
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards molecularly imprinted polymers that respond to and capture phosphorylated tyrosine epitopes using fluorescent bis-urea and bis-imidazolium receptors†
Evgeniia
Kislenko
 a,
Anıl
İncel
b,
Kornelia
Gawlitza
a,
Börje
Sellergren
b and
Knut
Rurack
*a
a,
Anıl
İncel
b,
Kornelia
Gawlitza
a,
Börje
Sellergren
b and
Knut
Rurack
*a
aChemical and Optical Sensing Division, Bundesanstalt für Materialforschung und -prüfung (BAM), Richard-Willstätter-Str. 11, D-12489 Berlin, Germany. E-mail: knut.rurack@bam.de
bDepartment of Biomedical Sciences, Faculty of Health and Society, Malmö University, SE-20506 Malmö, Sweden
First published on 16th October 2023
Abstract
Early detection of cancer is essential for successful treatment and improvement in patient prognosis. Deregulation of post-translational modifications (PTMs) of proteins, especially phosphorylation, is present in many types of cancer. Therefore, the development of materials for the rapid sensing of low abundant phosphorylated peptides in biological samples can be of great therapeutic value. In this work, we have synthesised fluorescent molecularly imprinted polymers (fMIPs) for the detection of the phosphorylated tyrosine epitope of ZAP70, a cancer biomarker. The polymers were grafted as nanometer-thin shells from functionalised submicron-sized silica particles using a reversible addition-fragmentation chain-transfer (RAFT) polymerisation. Employing the combination of fluorescent urea and intrinsically cationic bis-imidazolium receptor cross-linkers, we have developed fluorescent sensory particles, showing an imprinting factor (IF) of 5.0. The imprinted polymer can successfully distinguish between phosphorylated and non-phosphorylated tripeptides, reaching lower micromolar sensitivity in organic solvents and specifically capture unprotected peptide complements in a neutral buffer. Additionally, we have shown the importance of assessing the influence of counterions present in the MIP system on the imprinting process and final material performance. The potential drawbacks of using epitopes with protective groups, which can co-imprint with targeted functionality, are also discussed.
Introduction
Timely detection of cancer is crucial for successful treatment outcomes. Liquid biopsy, which involves analysing biomarkers in biological fluids using, for instance, immunoaffinity techniques, provides a non-invasive and convenient alternative to tissue biopsy.1–3 Detecting abnormal post-translational modifications (PTMs) of proteins, particularly phosphorylation of tyrosine residues, holds promise for cancer diagnosis.4–6 For example, ZAP70, a tyrosine-protein kinase, has been proposed as an important marker for early cancer detection and monitoring due to its pivotal role in phosphorylation.7Due to a large number of potential PTM sites on proteins and the rapid advances in identifying potential biomarkers, molecularly imprinted polymers (MIPs) offer an alternative to genomics and immunoassays for diagnostic screening, as MIPs have advantages such as versatility, tuneable functionality, robustness, and ease of handling. MIPs are polymeric materials designed to selectively bind target molecules in specific cavities formed during polymerisation.8–10 However, to broaden the scope of MIPs, the synthesis of new functional monomers and crosslinkers is essential for achieving successful imprinting.
While MIPs are widely employed for the capture and enrichment of phosphorylated proteins,8 using structurally simpler templates such as phenylphosphonic acid11–13 or derivatives of phosphorylated amino acids for imprinting,14–16 there is a need for faster testing and improved detection methods. Currently, the number of reported MIPs for sensing phosphorylated tyrosine is limited,17 even more so for phosphorylated peptides.18,19 Existing methods often rely on core–shell particles with limited sensitivity or fluorescence response, utilising quantum dots in the core and analyte binding in a (non-fluorescent) MIP shell17,18 or fluorescent probe monomers in the polymer shell grafted from a carrier particle core.19,20 Organic urea and imidazolium monomers are commonly used for phosphorylated tyrosine capture in MIPs.9,10,16,21 Urea groups form strong hydrogen bonds with phosphate anions,22,23 while imidazolium salts act as effective anion receptors, interacting through electrostatic and/or hydrogen bond interactions.24,25 Combining these units in a cooperative imprinting strategy can enhance MIP affinity through synergistic effects, particularly in polar media where hydrogen bond formation is challenging.26 A cooperative approach has also been demonstrated for fluorescent MIPs (fMIPs).27–29
This study focuses on the development of fMIPs for detecting phosphorylated epitopes. Preliminary investigations employed a model analyte, phenylphosphoric acid (H2PPA), to understand the interactions between the hydrogen bonding (neutral) fluorescent probe cross-linker, the dicationic assisting bis-imidazolium cross-linker and the anionic analyte/template. Moving from the model to a real analyte, a tripeptide (Y-pY-G) resembling the Y-Y-T epitope of the low-abundant phosphoprotein ZAP7030 was selected for MIP synthesis. The epitope includes 493Y, which is a highly-conserved autophosphorylation site for kinases.30 The cooperative imprinting strategy utilised two functional cross-linkers to ensure high affinity and minimise non-specific binding. Besides transducing the relevant binding event in an analytical assay, the fluorescent indicator cross-linker continuously probed the structure of the imprinted site in which it is contained, allowing for rational optimisation. The study also explored the influence of epitope protective groups on the imprinting process and material selectivity, demonstrating the ability to discriminate between closely related phosphopeptide sequences.
