
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulation of the innate immune system by fragmented heparin-conjugated lipids on lipid bilayered membranes in vitro†
Anna
Adler
 a,
Marlene
Fritsch
a,
Karin
Fromell
a,
Gero
Leneweit
bc,
Kristina N.
Ekdahl
ad,
Bo
Nilsson
a and
Yuji
Teramura
*aef
a,
Marlene
Fritsch
a,
Karin
Fromell
a,
Gero
Leneweit
bc,
Kristina N.
Ekdahl
ad,
Bo
Nilsson
a and
Yuji
Teramura
*aef
aDepartment of Immunology, Genetics and Pathology (IGP), Uppsala University, Dag Hammarskjölds väg 20, SE-751 85, Uppsala, Sweden
bABNOBA GmbH, Pforzheim, Germany
cCarl Gustav Carus-Institute, Association for the Promotion of Cancer Therapy, Niefern-Öschelbronn, Germany
dLinnaeus Center of Biomaterials Chemistry, Linnaeus University, SE-391 82 Kalmar, Sweden
eCellular and Molecular Biotechnology Research Institute (CMB), National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central Fifth, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: y.teramura@aist.go.jp; Tel: +81(0)29-861-6582
fMaster's/Doctoral Program in Life Science Innovation (T-LSI), University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
First published on 31st October 2023
Abstract
Surface modification with heparin is a powerful biomaterial coating strategy that protects against innate immunity activation since heparin is a part of the proteoglycan heparan sulfate on cell surfaces in the body. We studied the heparinization of cellular and material surfaces via lipid conjugation to a heparin-binding peptide. In the present study, we synthesized fragmented heparin (fHep)-conjugated phospholipids and studied their regulation of the innate immune system on a lipid bilayered surface using liposomes. Liposomes have versatile applications, such as drug-delivery systems, due to their ability to carry a wide range of molecules. Owing to their morphological similarity to cell membranes, they can also be used to mimic a simple cell-membrane to study protein–lipid interactions. We investigated the interaction of complement-regulators, factor H and C4b-binding protein (C4BP), as well as the coagulation inhibitor antithrombin (AT), with fHep-lipids on the liposomal surface. Herein, we studied the ability of fHep-lipids to recruit factor H, C4BP, and AT using a quartz crystal microbalance with dissipation monitoring. With dynamic light scattering, we demonstrated that liposomes could be modified with fHep-lipids and were stable up to 60 days at 4 °C. Using a capillary western blot-based method (Wes), we showed that fHep-liposomes could recruit factor H in a model system using purified proteins and assist in the degradation of the active complement protein C3b to iC3b. Furthermore, we found that fHep-liposomes could recruit factor H and AT from human plasma. Therefore, the use of fHep-lipids could be a potential coating for liposomes and cell surfaces to regulate the immune system on the lipid surface.
1. Introduction
Liposomes are synthetic lipid vesicles which have been extensively evaluated, for example to be used clinically as drug-delivery systems. Therapeutic liposomes are generally composed of phospholipids and cholesterol.1 Because of their structural similarity to cell membranes, liposomes can be used as models of simple cellular bilayered membranes. Therefore, liposomes are a useful tool for studying lipid–protein interactions without interference from the complex surface morphology of biological cell membranes.2,3Heparin is a linear polysaccharide belonging to the glycosaminoglycan family.4 It is mainly composed of repeating units of highly sulfated disaccharides.5 Heparin has been used for many years to modify surfaces of medical devices, this to improve their hemocompatibility by reducing surface-induced immune reactions, such as thrombosis and inflammation since heparin can regulate the coagulation and complement systems.6 Recently, cell-surface modification with heparin has also been used to improve graft survival during cell transplantation. This approach aims to mimic the cell-surface proteoglycan heparan sulfate, glycocalyx.7 The idea is that the heparin coating protects the cell surface by mimicking the glycocalyx of the cell, since transplanted cells are damaged or destroyed due to hemoincompatibility when they are infused into the bloodstream of the patient. These issues have been reported in the clinical transplantation of pancreatic islets8 and mesenchymal stem cells (MSCs).9 Our group found that cell destruction during the early stage of transplantation was caused by innate immune action and termed this phenomenon instant blood-mediated inflammatory reaction (IBMIR) or thromboinflammation.10–12
Our group has studied heparinization on cellular and material surfaces via lipid conjugation and heparin-binding peptides. Recently, we reported a single-step heparin coating of the cellular membrane using fragmented heparin (fHep)-conjugated phospholipids.13 When human MSCs (hMSCs) are infused into human blood, blood coagulation is activated due to tissue factor from hMSCs, which can trigger complement activation, followed by cell destruction. However, hMSCs coated with fHep-lipids have been demonstrated to bind to the serine protease inhibitor antithrombin (AT), which leads to reduced platelet aggregation and coagulation when the fHep-coated hMSCs were incubated in human whole blood ex vivo.13 AT is a 58 kDa glycoprotein consisting of a single chain, and has a plasma concentration of around 125 μg mL−1 in humans. As it can inhibit several coagulation cascade enzymes, such as thrombin and factor Xa, AT is one of the most important regulators of coagulation. The activity of AT increases in the presence of heparin.14
Heparin is also known to bind to other proteins, such as the fluid-phase complement-regulatory proteins factor H and C4b-binding protein (C4BP).15–18 Factor H is a 155 kDa plasma protein with a concentration of approximately 500 μg mL−1 in human plasma.18,19 Factor H is a major soluble complement inhibitor of the alternative pathway (AP) and regulates the pathway by binding to C3b, consequently inhibiting it from forming an alternative C3-convertase (C3bBb). It also prevents further complement activation by acting as a cofactor to factor I to inactivate C3b to iC3b, and it accelerates the decay of the AP C3 convertase.18 Factor H is composed of a single chain of 20 complement-control protein (CCP) domains.20–23 Haque et al. showed that factor H contains heparin-binding sites in CCP 6–8, 19–20, and possibly a weaker binding site in CCP 11–13.24 The C3b-binding sites within factor H are in CCP domains 1–4 and 19–20, with CCP1–4 having complement-regulatory activity.24,25 C4BP is a 540 kDa protein present in different forms in the plasma, with the main form consisting of one β-chain (46 kDa) and 7 α-chains (72 kDa) linked together by disulfide bonds and hydrophobic forces in a central core.20–22,26 C4BP is present in human plasma at a concentration of approximately 225 μg mL−1 and is the main fluid-phase regulator of the classical and lectin complement pathways.19 Therefore, it would be beneficial to use factor H and C4BP in human plasma as autoregulators for the complement system via fHep-lipids on the modified membrane surface, where factor H and C4BP are expected to interact with fHep-lipids. The strategy of hijacking the regulatory complement proteins of the human body to a surface is one of the strategies used by some bacteria to evade the immune system.19,27,28Streptococcus pneumoniae, Haemophilus influenzae, and Toxoplasma gondii are examples of pathogens that use this strategy to recruit host complement inhibitors to their surfaces to evade the immune system.19,29,30 Different factor H and C4BP recriuting-peptides have previously been used as a means to augment the hemocompatibility of various biomaterials.25,31–33
In the present study, the ability of fHep-lipids to bind factor H, C4BP, and AT was investigated in a model system with purified proteins using a quartz crystal microbalance with dissipation monitoring (QCM-D), which is often used for the analysis of molecular interactions.34 The liposomes were then modified with fHep-lipids and characterized before being incubated in pure factor H to investigate whether surface-bound factor H could help cleave C3b to iC3b. fHep-lipid-modified liposomes were also incubated in human EDTA plasma to study whether factor H and AT could be recruited onto the surface. We aimed to determine whether coating with fHep-lipids can function with anticoagulant activity and/or complement activity in vitro.
2. Materials and methods
2.1 Synthesis of heparin-conjugated lipids
Heparin-conjugated lipids were synthesized as described by Asawa et al.13 In short, unfractionated heparin was mixed with a solution of sulfuric acid and sodium nitrate to fragment the heparin. The pH was adjusted to 7.0 and the fHep solution was dialyzed against MQ-H2O. Next, the fHep solution was lyophilized. Maleimide–PEG–lipids were prepared by mixing N-hydroxysuccinimide–PEG–maleimide, triethylamine and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) in dichloromethane. Maleimide–PEG–lipids were obtained after precipitation with diethyl ether.35 After, a linker consisting of cysteine (C) and different numbers of lysines (K) were conjugated to the maleimide–PEG–lipids via the thiol–maleimide Michael addition reaction. The different peptide linkers used in this study were C, K1C, K2C, K4C and K8C, which introduced 1, 2, 3, 5 or 9 amine groups. The available amine groups on the KnC-lipids allowed for conjugation to an aldehyde group on fHep via Schiff base chemistry, followed by reduction by sodium cyanoborohydride (NaCNBH3).36 Succinic anhydride was then added to the fHep-lipids, converting any unreacted amine groups into carboxylic groups. The fHep-lipid solutions were then purified using spin columns, lyophilized, and the obtained fHep-lipid powder was stored at −20 °C until use (Fig. 1A–C). The following lipids were used in this study: fHep-C-lipids (10 kDa), fHep-K1C-lipids (17 kDa), fHep-K2C-lipids (23 kDa), fHep-K4C-lipids (34 kDa), and fHep-K8C-lipids (56 kDa). N-(Methyl polyoxyethylene oxycarbonyl)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (MeO-polyethylene glycol (PEG)5k-lipids, NOF Corporation, Tokyo, Japan) (5.7 kDa) was used as a control (Fig. 1D).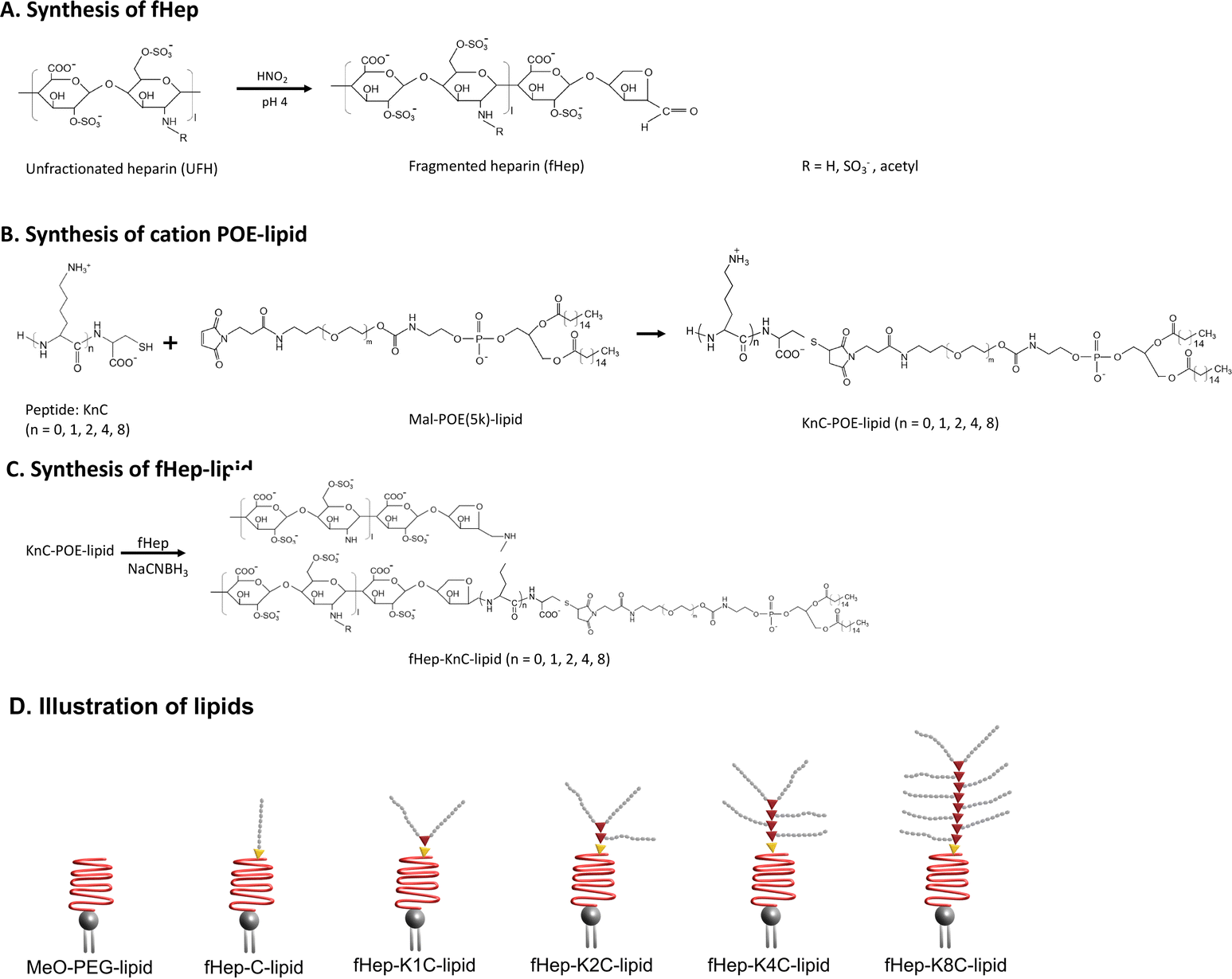 | ||
