
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-toxicity chemical solution deposition of ferroelectric Ca:HfO2†
Miguel
Badillo
 ab,
Sepide
Taleb
a,
Taraneh
Mokabber
a,
Jan
Rieck
b,
Rebeca
Castanedo
c,
Gerardo
Torres
c,
Beatriz
Noheda
*b and
Mónica
Acuautla
*a
ab,
Sepide
Taleb
a,
Taraneh
Mokabber
a,
Jan
Rieck
b,
Rebeca
Castanedo
c,
Gerardo
Torres
c,
Beatriz
Noheda
*b and
Mónica
Acuautla
*a
aEngineering and Technology Institute Groningen (ENTEG), Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, Groningen 9747AG, The Netherlands. E-mail: m.i.acuautla.meneses@rug.nl
bZernike Institute for Advanced Materials (ZIAM), Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, Groningen 9747AG, The Netherlands. E-mail: b.noheda@rug.nl
cCentro de Investigación y de Estudios Avanzados del I.P.N. (CINVESTAV), Unidad Querétaro, 76230, Querétaro, Mexico
First published on 7th December 2022
Abstract
So far, a few chemical solution routes for the fabrication of ferroelectric HfO2 films have been reported. Most of them employ precursors, solvents or additives that are considered difficult to handle, unstable, toxic, generally unfriendly with the environment and/or unsuitable for large scale industrial processes. In this work, we present a new effective chemical route for the preparation of ferroelectric doped-HfO2 films. The solution is prepared from simple, stable, and available precursors, handled in an open atmosphere and requires no restrictive processing conditions. We used 5 at% Ca as the dopant of HfO2 to induce a maximum remnant polarization of 9.3 and 11.1 μC cm−2 for 54 and 90 nm thick Ca:HfO2 films, respectively. The current-electric field loops show intense and distinctive ferroelectric switching peaks and the corresponding ferroelectric loops show excellent saturation, which speaks of good device quality with low leakage. Crystallization and the wake-up of ferroelectricity in Ca:HfO2 films were achieved by means of rapid thermal annealing at different temperatures and times in an Ar:O2 atmosphere. In comparison to thin films, thicker ones exhibited the highest remnant polarization at shorter annealing times, thus evidencing the need for precise control of thermal processing. The Ca:HfO2 films with a thickness of 50 nm displayed a good balance between leakage and retention, maintaining the ferroelectric response above 105 cycles at 1 kHz. The developed precursor solution is promising for its use in spray-coating and ink-jet printing techniques.
1 Introduction
Since its first report as a new lead-free ferroelectric material in 2011 by Böske et al.,1 doped-HfO2 (doped-hafnia), as well as its oxide sister ZrO2,2 has been regarded as a very promising functional oxide in a myriad of applications, including FeRAM,3 MEMs,4 NEMs,5 energy harvesting,6etc.7 Under standard conditions, HfO2 crystallizes into a stable monoclinic phase (m-). It goes through tetragonal (t-) and cubic phase (c-) structure transformations, which entails a large volume change, for increasing temperatures.8 While the t- and c-phases share the VIII coordination of the parent fluorite structure, the m-phase is strongly distorted with coordination VII. The reported polar orthorhombic (o-) and rhombohedral (r-) phases are metastable phases that provide bridging structures between the m- and the t-phase with intermediate volume.1,9,10 In order to achieve polar phases, the strategy is to provide stabilization of the tetragonal or cubic phases and then gradually increase the volume to reach the intermediate phases, this while avoiding the significantly more stable m-phase.11,12 It has been demonstrated that this can be achieved by different mechanisms.13One of the mechanisms for the stabilization of the polar phase of hafnia is chemical modification. Several doping elements have been used to stabilize hafnia's ferroelectric orthorhombic and rhombohedral phases. However, it has been experimentally and theoretically demonstrated that bigger ionic-radii elements best stabilize the polar phases in HfO2.12,14,15 Moreover, HfO2 can be mixed in all ratios with ZrO2. It has been found that Hf0.5Zr0.5O2 (HZO) can also attain good ferroelectricity.9,16,17 In 2017, using first-principles computations, Batra et al. concluded that Ca, Sr, Ba, Y, and elements from the lanthanide series (La and Gd), should produce the highest remnant polarization (Pr) values for ferroelectric HfO2.18 Similar theoretical conclusions were reached by other groups.12,19 Nevertheless, to the best of our knowledge, there is only one other experimental report on ferroelectric Ca-doped-HfO2 (Ca:HfO2) by Yifan Yao et al.20 Although the authors report a Pr value of 10.5 μC cm−2 for a 35 nm-thick film with 4.8 mol% Ca-doping, the effect of Ca-doping on the device leakage is unclear.
Ferroelectric HfO2 films with high Pr values have been fabricated by pulsed laser deposition (PLD),9 atomic layer deposition (ALD),21 sputtering deposition,22 and chemical solution deposition (CSD).14 Even though ALD and PLD have achieved the highest Pr values, sputtering and CSD are better at producing thicker ferroelectric HfO2 films, which holds promise for MEM devices.4 There are a few chemical techniques available for the synthesis of HfO2. Sol–gel involves the controlled hydrolysis, in an inert atmosphere, of air-sensitive metal precursors like HfCl4 and Hf-alkoxides.23,24 Compared to other techniques, the control of chemistry in the sol–gel method is exquisite, but counts with the disadvantage of more restrictive synthesis conditions. Moreover, precursors and solvents like HfCl4 and 2-methoxyethanol can release corrosive and toxic by-products. In the method of inorganic decomposition, substances like ammonia, H2O2, and nitric acid are used to produce Hf(NO3)4 in water from inorganic metal precursors.20 The metal nitrate is decomposed at moderate temperatures to deliver HfO2 and nitrogen compounds. Unfortunately, in addition to poor control of chemistry, the reagents and by-products can be highly corrosive to the processing equipment, and the purity of Hf(NO3)4 is hard to control. Another method is metallo-organic decomposition (MOD), where hafnium metallo-organic precursors, commonly hafnium 2,4-pentanedionate (also known as hafnium acetylacetonate or Hf(acac)4) are employed as the metal source.15 Because of full metal saturation of the coordination sphere of hafnium with acetylacetone ligands, Hf(acac)4 is an air-stable, non-hygroscopic, metal β-diketonate.25–27 In the MOD method, the corresponding metal precursor is dissolved by compatible solvents. The homogeneous mixtures are later heated at high temperature to decompose into HfO2 and various organic by-products that depend on the selection of the initial raw materials. Although MOD processes require higher decomposition temperatures, the toxicity and harmfulness of the precursor solution can be controlled by appropriate raw material selection.
In the literature concerning the fabrication of ferroelectric HfO2 films by MOD, the corresponding metal acetylacetonates are dissolved at moderate temperatures (100–160 °C) in either propionic acid4 or, most frequently, mixtures of propionic acid and propionic anhydride.15 Although propionic acid can be considered safe for humans in small quantities, it can be hazardous in the concentrated form. Besides, due to its volatility, propionic acid produces flammable, unpleasant, irritating fumes (CAS: 79-09-4). On the other hand, just as acetic anhydride is, propionic anhydride (CAS: 123-62-6) is a toxic, corrosive, flammable, reactive, and unstable compound that requires special handling conditions (such as avoiding uncontrolled contact with water, alcohols, and amines).28 Thus, the replacement of propionic anhydride with less dangerous precursors would represent a significant improvement that must be addressed. Likewise, limiting the use of propionic acid would also be of great importance for industrial processes. Moreover, mixtures of propionic acid and propionic anhydride typically require heating for dissolution. Nonetheless, the metal precursors might undergo undesired side chemical reactions that could make the process difficult to reproduce.29 Therefore, aiming for lower dissolution temperatures is also recommended. In this context, a simple procedure involving dissolution of Hf(acac)4 in acetic acid under mild conditions was recently reported by Zheng et al.30 However, despite a large Pr for Ce:HfO2, the reported ferroelectric loops are not saturated, thus further optimization might be required.30
Lactic acid (CAS: 50-21-5) is a carboxylic acid structurally similar to propionic acid and acetic acid, but it contains an extra O atom in the form of a –OH group. Due to the presence of a carboxy group, it is expected that lactic acid can coordinate to Hf4+ and displace the acetylacetonate molecules. Also, as is the case for propionic acid, lactic acid is regarded as a safe material for humans, and its associated risks come primarily from its acidity in concentrated form. The human body metabolizes D and L lactic acid, which is thus considered biodegradable. Currently, pure lactic acid is produced mainly by the petrochemical industry, but great strides have been made for its synthesis and separation from biogenic sources. Structurally, the extra –OH group in lactic acid makes it a viscous liquid through hydrogen bonding.31 Therefore, in contrast to propionic acid, lactic acid does not evaporate easily under environmental conditions (low flammability), and neither does it have a strong odor. Unfortunately, the viscous nature of pure lactic acid makes it inappropriate for chemical deposition processes, where rheological control is essential. Nevertheless, lactic acid is compatible, and sometimes even miscible, with other simple polar compounds like water and alcohols, which can be used to reduce its viscosity.31 In the past, we have used mixtures of lactic acid and triethylamine in alcoholic solvents for the effective dissolution of transition metal (Cu and Cd) compounds and ionic salts (SnCl2) at room or mild temperatures.32,33 Therefore, the mixture of additives has been helpful for dissolution of different precursor metal compounds and the corresponding synthesis of metal oxides.
In this paper, we report the use of a greener precursor solution to fabricate ferroelectric doped-HfO2. The new solution is based on Hf(acac)4, isopropanol as the main solvent, and small amounts of lactic acid and triethylamine as stabilizing agents. Moreover, the dissolution happens at temperatures below 100 °C and does not require special synthesis conditions. The precursor solution complies better with the definition of green chemistry in the 2000s by the International Union of Pure and Applied Chemistry (IUPAC): “The invention, design, and application of chemical products and processes to reduce or to eliminate the use and generation of hazardous substances”.34,35 Using spin-coating and rapid thermal annealing (RTA), ferroelectric Ca:HfO2 (5 mol%) metal–insulator–metal (MIM) capacitors were fabricated. We found a strong, saturated ferroelectric response with remnant polarization values of 9 and 11 μC cm−2 for Ca:HfO2 films of 54 and 90 nm, respectively. Due to the flexibility for adjustment of the solution chemistry, our HfO2 precursor solution is well suited for large-scale deposition techniques, such as spray-coating and ink-jet printing.
2 Experimental
2.1 Synthesis and thermal behavior of precursor solution
All metal precursors, solvents, and additives were used without purification steps. All the procedures for the preparation of the precursor solution were performed under normal air conditions. Hafnium 2,4-pentanedionate (Alfa-Aesar, 97%) and calcium 2,4-pentanedionate (Sigma-Aldrich, 99.95%) were weighed, in corresponding proportions, to produce a 5 mol% Ca-doped HfO2 precursor solution (concentration in the final Ca:HfO2 films was estimated to be around 5.86% by X-ray photoelectron spectroscopy analysis). The calcium doping concentration was based on the work of Yao et al.20 Ca(acac)2 was first added to a round flask. Then, measured volumes of isopropanol (IPA, Macron, 99.9%), triethylamine (TEA, Merck, 99%) and lactic acid (LA, Fischer-Scientific, 90%) were added to the flask. Dissolution of Ca(acac)2 took 30 minutes under stirring under room conditions. Hf(acac)4 powder was later added to the mixture. The dissolution took place in a few minutes. The molar ratios of all substances with respect to Hf were 1/4.3/8.4/214 for Hf(acac)4/TEA/LA/IPA, respectively. The estimated concentration of Hf in the solution was 0.056 mol L−1. The mixture was heated between 95–100 °C for 4 hours using a silicon oil bath to ensure complete dissolution of the salts. To avoid loss of solvents, reflux conditions were established with ice-chilled water. The refluxed solution was allowed to cool down to room temperature, and then filtered 2 times with 0.2 μm pore size filters (Whatman). The solution was stored in a tight glass container with a polypropylene cap. If protected from light, the solution remains transparent and colorless for several months. The preparation of the solution is discussed in more detail in Section 3.1.Thermal behavior of the solution was evaluated by thermogravimetric analysis and differential scanning calorimetry (DSC) using a DSC SDT 2960 TA Instrument. In alumina crucibles, samples of 15–20 mg were heated at 10 °C min−1 from room temperature to 800 °C under a dry-air flow of 100 ml min−1. Sapphire was used as a reference.
