
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEXAFS evidence for the spin–phonon coupling in the monoclinic PrNiO3 nickelate perovskite†
J. E.
Rodrigues
 *a,
A. D.
Rosa
a,
J.
López-Sánchez
bc,
E.
Sebastiani-Tofano
bc,
N. M.
Nemes
d,
J. L.
Martínez
b,
J. A.
Alonso
*b and
O.
Mathon
a
*a,
A. D.
Rosa
a,
J.
López-Sánchez
bc,
E.
Sebastiani-Tofano
bc,
N. M.
Nemes
d,
J. L.
Martínez
b,
J. A.
Alonso
*b and
O.
Mathon
a
aEuropean Synchrotron Radiation Facility (ESRF), 71 Avenue des Martyrs, 38000 Grenoble, France. E-mail: rodrigues.joaoelias@gmail.com; rodrigues.joaoelias@esrf.fr
bInstituto de Ciencia de Materiales de Madrid (ICMM), CSIC, Sor Juana Inés de la Cruz 3, E-28049 Madrid, Spain. E-mail: ja.alonso@icmm.csic.es
cSpanish CRG BM25-SpLine at the ESRF, 71 Avenue des Martyrs, 38000 Grenoble, France
dDepartamento Física de Materiales, Universidad Complutense de Madrid, E-28040 Madrid, Spain
First published on 18th November 2022
Abstract
An understanding of the electronic and structural changes across the temperature-induced phase transition in nickelates with a perovskite structure (RNiO3, with R being Y, Tl, rare-earths) is of key importance to shape these materials as devices for industrial applications in several fields, ranging from sensors, catalysts, and non-volatile memory devices. Particularly, PrNiO3 has received special attention because structural, electronic, and magnetic transitions coincide in this compound at temperatures of 125–130 K, which occur under differing pressure and temperature conditions in other nickelates. To draw a refined picture of the origin of these transitions, we investigated the structural changes taking place at a short-range order (local level) in PrNiO3 around the Ni atoms by means of X-ray absorption spectroscopy and at temperatures between 10 and 300 K. Ni K-edge extended X-ray-absorption fine structure (EXAFS) data below TN ≈ 130 K confirm the monoclinic phase (P21/n). At higher temperatures, we observed the convergence of the pair-unit distances Ni–Pr, suggesting the stabilization of the orthorhombic lattice (Pbnm). We derived the Einstein temperatures from the temperature-dependent EXAFS data, which provided an estimate of the Ni–O bond stiffness. We found an anomalous behaviour of the Debye–Waller factors σΓ2 of the Ni–O bond below the structural transition at 130 K. The anomalous temperature evolution of σΓ2 was modelled using a molecular field approximation for the scalar spin correlation function for the pair-bond Ni–O. This model suggests strong spin–phonon coupling and the softening of the lattice vibrations below TN ≈ 130 K in agreement with the magnetic and vibrational properties of this structure. The present results demonstrate that EXAFS is not only a powerful technique for depicting structural changes, but also for exploring the coupling behaviour between the spin configuration and phonons. The present approach provides new opportunities for such types of studies in related materials.
Introduction
Nickelates with a RNiO3 perovskite structure (R = Y, Tl, rare-earth elements) undergo an important insulator–metal transition (IMT), which can be easily tuned by temperature, external pressure, and the R cation radius size.1 The origin and systematics of this transition remain, however, highly debated despite its importance for fine-tuning their properties for industrial applications.2–5 While there is a general consensus that the bandgap closure mechanism involves orbital overlapping between adjacent Ni and O ions, the involvement of the R cation orbitals remains less constrained.6,7 Detailed information on the structural and concomitant electronic changes occurring during this transition is required, but is presently only available for a restricted amount of R cations. In this work, we have investigated the atomic and electronic rearrangements in PrNiO3 as a function of temperature across the structural transition temperature. PrNiO3 represents a special case in the group of nickelates: while it has strongly correlated 4f electrons, its R cation radius is intermediate between small- and medium-sized cations (e.g. R = Y and La). Besides, its structural phase transition, insulator–metal transition (TIM), and antiferromagnetic ordering (TN) temperatures coincide at 125–130 K.8 Therefore, understanding the behaviour of PrNiO3 allows identifying better systematic changes in the properties of nickelates over a wide range of R cation radii.Previous studies have proposed that the properties and the critical temperature of the insulator–metal transition in nickelates are directly affected by the cation size.1 For instance, LaNiO3 is metallic under ambient conditions because of its large R, forming a rhombohedral atomic structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c) that inhibits the insulating and antiferromagnetic state at low temperature. This contrasts with YNiO3 that has an insulating state with a highly distorted monoclinic lattice (P21/n) under ambient conditions due to the smaller cation size.9 Upon heating, YNiO3 metallizes at 600 K and transforms into the orthorhombic phase (Pbnm). In YNiO3, a rhombohedral phase similar to LaNiO3 was never detected. Instead, YNiO3 undergoes an antiferromagnetic ordering transition at around 143 K (TN) with a charge ordering between Ni3+δ and Ni3−δ. The itinerant behavior of the medium-sized rare-earths (R = Nd–Er and Nd0.7La0.3) falls between those of LaNiO3 and YNiO3. These medium-size R cation nickelates also exhibit the antiferromagnetic ordering state at low temperature. Studying the electronic and structural transitions occurring in R = Pr could, therefore, link the properties of the small and medium-size R cation nickelates and may allow establishing a refined picture of the systematic property changes as a function of R cation size.
c) that inhibits the insulating and antiferromagnetic state at low temperature. This contrasts with YNiO3 that has an insulating state with a highly distorted monoclinic lattice (P21/n) under ambient conditions due to the smaller cation size.9 Upon heating, YNiO3 metallizes at 600 K and transforms into the orthorhombic phase (Pbnm). In YNiO3, a rhombohedral phase similar to LaNiO3 was never detected. Instead, YNiO3 undergoes an antiferromagnetic ordering transition at around 143 K (TN) with a charge ordering between Ni3+δ and Ni3−δ. The itinerant behavior of the medium-sized rare-earths (R = Nd–Er and Nd0.7La0.3) falls between those of LaNiO3 and YNiO3. These medium-size R cation nickelates also exhibit the antiferromagnetic ordering state at low temperature. Studying the electronic and structural transitions occurring in R = Pr could, therefore, link the properties of the small and medium-size R cation nickelates and may allow establishing a refined picture of the systematic property changes as a function of R cation size.
So far, only a few studies have evaluated the atomic and electronic changes in PrNiO3 across the IMT at low temperature and ambient pressure and, therefore, large discrepancies remain on the proposed mechanisms. For example, Piamonteze et al. proposed that the insulating state in PrNiO3 is characterized by the local distortions of the [NiO6] octahedra based on one extended X-ray absorption fine structure (EXAFS) datum acquisition at 8 K.10 Acosta-Alejandro et al. suggested that the insulating low temperature state is due to an inhomogeneous local-atomic structure based on the X-ray absorption near edge structure (XANES) data across the structural phase transition.11 Both results were later enlightened by the high-resolution neutron diffraction results, demonstrating the occurrence of the monoclinic phase P21/n below 130 K. The latter confirmed the long-range charge ordering between Ni3+δ and Ni3−δ, as so-called charge disproportionation at the Ni sites.8 The magnetic transition has been so far only studied by neutron diffraction, showing that the magnetic moment abruptly decreases above TN. However, the mechanism has only been investigated in related systems, such as the LaMnO3 manganites using Raman spectroscopy that suggested a potential effect of the spin–phonon coupling.12 In this compound, the IMT is absent leaving the question if the spins couple with the lattice also in the PrNiO3 perovskite.
Despite these achievements, the interplay between the electronic and atomic structural changes behind the insulator–metal transition in PrNiO3 is not fully resolved. Therefore, we monitored the structural and electronic changes across the low temperature-induced structural phase transitions in PrNiO3 using synchrotron X-ray absorption spectroscopy (XAS) at Ni K-edge. Such a technique provides detailed information on both the local-atomic structure and the electronic properties around the absorber atom (Ni).13 In particular, EXAFS provides detailed insights into the local-atomic scale, such as the pair-bond distances, disorder, and coordination numbers around the absorber.14 We performed runs to cover the structural phase transition/insulator–metal transition/magnetic ordering in PrNiO3 under vacuum conditions in the temperature range of 10–300 K. Understating the underlying mechanisms for these transitions has a pivotal consequence for further improvements in devices used in different technological or industrial fields, ranging from sensors, catalysts, non-volatile memory devices. Here, we provide a careful analysis of the local-atomic structure in PrNiO3 at the onset of the aforementioned phase transition. Our results show strong evidence for the spin–phonon coupling and the softening of the lattice vibrations below TN ≈ 130 K.
Methods
Highly polycrystalline PrNiO3 nickelate was synthesized using the liquid-mixture method.15 From a solution with metal nitrates in citric acid, a black powder was obtained after a slow decomposition of the formed resin at 600 °C. A thermal treatment at 1000 °C under 200 bars under an O2 atmosphere was required for the stabilization of the perovskite single phase.The detailed crystal structure under room conditions of the PrNiO3 powder was then determined from synchrotron X-ray diffraction (SXRD) data at the beamline BM25 of the ESRF. For these measurements, we used an incident X-ray beam with a wavelength of λ = 0.4959 Å. The sample was contained in a quartz capillary of 0.5 mm diameter, which was rotating during data acquisition, in the angular 2θ interval between 5 and 45°, with 0.005° steps. A 2D photon-counting X-ray MAXIPIX detector was employed and the data were processed using the BINoculars software.16,17 The diffraction pattern was analysed by the Rietveld method, using the Fullprof refinement program.18 A pseudo-Voigt function generated the profile shape. The refined parameters included unit-cell constants, positional coordinates, and isotropic displacement factors for all the atoms.