Results and discussion
Synthesis of functional cross-linkers
The choice of functional monomers and cross-linkers is critical for successful imprinting. In this study, we focused on the sensing of the anionic phosphate group and selected a urea and a bis-imidazolium cross-linker as functional units. For phosphate recognition, we chose a fluorescent cross-linker fCL that carries a cleft-like urea binding motif integrated into a phenazine fluorophore (Scheme 1). This cross-linker has shown a strong affinity for phosphate groups and has been successfully used in previous work for imprinting protected phosphorylated tyrosine. It is ideal for binding of elongated molecules or side chains with a terminal phosphate group.19,20 The synthesis of fCL followed a modified reaction reported previously (for more details, see Section S1, ESI†).20 | ||
| Scheme 1 Structures of functional cross-linkers used in the present work. | ||
The bis-imidazolium salt bIm-Br (Scheme 1) and a related compound were previously used for synthesising MIPs for phosphate enrichment and phospholipid sensing.16,21,31 We reasoned that the bromide anion in bIm-Br could potentially interact with the urea cleft of fCL and compete with the phosphate anion for binding sites. To overcome this unfavourable interaction and improve the solubility of bis-imidazolium cross-linkers in a non-aqueous medium, we exchanged Br− with a weakly coordinating anion (WCA) such as hexafluorophosphate PF6−,32 resulting in bIm-PF6 (Scheme 1; for more details, see Section S1, ESI†).
Pre-polymerisation studies of fCL and bIm-Br/bIm-PF6
Pre-polymerisation studies were conducted to evaluate the influence of bis-imidazolium cross-linkers (bIm-Br or bIm-PF6) on the complex formation between fCL and the template during fMIP synthesis (for more details, see Table S3 and Section S2, ESI†). Fig. 1 and Fig. S2, ESI† present the results showing the interaction between fCL and bIm-Br or bIm-PF6. The addition of bIm-Br at low concentrations (0.1 eq.) induces a bathochromic shift in the absorption band of the dye, which increases with higher concentrations resulting in a 15 nm red shift for 1 eq. of bIm-Br (Fig. 1a and Fig. S2a, ESI†). A small red shift (5 nm) is also observed in the fluorescence emission spectra (Fig. 1b and Fig. S2b, ESI†). The fluorescence excitation spectra confirm these findings (Fig. S2c, ESI†). These observations can be explained by the formation of a complex between the bromide anion of bIm-Br and the urea groups of fCL through multiple hydrogen bonds.33,34 The small spherical Br− can fit inside the urea cleft of fCL (Fig. 1a, for more details, see Section S3, ESI†). In contrast, no bathochromic shift in absorption and emission is observed after adding bIm-PF6 to fCL (Fig. 1 and Fig. S2, ESI†). This can be attributed to the lower binding tendency of the hexafluorophosphate anion of bIm-PF6 to fCL due to its low hydrogen bond acceptor parameter33 and steric mismatch with the urea cleft (for more details, see Section S3, ESI†). | ||