2.2 Fabrication of Ca:HfO2 films by chemical solution deposition and spin-coating
Ca:HfO2 films were deposited by spin-coating on metalized silicon wafers. The structure of the substrate stack was Pt(100 nm)/Ti(5 nm)/SiO2(95 nm)/Si(100 orientation). Pt and Ti were deposited using an electron-beam evaporation method. The quality of the substrate is crucial for the performance of the films, thus we have included a procedure for wafer metallization, cutting, cleaning, and storage in the ESI.† Ca:HfO2 films were deposited at 4000 rpm for 30 s on 8 mm × 8 mm substrates. Each layer was dried immediately at 150 °C for 5 minutes, followed by pyrolysis at 300 °C for 5 minutes on a hot plate. Before deposition of a new layer, the previous layer was U.V./Ozone treated for 5 minutes (Ossila U.V./Ozone cleaner). The process was repeated 1, 3 or 5 times to produce films of 18, 54 and 90 nm, respectively. The viscosity of lactic acid was responsible for the deposition of relatively thick films in few repetitions.2.3 Crystallization of Ca:HfO2 films by rapid thermal annealing
Multi-stack Ca:HfO2 films were subject to Rapid Thermal Annealing (RTA) at 700 °C for 90 s, 800 °C for 60 s, or 800 °C for 90 s in an Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 atmosphere (1:1) at 0.5 atm. The RTA system uses an arrangement of 12 Phillips 500 W halogen lamps enclosed in two stainless-steel semi-boxes, each one focusing infrared radiation from below and above the samples. Temperature is precisely controlled by carefully tuned Novus 1200 controllers.36–38 The space for samples allows several samples to be heated at once. The annealing program for Ca:HfO2 films consisted of preconditioning at 150 °C for 5 minutes, followed by a heating ramp of 9.2 °C s−1 or 10.8 °C s−1 to 700 °C and 800 °C, respectively. The arrangement of the films during RTA processing is described in Fig. 1. The samples were placed on top of a TiO2-coated graphite plate. Above them, quartz windows are located, leaving free space above the surface of the films. The measuring thermocouple was placed close to the samples. Because of the different mechanisms of heating involved (radiation, conduction, and convection), the films received asymmetrical heating. The goal was to maximize the annealing of the Ca:HfO2 films while limiting the heating of the platinized silicon substrate. The cooling down of the samples happens naturally through heat loss to the environment through the system boundaries. Exemplification of the heating program, as well as the cooling off of the samples is depicted in Fig S1 of the ESI.†
:O2 atmosphere (1:1) at 0.5 atm. The RTA system uses an arrangement of 12 Phillips 500 W halogen lamps enclosed in two stainless-steel semi-boxes, each one focusing infrared radiation from below and above the samples. Temperature is precisely controlled by carefully tuned Novus 1200 controllers.36–38 The space for samples allows several samples to be heated at once. The annealing program for Ca:HfO2 films consisted of preconditioning at 150 °C for 5 minutes, followed by a heating ramp of 9.2 °C s−1 or 10.8 °C s−1 to 700 °C and 800 °C, respectively. The arrangement of the films during RTA processing is described in Fig. 1. The samples were placed on top of a TiO2-coated graphite plate. Above them, quartz windows are located, leaving free space above the surface of the films. The measuring thermocouple was placed close to the samples. Because of the different mechanisms of heating involved (radiation, conduction, and convection), the films received asymmetrical heating. The goal was to maximize the annealing of the Ca:HfO2 films while limiting the heating of the platinized silicon substrate. The cooling down of the samples happens naturally through heat loss to the environment through the system boundaries. Exemplification of the heating program, as well as the cooling off of the samples is depicted in Fig S1 of the ESI.†
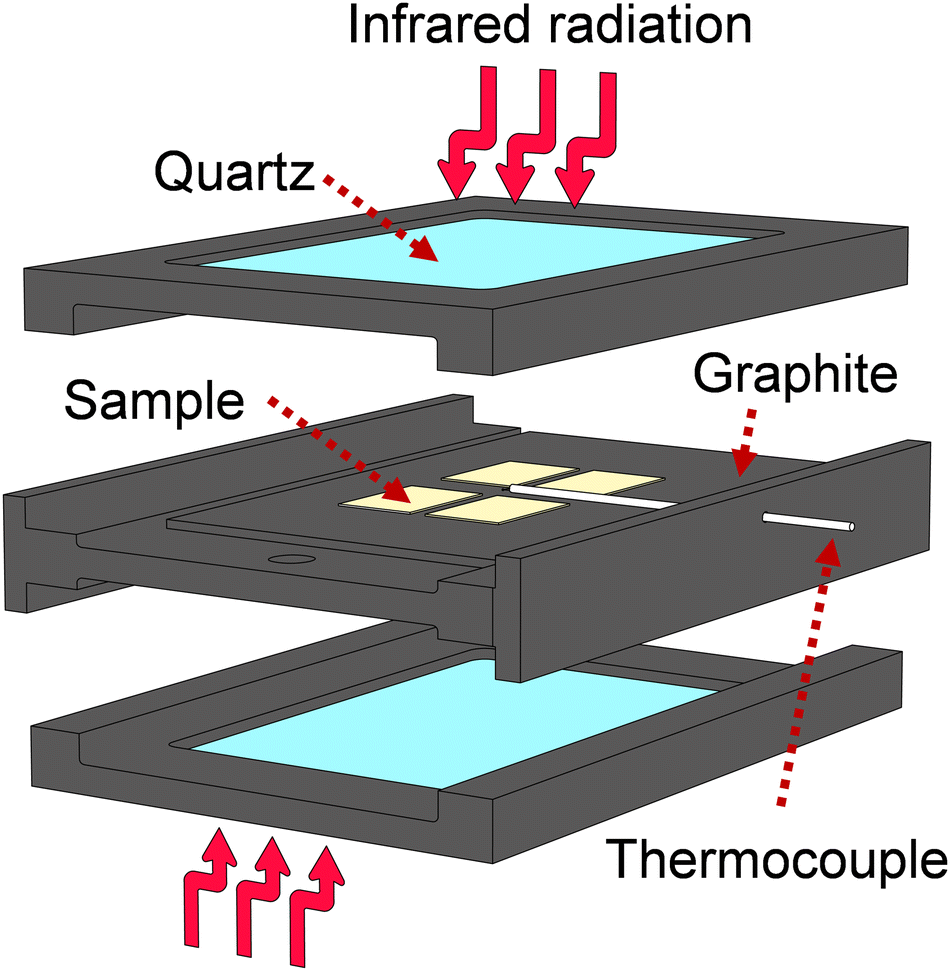 | ||
| Fig. 1 Scheme of samples inside the heating device during rapid thermal annealing. Different heating mechanisms take place due to contact of the samples with graphite (conduction), irradiation with I.R. light (radiation) and exchange of heat with the surrounding gas (convection). | ||
2.4 Characterization of films and MIM devices
X-ray diffraction measurements were performed in a Pananalytical X’Pert Pro MRD in grazing incidence geometry at an angle of θ = 0.55°. The surface topography of the films was evaluated with a Bruker's Dimensions Icon Atomic Force Microscope (AFM). Symmetric, Pt/Ca:HfO2/Pt, metal-insulator-metal (MIM) capacitors were fabricated by U.V. photolithography. Round, top Pt electrodes of 60 nm thickness and 50 μm radius were deposited by e-beam evaporation. Post-annealing of the MIM stack was performed on a hot plate at 100 °C for 5 minutes. A state-of-the-art aixACCT TF Analyzer 2000 was used for ferroelectric testing of the Ca:HfO2 films in the MIM stacks. Ferroelectric wake-up of the capacitor structures was achieved by application of 103 bipolar, rectangular pulses of 2.78 MV cm−1 amplitude at 1 kHz (15 and 25 V for 54 and 94 nm thin films, respectively). Dynamic Hysteresis Measurements (DHM) were performed with bipolar triangular pulses of 2.4 MV cm−1 amplitude at 1 kHz with an average of 3 repetitions. After wake-up conditioning, fatigue measurements at 1, 10, 50, and 100 kHz were made with the same rectangular scheme of 2.78 MV cm−1 pulses. Piezo Force Microscopy (PFM) imaging was performed with an Asylum Research Cypher SPM machine on an 18 nm Ca:HfO2 film with a bottom Pt electrode. A conductive Sb-doped Si tip with Co/Cr coating was used. An up-polarization square of 3.5 × 3.5 μm on an area of 5 × 5 μm was written. A DC bias of +10 V was first applied to the smaller area with a scanning frequency of 0.5 Hz. Then the full PFM image was collected with an AC signal of 1.5 V at a scanning frequency of 1 Hz. Moreover, to evaluate the evolution of wake-up ferroelectricity in Ca:HfO2, local switching spectroscopy (SS) PFM measurements were realized on a pristine point for 10 cycles with an AC signal of 2 V and DC sweep of ±8 V. Then, using the same location, a “PFM fatigue test” was carried out by applying first 2 cycles of DC poling at +10 V and −10 V for 5 seconds each, and repeating the local switching measurements.3 Results and discussion
3.1 Thermal characteristics of the precursor solution and its components
In metallo-organic decomposition (MOD), the final quality of the deposited metal oxide is determined by the thermal behavior of the precursor solution and its components. Therefore, several elements of the fabrication of Ca:HfO2 films were thermally analyzed under air-flow.Because of its good stability in open conditions, hafnium 2,4-pentanedionate (Hf(acac)4) is regularly employed in MOD chemistry to prepare HfO2 films. In Fig. 2(a), the thermogravimetric (TA, blue), differential thermogravimetric (DTA, black), and differential scanning calorimetry (DSC, red) analyses are shown for as-received Hf(acac)4. Initially, Hf(acac)4 is stable, but suffers an endothermic, 35% mass loss starting at 130 °C with maxima at 188 and 217 °C. A second, highly exothermic, significant mass loss is found at 428 °C. At a temperature of 800 °C, the mass of the residues accounts for 37.0% of the initial value. The theoretical yield for HfO2 after the full oxidation of Hf(acac)4 is 35.5% (considering a purity of 97%). Therefore, the observed residues might be composed of HfO2, other impurity oxides and carbonized organic matter. Because of the high yield, the endothermic event at around 200 °C is an organic decomposition step without loss of hafnium metal. In the past, the study of the decomposition of metal acetylacetonates in an N2 atmosphere has demonstrated the production of acetylacetone, acetone, CO2 and even methane as byproducts.25,39,40 On the contrary, it is expected that the highly exothermic event with maximum at 428 °C causes the complete oxidation of the initial compound and that most of the organic matter is finally released as CO2 and H2O.