| Fig. 1 Overview of fHep-lipid synthesis. (A) Unfractionated heparin is treated with HNO2 to fragment the heparin. (B) A linker with different numbers (n = 0, 1, 2, 4, 8) of lysine (K) and one cysteine (C), KnC, is conjugated to a maleimide-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE)-PEG5k-lipid via the thiol–maleimide Michael addition click reaction. (C) The fragmented heparin is then conjugated to the KnC-lipid via the available amine groups on the KnC-lipids. The amine groups allow for conjugation to an aldehyde group on fragmented heparin via Schiff chemistry, followed by reduction by sodium cyanoborohydride (NaCNBH3). (D) Illustration of the lipids used in this study. | ||
2.2 Evaluation of factor H, C4BP, and AT binding to heparin-conjugated lipids using QCM-D
The ability of heparin-conjugated lipids to bind complement-regulators, factor H and C4BP, as well as AT, was analyzed using QCM-D (QSense Pro, Biolin Scientific, Gothenburg, Sweden). Briefly, gold-plated quartz crystals (Q-sense QSX 301 Au, Biolin Scientific AB), which were cleaned by ozone irradiation, were incubated in 2 mM 1-dodecanethiol (Sigma-Aldrich, St. Louis, MO, USA) diluted in 99.9% ethanol at room temperature (20–25 °C) for 24 h to prepare a hydrophobic C12-self-assembled monolayer (SAM) surface. The sensor was rinsed with ethanol and dried with nitrogen gas to ensure the complete removal of ethanol from the sensor surfaces before use. All QCM-D experiments were performed in triplicate and were carried out at 22 °C. Phosphate-buffered saline (PBS, tablets, 0.14 M NaCl, 2.7 mM KCl, 10 mM phosphate buffer, pH 7.4, Medicago AB, Uppsala, Sweden) was used as the running buffer for priming and rinsing the sensors. The sensors were rinsed for 5 min before, between, and after sample addition. The flow rate was set to 20 μL min−1 for the injection of all samples, as well as for rinsing with running buffer. As the adsorbed protein layer is rigid (ΔD < 1), the Sauerbrey equation37 was used to calculate the mass of the proteins bound to the sensor surfaces using the resonance frequency change (Δf) at the 7th overtone. However, to accurately quantify the mass of adsorbed lipids to the sensor surface, a visco-elastic model should preferably be used as ΔD > 1, which indicates that the lipid layer is well hydrated with water molecules. Thus, the adsorbed water molecules result in a lower Δf, and if they are not accounted, it will result in an inaccurate estimation of the calculated lipid mass. In this study, we did not calculate the mass of the lipids, and only the mass of the proteins (ΔD < 1) adsorbed to the lipid layer was estimated using Sauerbrey's model.We analyzed factor H-binding to fHep-C-, fHep-K1C-, fHep-K2C-, fHep-K4C-, fHep-K8C-, and MeO-PEG-lipids as follows: 5 min rinsing with PBS, 30 min rinsing with 0.1 mg mL−1 fHep-/MeO-PEG-lipid in PBS, 5 min rinsing with PBS, 30 min rinsing with 1 mg mL−1 bovine serum albumin (BSA, A4503, Sigma-Aldrich Chemie GmbH, Steinheim, Germany), 5 min rinsing with PBS, 30 min rinsing with 50 μg mL−1 human factor H, purified in-house according to Hammer et al.,38 and, lastly, 10 min rinsing with PBS.
We also analyzed C4BP-binding to fHep-C-, fHep-K1C-, fHep-K2C-, fHep-K4C-, fHep-K8C-, and MeO-PEG-lipids as follows: 5 min rinsing with PBS, 30 min rinsing with 0.1 mg mL−1 fHep-/MeO-PEG-lipid in PBS, 5 min rinsing with PBS, 30 min rinsing with 1 mg mL−1 BSA (Sigma-Aldrich Chemie GmbH), 5 min rinsing with PBS, 30 min rinsing with 10 μg mL−1 human C4BP (A109, Complement Technology, Tyler, TX, USA), and, lastly, 10 min rinsing with PBS.
In addition, we studied the binding capacities of human factor H and AT to fHep-lipids. Human AT (0.5 g of human AT with an activity of 1000 IU [Baxter AG, Vienna, Austria]) was prepared by separation from human serum albumin using a Heparin-Sepharose column (Pharmacia Biotech, Uppsala, Sweden).39 A solution of 25 μg mL−1 human factor H in PBS, followed by 25 μg mL−1 human AT in PBS was flowed over different molar ratios (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 25:75, 50:50, 75:25, and 100:0 fHep-lipid:MeO-PEG-lipid) of fHep-K1C-lipid:MeO-PEG-lipids and fHep-K8C-lipid:MeO-PEG-lipids. The QCM-D program was set as follows: 5 min rinsing with PBS, 30 min rinsing with fHep-lipid:MeO-PEG-lipid in different molar ratios in PBS, 5 min rinsing with PBS, 30 min rinsing with 1 mg mL−1 BSA (Sigma-Aldrich Chemie GmbH), 5 min rinsing with PBS, 30 min rinsing with human factor H, 5 min rinsing with PBS, 30 min rinsing with human AT, and, finally, 10 min rinsing with PBS. A similar setup was also used as a control for 25 g mL−1 AT binding to 0.1 mg mL−1 fHep-K1C-lipid, fHep-K8C-lipid, and MeO-PEG-lipid, with the exception that the factor H step was removed and different molar ratios of the lipids were not used.
:100, 25:75, 50:50, 75:25, and 100:0 fHep-lipid:MeO-PEG-lipid) of fHep-K1C-lipid:MeO-PEG-lipids and fHep-K8C-lipid:MeO-PEG-lipids. The QCM-D program was set as follows: 5 min rinsing with PBS, 30 min rinsing with fHep-lipid:MeO-PEG-lipid in different molar ratios in PBS, 5 min rinsing with PBS, 30 min rinsing with 1 mg mL−1 BSA (Sigma-Aldrich Chemie GmbH), 5 min rinsing with PBS, 30 min rinsing with human factor H, 5 min rinsing with PBS, 30 min rinsing with human AT, and, finally, 10 min rinsing with PBS. A similar setup was also used as a control for 25 g mL−1 AT binding to 0.1 mg mL−1 fHep-K1C-lipid, fHep-K8C-lipid, and MeO-PEG-lipid, with the exception that the factor H step was removed and different molar ratios of the lipids were not used.
2.3 Liposome preparation
Liposomes containing 60 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, Avanti Polar Lipids Inc., Alabaster, AL, USA) and 40 mol% cholesterol (Sigma-Aldrich Chemie GmbH) were prepared using the thin-film hydration method, followed by manual extrusion. DPPC (10 mg) and cholesterol (3.5 mg) were dissolved in 99.9% ethanol solution (final volume, 1.35 mL). A rotary evaporator was used to evaporate ethanol and form a uniform lipid film. The lipid films were dried in vacuo for 24 h at 25 °C to ensure the complete removal of ethanol. The resulting lipid film was hydrated in MQ-H2O (1 mL) using a magnetic stirrer for 2 h at room temperature, and the liposome suspension was extruded through polycarbonate membranes (pore sizes: 1000, 400, and 200 nm; Nuclepore™ Track-Etched Membrane; Whatman™, Cytiva, Buckinghamshire, UK) using an Avanti Mini Extruder (Avanti Polar Lipids). The lipid suspension was passed through each filter 21 times. Dynamic light scattering (DLS, Zetasizer Pro, Malvern Panalytical, Worcestershire, UK) was used to characterize the liposomes (see Section 2.5). The total cholesterol concentration in the liposome solutions was determined using a chromogenic cholesterol kit (LabAssay™ Cholesterol, FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) according to the manufacturer's instructions, with the exception that the liposomes and cholesterol standard were diluted in 1 part sample in 1 part 20% sodium dodecyl sulfate solution (SDS, VWR Chemicals, Ohio, USA) and heated up at 95 °C for 15 min for solubilization. A total of 100 μL of the suspension was added to a 96-well microtiter plate, and the absorbance was measured at 600 nm (CLARIOstar Plus, BMG Labtech, Ortenberg, Germany).2.4 Modification of liposomes with fHep-lipids
To modify the surface of the liposomes with fHep-K8C-lipid or MeO-PEG-lipids, a solution of fHep-lipids was mixed with the liposomes. The liposome suspension (500 μL, [cholesterol] = 2.8 mg mL−1) was pelleted by ultracentrifugation (Beckman, Optima™ LE-80 K Ultracentrifuge with the Type 70.1 Ti rotor, Beckman Coulter, Bromma, Sweden) at 100000 × g for 60 min at 4 °C. The liposome pellet was resuspended in 500 μL of 1 mg mL−1 fHep-K8C-lipid in PBS or 1 mg mL−1 MeO-PEG-lipid in PBS as a control. The resultant mixed liposome suspension was incubated overnight at 37 °C at 20 rpm on a rotary wheel, and the unincorporated lipids were removed by ultracentrifugation at 100000 × g for 60 min at 4 °C. The liposome pellet was resuspended in 500 μL PBS and stored at 4 °C. The liposomes were characterized using DLS before and after modification (see Section 2.5).
2.5 Characterization and long-term stability of modified liposomes
The average particle size, polydispersity index (PDI), and zeta potential of the modified liposomes were determined using DLS (Zetasizer Pro, Malvern Panalytical). The modified liposomes were stored in PBS at 4 °C, and analyzed by monitoring the size, PDI, and zeta potential over a long-term period (90 days). All liposome samples (10 μL) were diluted in 1 mM NaCl aqueous solution (990 μL) before measurement. All measurements were performed at 25 °C. Data were analyzed using Malvern ZS Xplorer (version 2.1.1.1) software.2.6 Functional assay of modified liposomes using factor H, factor I, and C3b
To examine the interaction of fHep-lipid-modified liposomes with purified human factor H, the MeO-PEG-lipid- and fHep-K8C-lipid-modified liposome suspension (100 μL, [cholesterol] = 1 mg mL−1 in PBS) was incubated with factor H (100 μL, [factor H] = 100 μg mL−1 in PBS, A137, Complement Technology) for 60 min at 37 °C on a rotary wheel at 20 rpm. As a negative control, a solution of factor H mixed with 100 μL PBS without liposomes was used, and treated identically as the liposome-containing samples. Thereafter, the resultant solution was transferred into OptiSeal™ centrifuge tubes (8.9 mL, Beckman Coulter) for ultracentrifugation at 100000 × g for 60 min at 4 °C (Optima™ LE-80 K Ultracentrifuge with the Type 70.1 Ti rotor, Beckman Coulter) to remove unbound factor H. The pellet was resuspended into PBS (100 μL), and 15 μL of the sample was stored at −80 °C for further analysis of factor H using Wes (see Section 2.9). The remaining sample (85 μL) was incubated in a mixture solution (85 μL) of C3b (300 μg mL−1 in PBS, A114, Complement Technology) and factor I (40 μg mL−1 in PBS, A138, Complement Technology) for 15 min at 37 °C (final concentration: [C3b] = 150 μg mL−1, [factor I] = 20 μg mL−1). As a positive control, the interaction of fluid-phase factor H with C3b and factor I was analyzed; a mixed solution of factor H (50 μg mL−1), C3b (150 μg mL−1), and factor I (20 μg mL−1) in PBS (170 μL) was used. The reaction was stopped by diluting each sample (10 μL) in 0.1 × Wes Sample buffer (10 μL, ProteinSimple, Santa Clara, California, USA) and then heating it up at 95 °C for 5 min. The samples were then stored at −80 °C for Wes analysis for iC3b generation (see Section 2.9). The experiment was repeated thrice using three different liposome batches.