The Ni K-edge (at 8.333 keV) XAS run was performed on PrNiO3 aiming to determine the mechanism of the insulator–metal transition/magnetic ordering using EXAFS and was conducted in a temperature interval between 10 and 300 K at the beamline BM23 at the ESRF.19 For this purpose, a compacted pellet made of a mixture between PrNiO3 and cellulose, in appropriate proportions to achieve an edge jump of about 0.6, was prepared. The pellet was placed inside a liquid He cryostat under vacuum (work pressure: 10−7–10−6 mbar). The temperature was monitored using a Pt-based thermocouple placed close to the sample. XAS data at Ni K-edge were acquired using an unfocused beam collimated to 3 × 1 mm2 using slits. Harmonic rejection was achieved by setting two parallel silicon mirrors with an incident angle of 3 mrad. The ionization chambers filled with an appropriate gas mixture were used to determine the photon intensities before and after the sample. A third ionization was placed to collect the photon flux after a Ni foil, which was considered for monochromator angle-to-energy calibration.
Raw XAS data reductions were conducted using ATHENA software for the pre-edge background subtraction, edge jump normalization, and the extraction of the EXAFS oscillations. Then, the EXAFS analyses were performed using ARTEMIS software, based on FEFF's multiple-scattering path expansion.20,21 Based on the standard EXAFS equation, the FEFF program provides the amplitude, phase, and mean free path functions. Fourier-transform (FT) windows in both k- and R-spaces were set to k = 2–13.5 Å−1 and R = 1.1–5.9 Å, leading to 42 independent parameters. Seven single scattering paths (Γk) up to the fourth shell were considered. Fitted parameters encompassed the average distance and the Debye–Waller factor for each path, while the coordination number was kept fixed as extracted from neutron diffraction data to avoid correlation with the Debye–Waller factor. The amplitude reduction factor S02 was fixed to 0.8053, which was obtained from the fit of the reference Ni foil. A total of 21 parameters were used during the fitting procedure, which includes the background coefficients. The average R-factors of the fittings for all temperature points approached the value of 0.058(4), with an average E0 correction of ΔE ≈ −2.5(1.0) eV.
Magnetic measurements between 10 and 300 K were performed using a SQUID magnetometer (MPMS-3) from Quantum Design (San Diego, USA) under an applied magnetic field of 100 Oe. The sample was positioned in a residual vacuum under a He atmosphere (pressure of 10−5 torr) in the temperature range from 10 up to 300 K.
Results and discussion
The results of this paper are presented as follows: first, a brief crystal structure description under room conditions of the as-obtained PrNiO3 sample is provided. Then, a local structure study across the insulator–metal transition temperature (TN ≈ 125 K) is performed using EXAFS data collected in the temperature range of 10–300 K. The results from EXAFS analyses were used to model the Ni–O bond stiffness and the potential of the spin–phonon coupling contributions across the transitions.Crystal structure at room temperature
The diffraction data of the synthetic PrNiO3 sample acquired at room temperature (RT, 295 K) are represented in Fig. 1, together with the Rietveld refinement. They confirmed that the sample is mainly composed of PrNiO3 in the orthorhombic structure, as defined in the space group Pbnm (S.G. #62). In this structure, Pr is located at the low symmetry Wyckoff 4c (x, y, 1/4) sites; Ni atoms occupy a high-symmetry site at 4b (1/2, 0, 0), while two oxygen atoms exist including O1 and O2 that are located at low symmetry sites 4c and 8b (x, y, z), respectively. From the diffraction data, we have detected a minor impurity of NiO (bustenite; a = 4.146 Å) with a weight percentage of 2.46(4)% wt., which was included in the refinement. The refined unit-cell parameters of PrNiO3 under ambient conditions include a = 5.40337(7) Å, b = 5.36198(7) Å, c = 7.6045(1) Å, and V = 220.323(5) Å3. They are comparable with those described in the literature (e.g. a = 5.4161(5) Å, b = 5.3737(5) Å, c = 7.6226(7) Å; V = 221.85 Å3). In Fig. 1a the raw and fitted SXRD data together with the goodness of the fit is shown. In Fig. 1b a schematic view of the crystal structure is presented highlighting the slight tilting of the [NiO6] octahedra network in the orthorhombic structure along the c-axis.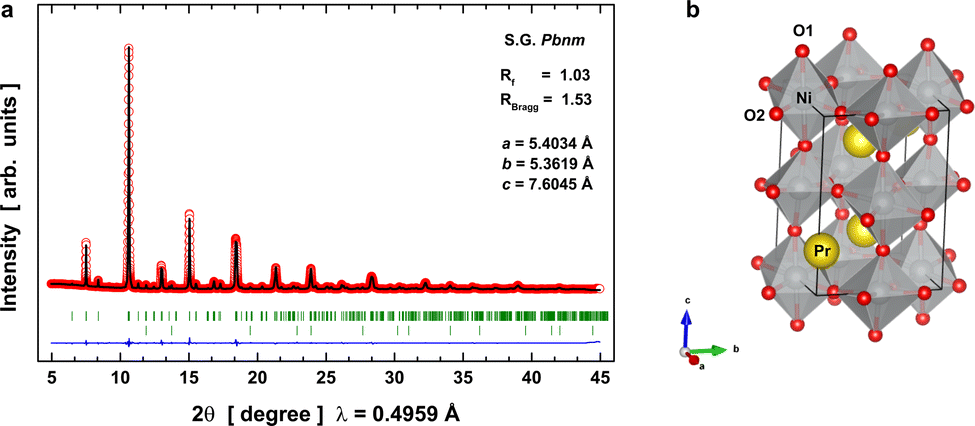 | ||
| Fig. 1 SXRD data refinement and crystal structure of PrNiO3 at room temperature. (a) Rietveld plot: observed (red circles) and calculated (black line) synchrotron X-ray diffraction pattern for PrNiO3 at 295 K shown together with expected peak positions (green line underneath the pattern) and the fit residual (blue line at the bottom). The second series of Bragg ticks correspond to the expected peak positions of NiO, which is present as a minor phase. (b) Crystal structure of orthorhombic PrNiO3: oxygen, nickel, and praseodymium ions are given in red, silver, and yellow, respectively. | ||
Evolution of the local structural environment across the IMT
EXAFS data were acquired up to 16 Å−1 in the k-space and across the structural transition at 130 K using a liquid–helium cryostat. This spectral dataset was used to monitor the local structure around the Ni absorber in PrNiO3 and the temperature evolution of the pair-bond distances (RΓ) and their associated Debye–Waller factors (σΓ2; or mean-square relative displacements, MSRDs) across the structural phase transition from P21/n → Pbnm beyond the Ni–O bonds. In the case of PrNiO3, this structural transition coincides with both insulator–metal and magnetic ordering transitions in the phase diagram.6 For fitting the EXAFS spectra, seven scattering paths were used by setting both k and R ranges to Δk = 11.5 Å−1 and ΔR = 4.8 Å, respectively. Details of the fitted parameters from the EXAFS data of PrNiO3 at 10 K are listed and compared with neutron diffraction data in Table 1, showing a reasonable agreement. In Fig. 2a, the experimental and fitted EXAFS oscillations [χ(k)] at 10 K are shown together with individual oscillations (contributions) from the single scattering paths Γk (k = 1–7). In Fig. 2b, both the modulus and real part of the Fourier-transform (FT) oscillations [χ(R)] are represented.| Attribution | Path | EXAFS at 10 K | Einstein's model | NPD at 10 K | |||
|---|---|---|---|---|---|---|---|
| R Γ [Å] | N Γ | σ Γ 2 [×10−3 Å2] | θ E [K] | σ stat 2 [×10−3 Å2] | d [Å] | ||
| Γ 1 | Nia–O1a | 1.911(2) | 2 | 4.4(3) | 553(2) | 0.1(1) | 1.915–1.921 |
| Γ 2 | Nia–O2a | 1.962(2) | 4 | 4.4(3) | 553(2) | 0.1(1) | 1.961–1.972 |
| Γ 3 | Nia–Pr1 | 3.202(5) | 2 | 3.0(5) | 267(3) | 0.0 | 3.149 |