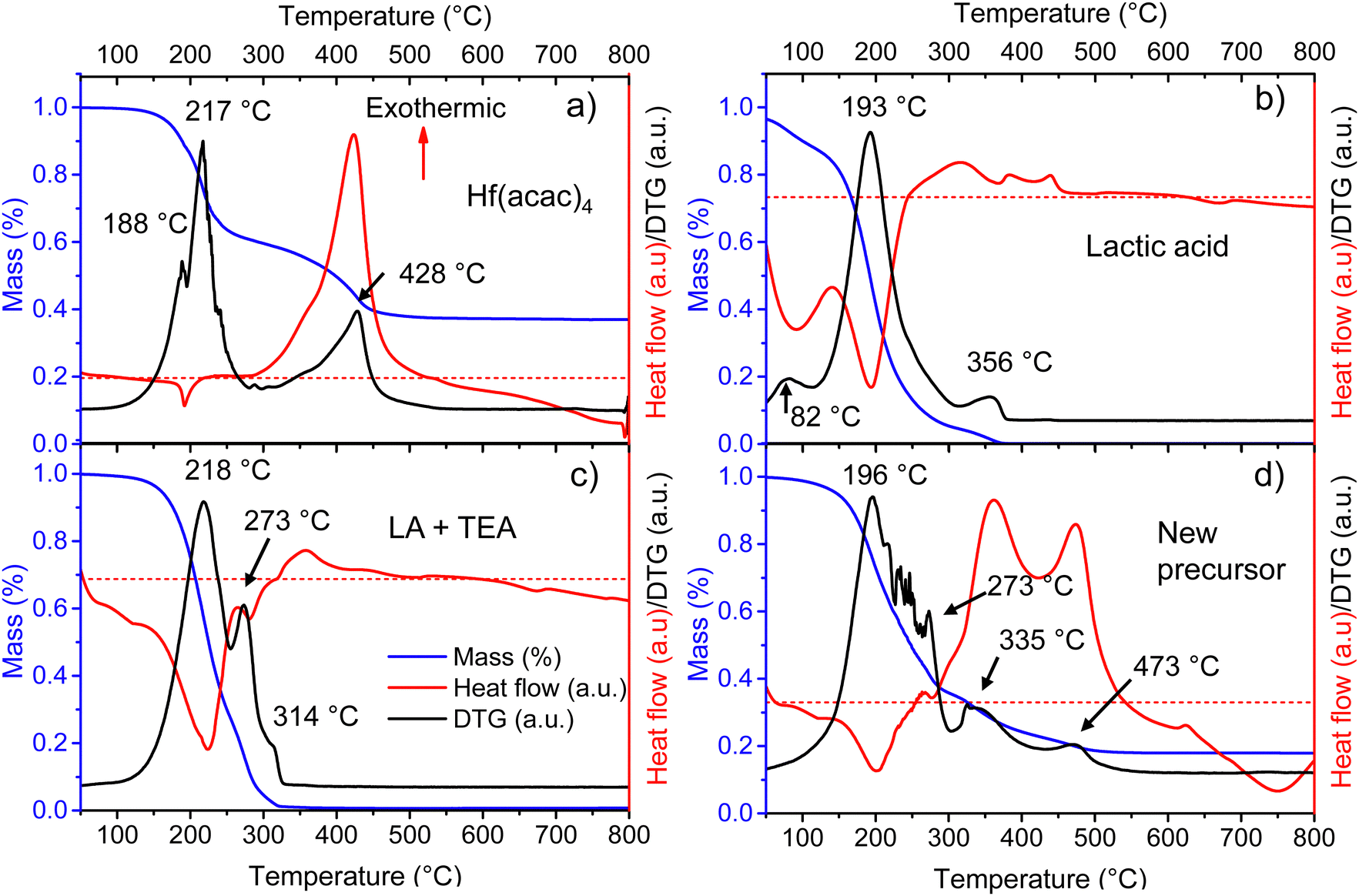 | ||
| Fig. 2 TA(blue)/DTA(black)/DSC(red) curves for (a) as received 2,4-pentanedionate hafnium precursor, (b) as-received lactic acid (90%), (c) evaporated IPA + LA + TEA at 120 °C for 5 hours, and (d) Ca:HfO2 evaporated precursor solution containing Hf(acac)4, Ca(acac)2, IPA, LA and TEA at 80 °C for a couple of hours. All measurements were carried out under an air flow of 100 ml min−1. The red dotted line establishes the zero value for the heat flow measurement. | ||
It is known that Hf4+ and Zr4+ can be stabilized in aqueous media by some carboxylic acids, which act as mono-, bi- or multidentate ligands.41,42 In a few solvents, apart from IPA, we observed that the dissolution of Hf(acac)4 was only achieved after adding lactic acid and triethylamine. While the amount and type of solvent can be varied, the minimum molar ratios needed for the total solubilization of the Hf precursor were 1:4.3:8.4, corresponding to Hf(acac)4/TEA/LA, respectively. This same ratio is also effective for the dissolution of the precursor of the doping metal element in small amounts (Ca(acac)2). The solution was put in reflux at 90–100 °C for 4 hours to ensure the total dissolution of any small salt nuclei. Reduced processing temperature ensures the stability of precursors.
To shed light on the thermal behavior of the new precursor solution, some of its components were analyzed independently. Three endothermic mass loss events were detected for as-received LA at 82, 193, and 356 °C (Fig. 2(b)). The events likely correspond to the evaporation of water and methanol, lactic acid, and lactide, respectively.43 All initial mass is lost after 370 °C. The LA used for this study is a racemic mixture of L and D lactic acid with a purity of 90%, where the remaining components are mainly water, and traces of methanol and lactide. Thermal analysis was also tried for triethylamine, but this was not possible due to its high volatility even at room temperature.
To evaluate the interaction between the organic additives a metal-free precursor solution was obtained by mixing the corresponding amounts of IPA, LA, and TEA, without adding the metal 2,4-pentanedionates. The mixture was then evaporated for a few hours at 120 °C on a hot plate in an open atmosphere to remove IPA. Thermal analysis of the viscous residues is shown in Fig. 2(c). Two endothermic mass loss events can be found at 218 and 273 °C. A third, very small mass loss event can also be detected at 314 °C. It is known that amines (A) and carboxylic acids (C) can form complexes involving 2, 3, or 4 molecules through hydrogen bonding. Depending on the conditions, complexes of A1:C2 are usually more stable. The formation of the complexes increases significantly the boiling points of the mixtures and decreases their volatility.31,44 For this study, the TEA and LA mixture corresponds to a molar ratio of almost 1:2 (A1:C2). In Fig. 2(c), the origin of the two peaks at 218 and 273 °C is most likely related to the separation and evaporation of a lactic acid + triethylamine complex. The mixture did not produce residues above 330 °C.
From a full, Hf and Ca-containing precursor solution, a small volume was separated and allowed to evaporate for a few hours at 120 °C on a hot plate in open conditions. The viscous, homogeneous, and slightly yellowish liquid residue was thermally characterized (Fig. 2(d)). Several endothermic, mass loss events, all related to evaporation, are found between 196 and 273 °C. The first mass loss, occurring at 196 °C is most likely related to the evaporation of LA, as was the case for pure lactic acid (Fig. 2(b)). The second evaporation, at 273 °C, might indicate the presence of an A1:C2 molecular complex as in Fig. 2(c). The other small mass loss events between 196 and 273 °C are probably related to the evaporation of released species from the total or partial replacement of acetylacetone groups surrounding the metal atoms. In Fig. 2(d), the following mass loss events at 335 and 473 °C are highly exothermic and none of them coincide with the exothermic decomposition temperature of pure Hf(acac)4 at 428 °C (Fig. 2(a)). Thus, it might indicate that the LA + TEA modified Hf precursor suffers from decomposition in two distinct steps, with a temperature difference of 133 °C. Due to the instability of nitrogenated organic-hafnium compounds, the first decomposition step at 335 °C could be related to pyrolysis of N-containing species (like products of TEA breakup) and LA.26,27 The second step at 473 °C might be due to total gasification to CO2 and H2O of carbonized oligomers produced by heating. No further mass loss is observed after 570 °C.