We also examined if the concentration of factor I affected iC3b generation. Here, 300 μL of fHep-K8C-/MeO-PEG-lipid-modified liposomes in PBS with [cholesterol] = 1.0 mg mL−1 were incubated in a final concentration of 50 μg mL−1 factor H (total volume 500 μL) for 60 min at 37 °C on a rotary wheel at 20 rpm. As a negative control, a solution of 50 μg mL−1 factor H mixed with PBS (final volume 500 μL) without liposomes was used and treated identically as the liposome-containing samples. Thereafter, the resultant solution was transferred into OptiSeal™ centrifuge tubes (8.9 mL, Beckman Coulter) for ultracentrifugation at 100000 × g for 60 min at 4 °C (Optima™ LE-80 K Ultracentrifuge with the Type 70.1 Ti rotor, Beckman Coulter) to remove unbound factor H. The pellet was resuspended into PBS (300 μL), and 15 μL of the sample was stored at −80 °C for further analysis of factor H using Wes (see Section 2.9). The liposomes were split into three different tubes (85 μL per tube) and were incubated in a mixture solution (85 μL) of C3b (150 μg mL−1 in PBS, A114, Complement Technology) and factor I (10/1/0.1 μg mL−1 in PBS, A138, Complement Technology) for 15 min at 37 °C (final concentration: [C3b] = 75 μg mL−1, [factor I] = 5/0.5/0.05 μg mL−1). As a positive control, the interaction of fluid-phase factor H with C3b and factor I was analyzed; a mixed solution of factor H (50 μg mL−1), C3b (75 μg mL−1), and factor I (5/0.5/0.05 μg mL−1) in PBS (final volume 170 μL) was used. The samples were then stored at −80 °C for Wes analysis for iC3b generation (see Section 2.9).
2.7 Blood collection
Human blood from three healthy donors was collected in EDTA Vacuette® tubes (Greiner Bio-One GmbH, Kremsmünster, Austria). Donors had not received any medication in the past ten days. Ethical approval was obtained from the regional ethics board (Uppsala diary number 2008/264). For EDTA plasma preparation, the blood was centrifuged at 2500 × g for 15 min at room temperature and the supernatant (plasma) was collected and stored at −80 °C until use.2.8 Incubation of modified liposomes in human EDTA plasma
fHep-K8C-/MeO-PEG-lipid-modified liposomes were incubated in human EDTA plasma to investigate whether the liposomes could interact with factor H and AT in the plasma. fHep-K8C-/MeO-PEG-liposomes (100 μL in PBS, [cholesterol] = 1.4 mg mL−1) were incubated in human EDTA plasma (400 μL) for 60 min at 37 °C on a rotary wheel at 20 rpm. PBS without liposomes (100 μL) in EDTA plasma (400 μL) was used as a negative control and treated in the same manner as the liposome-containing samples. The liposomes were separated from the unbound plasma proteins by ultracentrifugation. The resultant plasma was transferred into OptiSeal™ centrifuge tubes for ultracentrifugation at 100000 × g for 60 min at 4 °C (Optima™ LE-80 K Ultracentrifuge with the Type 70.1 Ti rotor, Beckman Coulter). This washing step was repeated thrice. The supernatant was removed by decantation, and the liposome pellet was resuspended in PBS to a final volume of 100 μL and stored in the −80 °C freezer until analysis. The experiment was repeated thrice using plasma from three different donors.
2.9 Analysis of factor H and AT recruitment onto the liposomes and iC3b generation
We performed immunoassays using Wes (ProteinSimple, Santa Clara, California, USA) to study the binding of factor H to the modified liposomes incubated in factor H solution, as described in Section 2.6. Goat anti-human factor H (4 μg mL−1, AF4779, R&D Systems, Minneapolis, MN, USA) was used as the primary antibody together with the Wes goat detection module (DM-006, ProteinSimple). The samples were diluted 1/5 in 0.1 × Wes sample buffer. Factor H (0.31 μg mL−1 in PBS) was used as a positive control.We also studied whether recruited factor H could work as a cofactor in cleaving C3b to iC3b in the presence of factor I using the prepared samples described in Section 2.8. A polyclonal rabbit anti-human C3c antibody (Q0368, Dako, Glostrup, Denmark) diluted 1/100 was used as the primary antibody, together with the Wes rabbit detection module (DM-001, ProteinSimple). The samples were diluted 1/10 in 0.1 × Wes sample buffer, and for the factor I titration experiment, the samples were diluted 1/5 in 0.1 × Wes sample buffer. Purified factor H, factor I, and C3b (A137, A138, A114, Complement Technology) were used as controls, which were diluted in 0.1 × Wes sample buffer to a final concentration of 10 μg mL−1. Native C3 was purified from plasma according to Hammer et al.,38 and iC3b, produced as described by Ekdahl et al. by incubating C3b with factor I with factor H as a cofactor,40 was used as a control. All controls were diluted to a final concentration of 10 μg mL−1. We investigated the binding of factor H to liposomes incubated in human EDTA plasma using a goat detection module (DM-006, ProteinSimple) and a goat anti-human factor H antibody (4 μg mL−1, AF4779, RnD) as the primary antibody. A standard curve of human factor H (A137, Complement Technology) ranging from 2.5 to 0.16 μg mL−1 was made to calculate the concentration of factor H in the samples. Liposome samples were diluted 1/3. To investigate AT binding to liposomes incubated in EDTA plasma, we used the Wes anti-rabbit detection module (DM-001, ProteinSimple) together with the primary rabbit anti-human AT III antibody (A-9522, Sigma Chemical Company, St. Louis, MO, USA) diluted 1/1000. Human AT (10 μg mL−1; Baxter AG) was used as a positive control. The liposome samples were diluted to 1/5.
All Wes analyses were performed according to standard settings, and the samples were prepared according to the manufacturer's protocol under reducing conditions with DTT. The Wes EZ Standard pack 1 (molecular weight spans: 12–230 kDa, PS-ST01EZ, ProteinSimple) was used for all analyses. All samples and controls were diluted in 0.1 × Wes sample buffer, and the primary antibodies were diluted in Wes antibody diluent 2. The samples were analyzed using Compass software (version 4.0.0), and a 4PL fit was used for the standard curve.
2.10 Statistical analysis
All experiments were repeated at least three times, except for the DLS data of the liposomes before and after modification with fHep-K8C-lipids. All data were analyzed using GraphPad Prism 9 for Mac OS version 9.4.1 (GraphPad Software, La Jolla, CA, USA). Data are presented as mean values ± SD or as representative images. Statistical calculations for the QCM-D experiments were performed using a one-way ANOVA, followed by Tukey's multiple comparison test. For the Wes data, statistical calculations were performed using the Wilcoxon matched-pairs signed-rank test. The calculated p-values were defined as follows: *p < 0.05, **p < 0.001, ***p < 0.001, and ****p < 0.0001.3. Results
3.1 Evaluation of the interaction of factor H and C4BP with different fHep-lipids