| Fig. 1 Normalised absorption (a) and fluorescence emission (λexc = 385 nm) (b) spectra of pre-polymerisation mixture of fCL (1.11 mM) in chloroform with 0.1 eq. and 1 eq. of bIm-Br and bIm-PF6 in chloroform; fCL – black line, fCL and 0.1 eq. of bIm-Br – dashed red line, fCL and 1 eq. of bIm-Br – dotted red line, fCL and 0.1 eq. of bIm-PF6 – dashed blue line and fCL and 1 eq. of bIm-PF6 – dotted blue line. | ||
To confirm the necessity of exchanging the bromide anion with a WCA, the competitive binding between bromide and the phosphate anion was investigated, utilising phenylphosphoric acid (H2PPA) as a model analyte.20 When bIm-Br (0.1 eq.) was added to a solution of fCL with 1 eq. of the tetrabutylammonium (TBA) salt of HPPA− at pre-polymerisation concentrations, the bathochromic shift of 18 nm in absorption due to the formation of a hydrogen-bonded complex between the urea group and the phosphate anion remained virtually unchanged (Fig. 2a and Fig. S3a, ESI†). However, adding an equimolar amount of bIm-Br resulted in a slight hypsochromic shift, indicating competition between the bromide and phosphate anions for hydrogen bond formation (Fig. 2a). As both anions bind via hydrogen bonds, such minor effects in absorption are to be expected. A stronger influence would yet be expected in fluorescence because bromide is known to have a certain heavy-atom character, often leading to the quenching of fluorophores through the heavy atom effect.35 Accordingly, the fluorescence response of fCL in the presence of template was more significantly affected by the presence of bIm-Br, with ca. 10% quenching observed upon adding 0.1 eq. of bIm-Br and strong quenching at equimolar concentrations, accompanied by a reversal of the initial red shift by 11 nm (Fig. 2b and Fig. S3b, ESI†). Bromide anions can thus interact with the fluorescent cross-linker fCL and compete with the phosphate anion for the binding sites of the fluorophore (Fig. 2), which makes the use of bIm-Br as a functional cross-linker in fMIP synthesis inadvisable.
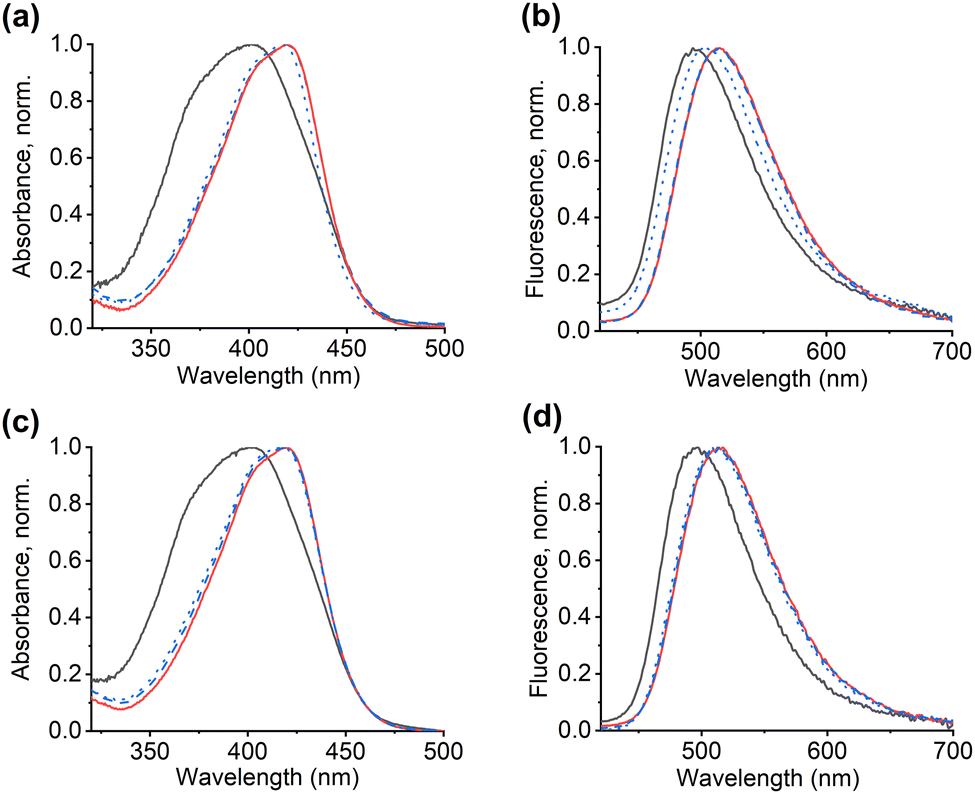 | ||
| Fig. 2 Normalised absorption spectra of pre-polymerisation mixture of fCL (1.11 mM) in chloroform with 1 eq. of HPPA-TBA as well as 0.1 eq. and 1 eq. of bIm-Br (a) and (b) and bIm-PF6 (c) and (d) in chloroform; fCL – black line, fCL and 1 eq. of HPPA-TBA – red line, fCL, 1 eq. of HPPA-TBA and 0.1 eq. of bIm-Br (a) and (b) or bIm-PF6 (c) and (d) – dashed blue line and fCL, 1 eq. of HPPA-TBA and 1 eq. of bIm-Br (a) and (b) or bIm-PF6 (c) and (d) – dotted blue line. | ||
In contrast, bIm-PF6 had a negligible effect on the complex formation with HPPA− (Fig. 2c, d and Fig. S4, ESI†). Based on these observations, bIm-PF6 was chosen as the cross-linker for fMIP synthesis due to its favourable characteristics and minimal interference with fCL-template interactions.