3.2 Phase stabilization by thermal processing
In Fig. 3, GIXRD diffractograms for different annealing temperatures at various holding times are shown. Moreover, the film thickness effect was also evaluated. For a Ca:HfO2 film of 54 nm, a temperature of 700 °C for 90 s stabilized the cubic phase of HfO2 (square symbols in Fig. 3(a)). The claim is based on the diffraction powder file shown in Fig. 3(h). However, although we will mostly refer to the cubic phase, it is difficult to establish if the tetragonal (Fig. 3(g)) or orthorhombic phases are present. This is because of the close resemblance of the three phases of HfO2 (all with a fluorite structure) with just slight variations of the crystalline parameters, producing Bragg reflection at very close angles.7 Therefore, most probably our Ca:HfO2 films are composed of a mixture of phases, including an orthorhombic phase. At an annealing temperature of 800 °C held for 60 s (Fig. 3(b)), the 54 nm Ca:HfO2 film crystallized into the cubic phase, but a small signal from the (111) plane at 31.7° of the monoclinic structure was also observed. A further increase of annealing time to 90 s at 800 °C made a few other m-phase reflections visible (Fig. 3(c)).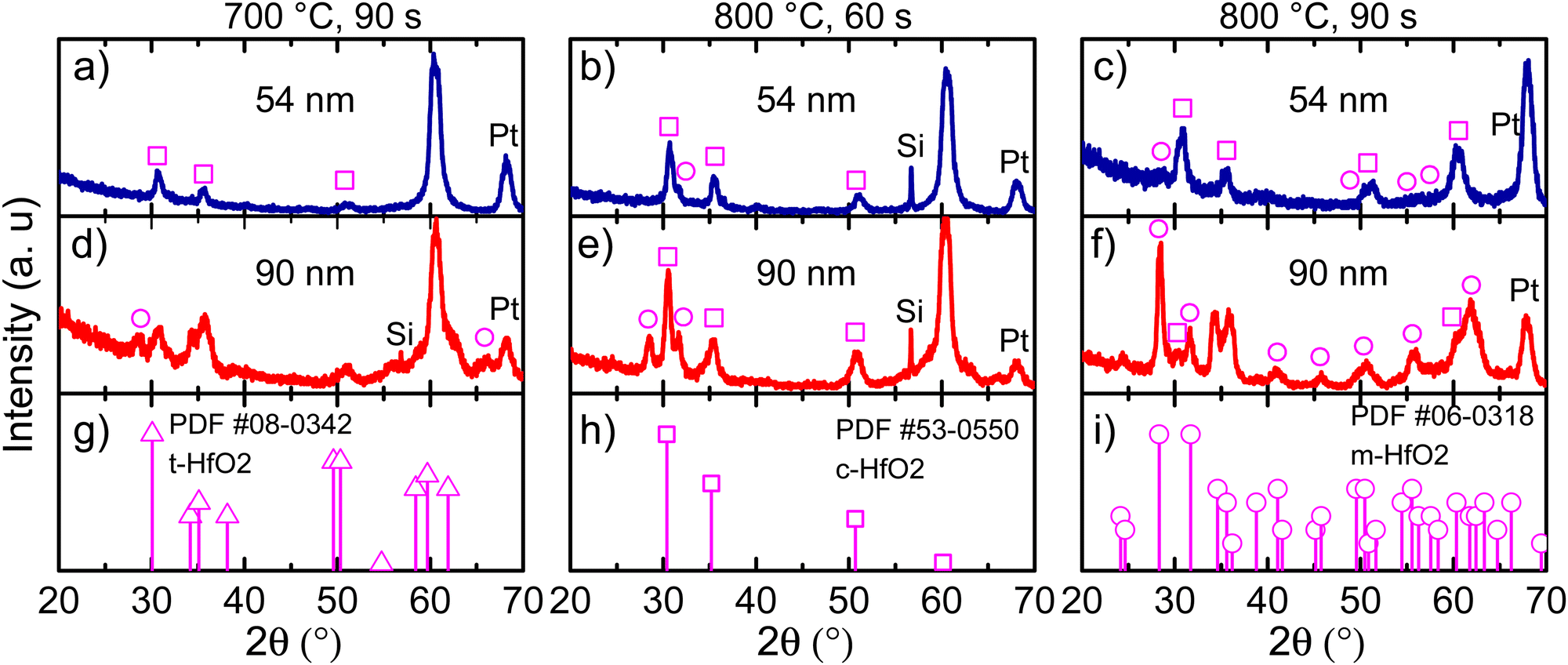 | ||
| Fig. 3 GIXRD data for 54 nm Ca:HfO2 films annealed at (a) 700 °C for 90 s, (b) 800 °C for 60 s, (c) 800 °C for 90 s, and for 90 nm Ca:HfO2 films annealed at (d) 700 °C for 90 s, (e) 800 °C for 60 s, and (f) 800 °C for 90 s. Powder diffraction files for tetragonal (triangles), cubic (squares), and monoclinic (circles) phases are shown in (g), (h) and (i), respectively. | ||
When the same rapid thermal annealing conditions are applied to thicker Ca:HfO2 films of 90 nm (Fig. 3(d)–(f)), the crystallization of the monoclinic phase tends to be favored. As shown in Fig. 3(d), annealing at 700 °C for 90 s produces diffraction patterns of mixed m-phase with c- and/or t-phases. When the temperature is increased to 800 °C and held for 60 s, the presence of a predominantly c-phase with an m-phase as the secondary phase becomes evident (Fig. 3(e)). Finally, for a thick film annealed for 90 s at 800 °C, the m-phase becomes the predominant crystalline structure (Fig. 3(f)). This is consistent with the expectation that a smaller crystalline size can stabilize the fluorite structure with respect to the m-phase.45 Therefore, it is expected that the increase in the annealing temperature or holding time and the augmentation of film thickness have affected the phase transformation in favor of a stable monoclinic structure.46 We have indeed observed this same trend in our work. An estimation of the crystallite size for films of predominantly c-phase produced values of around 8 nm (Scherrer's equation). Usually, the HfO2 crystallization into the ferroelectric orthorhombic phase has been ascribed to a transformation from t- to o-structure either during deposition or electric-field cycling.47 Even so, for HfO2 films obtained by CSD, transformation from c- to o-phase has also been reported.14 It is even possible to partially transform the m-phase to the o-phase under low-oxygen stoichiometric conditions and applied bias, which reduces the energy for transformation to the polar o-phase.10,48
Interestingly, the intensity of the (−111) Bragg reflection at 28.5° of the m-phase appears in different proportion to a powdered material (Fig. 3(i)). Growth in a preferential direction likely causes the effect. Moreover, for both 54 and 90 nm thick films, an intense diffraction plane appears at 60.3° upon annealing conditions of 700 °C for 90 s and 800 °C for 60 s. This angular position corresponds well to the (311) diffraction plane of the c-phase of HfO2, but its intensity is unusual and can also imply a preferential growth direction. No reflections of any other possible structures (given the multi-layer stacking Ca:HfO2/Pt/Ti/SiO2/Si of the films) such as Si, Pt, Ti, TiO2, SiO2 of CaO are expected to occur at this position. Similar observations have been made in a few other reports of HfO2 prepared by chemical solution deposition. In the work of Schenk et al.,49 the crystallization of La:HfO2 with preferred orientation was ascribed to heterogeneous nucleation of the cubic phase within the thickness of each deposited layer. This creates a multi-layer superstructure with density gradients, which helps to stabilize the higher symmetry phases of HfO2. Also, in the work of Starschich, although it was not explicitly mentioned, relatively intense peaks of the cubic phase at 50° (220) and 60° (311) were observed for Y:HfO2.15 From X-ray reflectivity measurements, the density of one of our Ca:HfO2 films annealed at 800 °C for 90 s was estimated to be 8.34 g cm−3. For La:HfO2 films fabricated by CSD, Schenk calculated a density of 8.0 g cm−3, which is only 80% of the expected value (9.6 g cm−3). Given the nature of CSD processes, the lower density is ascribed to porosity in the films caused by voids left during decomposition of organic residues during annealing.4
3.3 Quality of interfaces
For thin films applied in microelectronics, surface and interface morphology control is essential. Although typically Atomic Force Microscopy (AFM) is only useful for surface imaging, it can also provide a glimpse of the dynamics during HfO2 crystallization. As stated in Section 2.3, RTA was applied asymmetrically. The back side of the platinized silicon substrates were put in direct contact with TiO2-coated graphite plates, while the top surfaces were exposed to infrared and visible radiation through a quartz window. The goal of this arrangement was to maximize the heating of the HfO2 films while avoiding any possible change in the morphology of the bottom Pt electrode. The result of this treatment on the surface morphology can be observed in Fig. 4. For Ca:HfO2 films of 54 nm in thickness, the increasingly harsh annealing conditions produced the growth and fusion of grains on the surface. Initially, clear grain boundaries can be observed for the film annealed at 700 °C for 90 s (Fig. 4(a)). An increase in annealing temperature up to 800 °C for 60 s causes growth of the grains through coalescence (Fig. 4(b)). When the holding time is increased to 90 s at 800 °C, the grains are fused, and grain boundaries are less evident (Fig. 4(c)). In addition, very small, rounded particles appear to nucleate on the surface, coinciding with the appearance of the monoclinic phase in GIXRD (Fig. 3(c)). The Root Mean Square (RMS) roughness slightly increases from 0.67 to 0.69 and then to 0.78 nm, respectively. A similar trend is found for 90 nm thick Ca:HfO2 films. Nevertheless, for the thicker films, even from the beginning, the grains seem to be larger, and elongated crystals appear on the surface. For the film annealed at 800 °C for 90 s (Fig. 4(f)) the crystals are not elongated, but rather pyramids with a well-defined orientation. Again, GIXRD data showed the emergence of the monoclinic phase for these annealing conditions. In addition, the surface becomes heavily fused. The roughness of these thicker films increases from 1.2 nm to 1.5 nm, and 2.4 nm, respectively. Furthermore, to have an estimation of grain size behavior, the AFM images were analyzed by the watershed method with the open software Gwyddion.50 Values of 10.7, 12.1 and 14.9 nm were obtained for 54 nm films, and 10.9, 13.8 and 17.0 nm for 90 nm ones, which corresponds to 60 s at 700 °C, 60 s at 800 °C and 90 s at 800 °C for both cases. The trend in grain size clearly shows an increase with annealing conditions. Also, the grain growth rate is faster for the thicker films, which means that as the thickness of Ca:HfO2 films is increased, the control of grain size is restricted to narrower conditions. | ||
| Fig. 4 AFM images for surfaces of 54 nm Ca:HfO2 films annealed at (a) 700 °C for 90 s, (b) 800 °C for 60 s, (c) 800 °C for 90 s, and for 90 nm Ca:HfO2 films annealed at (d) 700 °C for 90 s, (e) 800 °C for 60 s, and (f) 800 °C for 90 s. | ||
Considering the observations made in GIXRD (Fig. 3), the observed nucleation of crystals on the surface, the increase in roughness, and the raise in grain size seem to be related to the nucleation and growth of the m-phase of HfO2. This result is due to the cell volume difference between the m-phase and the higher symmetry phases.20,51 Moreover, the pyramidal crystals pointing out of plane can be a confirmation of the preferential orientation of the crystallites, with the (−111) direction out of plane, as suggested by GIXRD analysis. In the next section we investigate how these differences affect the ferroelectric properties of the different Ca:HfO2 films.
3.4 Ferroelectric wake-up conditioning
Wake-up conditioning has been carried out to evaluate the ferroelectric characteristics of the films. As stated in Section 2.4, rectangular pulses of 2.78 MV cm−1 were applied at 1 kHz for 103–104 times. Top platinum electrodes of 100 μm in diameter were probed. Polarization and current density curves versus electric field are shown in Fig. 5 for Ca:HfO2 films with 54 nm thickness. After wake-up, films annealed at 700 °C for 90 s (Fig. 5(a) and (d)) show the behavior of a dielectric with linear polarization and a rectangular J–E behavior.52,53 However, upon closer evaluation, a small, pinched hysteresis loop can be found after wake-up at 104 cycles. The slight ferroelectric behavior of the film is evidenced better after endurance tests at 105 cycles (Fig. S2 in the ESI†). This observation means that an annealing temperature of 700 °C is enough to cause the nucleation of the stable ferroelectric o-phase. Similar observations have been made for other doped-HfO2 systems.20,51,54 Nonetheless, the ferroelectric wake-up effect becomes more evident for films annealed at 800 °C for 60 s (Fig. 5(b) and (e)). Polarization and current density loops start to be formed after 103 cycles, but a definite ferroelectric behavior, with switching currents peaks, is observed at 104 cycles. At this point, it is worth mentioning that a hysteretic polarization loop is not enough for some materials to prove ferroelectricity. Leakage and parasite currents can cause the appearance of an open loop, which can be interpreted as ferroelectric polarization.55 Likewise, high Pr values can be artificially obtained from leaky capacitors, thus this is not enough to prove ferroelectricity. Therefore, in J–E plots it is important to look for both spontaneous polarization of a ferroelectric material and low leakage of the dielectric. In some cases, high remnant polarization values are being taken as the only indication of the strength of a ferroelectric material. However, Pr values are calculated from integration of transient currents, and artificially high Pr values can easily be obtained from leaky capacitors.52,53,56 At least in the literature regarding doped-HfO2 films obtained by chemical solution methods, Pr values are seemingly often overestimated. We include a brief discussion of this in Section 3 of the ESI.† For Ca:HfO2 films annealed at 800 °C for 90 s (Fig. 5(c) and (d)) and cycled for 103 and 104 times, the ferroelectricity is robust after 103 wake-up cycles. The J–E graph shows strong switching peaks at +1.2 MV cm−1 and −1.4 MV cm−1. Moreover, due to the relatively large thickness of all the films, the current density versus filed characteristics remain rectangular, as expected for a capacitor, without indication of leakage. To the best of our knowledge, apart from the present work, Starschich is the only one who has shown well-saturated polarization loops for doped-HfO2 synthesized by CSD.14,15 | ||
| Fig. 5 Polarization and current density vs. electric field strength for 54 nm Ca:HfO2 films annealed at: (a) and (d) 700 °C for 90 s, (b) and (e) 800 °C for 60 s, and (c) and (f) 800 °C for 90 s. The films received wake-up conditioning at 1 kHz for 0 (pristine), 103, and 104 cycles at 2.78 MV cm−1. | ||
In Table 1, different ferroelectric indicators are shown for the Ca:HfO2 films that exhibited a wake-up effect. For films of 54 nm annealed at 800 °C, an increase in holding time from 60 to 90 s caused the coercive field (Ec+) to shift to higher values, from 0.8 to 1.2 MV cm−1. This was also the case for the remnant polarization (Pr+), which increased from 5.2 to 9.5 μC cm−2, respectively.