| Γ 4 | Nia–Pr2 | 3.339(5) | 2 | 3.0(5) | 267(3) | 0.0 | 3.291–3.343 |
| Γ 5 | Nia–Pr3 | 3.369(5) | 2 | 3.4(5) | 278(2) | 1.2(5) | 3.437 |
| Γ 6 | Nia–Ni1 | 3.921(8) | 6 | 8.0(9) | 315(2) | 5.4(5) | 3.806–3.816 |
| Γ 7 | Nia–Ni2 | 5.44(2) | 12 | 4.3(4) | — | — | 5.387–5.411 |
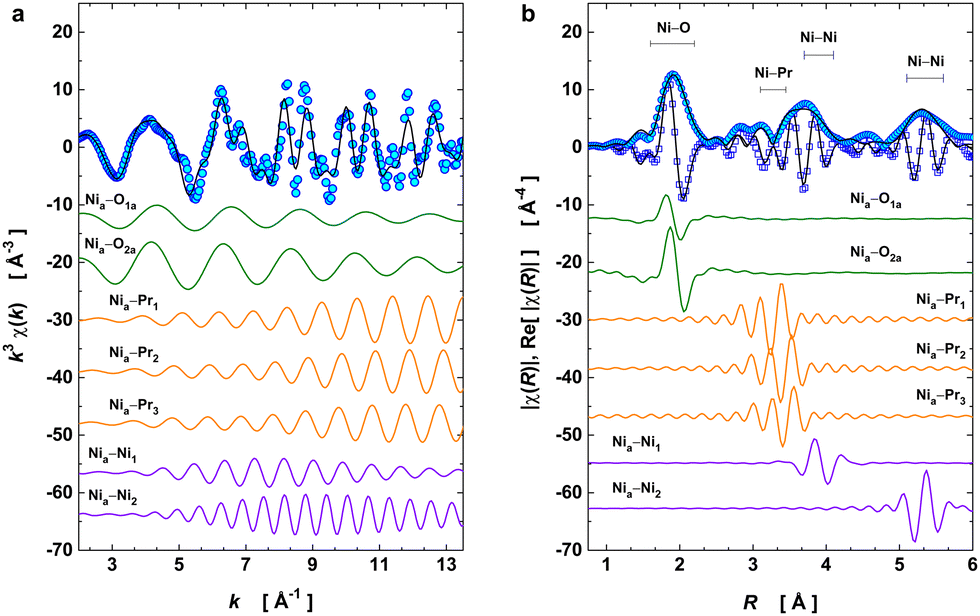 | ||
| Fig. 2 Ni K-edge EXAFS fitting at 10 K. (a) k3-weighted oscillation χ(k). (b) Fourier transform magnitude of k3χ(k) and the real part of χ(R). As shown in figure, the raw EXAFS signal can be reproduced by seven single scattering path contributions, namely: Ni–O (2 Å), Ni–Pr (3.3 Å), Ni–Ni (3.9 Å), and Ni–Ni (5.4 Å). Such a model provides a description of the average octahedral unit [NiO6], the distorted sublattice of Pr cations, and the lattice distortion along the c-axis (pair-unit Nia–Ni1), while Nia–Ni2 has a path encompassing the edges of the first-Brillouin zone. Experimental data points are represented by open symbols, black solid line denotes the best EXAFS fit. Coloured solid lines discriminate the individual single scattering paths which compose the EXAFS oscillations. | ||
To model the EXAFS signal at 10 K, we started from the monoclinic phase (at 10 K) as the input structural model to generate both single and multiple scattering paths using the FEFF code integrated with the ARTEMIS software package. This provided the amplitude, phase, and mean free path functions for each path used as input parameters in the fitting procedure. At 10 K, monoclinic PrNiO3 is characterized by the charge disproportionation between two Ni octahedra, here referred to as Ni1 and Ni2, corresponding to Ni+3+δ and Ni+3−δ, respectively. Each of the octahedra exhibits three sets of Ni–O distances (see Fig. S1 of the ESI†). Averaging these three distances reveals that the [Ni1O6] octahedron is smaller compared to [Ni2O6] by 0.071 Å. The EXAFS technique provides the resolving power to study such differences in distances.
Our FEFF calculations reveal that the most intense first peak at 2 Å in the modulus of the Fourier transform function |χ(R)| results from the contribution of these 6 single-scattering paths (see the ESI† for a detailed explanation). However, including all 6 paths in the model EXAFS model at 10 K (monoclinic phase) is not reasonable, because this would result in overfitting and, therefore, unreliable results. In general, in EXAFS analysis overlapping distances below 0.01–0.02 Å cannot be resolved, which is due to the limited data range in k- and R-spaces.
To provide a reliable model of Ni–O distances, we considered that the peak at 2 Å is composed of only two sets of averaged Ni–O distances, which will be referred to as Nia–O1a and Nia–O2a (subscript “a” denoting “average”). The first average distance was obtained from averaging all paths with RΓ < 1.89 Å, which are the 4 short Ni1–O1,3 (see Fig. S3 of the ESI†). The second average distance was calculated by averaging the remaining 8 long Ni-distances with RΓ > 1.89 Å. To ensure correct weighting of path contributions during fitting, we set the coordination numbers (CN) to 2 and 4 for Nia–O1a and Nia–O2a, respectively.
These structural input models allow studying the local structural variations due to the charge disproportionation in PrNiO3 below 130 K. This approach partially agrees with that used by Piamonteze et al.,10 which in addition included a third scattering path Ni–O with a pair-distance of 1.82 Å. However, this last path seems to be very short for a Ni–O bond distance, not agreeing with the structural data reported by Medarde et al. from neutron diffraction.8 The fitted path length for Nia–O1a reveals a value of 1.911(2) Å, which agrees well with a contribution from the small [Ni1O6] octahedra (Table 1). Finally, we have constrained the paths Nia–O1a and Nia–O2a (referred to as fitting paths Γ1 and Γ2 in Table 1) to have the same Debye–Waller factor, meaning that this parameter describes the average disorder at the level of the [NiO6] octahedron.
The broad peak at the radial interval of 3.2–3.4 Å was partly ascribed to the pair-units interaction between Ni and Pr, comprising three paths here referred to as Γ3, Γ4, and Γ5. These paths describe the sublattice composed by the R-sites (R = Pr). The lengths and their variations of these paths provide information on the octahedral tilting in PrNiO3,22 because the tilting process is strongly dependent on the radius of the R element. For instance, previous studies demonstrated a linear behaviour between the quasi-softmodes encompassing the pair-unit R–Ni and the tilt angle of RNiO3.23 Based on this observation, the temperature evolution of these paths features may be also be used as order parameters for describing structural phase transitions in nickelates. To reduce uncertainties of their fitted parameters, we have included constraints to their Debye–Waller factors following the crystallographic symmetry considerations: the paths Γ3 and Γ4 present the same disorder parameter, while path Γ5 shows an independent disorder exponent for accounting for a fourth pair-unit Ni–Pr that is missing in our model. Attempts to include it in the fitting failed probably due to the high disorder of this scattering path or possible Pr vacancies within the unit cell. Another possibility is that these oscillations are almost out-phase, which hinders a precise characterization of their features.
The contribution of pair-units Ni–Ni at 3.9 and 5.4 Å were included in the fit as paths Γ6 and Γ7, respectively. The path Γ6 is of particular importance because the length of R6 corresponds to half of the lattice parameter c for the orthorhombic lattice Pbnm. Hence, it can provide information about the possible distortions of the lattice and the suppression of the exchange interaction between the Ni magnetic moments within the first-Brillouin zone. For this path, the coordination number was kept equal to 6 with the same Debye–Waller factor. The path Γ7 encompasses a trajectory to the edges of the first-Brillouin zone, with a reasonable intensity in the Fourier-transform oscillations. For such a path, we have considered a coordination number fixed to 12 nickel atoms, with the same disorder parameter in each pair-unit Nia–Ni2.
With the model in Table 1, we have evaluated the local structure in PrNiO3 nickelate across the structural phase transition at 125–130 K that has the insulator–metal transition (TIM) and antiferromagnetic ordering transition (TN) occurring at the same temperature value. For this purpose, XAS spectra of PrNiO3 were recorded from 10 to 300 K with fine temperature steps in the vicinity of the structural transition. Fig. 3 exhibits the k3-weighted EXAFS oscillations k3χ(k) and the moduli of χ(R) and their real parts Re[χ(R)] at selected temperatures.