Heparin-conjugated lipids, fHep-C-lipid, fHep-K1C-lipid, fHep-K2C-lipid, fHep-K4C-lipid, and fHep-K8C-lipid (Fig. 1A–C), were synthesized as described previously.13 We analyzed the interaction of fHep-lipids with factor H or C4BP using QCM-D (Fig. 2), where hydrophobic C12-SAM coated gold sensors were used for the immobilization of fHep-lipids.13 The QCM-D analysis revealed that the fHep-lipids recruited more factor H, as evident from the factor H binding values; fHep-C-lipid 30.6 ± 30.6 ng cm−2, fHep-K1C-lipid 58.3 ± 41.8 ng cm−2, fHep-K2C-lipid 80.8 ± 27.2 ng cm−2, fHep-K4C-lipid 85.1 ± 25.6 ng cm−2, and fHep-K8C-lipid 140.8 ± 17.2 ng cm−2, whereas little factor H was bound to MeO-PEG-lipid (8.4 ± 6.8 ng cm−2) (Fig. 2A–C). More factor H bound to fHep-lipids, with an increase in the number of conjugated fHep molecules/chains compared to MeO-PEG-lipids (p-value = 0.0419). fHep-K8C-lipids bound more factor H than MeO-PEG-lipids (p-value = 0.0007) or fHep-C-lipids (p-value = 0.0270). Representative complete sensorgrams of factor H binding to MeO-PEG-lipids, fHep-C-lipid, fHep-K1C-lipid, fHep-K2C-lipid, fHep-K4C-lipid, and fHep-K8C-lipids are shown in Fig. S1A–F and S2A–F (ESI†). When analyzing the binding of C4BP to fHep-lipids by QCM-D, more C4BP bound to fHep-K8C-lipid (72.8 ± 31.3 ng cm−2), which was significantly higher than the amount of C4BP bound to MeO-PEG-lipid (2.1 ± 3.6 ng cm−2) (p-value = 0.0169) (Fig. 2D–F). Conversely, little binding of C4BP was observed onto other fHep-lipids-immobilized surfaces (fHep-C-lipid 19.3 ± 12.0 ng cm−2, fHep-K1C-lipid 28.7 ± 30.4 ng cm−2, fHep-K2C-lipid 25.2 ± 22.4 ng cm−2, and fHep-K4C-lipid 22.9 ± 15.6 ng cm−2). Representative complete sensorgrams of the binding of C4BP to MeO-PEG-lipids, fHep-C-lipid, fHep-K1C-lipid, fHep-K2C-lipid, fHep-K4C-lipid, and fHep-K8C-lipids are shown in Fig. S3A–F and S4A–F (ESI†). These results suggested that fragmented heparin conjugated to lipids retains both factor H- and C4BP-binding sites. The more heparin chains conjugated to the lipids, the more factor H and C4BP were bound, as demonstrated by the fact that fHep-K8C-lipids recruited the most factor H and C4BP in the QCM-D experiments. We also found similar results when we studied AT binding to fHep-lipids using QCM-D, where more AT was recruited to fHep-lipids with an increasing number of fragmented heparin chains.13 The fHep-K8C-lipid bound the most to factor H and C4BP, indicating that there were more binding sites available. However, even though fHep-K8C-lipids had almost double the number of fHep-chains conjugated to lipids, they did not recruit twice as much factor H as fHep-K4C-lipids (Fig. 2C). Presumably, not all binding sites were fully accessible because of the high density of heparin at the end of the lipid. For the binding of C4BP to heparin, the correlation with the heparin numbers is non-linear, as shown in Fig. 2F. Blom et al. characterized the heparin-binding site at C4BP with a cluster of four positively charged amino acids (Arg39, Arg64, Lys63, and Arg66). Only fHep-K8C-lipid provided a sufficiently high density of heparin charges to enable complete binding, which was not achieved for KnC with n = 0–4, which led to incomplete binding of C4BP.15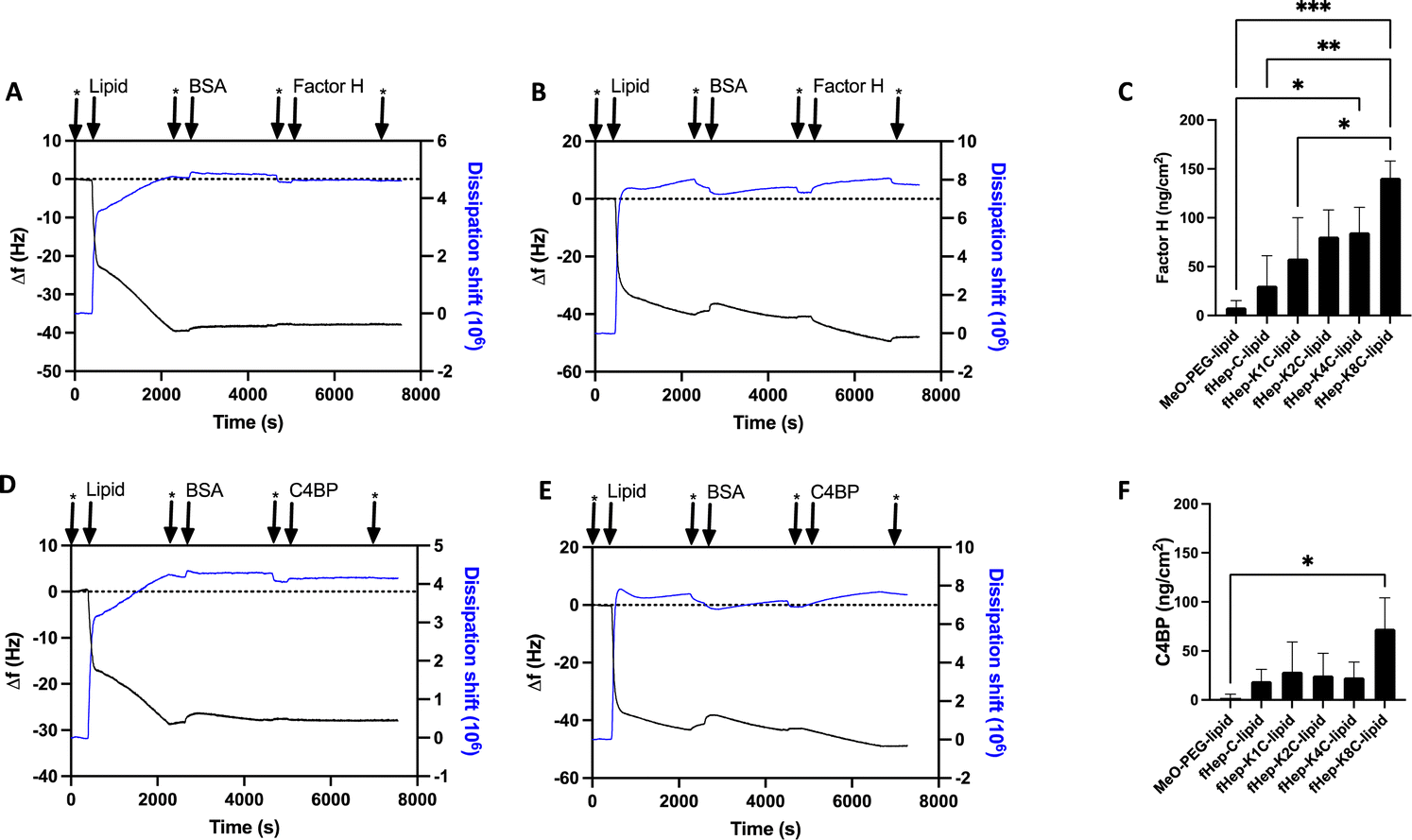 | ||
| Fig. 2 QCM-D analysis of interaction with purified factor H and C4BP. (A)–(C) Analysis of interaction between MeO-PEG-lipid or fragmented-heparin-conjugated lipids (fHep-lipids) with 50 μg mL−1 complement-regulator factor H. BSA (1 mg mL−1) was used for blocking unspecific binding to the sensors. Representative sensorgrams of: (A) MeO-PEG-lipid, (B) fHep-K8C-lipid. (C) Quantification of the amount of bound factor H to each of the lipids. (D)–(F) QCM-D analysis of interaction between 0.1 mg mL−1 MeO-PEG-lipid or fHep-lipids and 10 μg mL−1 complement-regulator C4b-binding protein (C4BP). Representative sensorgrams of: (D) MeO-PEG-lipid, (E) fHep-K8C-lipid. (F) Quantification of the amount of bound C4BP to each of the lipids. For all experiments, n = 3. The washing steps with PBS are indicated by an asterisk (*). The sensorgrams show the resonance frequency change (Δf) and the change in dissipation (ΔD) at the 7th overtone. | ||
3.2 Influence of MeO-PEG-lipids on the interaction of fHep-lipids with factor H and AT
We tested different ratios of fHep-lipids:MeO-PEG-lipids to investigate whether the surface density of fHep-lipids affected the binding of purified factor H and AT and whether factor H and AT could compete for the same binding site in fHep-lipids (Fig. 3). Herein, we mixed fHep-lipids with MeO-PEG5k-lipids, which had the same phospholipid and PEG molecular weight (5 kDa) but a non-functional methoxy group as the end group of the PEG chain. We focused on fHep-K1C-lipid and fHep-K8C-lipid in the present study because it would be easier to understand the reactions by comparing higher and lower numbers of conjugated heparin lipids. We used a mixture of fHep-K1C-lipid/fHep-K8C-lipid and MeO-PEG-lipids with different molar ratios (0:100, 25:75, 50:50, 75:25, and 100:0 fHep-lipid:MeO-PEG-lipid) for QCM-D analysis, and the biniding of factor H, followed by AT was analyzed. Binding of AT to lipids without factor H is shown in Fig. S5 (ESI†).
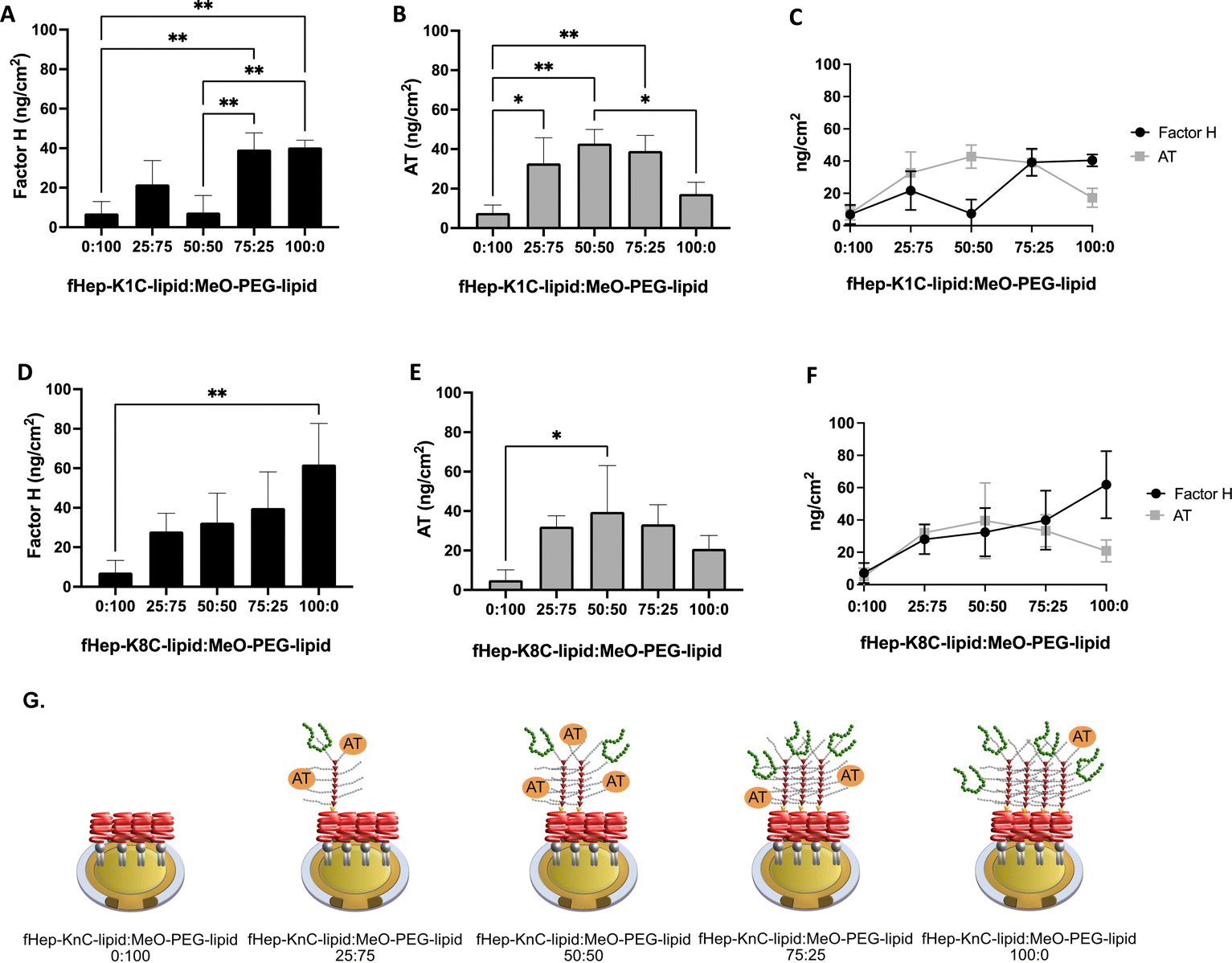 | ||
| Fig. 3 QCM-D analysis of interaction with purified factor H and AT. Analysis of different ratios of fragmented-heparin-conjugated lipids (fHep-lipids):MeO-PEG-lipids (0:100, 25:75, 50:50, 75:25, and 100:0), followed by flowing of 25 μg mL−1 factor H and then 25 μg mL−1 antithrombin (AT). (A) Quantification of the amount of factor H bound to different ratios of fHep-K1C-lipid:MeO-PEG-lipid, (B) quantification of the amount of AT bound to different ratios of fHep-K1C-lipid:MeO-PEG-lipid, (C) summary of the amount of factor H and AT bound to different ratios of fHep-K1C-lipid:MeO-PEG-lipid, (D) quantification of the amount of factor H bound to different ratios of fHep-K8C-lipid:MeO-PEG-lipid, (E) quantification of the amount of AT bound to different ratios of fHep-K8C-lipid:MeO-PEG-lipid, and (F) summary of the amount of factor H and AT bound to different ratios of fHep-K8C-lipid:MeO-PEG-lipid. For all experiments, n = 3. (G) Illustration of factor H (green) and AT binding (orange) to different ratios of fHep-KnC-lipids:MeO-PEG-lipids (0:100, 25:75, 50:50, 75:25, and 100:0). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :MeO-PEG-lipids.
The binding of purified factor H to fHep-K1C-lipid:MeO-PEG-lipid at different molar ratios was analyzed (Fig. 3A). There was more binding of factor H to fHep-K1C-lipid:MeO-PEG-lipid with a molar ratio of 75:25 (39.3 ± 8.5 ng cm−2) when compared to molar ratios of 0:100 (p-value = 0.0051) and 50:50 (p-value = 0.0057). More factor H was also bound to fHep-K1C-lipid:MeO-PEG-lipid with a molar ratio of 100:0 (40.5 ± 3.7 ng cm−2) when compared to molar ratios of 0:100 (p-value = 0.0041) and 50:50 (p-value = 0.0045). Conversely, low binding of factor H was observed for fHep-K1C-lipid:MeO-PEG-lipid with ratios of 0:100 (7.0 ± 6.0 ng cm−2), 25:75 (21.7 ± 12.1 ng cm−2), and 50:50 (7.4 ± 8.7 ng cm−2). Therefore, almost the same amount of factor H was bound to fHep-K1C-lipid:MeO-PEG-lipid at molar ratios of 75:25 and 100:0. The representative complete sensorgrams are shown in Fig. S6A–E and S7A–E (ESI†). For the binding of AT after the flowing of factor H to different molar ratios of fHep-K1C-lipid:MeO-PEG-lipid, a significant increase in AT binding was observed at molar ratios of 25:75 (32.7 ± 13.0 ng cm−2) (p-value = 0.0243 when compared to a molar ratio of 0:100), 50:50 (42.8 ± 7.2 ng cm−2) (p-value = 0.0026 when compared to a molar ratio of 0:100), and 75:25 (39.1 ± 7.9 ng cm−2) (p-value = 0.0058 when compared to a molar ratio of 0:100) (Fig. 3B and C).
:MeO-PEG-lipids.
