N. AlShareef, T. D. Anthopoulos and M. A. McLachlan, J. Mater. Chem. C, 2016, 4, 758–765 RSC.
- J. Buckeridge, C. R. A. Catlow, M. R. Farrow, A. J. Logsdail, D. O. Scanlon, T. W. Keal, P. Sherwood, S. M. Woodley, A. A. Sokol and A. Walsh, Phys. Rev. Mater., 2018, 2, 54604 CrossRef CAS.
- J. A. Marley and R. C. Dockerty, Phys. Rev., 1965, 140, 304–310 CrossRef CAS.
- G. Barbillon, T. Noblet and C. Humbert, Phys. Chem. Chem. Phys., 2020, 22, 21000–21004 RSC.
- W.-I. K. Chio, J. Liu, T. Jones, J. Perumal, U. S. Dinish, I. P. Parkin, M. Olivo and T.-C. Lee, J. Mater. Chem. C, 2021, 9, 12624–12632 RSC.
- EPA, Technical Fact Sheet – Dinitrotoluene (DNT), 2017.
Footnote |
| † Electronic supplementary information (ESI) available: Additional Raman spectra, nanoparticle synthesis and characterisation. See DOI: https://doi.org/10.1039/d2tc05069b |
| This journal is © The Royal Society of Chemistry 2023 |