Spectroscopic response behaviour of fCL
Prior to incorporating the functional dye fCL into the polymer network, its spectroscopic behaviour towards the template was reassessed considering the use of a bis-imidazolium-type co-cross-linker and chloroform as the solvent, different from our previous study, to achieve optimal binding between the two urea units and the anion.20 The template used was the sequence Y-pY-G, which shares structural similarity with the ZAP70 epitope Y-Y-T,36 and it was employed in its C-terminus protected form (Scheme 2a) to prevent competition with the phosphate anion for binding to the functional cross-linkers in the final MIP preparation. Additionally, the N-terminus was protected with a fluorenylmethyloxycarbonyl (Fmoc) group to enhance the solubility of the template in organic solvents. Furthermore, threonine (T) in the original epitope was replaced with glycine (G) to further improve solubility while preserving the autophosphorylation site.30 In line with the model analyte H2PPA, the singly deprotonated Fmoc-Y-pY-G-OMe was used for the spectroscopic experiments to ensure the formation of the hydrogen-bonded complex. TBA+ was selected as the counterion as it assists in improved anion imprinting (Scheme 2a).37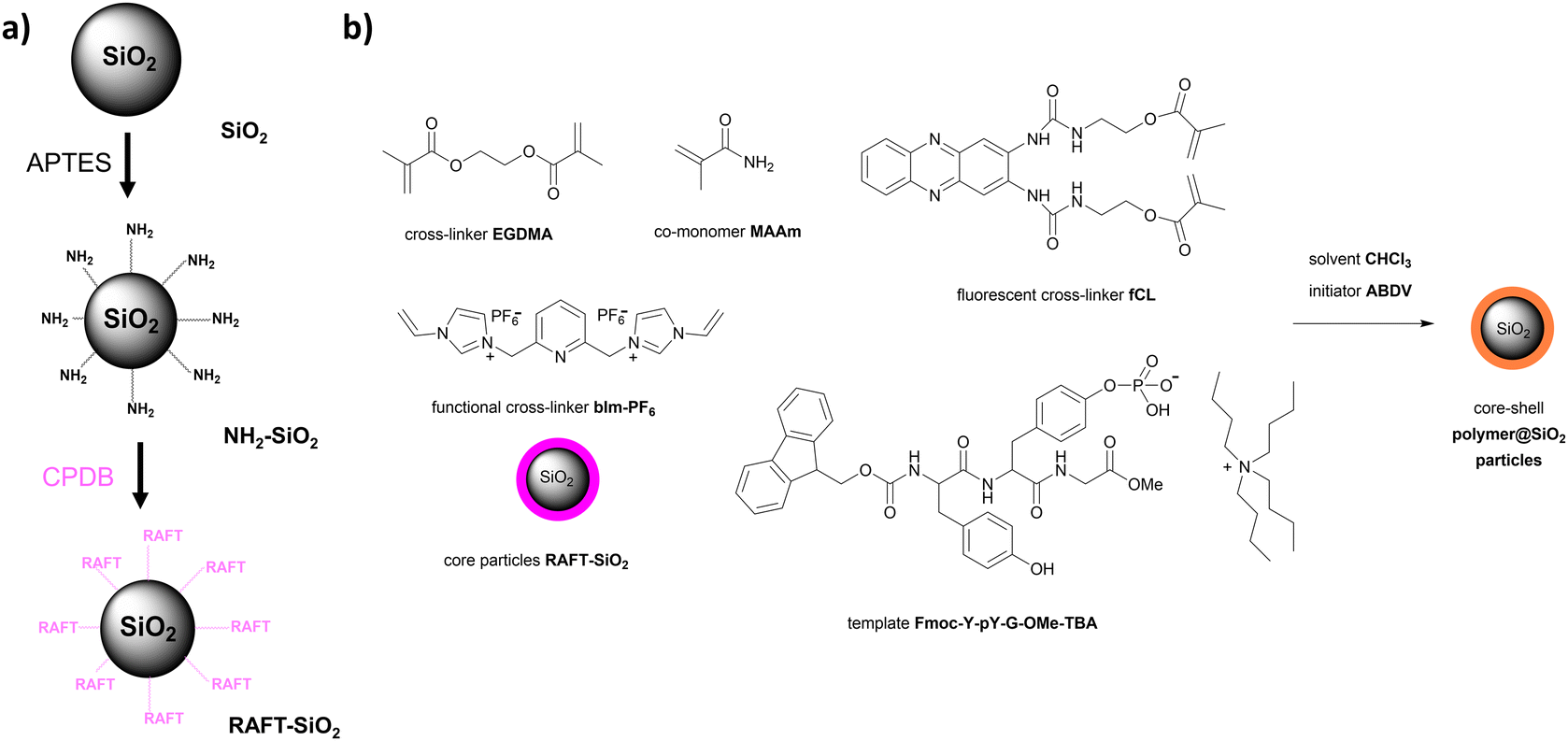 | ||
| Scheme 2 (a) MIP and NIP core–shell polymer@SiO2 particles synthesis and (b) functionalisation of SiO2 core particles. | ||
Titrations of fCL in chloroform (3.14 μM) were conducted with Fmoc-Y-pY-G-OMe-TBA that interacts with fCL through the formation of a hydrogen-bonded complex, evidenced by a hyper- and bathochromic shift in absorption (Fig. 3a) as well as the typical red shift and enhanced fluorescence emission (Fig. 3b).20 The binding constant was determined from the absorption spectra as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka = 4.8 ± 0.1 using a 1:1 binding model and the BindFit software (for more details, see Section S4, ESI†).38–40 To ensure sufficient complexation of the target analyte, affinity constants >103 M−1 are required for stochiometric imprinting.41 The obtained binding constants for fCL with Fmoc-Y-pY-G-OMe-TBA were satisfactory in this regard, enabling the synthesis of the fMIPs.
Ka = 4.8 ± 0.1 using a 1:1 binding model and the BindFit software (for more details, see Section S4, ESI†).38–40 To ensure sufficient complexation of the target analyte, affinity constants >103 M−1 are required for stochiometric imprinting.41 The obtained binding constants for fCL with Fmoc-Y-pY-G-OMe-TBA were satisfactory in this regard, enabling the synthesis of the fMIPs.
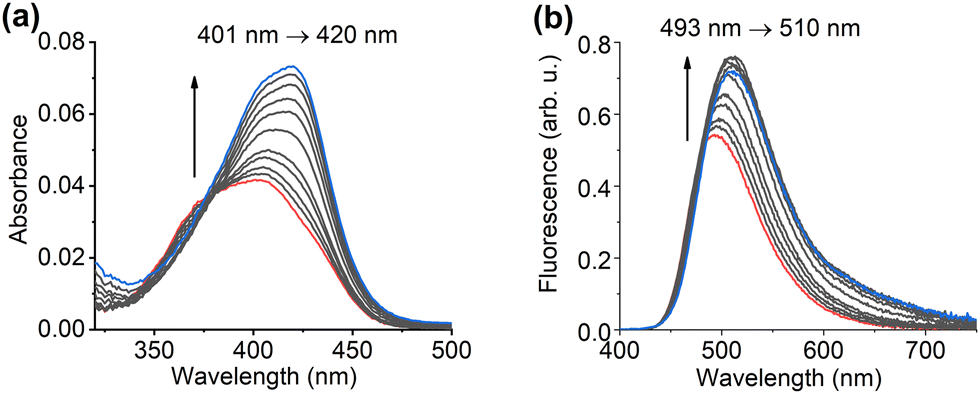 | ||