There was a tendency that the binding of purified factor H increased with an increase of the ratio of fHep-K8C-lipid, and the binding of factor H significantly increased to fHep-K8C-lipid:MeO-PEG-lipid with a molar ratio of 100:0 (61.9 ± 20.8 ng cm−2) when compared to a molar ratio of 0:100 (7.2 ± 6.2 ng cm−2) (p-value = 0.0079) (Fig. 3D). No significant difference in factor H binding was observed among the molar ratios of 0:100, 25:75, 50:50, and 75:25. The binding of purified AT following the flowing of factor H, a significant increase in AT-binding was observed at the molar ratio of 50:50 (39.6 ± 23.5 ng cm−2) when compared to a molar ratio of 0:100 (p-value = 0.0389) (Fig. 3E and F). Although there was no significant difference between the molar ratio of 50:50 and the molar ratio of 100:0 (20.9 ± 6.8 ng cm−2), it seemed that there was a decrease in AT-binding. Representative complete sensorgrams are shown in Fig. S8A–E and S9A–E (ESI†). The general trend observed in these experiments was that higher molar ratios of fHep-lipids used more factor H to be recruited to the surface, whereas the binding of AT decreased with an increase in factor H-binding beyond the maximum AT surface density at a 50:50 molar ratio (Fig. 3G). These results indicated that factor H either bound close to the binding site of AT on fragmented heparin or sterically hindered AT binding. This is a competitive test in a model system using purified proteins; therefore, this binding profile may not be present in a more complex system, such as human blood. Factor H-related proteins can possibly interact with fHep-lipids because of their structural similarity to factor H; therefore, we must consider this when evaluating the regulatory effect of fHep-lipids on the complement system.25
:MeO-PEG-lipids.
The binding of purified factor H to fHep-K1C-lipid:MeO-PEG-lipid at different molar ratios was analyzed (Fig. 3A). There was more binding of factor H to fHep-K1C-lipid:MeO-PEG-lipid with a molar ratio of 75:25 (39.3 ± 8.5 ng cm−2) when compared to molar ratios of 0:100 (p-value = 0.0051) and 50:50 (p-value = 0.0057). More factor H was also bound to fHep-K1C-lipid:MeO-PEG-lipid with a molar ratio of 100:0 (40.5 ± 3.7 ng cm−2) when compared to molar ratios of 0:100 (p-value = 0.0041) and 50:50 (p-value = 0.0045). Conversely, low binding of factor H was observed for fHep-K1C-lipid:MeO-PEG-lipid with ratios of 0:100 (7.0 ± 6.0 ng cm−2), 25:75 (21.7 ± 12.1 ng cm−2), and 50:50 (7.4 ± 8.7 ng cm−2). Therefore, almost the same amount of factor H was bound to fHep-K1C-lipid:MeO-PEG-lipid at molar ratios of 75:25 and 100:0. The representative complete sensorgrams are shown in Fig. S6A–E and S7A–E (ESI†). For the binding of AT after the flowing of factor H to different molar ratios of fHep-K1C-lipid:MeO-PEG-lipid, a significant increase in AT binding was observed at molar ratios of 25:75 (32.7 ± 13.0 ng cm−2) (p-value = 0.0243 when compared to a molar ratio of 0:100), 50:50 (42.8 ± 7.2 ng cm−2) (p-value = 0.0026 when compared to a molar ratio of 0:100), and 75:25 (39.1 ± 7.9 ng cm−2) (p-value = 0.0058 when compared to a molar ratio of 0:100) (Fig. 3B and C).
:MeO-PEG-lipids.
There was a tendency that the binding of purified factor H increased with an increase of the ratio of fHep-K8C-lipid, and the binding of factor H significantly increased to fHep-K8C-lipid:MeO-PEG-lipid with a molar ratio of 100:0 (61.9 ± 20.8 ng cm−2) when compared to a molar ratio of 0:100 (7.2 ± 6.2 ng cm−2) (p-value = 0.0079) (Fig. 3D). No significant difference in factor H binding was observed among the molar ratios of 0:100, 25:75, 50:50, and 75:25. The binding of purified AT following the flowing of factor H, a significant increase in AT-binding was observed at the molar ratio of 50:50 (39.6 ± 23.5 ng cm−2) when compared to a molar ratio of 0:100 (p-value = 0.0389) (Fig. 3E and F). Although there was no significant difference between the molar ratio of 50:50 and the molar ratio of 100:0 (20.9 ± 6.8 ng cm−2), it seemed that there was a decrease in AT-binding. Representative complete sensorgrams are shown in Fig. S8A–E and S9A–E (ESI†). The general trend observed in these experiments was that higher molar ratios of fHep-lipids used more factor H to be recruited to the surface, whereas the binding of AT decreased with an increase in factor H-binding beyond the maximum AT surface density at a 50:50 molar ratio (Fig. 3G). These results indicated that factor H either bound close to the binding site of AT on fragmented heparin or sterically hindered AT binding. This is a competitive test in a model system using purified proteins; therefore, this binding profile may not be present in a more complex system, such as human blood. Factor H-related proteins can possibly interact with fHep-lipids because of their structural similarity to factor H; therefore, we must consider this when evaluating the regulatory effect of fHep-lipids on the complement system.25
3.3 Characterization of modified liposomes
Next, we analyzed the liposomes before and after exogenous addition of fHep-K8C-lipids using DLS. The DLS data showed that the size of the liposomes increased by an average of 23 nm in diameter after modification with fHep-K8C-lipids (Fig. 4A), and the PDI values indicated the absence of any aggregation for the modified liposomes, as the value was <0.1 (Fig. 4B). The zeta potential shifted to the negative side from −5.8 ± 9.0 mV for non-modified liposomes to −22.0 ± 2.9 mV for MeO-PEG-lipid-modified liposomes, and −52.6 ± 3.4 mV for fHep-K8C-lipid-modified liposomes (Fig. 4C). This suggested that the liposomes could be modified via external modification, where liposome surfaces are modified by exogenously mixing fHep-lipids with liposomes.41 The long-term stability of the modified liposomes was also monitored over a period of 90 days, and no changes in size (Fig. 4D), PDI (Fig. 4E), or zeta potential (Fig. 4F) were observed over 60 days. However, after 90 days, the PDI for some of the modified liposome batches was >0.1, indicating that this may be the limit of liposome stability. Nevertheless, modification with fHep-lipids was stable over time, and most of the fHep-K8C-lipid did not detach from the bilayered membrane. The DLS data for the non-modified liposomes are shown in Fig. S10 (ESI†). We previously described a two-step coating method using a heparin-binding peptide-conjugated PEG-lipid, followed by heparinization, which was used to modify cell surfaces and prevent thromboinflammation in vitro.42 However, the advantage of fHep-lipids is that they are facile single-step coatings that can be used to modify the surface of both liposomes and cells for transplantation.13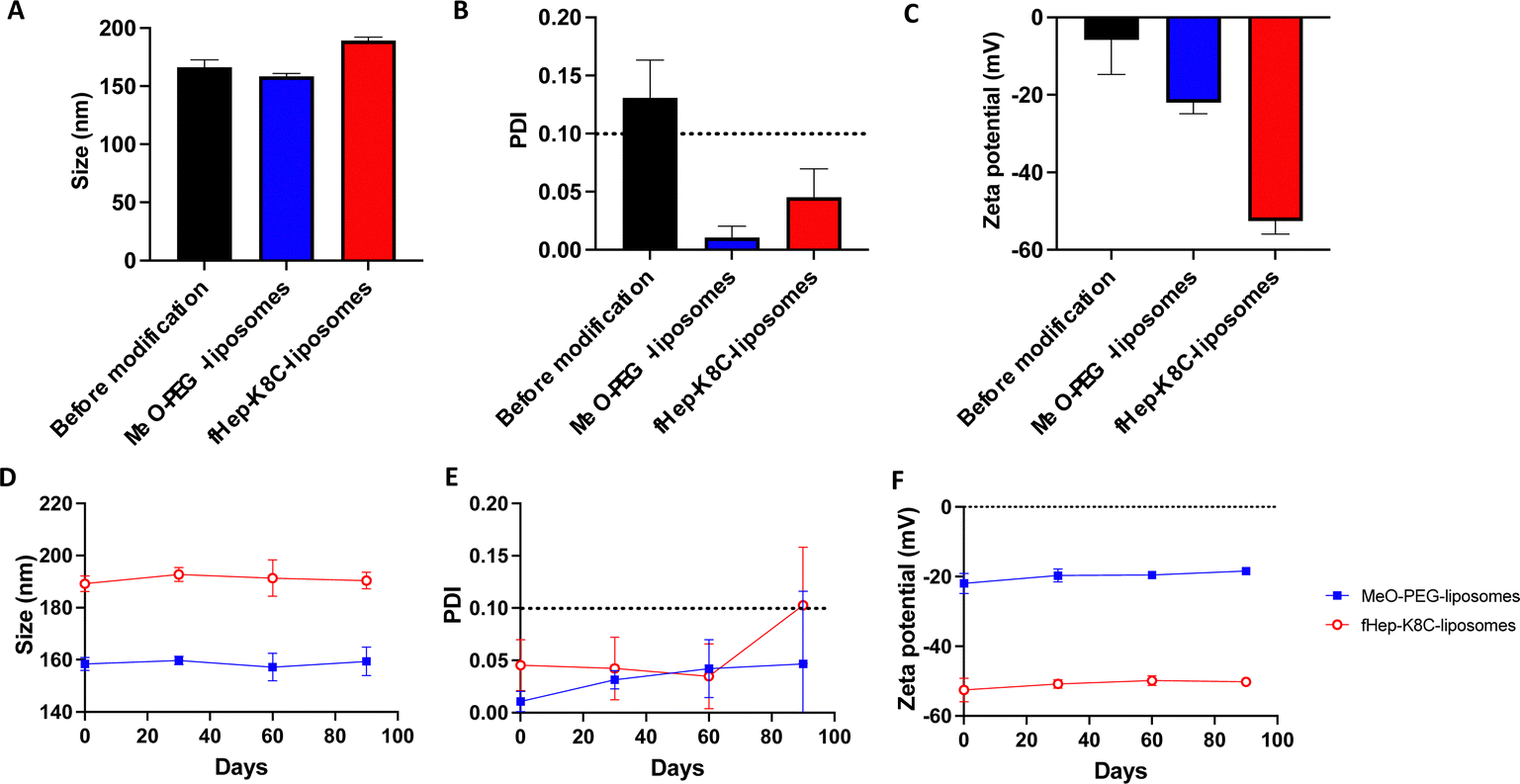 | ||
| Fig. 4 DLS analysis of modified liposomes. Analysis of liposomes before modification (black) and after modification with MeO-PEG-lipid (blue) or fHep-K8C-lipid (red) in 1 mM NaCl aqueous solution by measuring (A) size, (B) polydispersity index (PDI) and (C) zeta potential (n = 4). Long-term stability test (90 days) of liposomes modified with MeO-PEG-lipid and fHep-K8C-lipid stored in PBS at 4 °C. (D) Size, (E) PDI and (F) zeta potential (n = 4). | ||
3.4 Factor H recruitment and iC3b generation via immobilized fHep-lipids