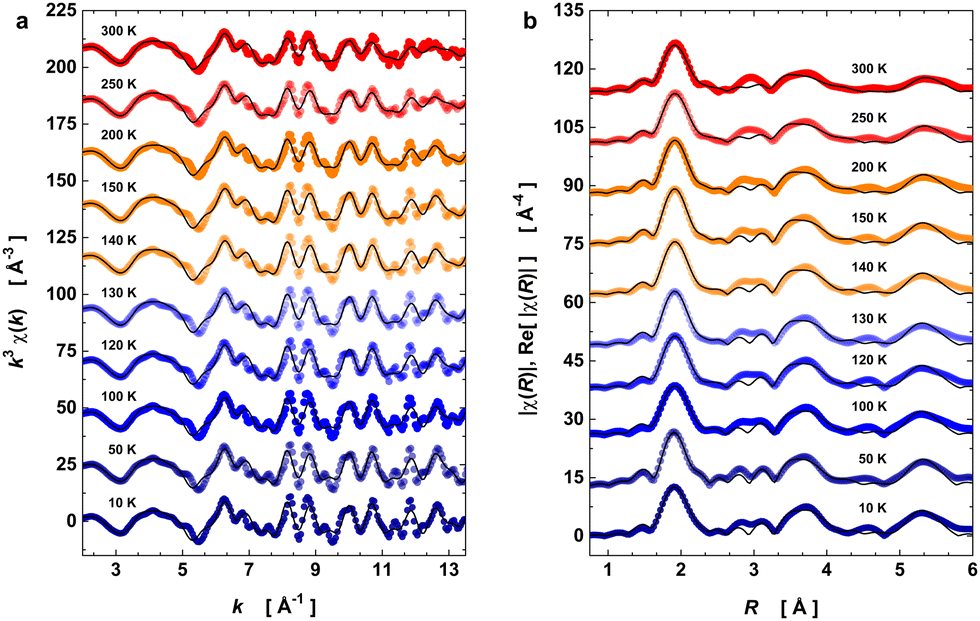 | ||
| Fig. 3 Temperature-dependent EXAFS data at Ni K-edge. (a) k3-weighted EXAFS oscillations in k space. (b) Modulus and real part of the Fourier transform oscillations χ(R) in R space. The open symbols denote the experimental data, whereas solid lines are the best EXAFS fit adjusted. The data acquisition was performed under heating from 10 up to 300 K. The temperature effect on the EXAFS data is visible through the damping of k3χ(k) and the peak broadening of |χ(R)|. | ||
The temperature-dependence of the fitted EXAFS parameters extracted from the best spectral fitting are presented in Fig. 4 together with a schematic representation of the scattering paths used for the EXAFS oscillations. We have seen an anomalous trend for the pair-unit distance (RΓ) and Debye–Waller factor (σΓ2) for almost all the studied paths across the structural transition at 125–130 K (i.e., as a departure from the expected Einstein behaviour in black lines). Our results show a slight change in the slope of the average Ni–O bond distance of the [NiO6] octahedra at 130 K (Fig. 4a) that is concomitant with a drop in the σΓ2 values of the first two paths (Fig. 4b). The fact that distances Nia–O1a and Nia–O2a are almost unaltered across TIM means that the charge disproportionation remains at the local level. At the Ni–Pr sublattice, variations along the RΓ and σΓ2 can also be noticed, as shown in Fig. 4c. The RΓ values of the three paths accounting for the Ni–Pr sublattice showed a pronounced anomaly at the onset of structural transition. Such an evolution is most obvious for the paths Γ4 and Γ5 that approached each other at around 130 K and for temperatures above it. This converges around at the structural transition temperature suggests that the Ni–Pr sublattice becomes more symmetric with partial suppression of the charge ordering between Ni3+δ and Ni3−δ, taking place only in the monoclinic phase P21/n and it is absent in the orthorhombic Pbnm.6,8 The evolution of the Debye–Waller factors with temperature for these three paths Γ3–5 are drawn in Fig. 4d. They exhibited a similar but subtler trend to the one seen for the pair-bonds Ni–O. In summary, this indicates that such a transition is accompanied by significant changes in the crystal structure at the local-scale within the unit-cell (<5 Å).
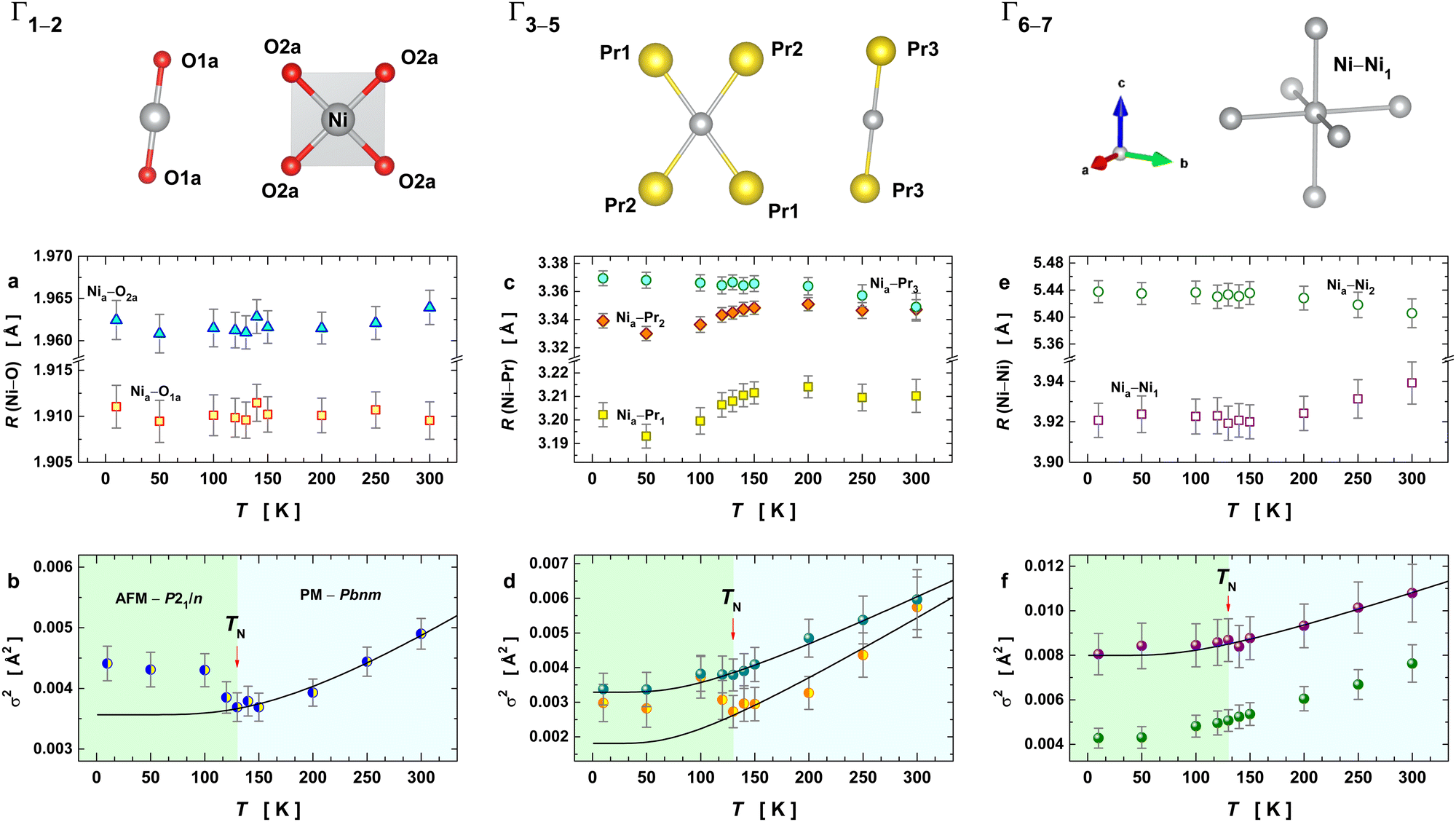 | ||
| Fig. 4 Temperature dependence of the structural parameters extracted from the fits to the EXAFS data. A schematic representation of the single scattering paths corresponding to the data shown below is given on the top of the figure. Symbols in the individual plots represent EXAFS parameters extracted from the best fit together with their respective error bars, while the black solid lines denote Einstein's model fitting for temperatures above 130 K (a) pair-unit distances Nia–O1a and Nia–O2a and (b) their Debye–Waller factors (Γ1 and Γ2 have the same value for DW). (c) Pair-unit distances Nia–Pr1, Nia–Pr2, and Nia–Pr3 and (d) their Debye–Waller factors (Γ3 and Γ4 are described by the same DW, while Γ5 has an independent DW value). (e) Pair-unit distances Nia–Ni1 and Nia–Ni2 and (f) their Debye–Waller factors. | ||
Fig. 4e displays the temperature evolution of the two Ni–Ni cation distances for the paths Γ6,7 (pairs Nia–Ni1 and Nia–Ni2). The RΓ values of Γ6,7 behave almost in an opposite way to each other until 130 K, showing a tendency to converge at higher temperatures. The corresponding Debye–Waller factors and their temperature evolution are depicted in Fig. 4f. They revealed a small anomaly at the structural transition temperature at 130 K. This anomaly lies within the uncertainty of σΓ2, making any conclusion about the variation of this parameter unprecise. However, it is evident that the anomalous behaviour of the disorder parameters is more pronounced when the neighbouring shell is closest to the absorber atom (Ni), the case of the paths Γ1 and Γ2. Therefore, this anomaly has a local origin that encompasses paths up to the second shell (RΓ < 3.4 Å) and it could be associated with the local actions of the spin interaction between Ni and its neighbourhood.
Evaluation of bond disorder and vibrational properties in orthorhombic PrNiO3
We used the temperature-dependence of fitted EXAFS parameters to evaluate the lattice dynamics of PrNiO3. It details the dynamic part of the Debye–Waller factor for the paths Γk (k = 1–6), which was fitted to Einstein's model, as described below: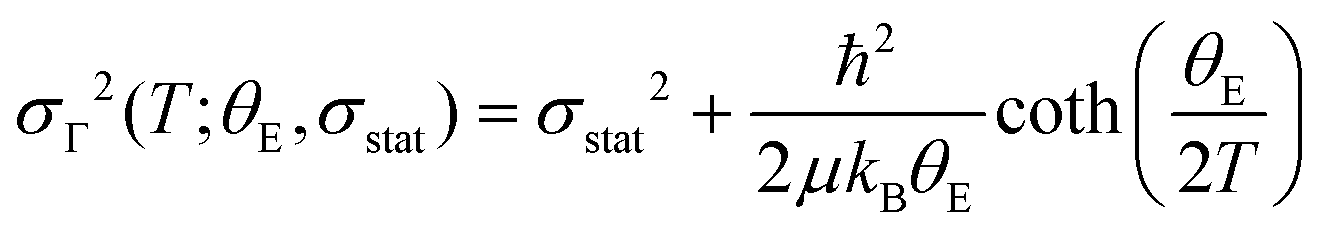 | (1) |
At the onset of the structural phase transition at 130 K and below, a departure from the Einstein-like trend was clearly noticed in Fig. 4b and d for the shells Ni–O (Γ1,2) and Ni–Pr (Γ3,4), respectively. For the paths Ni–Pr (Γ5) and Ni–Ni (Γ6), it is more difficult to say if this trend really takes place below the transition temperature due to the error bars, see Fig. 4d and f, respectively. Therefore, the fitting of σΓ2 using Einstein's model was conducted for temperatures above 130 K and the resulting Einstein temperature of each path is listed in Table 1.