| Annealing temperature (°C) | Time (s) | Thickness (nm) | Current peak max (+) (μA cm−2) | Current peak max (−) (μA cm−2) | E c+ (MV cm−1) | E c− (MV cm−1) | P r+ (μC cm−2) | P r− (μC cm−2) |
|---|---|---|---|---|---|---|---|---|
| 800 | 60 | 54 | 157 | 163 | 0.8 | 1.0 | 5.2 | 4.8 |
| 800 | 90 | 54 | 343 | 381 | 1.2 | 1.4 | 9.5 | 9.2 |
| 800 | 60 | 90 | 523 | 559 | 1.1 | 1.4 | 11.1 | 10.5 |
For thicker Ca:HfO2 films of 90 nm, a purely dielectric behavior was found for the films annealed at 700 and 800 °C for 90 s and cycled for 103 and 104 times at 2.78 MV cm−1 (Fig. 6(a), (d), (c) and (f), respectively). However, the sample annealed at 800 °C for 60 s showed a clear ferroelectric behavior with a well-saturated polarization loop and strong switching peaks (Fig. 6(b) and (c)). For this thicker film, the shorter annealing time restricted the growth of the grain size. In Table 1, it is seen that the Ec+ of this film was 1.1 MV cm−1 and it attained a slightly higher Pr+ value of 11.1 μC cm−2 compared to the thinner saturated film (9.5 μC cm−2, annealed for 90 s). The study of these thicker samples can help to highlight the following points: (i) although CSD methods have been more successful in producing thick, ferroelectric, and piezoelectric HfO2 films, a layer-by-layer annealing process has often had to be adopted.49 Our current CSD method could benefit from such an approach but, for the moment, we consider that good results can be obtained with fewer processing steps. (ii) The annealing step is one of the most important steps towards achieving control of ferroelectric HfO2. Wang et al. found that minor changes in RTA processing conditions extensively affect the ferroelectricity of the films.54 A gradual increase in ferroelectricity with increasing both annealing temperature and holding time was found for our Ca:HfO2 films of 54 nm. This is, the film annealed at 700 °C for 90 s did not show significant ferroelectric behavior, but the film annealed at 800 °C for 60 s achieved a Pr+ of 5.2 μC cm−2 and the film heat-treated at 800 °C for 90 s attained a Pr+ of 9.5 μC cm−2. For thicker films (90 nm), ferroelectricity appeared in narrower annealing conditions. In addition, the annealing time needed to achieve ferroelectricity was 30 s shorter for the thicker Ca:HfO2 film than for the thinner one. These effects are due to the propensity of HfO2 to crystallize into the monoclinic phase when its vertical direction growth is not restricted, which favors the growth of the crystallites.49 This is important for piezoelectric applications, where the control of ferroelectricity in thick HfO2 films is a key point.4
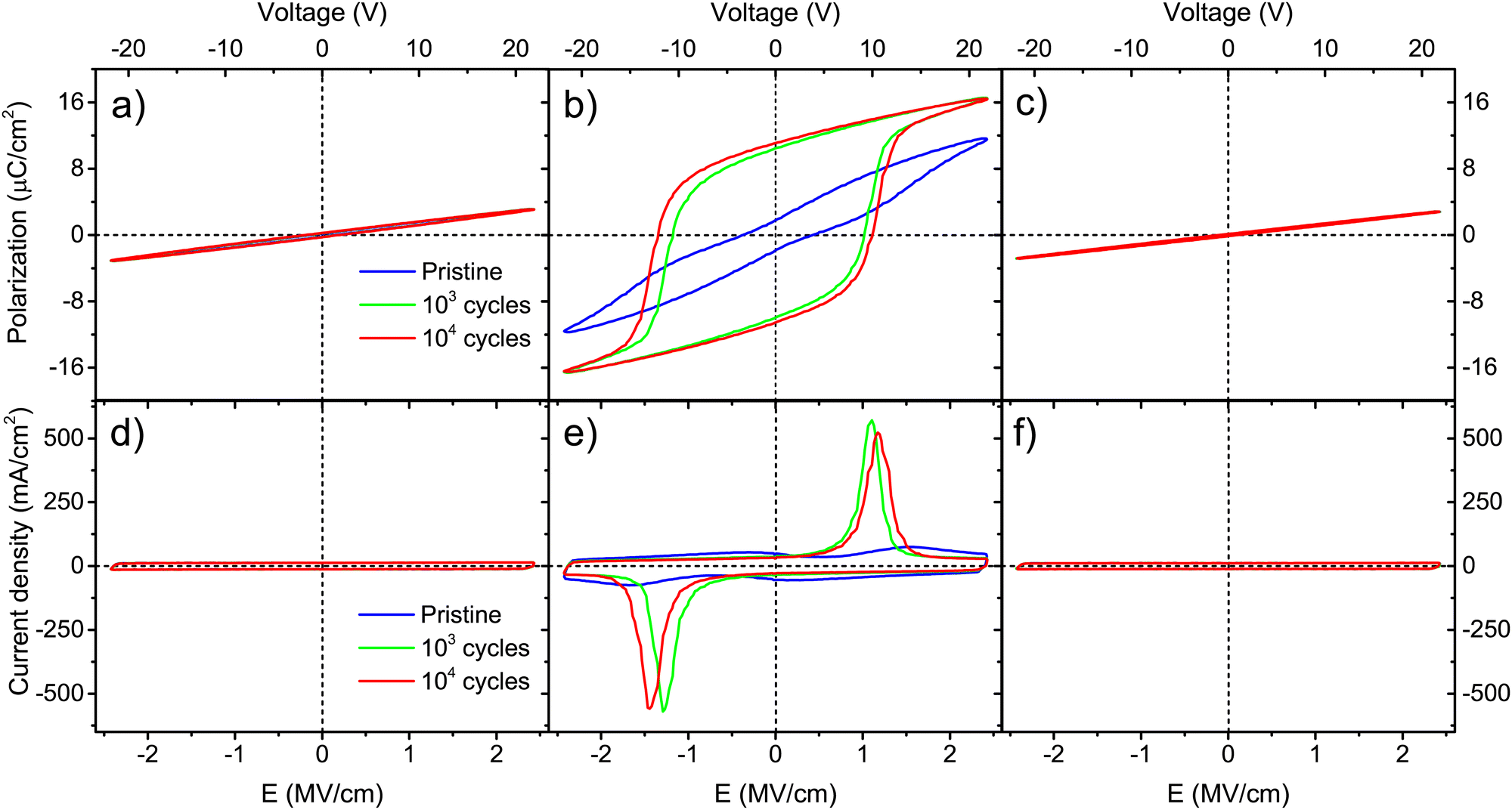 | ||
| Fig. 6 Polarization and current vs. electric field strength for 90 nm Ca:HfO2 films annealed at (a) and (d) 700 °C for 90 s, (b) and (e) 800 °C for 60 s, and (c) and (f) 800 °C for 90 s. The films received wake-up conditioning at 1 kHz for 0 (pristine), 103, and 104 cycles at 2.78 MV cm−1. | ||
The ferroelectricity of the Ca:HfO2 films can be related to the results obtained by GIXRD (Fig. 3) and AFM analysis (Fig. 4). It has been commonly reported that, under certain conditions, the o-phase can be stabilized just before it transforms into the m-phase.54 All 54 nm Ca:HfO2 films crystallized into the c- or t-phase. However, a small increase in m-phase was also observed for higher annealing temperatures and holding times. The m-phase is non-polar and has a lower dielectric constant compared to higher symmetry phases, thus its appearance is regularly avoided. For thicker Ca:HfO2 films, evidence of the m-phase was observed at all annealing conditions, but it became predominant at 800 °C for 90 s. The m-phase presence in AFM images was evidenced by the appearance of pyramid-shaped crystals on the surface of Ca:HfO2 films (Fig. 4(f)). During electrical field cycling, amorphous and crystalline HfO2 is subjected to stress and strain. Due to the presence of dopants stabilizing high symmetry phases, the intermediate ferroelectric orthorhombic phase can be achieved.19 According to Lederer et al., initially amorphous or semicrystalline Hf-based films react differently to the applied field. Semicrystalline films show better ferroelectric properties after wake-up than fully crystallized ones.57 Our Ca:HfO2 films show the behavior of a semicrystalline film. For the ferroelectric film of 90 nm, despite the presence of a small fraction of m-phase, a good, saturated response was achieved. Thus, it seems possible that a higher, tailored Pr value could be obtained at slightly shorter holding times at 800 °C.
To the best of our knowledge, only one other report deals with ferroelectric HfO2 stabilized with calcium doping.20 In their work, Yao et al. stabilized the higher symmetry phases of HfO2 by varying Ca concentrations through a different precursor solution and annealing conditions. They report a maximum Pr value of 10.5 μC cm−2 for a 35 nm Ca:HfO2 film doped with 4.8 mol% Ca and cycled 104 times at 100 Hz. They clearly evidence the wake-up of ferroelectricity in Ca:HfO2. However, the smaller thickness of those films is likely responsible for the lack of saturation of the polarization loops and the larger coercive fields (2 MV cm−1, instead of our maximum 1.4 MV cm−1).
According to theoretical and experimental studies, the polar o-phase of HfO2 can only be stabilized by the presence of oxygen vacancies.58,59 This decreases the energy difference between the more stable m-phase and the higher symmetry phases. When the effect of oxygen vacancies in phase stabilization of HfO2 is studied, the polar o-phase sits in between the m- and t-phase, where m- has the lowest oxygen vacancies concentration.10 Coherently with the latter, it has been reported that the redistribution of defects, particularly oxygen vacancies, by application of wake-up cycling, can help to stabilize the ferroelectric orthorhombic structure through field-induced phase transformation.60 Often, the oxygen vacancies (which in this material are known to be positively charged) are generated near the electrode interfaces, or migrate there upon the application of the electric field. The defects at any location can create pinning of domains, but most commonly they induce a built-in bias field that suppresses the polarization of ferroelectric dipoles. During field cycling, a redistribution or recombination (oxygen vacancies and oxygen interstitials for example) of defects occurs, lowering the built-in field and allowing for the polarization of dipoles to the same direction. This is why pinched hysteresis loops can appear for pristine samples, but upon defect diffusion and recombination they open up to form more typical ferroelectric loops.20,48,51
3.5 Ferroelectricity in thinner Ca:HfO2 layers
One advantage of HfO2-based ferroelectrics is that, contrary to other ferroelectrics, it can sustain a remnant polarization even at small thicknesses. Also, HfO2 exhibits larger coercive fields compared to other ferroelectrics, which means that higher voltages are required to disrupt its polarization. Moreover, hafnia tends to produce a stronger ferroelectric behavior with a decrease in thickness. Indeed, the highest remnant polarization values in the literature have been obtained for films of a few nanometers. Thin films with strong ferroelectricity are of particular interest for high-density FeRAM devices, which are usually obtained by physical deposition methods like PLD and ALD. Nonetheless, such methods are also more complicated than CSD.We evaluated our chemical processing for the deposition of ferroelectric thin films of HfO2. Following the same procedures, we deposited a thin 18 nm Ca:HfO2 layer on platinized silicon and annealed it at 800 °C for 90 s (roughness of 0.56 nm). The top electrodes were also deposited and measured but they were in short circuit. More likely, this issue arises from some contamination during deposition under room conditions. Therefore, we investigated the local ferroelectric properties using instead PFM, which utilizes the electrically conductive tip of an AFM (radius of 10–30 nm) in contact mode as the top electrode. As discussed in the previous section, HfO2 based ferroelectrics often require AC and/or DC conditioning to show ferroelectric behavior. To induce wake-up of the films using PFM equipment, a DC +10 V bias has been applied to the tip while scanning a squared region of 3.5 μm × 3.5 μm at a frequency of 0.5 Hz. Later, a larger area of 5 μm × 5 μm was PFM-scanned with an AC signal of 1.5 V. As presented in Fig. 7, both phase and amplitude images were obtained. Clearly, the phase signal shows the switching of ferroelectric domains with applied DC poling to the 3.5 μm × 3.5 μm area. The brighter color represents polarization out of plane (up), indicating that the original polarization was pointing down. Moreover, the amplitude image measures the piezoelectric response of the films. The areas with polarization pointing up and down are expected to have the same piezoelectric response, while no piezoelectric response is expected at the domain wall.23 This is indeed consistent with the observations.