Next, we aimed to investigate factor H recruitment on the modified liposome surface using the Wes (ProteinSimple) immunoassay, where the amount of bound factor H on the liposome surface could be quantified (Fig. 5). The liposomes were incubated in purified factor H, and unbound proteins were removed by ultracentrifugation. More factor H was detected on the surface of fHep-K8C-lipid-modified liposomes than on the surface of MeO-PEG lipid-modified liposomes. Although there was non-specific binding of factor H on MeO-PEG-lipid-modified liposomes, factor H-binding was more pronounced for the fHep-K8C-lipid on the membrane (Fig. 5A–C). We also found that the factor H bound to the modified liposomes enabled the factor I-mediated degradation of C3b to iC3b, as indicated by the generated 40k-fragment and the reduction in the α-chain band at around 110 kDa (Fig. 6A). C3b bound to factor H undergoes conformational changes, resulting in a 15-fold increase in its affinity for factor I.43,44 As described above, the heparin-binding sites of factor H have been shown to exist within CCP 6–8, 19–20 and CCP 11–13.24 In our study, we could not determine to which of the factor H CCP domain(s) the fragmented heparin bound to. However, we found that factor H recruited onto the liposome surface was still active, that is, it assisted in the degradation of C3b to iC3b, suggesting that the N-terminal CCP1–4 domains are still available and not rigidly bound to the fHep-lipids.24,25 Interestingly, factor H also bound to MeO-PEG-liposomes, which was unexpected because we did not observe strong binding in the QCM-D study. Presumably, the PEG density on the liposomes was different from that on the QCM sensor chip surface, which enabled factor H to interact with the PEG chains directly or be caught between PEG chains. We also changed the concentration of factor I (5, 0.5 and 0.05 μg mL−1) for this study, and found that the fHep-K8C-liposomes pre-incubated with factor H were more effective at cleaving C3b to iC3b (Fig. 6B), indicating that the more the factor H that is found on the liposome surface, the more effective it is at degrading C3b.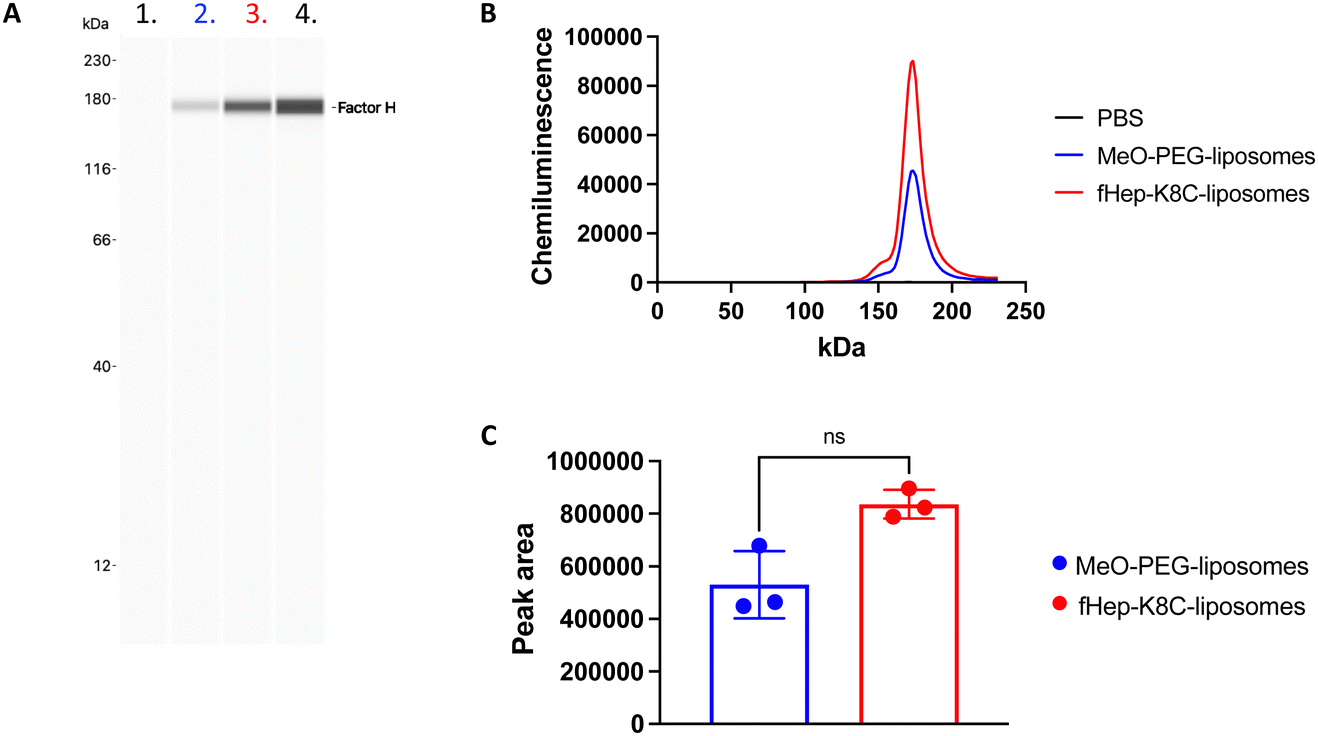 | ||
| Fig. 5 Wes immunoassay of modified liposomes. Measurements of factor H recruitment to the surface of 100 μL 1.0 mg mL−1 cholesterol MeO-PEG-lipid- (blue) and fHep-K8C-lipid- (red) modified liposomes incubated in 100 μL 50 μg mL−1 factor H for 60 min at 37 °C at 20 rpm, followed by the removal of unbound factor H using ultracentrifugation. A sample with only PBS and no liposomes, treated the same way as the liposome-containing samples, was used as a negative control. A goat-anti-human factor H antibody was used as the detection antibody. (A) Representative Wes virtual blot, lane 1: negative control with the PBS sample, lane 2: MeO-PEG-liposomes, lane 3: fHep-K8C-liposomes, and lane 4: positive control with 0.31 μg mL−1 factor H. (B) Representative chromatogram of the peak area of recruited factor H, and (C) comparison of peak area of factor H recruited to MeO-PEG-liposomes and fHep-K8C-liposomes (n = 3). | ||
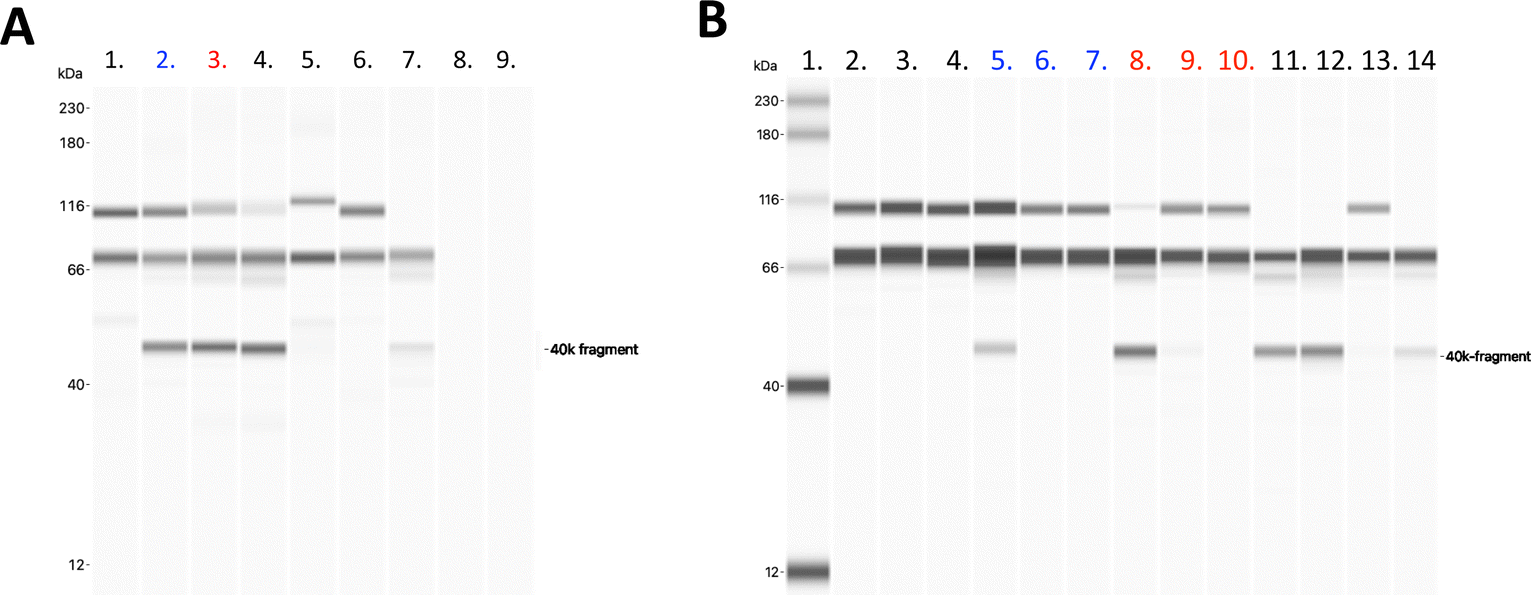 | ||
| Fig. 6 (A) Wes immunoassay of modified liposomes for iC3b. Measurements of iC3b generation by MeO-PEG-liposomes and fHep-K8C-liposomes after being incubated in 50 μg mL−1 factor H, followed by incubation in 150 μg mL−1 C3b and 20 μg mL−1 factor I for 15 min at 37 °C. Additionally, a sample with PBS and no liposomes, treated the same way as the liposome-containing samples, was used as a negative control. A sample with no liposomes and 50 μg mL−1 factor H, 150 μg mL−1 C3b, and 20 μg mL−1 factor I, which had not been subjected to ultracentrifugation, was used as a positive control. A polyclonal rabbit anti-human C3c antibody was used as the primary antibody. Representative Wes virtual blot, lane 1: negative control with the PBS sample, lane 2: MeO-PEG-liposome, lane 3: fHep-K8C-liposome, lane 4: positive control with factor H, C3b, and factor I, lane 5: 10 μg mL−1 native C3, lane 6: 10 μg mL−1 C3b, lane 7: 10 μg mL−1 iC3b, lane 8: 10 μg mL−1 factor H, and lane 9: 10 μg mL−1 factor I. For all experiments, n = 3. (B) Wes immunoassay measurements of iC3b generation by MeO-PEG-liposomes and fHep-K8C-liposomes after being incubated in 50 μg mL−1 factor H, followed by incubation in 75 μg mL−1 C3b and 5/05/0.05 μg mL−1 factor I for 15 min at 37 °C. Additionally, a sample with PBS and no liposomes, treated the same way as the liposome-containing samples, was used as a negative control. A sample with no liposomes and 50 μg mL−1 factor H, 75 μg mL−1 C3b, and 5/05/0.05 μg mL−1 factor I, which had not been subjected to ultracentrifugation, was used as a positive control. A polyclonal rabbit anti-human C3c antibody was used as the primary antibody. Representative Wes virtual blot, lane 1: 12–230 kDa biotinylated ladder, lane 2–4: 5/0.5/0.05 μg mL−1 factor I plus C3b incubated with the negative control with the PBS sample, lane 5–7: 5/0.5/0.05 μg mL−1 factor I plus C3b incubated with factor H-incubated MeO-PEG-liposomes, lane 8–10: 5/0.5/0.05 μg mL−1 factor I plus C3b incubated with factor H-incubated fHep-K8C-liposome, lane 11–13; 5/0.5/0.05 μg mL−1 factor I plus C3b incubated with 50 μg mL−1 factor H, as a positive control, lane 14; 10 μg mL−1 iC3b (n = 1). | ||
3.5 Factor H and AT recruitment to liposomes incubated in human plasma
We then attempted to detect factor H on the modified liposome surfaces after exposure to human EDTA plasma (from three donors) using the Wes immunoassay (Fig. 7A–C). There was a strong binding of factor H detected on fHep-K8C-lipid-modified liposomes (0.99 ± 0.54 μg mL−1 on liposomes with [cholesterol] = 1.4 mg mL−1) when compared to MeO-PEG-lipid-modified liposomes (0.29 ± 0.21 μg mL−1), where non-specific binding of factor H was observed on MeO-PEG-lipid-modified liposomes, as described in the previous section. However, factor H-binding on the membrane was more pronounced by fHep-K8C-lipid as well, even in the presence of human plasma. For all donors, there was a clear trend that more factor H was recruited to fHep-K8C-lipid-modified liposomes than to MeO-PEG lipid-modified liposomes (Fig. 7C). When we studied AT-binding on modified liposomes after exposure to human EDTA plasma using the Wes immunoassay, AT was also specifically detected on fHep-K8C-lipid-modified liposomes but not on MeO-PEG-lipid-modified liposomes (Fig. 7D and E). In Fig. 7D, an additional band was detected at approximately 90 kDa, in addition to the AT band (60 kDa). We speculate that the 90 kDa band could be a complex of AT with thrombin (37 kDa). These results clearly indicated that fHep-K8C-lipid-modified liposomes were able to specifically recruit both factor H and AT onto the surface of human plasma. This is in line with the results from the QCM-D experiments (Fig. 2 and 3) as well as the results from the binding assay with purified factor H (Fig. 5).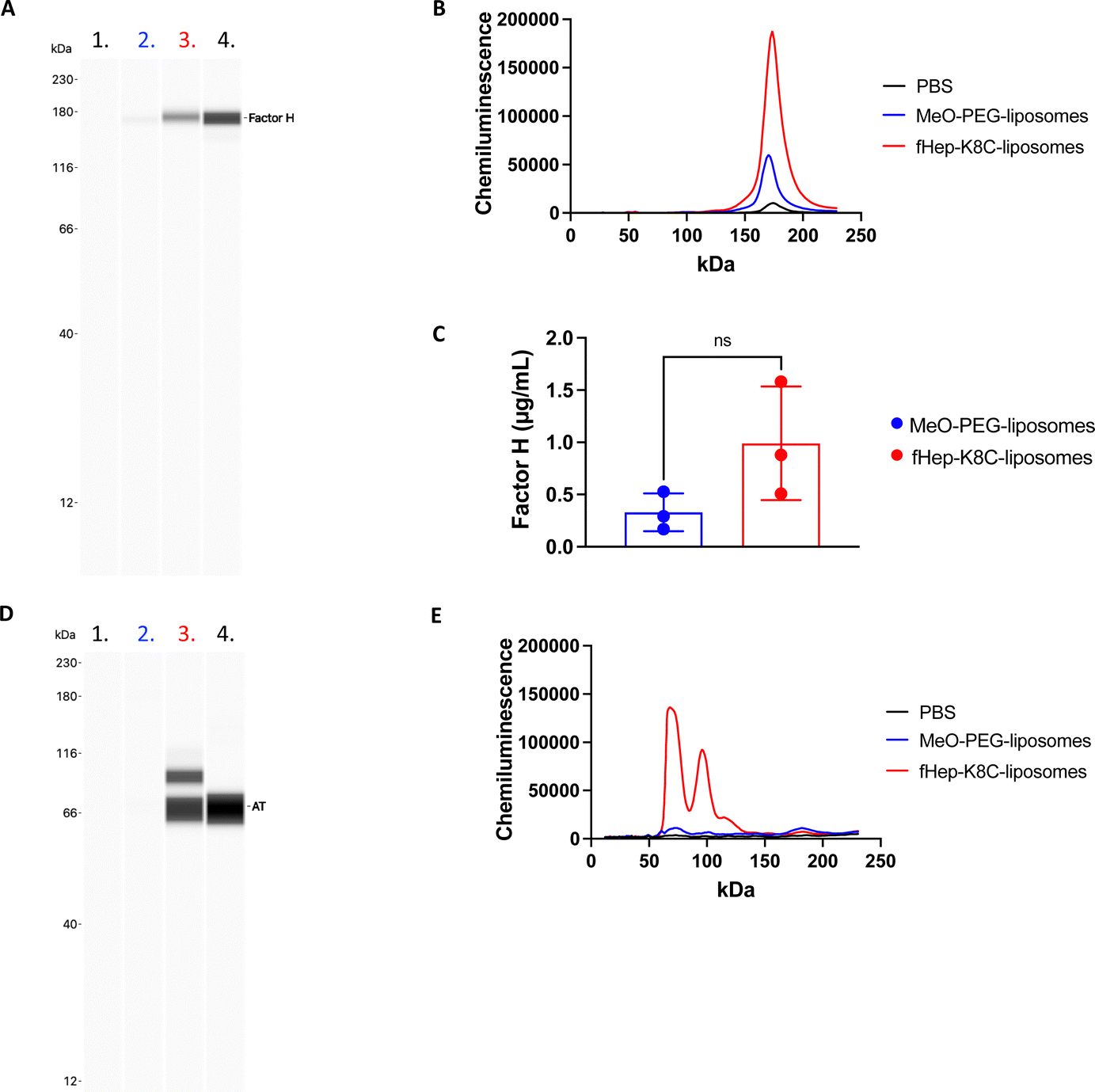 | ||
| Fig. 7 Wes immunoassay of modified liposomes for factor H and AT. Measurements of factor H recruitment to the surface of MeO-PEG-lipid- (blue) and fHep-K8C-lipid- (red) modified liposomes incubated in human EDTA plasma (from three different donors) for 60 min at 37 °C at 20 rpm, followed by the removal of unbound plasma proteins using ultracentrifugation. A sample with only PBS and no liposomes, treated the same way as the liposome-containing samples, was used as a negative control. A goat-anti-human factor H antibody was used as the detection antibody. (A) Representative Wes virtual blot, lane 1: negative control with the PBS sample, lane 2: MeO-PEG-liposomes, lane 3: fHep-K8C-liposomes, and lane 4: positive control with 0.625 μg mL−1 factor H. (B) Representative chromatogram of the peak area of recruited factor H, and (C) quantification of the peak area of factor H recruited to MeO-PEG-liposomes and fHep-K8C-liposomes (n = 3). (D) and (E) Wes immunoassay measurements of antithrombin (AT) recruitment to the surface of MeO-PEG-lipid and fHep-K8C-lipid-modified liposomes incubated in human EDTA plasma for 60 min at 37 °C at 20 rpm, followed by the removal of unbound plasma proteins using ultracentrifugation. A sample with PBS and no liposomes, treated the same way as the liposome-containing samples, was used as a negative control. A rabbit anti-human ATIII antibody was used as the detection antibody. (D) Representative Wes virtual blot, lane 1: negative control with the PBS sample, lane 2: MeO-PEG-liposomes, lane 3: fHep-K8C-liposomes, and lane 4: positive control with 10 μg mL−1 AT. (E) Representative chromatogram of the peak area of recruited AT. For all experiments, n = 3. | ||