| Fig. 3 Absorption (a) and fluorescence emission (λexc = 385 nm) (b) spectra of fCL (3.14 μM) in chloroform upon addition of an increasing amount of Fmoc-Y-pY-G-OMe-TBA (0–63.1 eq.) in chloroform; fCL – red line, fCL and 63.1 eq. of Fmoc-Y-pY-G-OMe-TBA – blue line. | ||
Synthesis and functionalisation of silica core particles
As highlighted in the introduction, the core–shell particle format, consisting of a carrier particle (core) and a responsive layer (shell), is particularly suitable for analytical applications involving both the capture and detection of an analyte. Accordingly, in this study, a thin MIP shell was grafted from submicron-sized silica (SiO2) particles. Thin polymer shells offer rapid diffusion of the analyte from the surrounding medium into the MIP, which is crucial for sensing applications. Additionally, they provide binding cavities that are more homogenous and closer to the surface compared to those of monolithic polymers.SiO2 particles were chosen as the core particles, synthesised through a modified Stöber procedure,42 resulting in high monodispersity. These particles could be functionalised in a straightforward manner using silane chemistry, facilitating subsequent shell growth via controlled living reversible addition-fragmentation chain-transfer (RAFT) polymerisation, a preferred method for such architectures.20,43
The SiO2 particles prepared here had an average diameter of 318 ± 24 nm (see Fig. S10 and Section S5, ESI† for more details). They were functionalised through two condensation reactions with amino groups using (3-aminopropyl)triethoxysilane (APTES) and the RAFT agent 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPDB), resulting in the formation of RAFT-SiO2 particles (Scheme 2b). Detailed synthetic and characterisation information can be found in Sections S1, S5 and S6, ESI.†
The successful functionalisation of the particles was confirmed through zeta potential measurements, thermogravimetric analysis (TGA) and elemental analysis; see Fig. S13 and Section S6 for more details, ESI.†
MIP and NIP core–shell polymer@SiO2 particles synthesis
After preliminary experiments, fluorescent core–shell polymer@SiO2 MIP and NIP particles were synthesised for the detection of the Y-pY-G sequence, structurally similar to the ZAP70 epitope Y-Y-T.30,36 The synthesis involved the use of the fluorescent cross-linker fCL, the template Fmoc-Y-pY-G-OMe-TBA, the structural cross-linker ethylene glycol dimethacrylate (EGDMA), the structural co-monomer methacrylamide (MAAm) and the functional cross-linker bIm-PF6 (Scheme 2a).EGDMA, a hydrophilic cross-linker, was employed to achieve highly cross-linked and rigid shells during polymerisation, promoting the formation of specific binding cavities for improved analyte recognition. It was used in excess compared to the functional