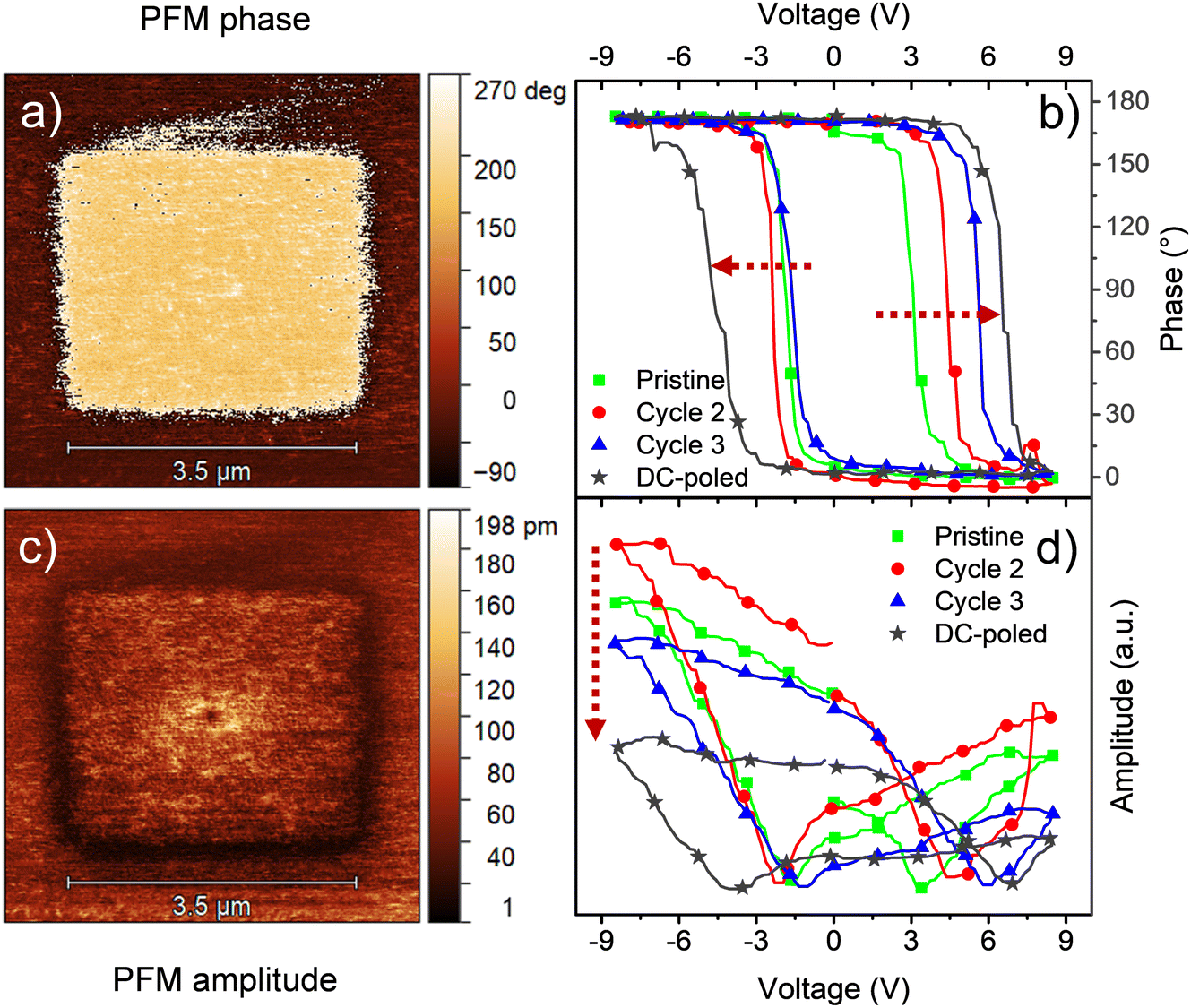 | ||
| Fig. 7 PFM (a) phase and (c) amplitude imaging, and local PFM measurements for (b) phase and (d) amplitude for an 18 nm Ca:HfO2 film annealed at 800 °C for 90 s. | ||
Local switching spectroscopy (SS) measurements were made on a point on the pristine area. During the very first local SS measurement (green lines in Fig. 7(b) and (d)), the phase exhibits a narrow loop with 180° phase difference and the amplitude presents a butterfly loop, as expected for a piezoelectric Ca:HfO2 film. However, the butterfly loop is highly asymmetric, and it does not close after the first cycle. For a second cycle (red line), the phase loop moves to larger coercive voltages and the piezoelectric signal increases but the ends do not meet (first and last measurement points in the loop). These measurements, thus, also show that ferroelectric response of Ca:HfO2 increases with electrical processing, in agreement with the wake-up effect discussed in Section 3.4. For the third SS cycle (blue line), the ends of the amplitude loop meet, but the signal is also reduced. Moreover, the asymmetry is deepened for the positive bias and the coercive voltage also rises. In contrast, a SS loop obtained after DC poling at ±10 V displays a significant increase in the coercive voltage with respect to the non-poled area, but its amplitude diminishes in intensity and becomes very asymmetric (gray line). Thus, the material under the PFM tip can be considered fatigued. The observed asymmetry in the loops is likely related to the difference in work function between the bottom Pt electrode of the sample and the Co/Cr coating of the PFM scanning tip. Nevertheless, other effects such as a built-in field, or interfacial effects as charge accumulation during the SS measurement cannot be disregarded.61
3.6 Fatigue measurements
One of the major challenges for the development of ferroelectric HfO2 is to increase its endurance. Following the same protocol of rectangular pulses for the ferroelectric wake-up of our Ca:HfO2 films, we have performed fatigue measurements at different frequencies for up to 107 cycles with electric fields of 2.78 MV cm−1. As stated in Section 2.4, all Ca:HfO2 micro-capacitors were initially woken-up at a frequency of 1 kHz with application of 103 bipolar cycles. In Fig. 8, endurance results are shown. For both, 54 nm and 90 nm films, we found that pristine Ca:HfO2 capacitors are barely ferroelectric with a small Pr value. However, they soon become ferroelectric after the application of field cycling at 1 kHz 103 times. Upon further cycling at 1 kHz, the Pr value slightly increases. Nonetheless, the Pr peaks at 104 cycles and then the polarization starts to decrease. Remarkably, ferroelectric fatigue (decrease in switching currents) sets in faster for the thicker Ca:HfO2 film (Fig. 8(b)), but the thinner film becomes dielectrically fatigued (increase in leakage) already at around 105 cycles. These effects are more evident when looking at Fig. 9. After 105 cycles, the 54 nm Ca:HfO2 film shows a decrease in Pr due to reduced switching currents. Also, the maximum of the switching currents are displaced to larger Ec values, which can be related to the creation of new defects (mainly oxygen vacancies) that, if sufficiently high, create conductive filaments where current flows more freely.62 In addition, if there is a transformation from the o- to the m-phase, this would create regions of lower dielectric permittivity, thus affecting the distribution of the electric field. Therefore, creation of non-polar dead-layers within the film or at the electrodes, and pinning of domains by defect generation can increase Ec while lowering the Pr.48 At 106 cycles, there is no evidence of switching currents. Instead, the exponential current at high field strength is evidence of dielectric fatigue through a surge in leakage. Although the leakage might look comparatively small, it produces an extrinsic Pr value of 8 μC cm−2, which is an artifact and does not arise from ferroelectric switching (green lines in Fig. 9(a)and (c)). Besides, a “banana” loop appears in the corresponding polarization figure, which is evidence of a leaky dielectric.55 A similar trend is observed for the thicker Ca:HfO2 film of 90 nm (Fig. 9(b) and (d)). Nonetheless, the ferroelectric fatigue is severe even at 105 cycles, for which switching currents are barely noticeable. Further cycling produces the markers of a slightly leaky dielectric, which also causes the polarization loop to look slightly open. The higher resilience of the thicker Ca:HfO2 against leakage is expected due to better shielding of weak/hot spots. In this case, it is easy to distinguish ferroelectric from dielectric fatigue, a distinction that gets increasingly complicated for leaky ferroelectrics, often leading to overestimation of the Pr values (see the discussion in Section 3 of the ESI†). Although in the work of Yao et al. only 35 nm Ca:HfO2 films were tested at a frequency of 10 kHz, and we found a similar endurance behavior for our thin Ca:HfO2 films.20 | ||
| Fig. 8 Fatigue measurements at varying frequencies for Ca:HfO2 films of (a) 54 nm annealed at 800 °C for 90 s, and (b) 90 nm annealed at 800 °C for 60 s. All measured capacitors were woken-up at a frequency of 1 kHz. | ||
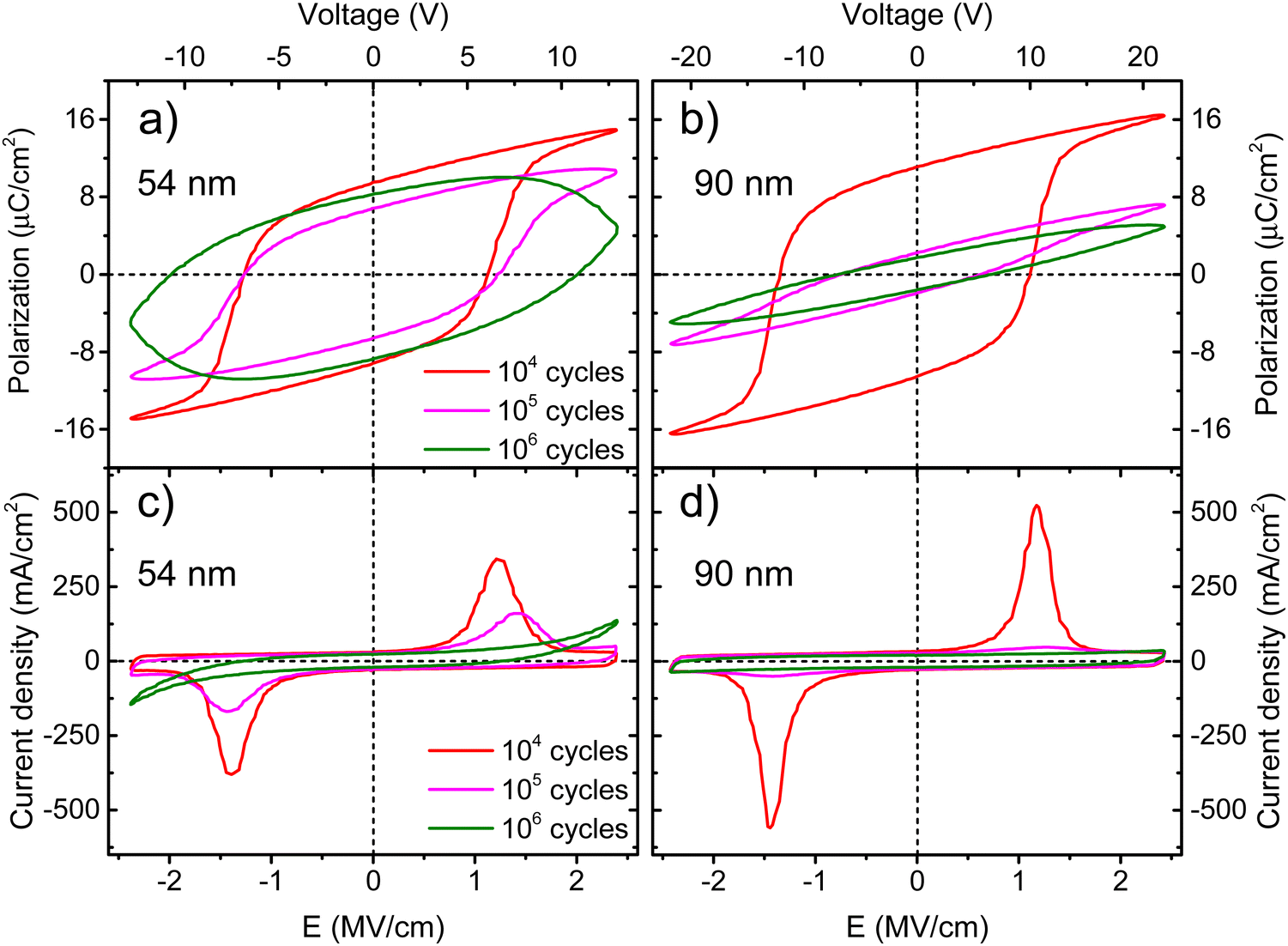 | ||
| Fig. 9 Polarization and current density vs. electric field strength for 54 nm Ca:HfO2 films ((a) and (c)) annealed at 800 °C for 90 s, and 90 nm Ca:HfO2 films ((b) and (d)) annealed at 800 °C for 60 s. The films were fatigued at 1 kHz for an increasing number of cycles at 2.78 MV cm−1. | ||
It is well known in ferroelectric HfO2 literature that endurance depends on the applied frequency, the electric field's strength, and the shape of the pulses.62 Fatigue tests at higher frequencies cause less ferroelectric wear down for the same number of cycles because the material is exposed to high electric fields for shorter periods. Despite this, our Ca:HfO2 films show ferroelectric fatigue at less than 106 cycles for all tested frequencies (Fig. 8). Furthermore, fatigue has been associated with increased leakage, generating new charge defects and pinning of the domain walls.48 The latter holds for our thinner Ca:HfO2 film. However, the thicker film fatigues ferroelectrically faster than the thinner one, as a transformation from the o- to m-phase can take place during field cycling.63 Therefore, if possible, a way to delay the o- to m-phase transition should be found for increasing the endurance of doped-HfO2.