In the following, we compare the extracted Einstein-temperatures for individual paths to known vibrational Raman modes for better understanding the experimental observations. This comparison is based on the nature of the Debye–Waller factor extracted from EXAFS, which represents a mean-square relative atomic displacement along the direction of the scattering path (i.e. parallel), being thus connected to some of the vibrational modes of the system. For the orthorhombic PrNiO3, 24 Raman (ΓRaman = 7A1g ⊕ 7B1g ⊕ 5B2g ⊕ 5B3g) and 25 infrared (ΓInfrared = 7B1u ⊕ 9B2u ⊕ 9B3u) active phonons are predicted, such that most of them appear as accidental degeneracies or with very low intensities.7 However, they are assigned as symmetric and asymmetric stretching modes, bending modes, librations (rigid rotations of [NiO6] units), and translations (Pr movements against the [NiO6] units), appearing at high down to low-wavenumbers, respectively, in both Raman and infrared spectra.6,7,26 A direct correlation of the Debye–Waller factor and the vibrational modes by comparing them with the θE values has to be done with caution, because σΓ2 projects the normal modes along the radial direction (i.e. parallel to the scattering path).
In PrNiO3, the translational Raman modes appear in the range of 100–200 cm−1. The paths Nia–Pr (Γ3–5) have an θE in the range of 185–193 cm−1 and therefore they are likely related to the displacements of Pr against Ni. The path Nia–Ni (Γ6) has a θE of 315(2) K which could be assigned to some extent to the libration and/or bending Raman mode at 219 cm−1. The paths Nia–Oa (Γ1,2) with θE of 553(2) K (385 cm−1) with an intrinsic disorder of 0.1(1) × 10−3 Å2 may be linked to the asymmetric stretching of [NiO6] units. The reasons for that are as follows: (i) the symmetric stretching vibrations of PrNiO3 occur above 400 cm−1 and (ii) the uneven path distances are most likely describing the out-of-phase displacements of apical and equatorial oxygens from the Ni absorber.7
Bond stiffness of Ni–O and Ni–Pr for orthorhombic PrNiO3
Using the Einstein temperature, an estimate of the bond stiffness from the scattering paths (for the shells Ni–O and Ni–Pr) in orthorhombic PrNiO3 can be obtained from a harmonic approximation that defines an average force constant (κE),27,28i.e.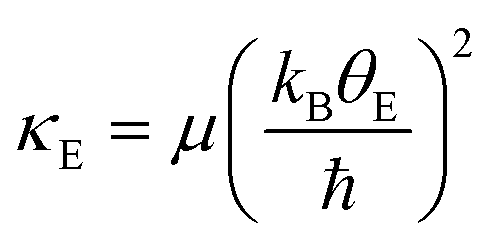 | (2) |
The estimated force constant for the pair-bond Ni–O is around 6.8(4) eV Å−2 (1.1 mdyn Å−1), which agrees with typical force constant calculated for bonds M–X (M = Ni, Co, Mn; X = P, O) in both tetrahedral and octahedral environments.29,30 The estimation of the force constant for the pair-unit Ni–Pr is less reliable, because this is not a chemical bond as in the case of Ni–O. Regardless of that, the average force constant as calculated by eqn (2) for the pair-unit Ni–Pr lies in the range of 5.2–5.7 eV Å−2 (0.8–0.9 mdyn Å−1). This result shows a higher stiffness of the octahedral environment [NiO6] than the sublattice containing Pr, as a result of the strong covalent bonding between Ni and O ions. A more precise evaluation of the Pr sublattice would be performed by collecting EXAFS data at the Pr L3-edge, which is beyond the scope of this work.
Spin–phonon coupling contributions in monoclinic PrNiO3
Similar anomalous temperature-evolutions of the Debye–Waller factor σΓ2 extracted from EXAFS as observed here (i.e., the departure from the Einstein-like trend at 130 K) have been reported in orthoferrite at the temperature induced magnetic ordering transition and at critical temperatures for layered superconductors. DyFeO3 orthoferrite has the same orthorhombic crystal structure (Pbnm) of PrNiO3 under room conditions.31 This orthoferrite is a magnetic system with a spin reorientation transition (SRT) in the range of 50–100 K. Panchwanee et al. reported a slight compression of the bond Fe–O above SRT and an anomaly in σΓ2 similar to that seen for the bond Ni–O in PrNiO3 at magnetic ordering temperature TN ≈ 130 K, the anomaly being absent in the paramagnetic YFeO3 orthoferrite. To our knowledge, both results showing the sensitivity of the EXAFS Debye–Waller factors to magnetic transitions are novel. Other prominent examples are the iron-based superconductors, as the case of Lix(NH3)yFe2Se232 or Ba(Fe1−xCox)2As233 that shows only slight bond-distance changes with temperature for the pair-unit Fe–Fe, but its associated Debye–Waller factor has a sharp and well-defined drop at TC. For the superconductor based on La1.85Sr0.15CuO4, two paths of Cu–O were considered to describe the planar [CuO4] unit, where both bond-distance and Debye–Waller factor depicted anomalies at the onset of the critical temperature.34In contrast to the view EXAFS studies reporting vibrational anomalies at the onset of the magnetic transitions, these anomalies have been widely reported from vibrational spectroscopy, including Raman and infrared techniques.12,35,36 For instance, Granado et al. probed the temperature dependence of the Raman modes around the magnetic ordering temperatures in doped and undoped La1−xMnxO3 perovskite manganites, showing a well-distinguished softening below TN of the stretching mode of the [MnO6] units.12 These results were extended for different members of the manganite family, including RMnO3 (R = Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Y), where the role of the R cation size was established for the phonon softening behaviour: this effect is almost negligible for small R cations.23 Since the Pr atom has an intermediate cation size among the rare-earth elements, the softening was quite evident below TN. Later, this phonon anomaly at the onset of the magnetic ordering temperature was reported in different systems. Rodrigues et al. showed that this phonon softening also occurs in Ising-like spin-glasses (i.e. a system with short-range magnetic order), as the case of Fe2TiO5, using low-temperature Raman data.37 Calder et al. reported a large softening of the symmetric stretching in 5d NaOsO3 osmate, which was the largest shift (≈40 cm−1) already recorded in the literature.38 Radionov et al. detected an anomalous behaviour at the magnetic ordering temperature in the magnetoelectric LiNiPO4 single-crystals using polarized infrared reflectance.39 A recent paper on RNiO3 nickelates (R = Y, Er, Ho, Dy, Sm, Nd) revealed phonon anomalies around both magnetic ordering and insulator-metal transition temperatures by means of Raman spectroscopy.7