4. Discussion
Liposome coatings are used to reduce protein adsorption on liposomal surfaces, thus trying to avoid the rapid clearance of liposomes. Consolidated identities, such as lipid composition, surface charge, and liposome size, influence the proteins that are adsorbed to the surface, thereby giving liposomes their biological identity.45,46 One approach to suppress the activation of the immune system against liposomes is to recruit specific proteins, such as complement regulators, to the liposomal surface.Currently, the gold standard coating for clinically used liposomes is PEG coating.47,48 Although PEGylation is not sufficient to regulate immunity, as there are no regulators of PEGylated liposomes. In 2000, Dams et al. published a paper showing that PEGylated liposomes are rapidly removed from the circulation upon repeated injections in rats by the generation of anti-PEG antibodies, which was called the accelerated blood clearance (ABC) effect.49–51 The ABC effect has been described in several other animal models.52,53 Recently, this phenomenon has been observed in humans.54 In our system, fHep is conjugated to a PEG–lipid, where it can recruit specific proteins, such as factor H, C4BP, and AT, which contain heparin-binding domains. When using fHep-lipids for coating transplanted cells, the ABC effect, in theory, should not be an issue unless the patient has pre-existing anti-PEG antibodies,55 since the coated transplanted cells are intended to be injected into the body only once. In this paper we showed that fHep-K8C-lipids can successfully be used for the stable coating of liposomes (60 days). Therefore, they have the potential to be used as a coating strategy for liposomal drug-delivery systems to suppress immune activation due to the surface recruitment of factor H and C4BP. However, considering the ABC-effect in the development of immunologically inert liposomal drug-delivery systems, it would be interesting to investigate fHep-conjugation with different polymer lipids, for example, poly(2-methacryloyloxyethyl phosphorylcholine)-conjugated lipids.56–59 To support these in vitro findings of fHep-lipids, conducting in vivo experiments is an essential next step to further evaluate the hemocompatibility and potential of the fHep-lipids to recruit complement regulators to suppress innate immunity.
5. Conclusions
The factor H-, C4BP-, and AT-binding capacity of fHep-lipids increased with increasing numbers of conjugated heparins and higher surface densities of fHep-lipids. Liposomes were successfully coated with fHep-K8C-lipids and their size, PDI, and zeta potential were stable over time (60 days). Factor H bound to fHep-K8C-lipid-modified liposomes enabled factor I-mediated degradation of C3b to iC3b. Finally, fHep-K8C-liposomes can recruit factor H and AT from human plasma. Therefore, in addition to being a promising coating strategy of therapeutic cells to suppress the coagulation system by recruiting AT to the cell surface after injection, our in vitro findings suggest that coating with fHep-lipids could also be a potential liposome and cell-surface coating strategy to regulate the activation of the immune system on the coated surface by recruitment of complement regulators factor H and C4BP.Author contributions
A. A. and Y. T. conceptualized and administered the project. Y. T. supervised the project. The QCM-D investigations were performed by A. A., M. F. and K. F. Liposome production, characterization and Wes investigations were performed by A. A. A. A. was responsible for the data curation and performed the formal analysis. B. N, Y. T. and G. L. provided the resources and received the financial support for the project. A. A. and Y. T. and wrote the original draft. M. F., K. F., K. N. E., G. L. and B. N. reviewed and edited the manuscript. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the Bilateral Joint Research Project (Japan–Sweden) of the Japan Society for the Promotion of Science (JSPS) and STINT, a Grant-in-Aid for Scientific Research (B) (18H03528, 22H03966) a Grant-in-Aid for Scientific Research for Fostering Joint International Research (18KK0305) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, StemTherapy, and grants 2018-04199 from the Swedish Research Council. The research project RELIEF has received funding from the Eurostars-2 joint program (project ID: E! 113670) with co-funding from the European Union Horizon 2020 Research and Innovation Program and further co-funding for the French, German, Dutch, Swedish partners from BPIfrance, the German Federal Ministry of Education and Research, RVO, VINNOVA. We also thank Dr Kenta Asawa (U Tokyo) for useful discussions regarding heparin synthesis.References
- T. M. Allen and P. R. Cullis, Liposomal drug delivery systems: from concept to clinical applications, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Liposome: classification, preparation, and applications, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- R. H. Fang, B. T. Luk, C. M. Hu and L. Zhang, Engineered nanoparticles mimicking cell membranes for toxin neutralization, Adv. Drug Delivery Rev., 2015, 90, 69–80 CrossRef CAS PubMed.
- R. J. Linhardt and T. Toida, Role of glycosaminoglycans in cellular communication, Acc. Chem. Res., 2004, 37, 431–438 CrossRef CAS PubMed.
- D. A. Simon Davis and C. R. Parish, Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity, Front. Immunol., 2013, 4, 470 Search PubMed.
- R. Biran and D. Pond, Heparin coatings for improving blood compatibility of medical devices, Adv. Drug Delivery Rev., 2017, 112, 12–23 CrossRef CAS PubMed.
- S. Cabric, J. Sanchez, T. Lundgren, A. Foss, M. Felldin, R. Kallen, K. Salmela, A. Tibell, G. Tufveson, R. Larsson, O. Korsgren and B. Nilsson, Islet surface heparinization prevents the instant blood-mediated inflammatory reaction in islet transplantation, Diabetes, 2007, 56, 2008–2015 CrossRef CAS PubMed.
- E. A. Ryan, B. W. Paty, P. A. Senior, D. Bigam, E. Alfadhli, N. M. Kneteman, J. R. Lakey and A. M. Shapiro, Five-year follow-up after clinical islet transplantation, Diabetes, 2005, 54, 2060–2069 CrossRef CAS PubMed.
- G. Moll, I. Rasmusson-Duprez, L. von Bahr, A. M. Connolly-Andersen, G. Elgue, L. Funke, O. A. Hamad, H. Lonnies, P. U. Magnusson, J. Sanchez, Y. Teramura, K. Nilsson-Ekdahl, O. Ringden, O. Korsgren, B. Nilsson and K. Le, Blanc, Are therapeutic human mesenchymal stromal cells compatible with human blood?, Stem Cells, 2012, 30, 1565–1574 CrossRef CAS PubMed.
- L. Moberg, H. Johansson, A. Lukinius, C. Berne, A. Foss, R. Kallen, O. Ostraat, K. Salmela, A. Tibell, G. Tufveson, G. Elgue, K. Nilsson Ekdahl, O. Korsgren and B. Nilsson, Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation, Lancet, 2002, 360, 2039–2045 CrossRef CAS PubMed.
- B. Nilsson, K. N. Ekdahl and O. Korsgren, Control of instant blood-mediated inflammatory reaction to improve islets of Langerhans engraftment, Curr. Opin. Organ Transp., 2011, 16, 620–626 CrossRef CAS PubMed.
- B. Nilsson, Y. Teramura and K. N. Ekdahl, The role and regulation of complement activation as part of the thromboinflammation elicited in cell therapies, Mol. Immunol., 2014, 61, 185–190 CrossRef CAS PubMed.
- K. Asawa, K. Ishihara, K. N. Ekdahl, B. Nilsson and Y. Teramura, Cell surface functionalization with heparin-conjugated lipid to supress blood activation, Adv. Funct. Mater., 2021, 31, 2008167 CrossRef CAS.