cross-linkers as well as the template to ensure effective cross-linking. MAAm, a neutral co-monomer, was included based on its demonstrated enhancement of the imprinting process in similar MIP systems.20 The deprotonated phosphate group in Fmoc-Y-pY-G-OMe-TBA could form the desired hydrogen-bonded complex with both urea moieties of fluorophore fCL and the H2 protons of bIm-PF6 (see Fig. S1, ESI†). Furthermore, the presence of positive charges in bIm-PF6 could enhance imprinting by replacing the TBA cation as the counterion in the ternary ensemble of fCL:template anion:counter-cation.
It is important to note that unlike the displacement of the template to a minor extent by Bis-Im2+, the exchange of a TBA counterion with Bis-Im2+ would not hinder imprinting but could instead result in more rigid cavities, as the Bis-Im2+ counterion would be fixed in the network alongside the [fCL⊂pY(R1R2)]−/Bis-Im2+/PF6− complex (see discussion below and Section S12, ESI†). Therefore, employing both functional cross-linkers simultaneously during MIP synthesis has the potential to yield more precisely tailored cavities and enhance the analytical response.
To investigate the impact of polymer network composition on imprinting, two batches of particles with different monomer concentrations (M1bIm and M2bIm) were synthesised (Table 1). M1bIm was produced using lower amounts of polymerisation components and a higher quantity of initiator, while M2bIm was prepared with a higher monomer concentration and a lower amount of initiator. To ensure complete solubilisation of the functional cross-linkers in chloroform and prevent dimerization, the concentrations of fCL and bIm-PF6 were kept low. A stochiometric imprinting ratio of 1:0.5:1 (fCL:bIm-PF6:template) was maintained to ensure precise and effective imprinting.41
| Sample | Fmoc-Y-pY-G-OMe (mg) | fCL (mg) | MAAm (mg) | bIm-PF6 (mg) | EGDMA (μL) | ABDV (mg) | CHCl3 (mL) | RAFT-SiO2 (mg) |
|---|---|---|---|---|---|---|---|---|
| M1bIm | 1.01 | 0.55 | 1.83 | 0.31 | 20.6 | 2 | 2 | 20 |
| N1bIm | — | 0.55 | 1.83 | 0.31 | 20.6 | 2 | 2 | 20 |
| M2bIm | 2.8 | 1.52 | 5.06 | 0.85 | 56.12 | 0.78 | 2 | 20 |
| N2bIm | — | 1.52 | 5.06 | 0.85 | 56.12 | 0.78 | 2 | 20 |
As control materials, non-imprinted polymers (NIPs), referred to as N1bIm and N2bIm, were synthesised simultaneously using a procedure identical to MIP preparation but without adding the analyte. The pre-polymerisation mixtures for both MIP and NIP batches were sampled directly from the polymerisation reaction prior to the addition of the initiator and particles. This allowed for examining the influence of cross-linkers and co-monomers and assessing the stability of the complex between the template and fluorescent probe fCL.