3.7 Implications of the new chemical route for ferroelectric hafnia synthesis
In most reports involving the fabrication of ferroelectric films of HfO2 or ZrO2 by chemical solution deposition, the metal acetylacetonates are dissolved in propionic acid or its mixture with propionic anhydride (the last requiring well-controlled ambient conditions, typically in an N2 glovebox). For such reports, it has not been explained how the dissolution of the original Zr or Hf acetylacetonates takes place, but it is most probably due to the formation of metal propionates and of acetates derived from decomposition of the acetylacetonates at high reflux temperatures.29 The carboxy groups in the carboxylates are coordinated to the metal center.64 The decomposition of such modified precursors proceeds through different mechanisms than those of the pure metal acetylacetonates.29 Depending on the modified precursor, the synthesis of the metal oxides can result in advantages or disadvantages for the final device fabrication and performance.15,65It is known from ALD and chemical vapor deposition (CVD) precursor studies that nitrogen in nitrogenated organic compounds can coordinate directly to the hafnium atom to form volatile substances. Nevertheless, these precursors are unstable and highly prone to hydrolysis,26,27 making them difficult to handle. Instead, some researchers have shown that heteroleptic precursors (consisting of more than one type of ligand bound to the metal atom) can have higher stability than homoleptic ones.26 Therefore, given the thermal behavior of our modified Hf precursor, we might be dealing with a heteroleptic precursor in liquid form. However, this needs further analysis. A deeper understanding could also result in the increase of substances that are capable of dissolving hafnium precursors in less harsh and restricted conditions.
Our results show that the new chemical route offers less toxicity, higher flexibility, and more adaptability, being useful for larger scale deposition techniques, where control of the rheology of solution is mandatory.66 Although triethylamine is toxic compared to the other components of the new precursor solution, it is used in minimal amounts (3.4% by volume). This low content limits the toxicity of the solution as a whole. Besides, less toxic amines or substitutes could be researched to replace triethylamine.41 Furthermore, the new solution possesses higher flexibility because the dissolution of the precursor salt is achieved by additives and not by the solvent. Thus, different solvents can be used. The latter also provides a new chemical route with better adaptability. Viscosity, reactivity, and rheology can be tuned by using different amounts and types of solvents; thus, different fabrication techniques can be applied. Moreover, other additives can be used in the new solution. We have tried this for glycerol, citric acid, nitric acid, and monoethanolamine, which did not affect the solubility of the precursor. Furthermore, it is also essential to use an easy-to-handle, non-toxic, and simple solutions to avoid corrosion, clogging, or damage of the equipment components. The synthesis of ferroelectric hafnia films by ultrasonic spray-coating is in progress.
Finally, future and extensive research can be carried out on the proposed precursor solution by studying different parameters such as drying temperatures,15 heating ramps,54 diverse dopants, layer-by-layer annealing,67 thickness constraint,49 wake-up free synthesis,54,67etc. Therefore, there is plenty of promising research to improve this low-toxicity chemical solution route for ferroelectric HfO2 synthesis.
4 Conclusions
A new precursor chemical solution for the fabrication of ferroelectric doped-HfO2 films, particularly Ca:HfO2, has been introduced in this work. Compared to other routes reported in the literature, our solution has low toxicity. Lactic acid and triethylamine were used as solubilizing additives in small amounts in an isopropanol solvent. However, isopropanol can be interchanged with other alcohols. It was demonstrated upon thermal analysis that the modified precursor does not follow the same decomposition path as the starting metal compound. Therefore, our solution follows a different decomposition route. Moreover, different additives can be used to tailor the properties of the solution and make it suitable for the fabrication of HfO2 films by other, larger scale deposition techniques like spray coating and ink-jet printing. We found that high, saturated ferroelectricity with no indication of high leakage can be induced in Ca:HfO2 (5 at%) by rapid thermal annealing under an Ar:O2 atmosphere (1:1). The temperature and time of annealing are critical in determining the ferroelectric response of HfO2 films. While thin films of 54 nm show an increasingly higher ferroelectric response with increasing temperature and time, thick films (90 nm) exhibit ferroelectric behavior only under a narrow window at a shorter time (30 s less than the thinner film at 800 °C). From GIXRD and AFM measurements, this difference seems to arise from the nucleation and growth of the non-polar monoclinic phase within the orthorhombic phase. In thick films, the monoclinic phase can grow more unrestrictedly. This also affects the difference in ferroelectric endurance between thin and thick Ca:HfO2 films. Although the initial Pr and dielectric endurance of 90 nm Ca:HfO2 films at different frequencies were high (9.5 and 11.1 μC cm−2), they also suffered from loss of the ferroelectric response much faster. Despite the loss of switching currents at above 105 fatigue cycles, it is remarkable that 90 nm Ca:HfO2 films prepared by our accessible chemical method exhibited strong and well saturated ferroelectricity. Our proposed chemical solution route could be effectively used for studies with other dopants and processing conditions. Moreover, it can also be used for fabrication of ZrO2 based films.
Author contributions
Miguel Badillo conceived the idea of low-toxicity HfO2 by CSD, fabricated the films, and coordinated, performed or participated in the measurements and in the analysis of data. Sepide Taleb and Taraneh Mokabber collaborated on the execution of experiments and particularly in ferroelectric testing of devices. Jan Rieck performed the PFM measurements and analysis. Rapid thermal annealing was performed by Prof. Rebeca Castanedo and Prof. Gerardo Torres in Mexico. They adapted their custom-made RTA system for the special requirements of HfO2 processing. Prof. Mónica Acuautla and Prof. Beatriz Noheda provided the materials, infrastructure, lab-space, and scientific guidance during the realization of experiments, measurements, and analysis. All authors participated in the writing and revision of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the start-up grant of Mónica Acuautla of the FSE at the University of Groningen, the Netherlands, and by a postdoctoral fellowship granted to Miguel Badillo (CVU 356403) by The National Council for Science and Technology (CONACYT), México. Jan Rieck acknowledges funding from EU's Horizon 2020, MSCA-ITN-2019 MANIC (grant agreement no. 861153). We want to thank Jacob Baas, Henk Bonder and Joaquín Márquez for their technical support. We also thank Cristian Ferreyra and Francisco Flores for the interesting discussion on electrical properties and PFM measurements. Special thanks are given to Rebeca Torres for the drawing of Fig. 1. Finally, thanks are given for the NanoLab facilities and its staff at the University of Groningen.References
- T. S. Böscke, J. Müller, D. Bräuhaus, U. Schröder and U. Böttger, Appl. Phys. Lett., 2011, 99, 102903 CrossRef.
- S. Starschich and U. Böttger, J. Appl. Phys., 2018, 123, 044101 CrossRef.
- T. Francois, C. Pellissier, S. Slesazeck, V. Havel, C. Richter, A. Makosiej, B. Giraud, E. T. Breyer, M. Materano, P. Chiquet, M. Bocquet, L. Grenouillet, E. Nowak, U. Schroeder, F. Gaillard, J. Coignus, P. Blaise, C. Carabasse, N. Vaxelaire, T. Magis, F. Aussenac and V. Loup, Demonstration of BEOL-compatible ferroelectric Hf0.5Zr0.5O2 scaled FeRAM co-integrated with 130nm CMOS for embedded NVM applications, IEEE, San Francisco, 2019 Search PubMed.
- T. Schenk, N. Godard, A. Mahjoub, S. Girod, A. Matavz, V. Bobnar, E. Defay and S. Glinsek, Phys. Status Solidi RRL, 2020, 14, 1900626 CrossRef CAS.
- S. Kim, J. Y. Park, S. Go, H. S. Kwon, W. Y. Choi and S. Kim, Solid State Electron., 2021, 175, 107956 CrossRef CAS.
- S. Kirbach, K. Kuhnel and W. Weinreich, Piezoelectric Hafnium Oxide Thin Films for Energy-Harvesting Applications, IEEE, Cork, 2018, vol. 2018-July Search PubMed.
- H. Chen, X. Zhou, L. Tang, Y. Chen, H. Luo, X. Yuan, C. R. Bowen and D. Zhang, Appl. Phys. Rev., 2022, 9, 011307 CAS.
- O. Ohtaka, H. Fukui, K. Funakoshi, W. Utsumi, T. Irifune and T. Kikegawa, High Press. Res., 2002, 22, 221–226 CrossRef.