In all of the papers mentioned above and others cited elsewhere,40–42 the phonon anomaly is in general explained by considering the contribution of four physical effects: (i) lattice expansion/contraction described by the volume change and Grüneisen parameter; (ii) intrinsic anharmonicity of the phonon mode according to the Balkanski's model;43 (iii) phonon renormalization due to the electronic states at the spin ordering temperature; and (iv) modulation of the exchange integral as a consequence of the spin–phonon coupling. The latter effect may be stronger when the magnetic ordering appears concomitantly with the insulator–metal transition, being the case of both PrNiO3 and NaOsO3. In PrNiO3, one may expect that all four effects could occur, because the structural phase transition, magnetic ordering, and insulator–metal transition take place at T ≈ 130 K.6 In contrast to PrNiO3, systems that exhibit neither concomitant structural transition nor magnetic ordering, only intrinsic anharmonicity is seen by Raman or infrared spectroscopies.43,44 This suggests that phonon anomalies can be modelled as a first-order perturbation from the expected phonon anharmonicity. Based on this assumption and to account for any of the four physical effects mentioned above, one may propose a perturbative term (ΔθE) on the Einstein temperature and a modified equation of the Einstein′s model:
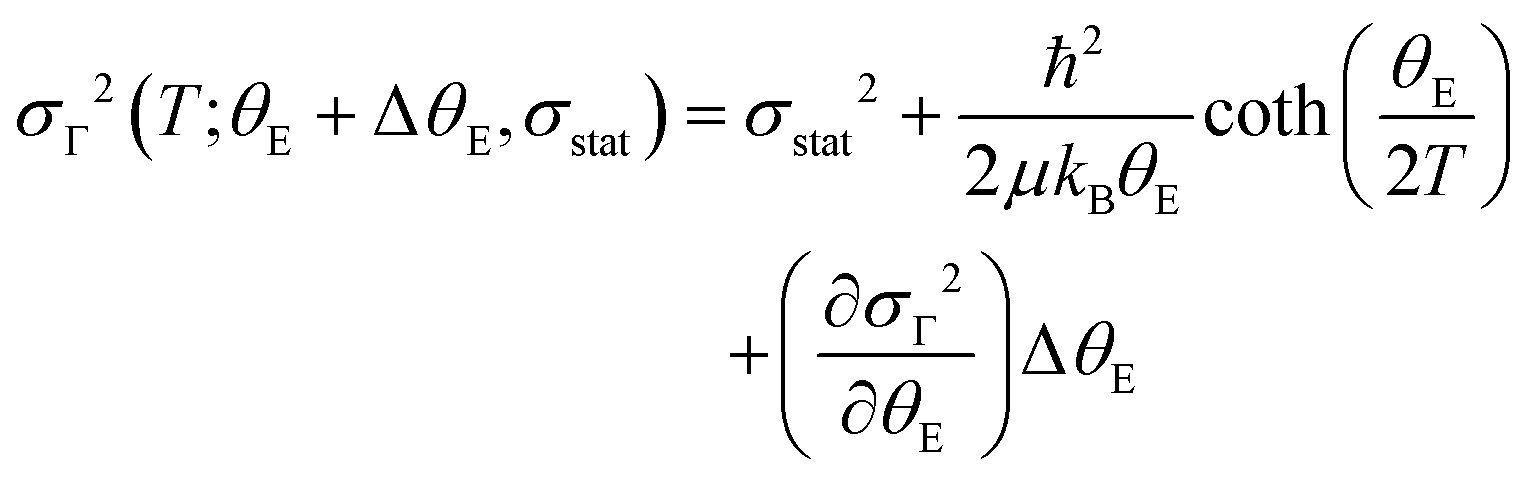 | (3) |
The quantity ΔθE can be extracted from the experimental data in Fig. 4 by estimating the difference σΓ2(T; ΔθE + θE, σstat) − σΓ2(T; θE, σstat) and the first-order derivative the non-perturbated Einstein's model 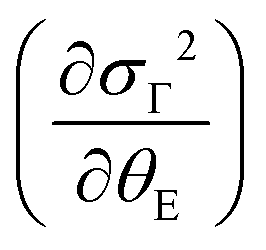 . The former data extraction was performed using a Python-SciPy code using the method interpolate (linear procedure), while the latter depends on the non-perturbated Einstein temperature that is the same value obtained by Einstein's model for T > TN. We have obtained the quantity ΔθE from the Debye–Waller factors only for the shells Nia–Oa (Γ1,2). Some assumptions can be considered, as follows: (i) contributions from the bond-distances Ni–O and, consequently, their anharmonicities were disregarded in the temperature range of 10–200 K, meaning that ΔθE only departs from a constant value of θE = 553(2) K. Indeed, the anharmonic effects take place at temperatures above 200–300 K in most of the known compounds, including oxides; (ii) PrNiO3 seems to present a common mechanism for both TN and TIM, meaning that the renormalization of the electronic states is intimately connected to the modulation of the exchange integral Jij. Then, the perturbation ΔθE may be estimated by the term (ΔθE)sp–ph that is dependent on the scalar spin correlation function 〈
. The former data extraction was performed using a Python-SciPy code using the method interpolate (linear procedure), while the latter depends on the non-perturbated Einstein temperature that is the same value obtained by Einstein's model for T > TN. We have obtained the quantity ΔθE from the Debye–Waller factors only for the shells Nia–Oa (Γ1,2). Some assumptions can be considered, as follows: (i) contributions from the bond-distances Ni–O and, consequently, their anharmonicities were disregarded in the temperature range of 10–200 K, meaning that ΔθE only departs from a constant value of θE = 553(2) K. Indeed, the anharmonic effects take place at temperatures above 200–300 K in most of the known compounds, including oxides; (ii) PrNiO3 seems to present a common mechanism for both TN and TIM, meaning that the renormalization of the electronic states is intimately connected to the modulation of the exchange integral Jij. Then, the perturbation ΔθE may be estimated by the term (ΔθE)sp–ph that is dependent on the scalar spin correlation function 〈![[S with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0053_20d1.gif) i·j〉, i.e.
i·j〉, i.e.
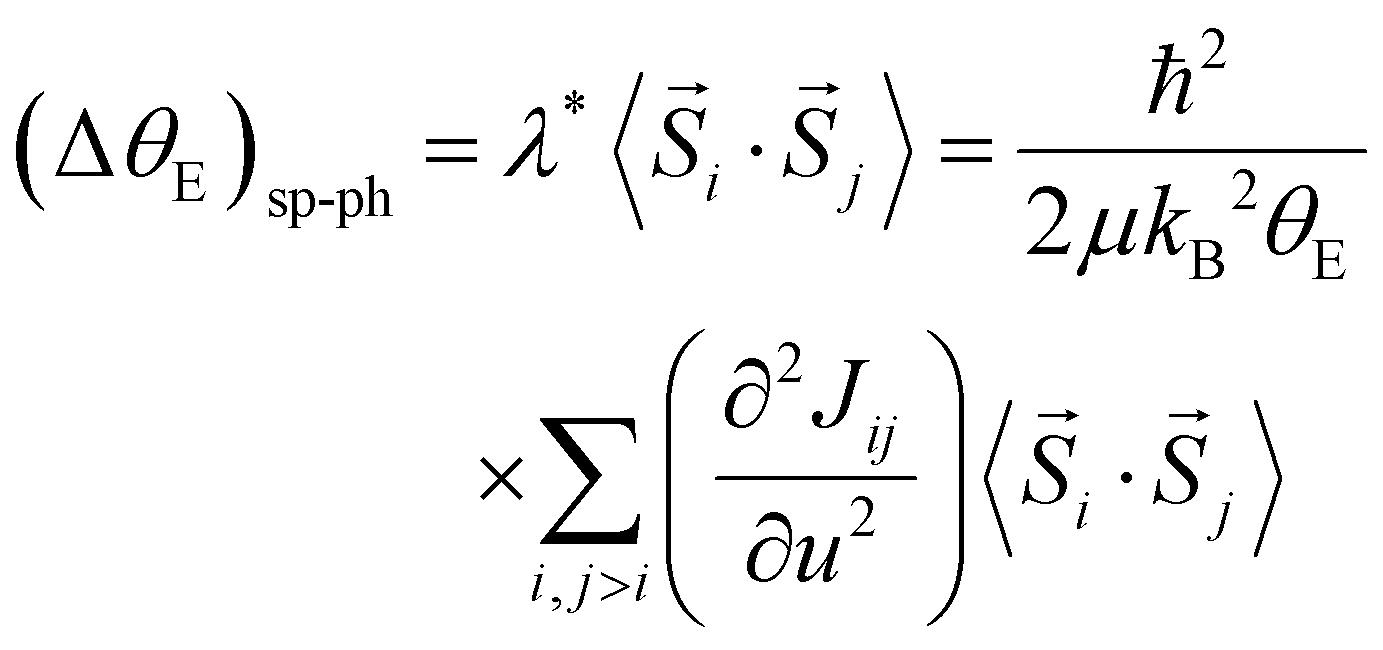 | (4) |
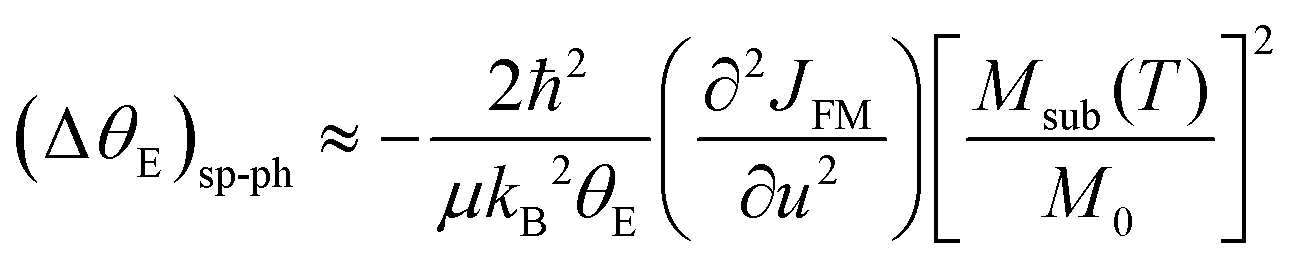 | (5) |
In Fig. 5, we show (a) the extracted quantity θE + ΔθE together with (b) the average magnetization of Ni3+ (μNi) in PrNiO3 from neutron diffraction in Gawryluk et al.,6 (c) the first-derivative of the DC magnetic susceptibility (M/H) under heating in a magnetic field of 100 Oe, and (d) the resistivity measurements in Mroginski et al.26 For the sake of comparison, Fig. 5b also has the average magnetization of Ni3+ in LuNiO3 from the neutron diffraction data.47 The magnetic susceptibility exhibits a strong paramagnetic signal from Pr3+; however, the magnetic ordering temperature in PrNiO3 is clearly seen in 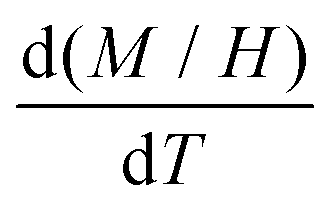 at TN ≈ 125 K. This value agrees with the insulator–metal transition temperature at 125 K as obtained from electrical resistivity data, then it corroborates that TN = TIM. The magnetization μNi is indeed quite similar to the extracted (ΔθE)sp-ph, i.e. the quadratic scaling between those quantities is consistent with eqn (5). The magnetization μNi of LuNiO3 with TN ≈ 125 K also scales well to the quantity ΔθE. Both observations strongly suggest that the anomaly seen in σΓ2 is related to a spin–phonon interaction. Physically, such a phenomenon takes place when the active vibrational modes are affected by an interaction among the magnetic ions within the crystal structure, as shown in eqn (4). Beyond this finding, we obtained a value for the second-order derivative
at TN ≈ 125 K. This value agrees with the insulator–metal transition temperature at 125 K as obtained from electrical resistivity data, then it corroborates that TN = TIM. The magnetization μNi is indeed quite similar to the extracted (ΔθE)sp-ph, i.e. the quadratic scaling between those quantities is consistent with eqn (5). The magnetization μNi of LuNiO3 with TN ≈ 125 K also scales well to the quantity ΔθE. Both observations strongly suggest that the anomaly seen in σΓ2 is related to a spin–phonon interaction. Physically, such a phenomenon takes place when the active vibrational modes are affected by an interaction among the magnetic ions within the crystal structure, as shown in eqn (4). Beyond this finding, we obtained a value for the second-order derivative 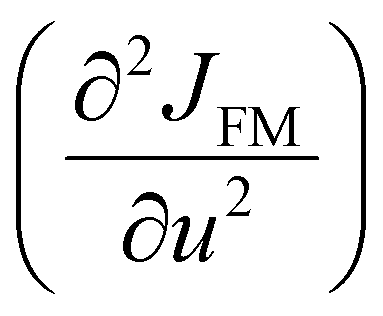 of ≈13 mRy Å−2, that is close to the one reported for undoped LaMnO3 (≈ 16 mRy Å−2).12 This result demonstrates that the parameter σΓ2 for the shells Nia–Oa (Γ1,2) represents the stretching modes, which only involve oxygen displacements, while the central atom (Ni or Mn) is fixed when they are Raman-active modes.