- A. R. Rezaie and H. Giri, Anticoagulant and signaling functions of antithrombin, J. Thromb. Haemost., 2020, 18, 3142–3153 CrossRef CAS PubMed.
- A. M. Blom, Structural and functional studies of complement inhibitor C4b-binding protein, Biochem. Soc. Trans., 2002, 30, 978–982 CrossRef CAS PubMed.
- M. K. Pangburn, M. A. Atkinson and S. Meri, Localization of the heparin-binding site on complement factor H, J. Biol. Chem., 1991, 266, 16847–16853 CrossRef CAS PubMed.
- S. J. Perkins, K. W. Fung and S. Khan, Molecular Interactions between Complement Factor H and Its Heparin and Heparan Sulfate Ligands, Front. Immunol., 2014, 5, 126 Search PubMed.
- V. P. Ferreira, M. K. Pangburn and C. Cortes, Complement control protein factor H: the good, the bad, and the inadequate, Mol. Immunol., 2010, 47, 2187–2197 CrossRef CAS PubMed.
- E. S. Hovingh, B. van den Broek and I. Jongerius, Hijacking Complement Regulatory Proteins for Bacterial Immune Evasion, Front. Microbiol., 2016, 7, 2004 Search PubMed.
- L. A. Trouw, A. A. Bengtsson, K. A. Gelderman, B. Dahlback, G. Sturfelt and A. M. Blom, Correction: C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack, J. Biol. Chem., 2020, 295, 9263 CrossRef CAS PubMed.
- L. A. Trouw, A. A. Bengtsson, K. A. Gelderman, B. Dahlback, G. Sturfelt and A. M. Blom, C4b-binding protein and factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack, J. Biol. Chem., 2007, 282, 28540–28548 CrossRef CAS PubMed.
- A. M. Blom, B. O. Villoutreix and B. Dahlback, Complement inhibitor C4b-binding protein-friend or foe in the innate immune system?, Mol. Immunol., 2004, 40, 1333–1346 CrossRef CAS PubMed.
- L. Kask, B. O. Villoutreix, M. Steen, B. Ramesh, B. Dahlback and A. M. Blom, Structural stability and heat-induced conformational change of two complement inhibitors: C4b-binding protein and factor H, Protein Sci., 2004, 13, 1356–1364 CrossRef PubMed.
- A. Haque, C. Cortes, M. N. Alam, M. Sreedhar, V. P. Ferreira and M. K. Pangburn, Characterization of Binding Properties of Individual Functional Sites of Human Complement Factor H, Front. Immunol., 2020, 11, 1728 CrossRef CAS PubMed.
- C. Bechtler, S. Koutsogiannaki, E. Umnyakova, A. Hamid, A. Gautam, Y. Sarigiannis, R. B. Pouw, C. Lamers, S. Rabbani, C. Q. Schmidt, J. D. Lambris and D. Ricklin, Complement-regulatory biomaterial coatings: activity and selectivity profile of the factor H-binding peptide 5C6, Acta Biomater., 2023, 155, 123–138 CrossRef CAS PubMed.
- L. Kask, A. Hillarp, B. Ramesh, B. Dahlback and A. M. Blom, Structural requirements for the intracellular subunit polymerization of the complement inhibitor C4b-binding protein, Biochemistry, 2002, 41, 9349–9357 CrossRef CAS PubMed.
- J. D. Lambris, D. Ricklin and B. V. Geisbrecht, Complement evasion by human pathogens, Nat. Rev. Microbiol., 2008, 6, 132–142 CrossRef CAS.
- S. R. Moore, S. S. Menon, C. Cortes and V. P. Ferreira, Hijacking Factor H for Complement Immune Evasion, Front. Immunol., 2021, 12, 602277 CrossRef CAS PubMed.
- A. M. Blom, L. Kask and B. Dahlback, CCP1-4 of the C4b-binding protein alpha-chain are required for factor I mediated cleavage of complement factor C3b, Mol. Immunol., 2003, 39, 547–556 CrossRef CAS PubMed.
- P. M. Sikorski, A. G. Commodaro and M. E. Grigg, Toxoplasma gondii Recruits Factor H and C4b-Binding Protein to Mediate Resistance to Serum Killing and Promote Parasite Persistence in vivo, Front. Immunol., 2019, 10, 3105 CrossRef CAS PubMed.
- P. H. Nilsson, K. N. Ekdahl, P. U. Magnusson, H. Qu, H. Iwata, D. Ricklin, J. Hong, J. D. Lambris, B. Nilsson and Y. Teramura, Autoregulation of thromboinflammation on biomaterial surfaces by a multicomponent therapeutic coating, Biomaterials, 2013, 34, 985–994 CrossRef CAS PubMed.
- Y. Q. Wu, H. Qu, G. Sfyroera, A. Tzekou, B. K. Kay, B. Nilsson, K. Nilsson Ekdahl, D. Ricklin and J. D. Lambris, Protection of nonself surfaces from complement attack by factor H-binding peptides: implications for therapeutic medicine, J. Immunol., 2011, 186, 4269–4277 CrossRef CAS PubMed.
- A. E. Engberg, K. Sandholm, F. Bexborn, J. Persson, B. Nilsson, G. Lindahl and K. N. Ekdahl, Inhibition of complement activation on a model biomaterial surface by streptococcal M protein-derived peptides, Biomaterials, 2009, 30, 2653–2659 CrossRef CAS PubMed.
- T. Oh, T. Uemura, M. Nagao, Y. Hoshino and Y. Miura, A QCM study of strong carbohydrate-carbohydrate interactions of glycopolymers carrying mannosides on substrates, J. Mater. Chem. B, 2022, 10, 2597–2601 RSC.
- Y. Teramura and H. Iwata, Surface modification of islets with PEG-lipid for improvement of graft survival in intraportal transplantation, Transplantation, 2009, 88, 624–630 CrossRef PubMed.
- S. A. Irvine, T. W. Steele, R. Bhuthalingam, M. Li, S. Boujday, M. Prawirasatya, K. G. Neoh, F. Y. Boey and S. S. Venkatraman, Quantification of aldehyde terminated heparin by SEC-MALLS-UV for the surface functionalization of polycaprolactone biomaterials, Colloids Surf., B, 2015, 132, 253–263 CrossRef CAS.
- G. Sauerbrey, Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung, Z. Physik, 1959, 206–222 CrossRef CAS.
- C. H. Hammer, G. H. Wirtz, L. Renfer, H. D. Gresham and B. F. Tack, Large scale isolation of functionally active components of the human complement system, J. Biol. Chem., 1981, 256, 3995–4006 CrossRef CAS.
- J. E. Klinth, R. Larsson, P. O. Andersson and K. N. Ekdahl, A novel application of multi-wavelength TIRF spectroscopy for real time monitoring of antithrombin interactions with immobilized heparin, Biosens. Bioelectron., 2006, 21, 1973–1980 CrossRef CAS PubMed.
- K. N. Ekdahl, U. R. Nilsson and B. Nilsson, Inhibition of factor I by diisopropylfluorophosphate. Evidence of conformational changes in factor I induced by C3b and additional studies on the specificity of factor I, J. Immunol., 1990, 144, 4269–4274 CrossRef CAS.
- H. Suzuki, A. Adler, T. Huang, A. Kuramochi, Y. Ohba, Y. Sato, N. Nakamura, V. A. Manivel, K. N. Ekdahl, B. Nilsson, K. Ishihara and Y. Teramura, Impact of spontaneous liposome modification with phospholipid polymer-lipid conjugates on protein interactions, Sci. Technol. Adv. Mater., 2022, 23, 845–857 CrossRef CAS PubMed.
- S. Asif, K. N. Ekdahl, K. Fromell, E. Gustafson, A. Barbu, K. Le Blanc, B. Nilsson and Y. Teramura, Heparinization of cell surfaces with short peptide-conjugated PEG-lipid regulates thromboinflammation in transplantation of human MSCs and hepatocytes, Acta Biomater., 2016, 35, 194–205 CrossRef CAS PubMed.
- R. G. DiScipio, Ultrastructures and interactions of complement factors H and I, J. Immunol., 1992, 149, 2592–2599 CrossRef CAS.
- M. Martin, J. Leffler, K. I. Smolag, J. Mytych, A. Bjork, L. D. Chaves, J. J. Alexander, R. J. Quigg and A. M. Blom, Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes, Cell Death Differ., 2016, 23, 903–911 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. Chan, Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- F. Giulimondi, L. Digiacomo, D. Pozzi, S. Palchetti, E. Vulpis, A. L. Capriotti, R. Z. Chiozzi, A. Lagana, H. Amenitsch, L. Masuelli, G. Peruzzi, M. Mahmoudi, I. Screpanti, A. Zingoni and G. Caracciolo, Interplay of protein corona and immune cells controls blood residency of liposomes, Nat. Commun., 2019, 10, 3686 CrossRef CAS PubMed.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Liposomal Formulations in Clinical Use: An Updated Review, Pharmaceutics, 2017, 9, 12 CrossRef.
- R. Talens-Visconti, O. Diez-Sales, J. V. de Julian-Ortiz and A. Nacher, Nanoliposomes in Cancer Therapy: Marketed Products and Current Clinical Trials, Int. J. Mol. Sci., 2022, 23, 4249 CrossRef CAS PubMed.
- T. Ishida and H. Kiwada, Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes, Int. J. Pharm., 2008, 354, 56–62 CrossRef CAS PubMed.
- P. Laverman, M. G. Carstens, O. C. Boerman, E. T. Dams, W. J. Oyen, N. van Rooijen, F. H. Corstens and G. Storm, Factors affecting the accelerated blood clearance of polyethylene glycol-liposomes upon repeated injection, J. Pharmacol. Exp. Ther., 2001, 298, 607–612 CAS.
- E. T. Dams, P. Laverman, W. J. Oyen, G. Storm, G. L. Scherphof, J. W. van Der Meer, F. H. Corstens and O. C. Boerman, Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes, J. Pharmacol. Exp. Ther., 2000, 292, 1071–1079 CAS.
- T. Suzuki, M. Ichihara, K. Hyodo, E. Yamamoto, T. Ishida, H. Kiwada, H. Kikuchi and H. Ishihara, Influence of dose and animal species on accelerated blood clearance of PEGylated liposomal doxorubicin, Int. J. Pharm., 2014, 476, 205–212 CrossRef CAS PubMed.
- T. Ishida, M. Ichihara, X. Wang, K. Yamamoto, J. Kimura, E. Majima and H. Kiwada, Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes, J. Controlled Release, 2006, 112, 15–25 CrossRef CAS PubMed.
- G. T. Kozma, T. Shimizu, T. Ishida and J. Szebeni, Anti-PEG antibodies: properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals, Adv. Drug Delivery Rev., 2020, 154–155, 163–175 CrossRef CAS PubMed.
- P. Zhang, F. Sun, S. Liu and S. Jiang, Anti-PEG antibodies in the clinic: current issues and beyond PEGylation, J. Controlled Release, 2016, 244, 184–193 CrossRef CAS PubMed.
- A. Adler, Y. Inoue, Y. Sato, K. Ishihara, K. N. Ekdahl, B. Nilsson and Y. Teramura, Synthesis of poly(2-methacryloyloxyethyl phosphorylcholine)-conjugated lipids and their characterization and surface properties of modified liposomes for protein interactions, Biomater. Sci., 2021, 5854–5867 RSC.
- A. Adler, Y. Inoue, K. N. Ekdahl, T. Baba, K. Ishihara, B. Nilsson and Y. Teramura, Effect of liposome surface modification with water-soluble phospholipid polymer chain-conjugated lipids on interaction with human plasma proteins, J. Mater. Chem. B, 2022, 10, 2512–2522 RSC.
- W. Lin, R. Goldberg and J. Klein, Poly-phosphocholination of liposomes leads to highly-extended retention time in mice joints, J. Mater. Chem. B, 2022, 10, 2820–2827 RSC.
- S. Seetasang and Y. Xu, Recent progress and perspectives in applications of 2-methacryloyloxyethyl phosphorylcholine polymers in biodevices at small scales, J. Mater. Chem. B, 2022, 10, 2323–2337 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01721d |
| This journal is © The Royal Society of Chemistry 2023 |