- Y. Wei, P. Nukala, M. Salverda, S. Matzen, H. J. Zhao, J. Momand, A. S. Everhardt, G. Agnus, G. R. Blake, P. Lecoeur, B. J. Kooi, J. Íñiguez, B. Dkhil and B. Noheda, Nat. Mater., 2018, 17, 1095–1100 CrossRef CAS PubMed.
- K. Z. Rushchanskii, S. Blügel and M. Ležaić, Phys. Rev. Lett., 2021, 127, 087602 CrossRef CAS PubMed.
- S. Mueller, J. Mueller, A. Singh, S. Riedel, J. Sundqvist, U. Schroeder and T. Mikolajick, Adv. Funct. Mater., 2012, 22, 2412–2417 CrossRef CAS.
- U. Schroeder, E. Yurchuk, J. Müller, D. Martin, T. Schenk, P. Polakowski, C. Adelmann, M. I. Popovici, S. V. Kalinin and T. Mikolajick, Jpn. J. Appl. Phys., 2014, 53, 08LE02 CrossRef CAS.
- Ferroelectricity in Doped Hafnium Oxide: Materials, Properties and Devices, ed. U. Schröder, C. Seong Hwang and H. Funakubo, Woodhead Publishing, Duxford, 1st edn, 2019 Search PubMed.
- S. Starschich and U. Boettger, J. Mater. Chem. C, 2017, 5, 333–338 RSC.
- S. Starschich, D. Griesche, T. Schneller and U. Böttger, ECS J. Solid State Sci. Technol., 2015, 4, P419–P423 CrossRef CAS.
- T. Shiraishi, K. Katayama, T. Yokouchi, T. Shimizu, T. Oikawa, O. Sakata, H. Uchida, Y. Imai, T. Kiguchi, T. J. Konno and H. Funakubo, Appl. Phys. Lett., 2016, 108, 262904 CrossRef.
- M. Hyuk Park, H. Joon Kim, Y. Jin Kim, T. Moon and C. Seong Hwang, Appl. Phys. Lett., 2014, 104, 072901 CrossRef.
- R. Batra, T. D. Huan, G. A. Rossetti and R. Ramprasad, Chem. Mater., 2017, 29, 9102–9109 CrossRef CAS.
- H. Yang, K. Park, H. J. Lee, J. Jo, H. Park, N. Park, J. Park and J. H. Lee, Inorg. Chem., 2020, 59, 5993–5999 CrossRef CAS PubMed.
- Y. Yao, D. Zhou, S. Li, J. Wang, N. Sun, F. Liu and X. Zhao, J. Appl. Phys., 2019, 126, 154103 CrossRef.
- U. Schroeder, C. Richter, M. H. Park, T. Schenk, M. Pešić, M. Hoffmann, F. P. G. G. Fengler, D. Pohl, B. Rellinghaus, C. Zhou, C.-C. Chung, J. L. Jones and T. Mikolajick, Inorg. Chem., 2018, 57, 2752–2765 CrossRef CAS PubMed.
- T. Mimura, T. Shimizu and H. Funakubo, Appl. Phys. Lett., 2019, 115, 032901 CrossRef.
- S. Kim and J. Hong, Ceram. Int., 2017, 43, S158–S161 CrossRef CAS.
- S. Nakayama, H. Funakubo and H. Uchida, Jpn. J. Appl. Phys., 2018, 57, 4–9 Search PubMed.
- G. V. Bazuev and L. D. Kurbatova, Russ. Chem. Rev., 1993, 62, 981–989 CrossRef.
- T. Blanquart, J. Niinistö, M. Ritala and M. Leskelä, Chem. Vap. Deposition, 2014, 20, 189–208 CrossRef CAS.
- M. G. Walawalkar, A. Kottantharayil and V. R. Rao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2009, 39, 331–340 CrossRef CAS.
- Handbook of Reactive Chemical Hazards, ed. P. G. Urben and M. J. Pitt, Elsevier, Amsterdam, 8th edn, 2017 Search PubMed.
- S. Petit, S. Morlens, Z. Yu, D. Luneau, G. Pilet, J. L. Soubeyroux and P. Odier, Solid State Sci., 2011, 13, 665–670 CrossRef CAS.
- S. Zheng, Z. Zhao, Z. Liu, B. Zeng, L. Yin, Q. Peng, M. Liao and Y. Zhou, Appl. Phys. Lett., 2020, 117, 0–6 Search PubMed.
- A. Kumar, A. Thakur and P. S. Panesar, Rev. Environ. Sci. Bio/Technol., 2019, 18, 823–853 CrossRef.
- C. J. Diliegros Godines, C. G. Torres Castanedo, R. Castanedo Pérez, G. Torres Delgado and O. Zelaya Ángel, Sol. Energy Mater. Sol. Cells, 2014, 128, 150–155 CrossRef CAS.
- M. A. Badillo-Ávila, R. Castanedo-Pérez, G. Torres-Delgado, J. Márquez-Marín and O. Zelaya-Ángel, Mater. Sci. Semicond. Process., 2018, 74, 203–209 CrossRef.
- P. Tundo, P. Anastas, D. S. Black, J. Breen, T. J. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207–1228 CrossRef CAS.
- J. Fu and Y. Wu, Chem. – Eur. J., 2021, 27, 9967–9987 CrossRef CAS PubMed.
- V. D. Moreno-Regino, C. G. Torres-Castanedo, J. S. Arias-Cerón, M. A. Badillo-Ávila, J. Márquez-Marín, R. Castanedo-Pérez, O. Zelaya-Ángel and G. Torres-Delgado, J. Alloys Compd., 2019, 803, 1168–1177 CrossRef CAS.
- B. Franco-Linton, R. Castanedo-Pérez, G. Torres-Delgado, J. Márquez-Marín and O. Zelaya-Ángel, Mater. Sci. Semicond. Process., 2016, 56, 302–306 CrossRef CAS.
- G. Martinez-Saucedo, R. Castanedo-Perez, G. Torres-Delgado, A. Mendoza-Galvan and O. Z. Angel, Mater. Sci. Semicond. Process., 2017, 68, 133–139 CrossRef CAS.
- J. Von Hoene, R. G. Charles and W. M. Hickam, J. Phys. Chem., 1958, 62, 1098–1101 CrossRef CAS.
- H. M. Ismail, Powder Technol., 1995, 85, 253–259 CrossRef CAS.
- T. Qiang, J. Zhao and J. Li, Microporous Mesoporous Mater., 2018, 257, 162–174 CrossRef CAS.
- A. Tonosaki and T. Nishide, Appl. Phys. Express, 2010, 3, 125801 CrossRef.
- A. Komesu, P. F. Martins Martinez, B. H. Lunelli, J. Oliveira, M. R. Wolf Maciel and R. Maciel Filho, J. Chem., 2017, 2017, 1–7 CrossRef.
- N. S. Nhlapo, W. W. Focke and E. Vuorinen, Thermochim. Acta, 2012, 546, 113–119 CrossRef CAS.
- G. Sharma, S. V. Ushakov and A. Navrotsky, J. Am. Ceram. Soc., 2018, 101, 31–35 CrossRef CAS.
- H. Liang, J. Xu, D. Zhou and S. Ren, J. Alloys Compd., 2021, 861, 158241 CrossRef CAS.
- P. Jiang, Q. Luo, X. Xu, T. Gong, P. Yuan, Y. Wang, Z. Gao, W. Wei, L. Tai and H. Lv, Adv. Electron. Mater., 2021, 7, 2000728 CrossRef CAS.
- M. Pešić, F. P. G. Fengler, L. Larcher, A. Padovani, T. Schenk, E. D. Grimley, X. Sang, J. M. LeBeau, S. Slesazeck, U. Schroeder and T. Mikolajick, Adv. Funct. Mater., 2016, 26, 4601–4612 CrossRef.
- T. Schenk, A. Bencan, G. Drazic, O. Condurache, N. Valle, B. El Adib, N. Aruchamy, T. Granzow, E. Defay and S. Glinsek, Appl. Phys. Lett., 2021, 118, 1–16 CrossRef.
- P. Klapetek, D. Nečas and C. Anderson, Gwyddion user guide, https://gwyddion.net.
- Z. Li, D. Zhou, J. Wang, N. Sun and W. Zhang, J. Appl. Phys., 2022, 131, 074101 CrossRef CAS.
- L. Jin, F. Li and S. Zhang, J. Am. Ceram. Soc., 2014, 97, 1–27 CrossRef CAS.
- T. Schenk, E. Yurchuk, S. Mueller, U. Schroeder, S. Starschich, U. Böttger and T. Mikolajick, Appl. Phys. Rev., 2014, 1, 0–14 CAS.
- J. Wang, D. Zhou, W. Dong, Y. Yao, N. Sun, F. Ali, X. Hou and F. Liu, Adv. Electron. Mater., 2021, 7, 2000585 CrossRef CAS.
- J. F. Scott, J. Phys.: Condens. Matter, 2008, 20, 021001 CrossRef.
- T. Schenk, U. Schroeder and T. Mikolajick, IEEE Trans. Ultrason. Eng., 2015, 62, 596–599 Search PubMed.
- M. Lederer, S. Abdulazhanov, R. Olivo, D. Lehninger, T. Kämpfe, K. Seidel and L. M. Eng, Sci. Rep., 2021, 11, 1–7 CrossRef PubMed.
- Y. Zhou, Y. K. Zhang, Q. Yang, J. Jiang, P. Fan, M. Liao and Y. C. Zhou, Comput. Mater. Sci., 2019, 167, 143–150 CrossRef CAS.
- M. Hoffmann, U. Schroeder, T. Schenk, T. Shimizu, H. Funakubo, O. Sakata, D. Pohl, M. Drescher, C. Adelmann, R. Materlik, A. Kersch and T. Mikolajick, J. Appl. Phys., 2015, 118, 072006 CrossRef.
- S. Starschich, S. Menzel and U. Böttger, Appl. Phys. Lett., 2016, 108, 032903 CrossRef.
- L. Collins and U. Celano, ACS Appl. Mater. Interfaces, 2020, 12, 41659–41665 CrossRef CAS PubMed.
- S. Starschich, S. Menzel and U. Böttger, J. Appl. Phys., 2017, 121, 154102 CrossRef.
- S. S. Fields, S. W. Smith, P. J. Ryan, S. T. Jaszewski, I. A. Brummel, A. Salanova, G. Esteves, S. L. Wolfley, M. D. Henry, P. S. Davids and J. F. Ihlefeld, ACS Appl. Mater. Interfaces, 2020, 12, 26577–26585 CrossRef CAS PubMed.
- Z. Czech and M. Wojciechowicz, Eur. Polym. J., 2006, 42, 2153–2160 CrossRef CAS.
- S. Starschich, D. Griesche, T. Schneller, R. Waser and U. Böttger, Appl. Phys. Lett., 2014, 104, 202903 CrossRef.
- N. Godard, M. A. Mahjoub, S. Girod, T. Schenk, S. Glinšek and E. Defay, J. Mater. Chem. C, 2020, 8, 3740–3747 RSC.
- S. Samanta, G. Anoop, H. Joh, W. Seol, S. M. Park, S. Unithrattil, J. Y. Lee, T. Y. Kim, H. Kim, J. Yeom, S. Hong and J. Y. Jo, Adv. Mater. Interfaces, 2021, 8, 2100907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04182k |
| This journal is © The Royal Society of Chemistry 2023 |