of ≈13 mRy Å−2, that is close to the one reported for undoped LaMnO3 (≈ 16 mRy Å−2).12 This result demonstrates that the parameter σΓ2 for the shells Nia–Oa (Γ1,2) represents the stretching modes, which only involve oxygen displacements, while the central atom (Ni or Mn) is fixed when they are Raman-active modes.
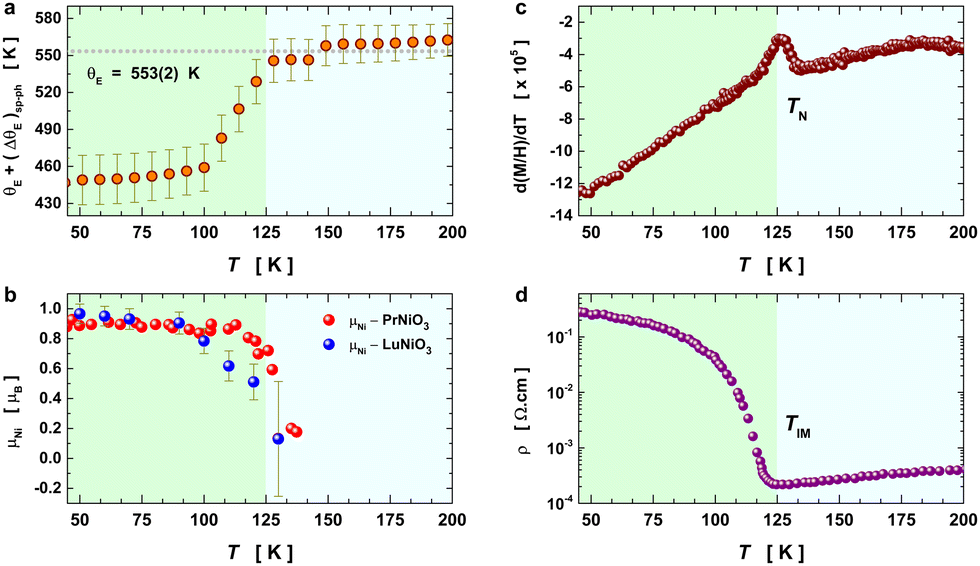 | ||
| Fig. 5 Temperature variation of the Einstein temperature, staggered magnetization, magnetic susceptibility, and resistivity in PrNiO3 across the IMT at TN ≈ 130 K. (a) Extracted quantity θE + (ΔθE)sp−ph from the Debye–Waller factors of the first shell Nia–Oa (Γ1,2). (b) Staggered magnetization of Ni ions in PrNiO3 and LuNiO3 as extracted from neutron powder diffraction.6,47 Both compounds show magnetic transition at TN ≈ 125–130 K. (c) First-derivative of the DC magnetic susceptibility (M/H) under heating in a magnetic field of 100 Oe, which display a magnetic transition at 125 K. (d) Resistivity measurements of PrNiO3 as reported in Mroginski et al.,26 which also exhibit the insulator–metal transition at 125 K. | ||
In the following, we discuss the likelihood of spin–phonon coupling in PrNiO3 based on the magnetic structure and the phonons. The magnetic moment of Ni arises from a collinear magnetic structure (S.G. P21/n) with two Ni magnetic moments at distinct Wyckoff sites. The magnetic propagation vector, as derived from neutron diffraction, was found to be 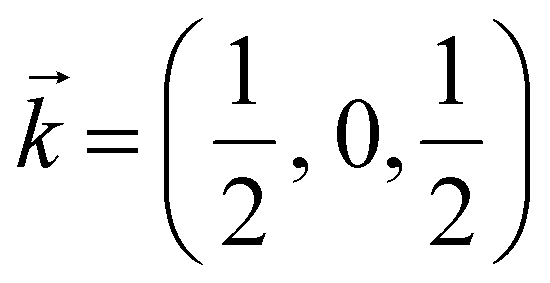 and the spin operator may be projected along both a- and c-axes.47,48 For the vibrational modes mainly concerning the stretching modes of the apical (O1; parallel to c-axis) and equatorial (O2; ab-plane) oxygens, one may expect a stronger coupling between the magnetic propagation vector for Ni ions and the eigenvectors which describe the normal modes. Therefore, this effect is seen through the softening of the Einstein temperature below TN, as predicted by eqn (5).
and the spin operator may be projected along both a- and c-axes.47,48 For the vibrational modes mainly concerning the stretching modes of the apical (O1; parallel to c-axis) and equatorial (O2; ab-plane) oxygens, one may expect a stronger coupling between the magnetic propagation vector for Ni ions and the eigenvectors which describe the normal modes. Therefore, this effect is seen through the softening of the Einstein temperature below TN, as predicted by eqn (5).
The Ni K-edge EXAFS data of PrNiO3 across TN ≈ 130 K unveiled a new ingredient for describing its itinerant behaviour, which is the spin–phonon coupling. Our results establish the role of the lattice dynamics together with orbital overlapping between Ni 3d and O 2p for describing the insulator-metal transition in PrNiO3 nickelate. Additional EXAFS measurements on nickelates with differing R cations are required to untangle the perturbation ΔθE from spin–phonon coupling and phonon renormalization due to the electronic states for drawing a complete picture of the underlying physics. It would be mandatory to probe it in other nickelates with TN and TIM well separated in the phase diagram, as in the case of R = Lu, Ho, Tl.
Conclusions
In this work, high-purity PrNiO3 nickelate has been successfully synthesized using the liquid-mixture method combined with thermal treatments under 200 bars under an O2 atmosphere. Synchrotron X-ray diffraction data confirmed the occurrence of the orthorhombic lattice (Pbnm) under room conditions, but with small impurities of NiO (<2.5% wt). Low-temperature EXAFS oscillations at Ni K-edge unveiled the very local units of the PrNiO3 lattice, which were modelled in agreement with the monoclinic crystal structure (P21/n) for T < 125–130 K. Above this temperature, a convergence of the pair-unit distances Ni–Pr was clearly identified, denoting the stabilization of the orthorhombic lattice (Pbnm). The lattice dynamics of the PrNiO3 nickelate across the structural transition were monitored using temperature-dependent EXAFS data in the range of 10–300 K (under isobaric conditions). From the thermal evolution of the Debye–Waller factors above 130 K, we extracted the Einstein temperatures of individual atomic pair-units and estimated the Ni–O bond stiffness. The thermal evolution of the Debye–Waller factors and bond-length evolution of the Ni–O bonds and Ni–Pr pair showed strong anomalies across the IMT. We modelled the evolution of the Ni–O bond Debye–Waller factors by means of molecular field approximation for the scalar spin correlation function. This model suggested a continuous increase of the spin–phonon coupling below the IMT transition temperature of 130 K, which was confirmed by the magnetic and vibrational properties of this compound. Our EXAFS results clearly showed that the insulator–metal transition in the PrNiO3 nickelate has an important contribution from the lattice dynamics. The approach presented here may be further extended and applied to similar systems that show magnetic or insulator–metal transitions to provide a physical description of the underlying mechanisms.Author contributions
Conceptualization, J. E. R., J. A. A., A. D. R., and O. M.; methodology, J. E. R, J. L-S., E. S-T, J. L. M., N. M. N., and O. M.; software, J. E. R.; formal analysis, J. E. R.; resources, O. M.; writing – original draft preparation, J. E. R.; writing – review and editing, J. E. R., A. D. R, J. L. M., and J. A. A; funding acquisition, J. A. A., J. L. M., and O. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially funded by the Spanish Ministry of Science and Innovation, MCIN/AEI/10.13039/501100011033/, with the project numbers: PID2021-122477OB-I00, TED2021-129254B-C22, PIE: 2021-60-E-030, and PIE: 2010-6-OE-013. All the authors acknowledge the European Synchrotron (ESRF, Grenoble) for making all the facilities available for X-ray diffraction (BM25) and X-ray absorption experiments (BM23). J. E. R. thanks the anonymous reviewers for their pertinent and useful comments.References
- G. Catalan, Phase Transitions, 2008, 81, 729–749 CrossRef CAS.