Lower monomer and fCL concentrations in the pre-polymerisation mixtures of M1bIm and N1bIm resulted in reduced absorption and fluorescence intensities compared with M2bIm and N2bIm. Absorption and fluorescence spectra of N1bIm particles (Fig. S14a and c, ESI†) were similar to the free dye but slightly red-shifted compared to diluted conditions (Fig. 3). Similar spectra were observed for N2bIm (Fig. S14b and d, ESI†). Upon template addition, both batches exhibited a red shift and intensity increase in absorption and fluorescence (Fig. S14, ESI†), indicating the successful formation of the complex between fCL and Fmoc-Y-pY-G-OMe-TBA. The red shift in the pre-polymerisation mixture with an equimolar amount of template was similar to diluted conditions (cf.Fig. 3vs. Fig. S14, ESI†). Although a higher enhancement was observed for M1bIm compared to N1bIm, M2bIm showed a more pronounced red shift than N2bIm (Fig. S14, ESI†). However, the intensity effect may be only apparent, possibly due to different changes in absorbance at the excitation wavelength that could not be measured under the present conditions.
Following the synthesis and washing of the core–shell polymer@SiO2 particles, the formation of the polymer network was evaluated using TEM (Fig. 4; Table S4 and Section S5, ESI†). All core–shell polymer@SiO2 particles exhibit an even shell, indicating successful polymerisation. The MIP shell thickness (Fig. 4a and c) was slightly lower and more polydisperse compared to the corresponding NIPs (Fig. 4b and d). The presence of the template may influence the reactivity rates of the monomers and co-monomers due to hydrogen bonding and/or electrostatic interaction.44M1bIm and N1bIm (Fig. 4a and b) had thinner shells than the second particle batch (Fig. 4c and d), reflecting the difference in initial monomer and initiator concentrations. The successful removal of the template was also confirmed by absorption measurements of MIP and NIP suspensions at identical concentrations of 0.5 mg mL−1 in chloroform, which yielded identical spectra (Fig. S15, ESI†). The concentration of the dye in the polymer layer was estimated using the absorption spectra of MIP and NIP suspensions and the molar absorption coefficient of fluorophore fCL in chloroform at 401 nm of ε401 = 13638 ± 573 M−1 cm−1 (for more details, see Sections S8 and S9, ESI†). The results showed that MIP particles incorporated a lower amount of fCL compared to non-imprinted particles, supporting the idea that the polymerisation rate depends on the presence of the template and is influenced by the complexation process between monomers and template (Table S5, ESI†).44 The concentration of the dye in the polymer shells was comparable for both MIPs and both NIPs, indicating that changing the polymerisation ingredients influenced the shell thickness but not the dye concentration.
 | ||
| Fig. 4 TEM images of M1bIm (a), N1bIm (b), M2bIm (c) and N2bIm (d) core–shell polymer@SiO2 particles. Scale bar = 50 nm. | ||
Analytical performance of MIP and NIP particles in chloroform
To evaluate the response of the MIP particles, titration experiments were conducted using the same solvent as the one used for polymerisation to ensure that the polymer shell's swelling behaviour remained unaltered, and all the imprinted sites were accessible for analyte rebinding. Absorption and fluorescence spectra were measured upon the addition of Fmoc-Y-pY-G-OMe-TBA in chloroform at a particle concentration of 0.5 mg mL−1 (Fig. 5). | ||
| Fig. 5 Fluorescence emission (λexc = 385 nm) spectra of M1bIm (a), N1bIm (b), M2bIm (c) and N2bIm (d) core–shell polymer@SiO2 particles (0.5 mg mL−1) in chloroform upon addition of an increasing amount of Fmoc-Y-pY-G-OMe-TBA (0–69.8 μM) in chloroform; M1bIm, N1bIm, M2bIm and N2bIm – red line, M1bIm, N1bIm, M2bIm and N2bIm and 69.8 μM of Fmoc-Y-pY-G-OMe-TBA – blue line. | ||
The binding affinity of Fmoc-Y-pY-G-OMe-TBA to the M1bIm and M2bIm core–shell polymer@SiO2 particles was evaluated using the corrected absorption spectra (Fig. S19, ESI†). Binding constants of logKa = 5.4 ± 0.3 and 5.4 ± 0.2 for M1bIm and M2bIm, respectively, were calculated using a 1:1 binding model (Fig. S20, ESI†). The binding constants were higher compared to the interaction of fCL and the tripeptide epitope in a diluted state (logKa = 4.8 ± 0.1), indicating an increase in the affinity of the analyte towards the integrated fCL in the polymer network.