- J. Liu, E. Jia, L. Wang, K. A. Stoerzinger, H. Zhou, C. S. Tang, X. Yin, X. He, E. Bousquet, M. E. Bowden, A. T. S. Wee, S. A. Chambers and Y. Du, Adv. Sci., 2019, 6(19), 1901073 CrossRef CAS PubMed.
- E. Yadav, S. Harisankar, K. Soni and K. R. Mavani, Vib. Spectrosc., 2021, 112, 103185 CrossRef CAS.
- L. Korosec, M. Pikulski, T. Shiroka, M. Medarde, H. Luetkens, J. A. Alonso, H. R. Ott and J. Mesot, Phys. Rev. B, 2017, 95, 1–5 CrossRef.
- F. Serrano-Sánchez, J. L. Martínez, F. Fauth and J. A. Alonso, Dalton Trans., 2021, 50, 7085–7093 RSC.
- D. J. Gawryluk, Y. M. Klein, T. Shang, D. Sheptyakov, L. Keller, N. Casati, P. Lacorre, M. T. Fernández-Díaz, J. Rodríguez-Carvajal and M. Medarde, Phys. Rev. B, 2019, 100, 205137 CrossRef CAS.
- I. Ardizzone, J. Teyssier, I. Crassee, A. B. Kuzmenko, D. G. Mazzone, D. J. Gawryluk, M. Medarde and D. van der Marel, Phys. Rev. Res., 2021, 3, 033007 CrossRef CAS.
- M. Medarde, M. T. Fernández-Díaz and P. Lacorre, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 2–5 CrossRef.
- A. Y. Ramos, C. Piamonteze, H. C. N. Tolentino, N. M. Souza-Neto, O. Bunau, Y. Joly, S. Grenier, J. P. Itié, N. E. Massa, J. A. Alonso and M. J. Martinez-Lope, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 1–5 Search PubMed.
- C. Piamonteze, in AIP Conference Proceedings, AIP, 2003, vol. 652, pp. 450–455.
- M. Acosta-Alejandro, J. M. De León, M. Medarde, P. Lacorre, K. Konder and P. A. Montano, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1–5 CrossRef.
- E. Granado, A. García, J. A. Sanjurjo, C. Rettori, I. Torriani, F. Prado, R. D. Sánchez, A. Caneiro and S. B. Oseroff, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 11879–11882 CrossRef CAS.
- C. T. Chantler, X-ray absorption spectroscopy definitions, International Tables for Crystallography, X-ray Absorption Spectroscopy and Related Techniques, 2021, vol. I Search PubMed.
- P. A. Lee, P. H. Citrin, P. Eisenberger and B. M. Kincaid, Rev. Mod. Phys., 1981, 53, 769–806 CrossRef CAS.
- J. A. Alonso, M. J. Martínez-Lope and M. A. Hidalgo, J. Solid State Chem., 1995, 116, 146–156 CrossRef CAS.
- C. Ponchut, J. M. Rigal, J. Clément, E. Papillon, A. Homs and S. Petitdemange, J. Instrum., 2011, 6, C01069 Search PubMed.
- S. Roobol, W. Onderwaater, J. Drnec, R. Felici and J. Frenken, J. Appl. Crystallogr., 2015, 48, 1324–1329 CrossRef CAS PubMed.
- J. Rodríguez-Carvajal, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 192, 55–69 CrossRef.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, J. Synchrotron Radiat., 2015, 22, 1548–1554 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- B. Ravel and M. Newville, ATHENA and ARTEMIS, International Tables for Crystallography, X-ray Absorption Spectroscopy and Related Techniques, 2020, vol. I Search PubMed.
- M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 725–738 CrossRef CAS PubMed.
- N. D. Todorov, M. V. Abrashev and V. G. Ivanov, J. Phys.: Condens. Matter, 2012, 24, 175404 CrossRef CAS PubMed.
- P. Fornasini and R. Grisenti, J. Synchrotron Radiat., 2015, 22, 1242–1257 CrossRef CAS PubMed.
- G. Dalba and P. Fornasini, J. Synchrotron Radiat., 1997, 4, 243–255 CrossRef CAS PubMed.
- M. A. Mroginski, N. E. Massa, H. Salva, J. A. Alonso and M. J. Martínez-Lope, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 5304–5311 CrossRef CAS.
- A. Nakatsuka, A. Yoshiasa, K. Fujiwara and O. Ohtaka, J. Mineral. Petrol. Sci., 2018, 113, 280–285 CrossRef CAS.
- J. E. F. S. Rodrigues, J. Gainza, F. Serrano-Sánchez, C. Marini, Y. Huttel, N. M. Nemes, J. L. Martínez and J. A. Alonso, Chem. Mater., 2022, 34, 1213–1224 CrossRef CAS.
- H. G. M. Edwards, J. Mol. Struct., 1987, 156, 137–142 CrossRef CAS.
- R. X. Silva, H. Reichlova, X. Marti, D. A. B. Barbosa, M. W. Lufaso, B. S. Araujo, A. P. Ayala and C. W. A. Paschoal, J. Appl. Phys., 2013, 114(19), 194102 CrossRef.
- A. Panchwanee, I. Schiesaro, S. Mobilio, S. S. K. Reddy, C. Meneghini, E. Welter and V. Raghavendra Reddy, J. Phys.: Condens. Matter, 2019, 31, 345403 CrossRef CAS PubMed.
- E. Paris, L. Simonelli, T. Wakita, C. Marini, J. H. Lee, W. Olszewski, K. Terashima, T. Kakuto, N. Nishimoto, T. Kimura, K. Kudo, T. Kambe, M. Nohara, T. Yokoya and N. L. Saini, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- M. Y. Hacisalihoglu, E. Paris, B. Joseph, L. Simonelli, T. J. Sato, T. Mizokawa and N. L. Saini, Phys. Chem. Chem. Phys., 2016, 18, 9029–9035 RSC.
- A. Bianconi, N. L. Saini, A. Lanzara, M. Missori, T. Rossetti, H. Oyanagi, H. Yamaguchi, K. Oka and T. Ito, Phys. Rev. Lett., 1996, 76, 3412–3415 CrossRef CAS PubMed.
- B. S. Araújo, A. M. Arévalo-López, C. C. Santos, J. P. Attfield, C. W. A. Paschoal and A. P. Ayala, J. Phys. Chem. Solids, 2021, 154, 110044, DOI:10.1016/j.jpcs.2021.110044.
- L. D. Casto, A. J. Clune, M. O. Yokosuk, J. L. Musfeldt, T. J. Williams, H. L. Zhuang, M. W. Lin, K. Xiao, R. G. Hennig, B. C. Sales, J. Q. Yan and D. Mandrus, APL Mater., 2015, 3(4), 041515 CrossRef.
- J. E. F. S. Rodrigues, W. S. Rosa, M. M. Ferrer, T. R. Cunha, M. J. Moreno Zapata, J. R. Sambrano, J. L. Martínez, P. S. Pizani, J. A. Alonso, A. C. Hernandes and R. V. Gonçalves, J. Alloys Compd., 2019, 799, 563–572 CrossRef CAS.
- S. Calder, J. H. Lee, M. B. Stone, M. D. Lumsden, J. C. Lang, M. Feygenson, Z. Zhao, J. Q. Yan, Y. G. Shi, Y. S. Sun, Y. Tsujimoto, K. Yamaura and A. D. Christianson, Nat. Commun., 2015, 6, 1–6 Search PubMed.
- M. S. Radionov, S. A. Klimin, A. Y. Glamazda and A. V. Peschanskii, Low Temp. Phys., 2022, 48, 246–252 CrossRef CAS.
- B. S. Araújo, A. M. Arévalo-López, C. C. Santos, J. P. Attfield, C. W. A. Paschoal and A. P. Ayala, J. Appl. Phys., 2020, 127(11), 114102 CrossRef.
- M. C. Weber, M. Guennou, D. M. Evans, C. Toulouse, A. Simonov, Y. Kholina, X. Ma, W. Ren, S. Cao, M. A. Carpenter, B. Dkhil, M. Fiebig and J. Kreisel, Nat. Commun., 2022, 13(1), 443 CrossRef CAS PubMed.
- J. Son, B. C. Park, C. H. Kim, H. Cho, S. Y. Kim, L. J. Sandilands, C. Sohn, J. G. Park, S. J. Moon and T. W. Noh, npj Quantum Mater., 2019, 4, 1–8 CrossRef CAS.
- M. Balkanski, R. F. Wallis and E. Haro, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 1928–1934 CrossRef CAS.
- P. Fornasini, J. Phys.: Condens. Matter, 2001, 13, 7859–7872 CrossRef CAS.
- W. Baltensperger and J. S. Helman, Helv. Phys. Acta, 1968, 41, 668–673 Search PubMed.
- D. J. Lockwood and M. G. Cottam, J. Appl. Phys., 1988, 64, 5876–5878 CrossRef CAS.
- F. Serrano, M. T. Fernandez-Diaz, J. L. Martinez and J. A. Alonso, Dalton Trans., 2022, 51(6), 2278–2286 RSC.
- J. Mesot, M. Medarde, S. Rosenkranz, P. Fischer, P. Lacorre and K. Gobrecht, High Pressure Res., 1995, 14, 35–40 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03063b |
| This journal is © The Royal Society of Chemistry 2023 |