During the titration, a red shift of ca. 10 nm and an enhancement in fluorescence emission were observed upon template addition to the MIP particles. The intensity reached saturation at ca. 70 μM of the analyte (Fig. 5a and c). The fluorescence response was similar for both MIPs, indicating comparable accessibility of fCL to the template despite the different shell thicknesses. The bathochromic shift was smaller compared to dilute conditions, which can be explained by the presence of a highly cross-linked polymer network in which it is more difficult to achieve a fully relaxed conformation and an optimal arrangement of fCL and Fmoc-Y-pY-G-OMe-TBA.
The fluorescence response of the NIP particles to template addition was considerably smaller, indicating a highly cross-linked network with no specific binding sites for the template (Fig. 5b and d). The fluorescence change in NIP particles may be attributed to non-specific interaction between the analyte and the dye in the outer polymer shell. The fluorescence emission spectra of N1bIm and N2bIm were red shifted compared to the corresponding imprinted particles (Fig. 5). This may be due to an even denser polymer network, which, because the structural monomer and cross-linker are more polar than chloroform, may lead to better dipolar stabilisation of an excited charge-transfer state, as it is the relevant emitting state in fCL.45
The fluorescence intensity enhancement of both MIPs was higher compared to the corresponding NIPs (cf.Fig. 6a, b vs. c and d). The imprinting factor (IF), calculated as the ratio of intensity change between MIP and NIP at the maxima of the fluorescence band, was 5.0 and 4.5 for M1bIm and M2bIm, respectively (Fig. 6). The limit of detection for M1bIm was determined to 11.2 μM (for more details, see Section S15, ESI†).
 | ||
| Fig. 6 Relative fluorescence emission (λexc = 385 nm) intensity change at λ = 503 nm for M1bIm (a), M2bIm (b), N1bIm (c) and N2bIm (d) core–shell polymer@SiO2 particles (0.5 mg mL−1) in chloroform upon addition of an increasing amount of analytes (0–69.8 μM) in chloroform; Fmoc-Y-pY-G-OMe-TBA – black ■ with line, Fmoc-pY-OEt-TBA – red ●, HPPA-TBA – blue ▲, HPPA-THA – green ▼ and Fmoc-Y-Y-G-OMe – purple ♦. Measurement uncertainties as indicated for selected data points. ΔF/Fmin = (Fx − Fmin)/Fmin. CT – concentration of template. | ||
To assess the discrimination factor (DF) between the targeted analyte and competitor molecules, titrations were performed using Fmoc-Y-Y-G-OMe as a related compound and competitors, including Fmoc-pY-OEt-TBA, HPPA-TBA and HPPA-THA (Scheme 3). M1bIm and M2bIm exhibited minimal response when titrated with Fmoc-Y-Y-G-OMe, which agrees well with the absence of a response upon titration of fCL with the competitor (Fig. 6a, b and Fig. S22e, f, cf. Fig. S23, ESI†).
 | ||
| Scheme 3 Structures of target analyte and related molecules. | ||
Both M1bIm and M2bIm show no discrimination between the targeted tripeptide and Fmoc-protected phosphorylated tyrosine (Fig. 6a, b and Fig. S24, ESI†), suggesting that the presence of the Fmoc protective group, which is imprinted during polymerisation, plays a role in inducing a similar or slightly stronger response. Comparison with the non-imprinted control materials confirmed that the smaller protected amino acid molecule could more easily diffuse into the highly cross-linked polymer shell and may more easily adopt an optimum fit into the cleft, inducing a slightly enhanced response (Fig. 6c, d and Fig. S24, ESI†).
Further titration of M1bIm and M2bIm with HPPA-TBA, a competitor molecule, resulted in a weaker response compared to Fmoc-Y-pY-G-OMe-TBA or Fmoc-pY-OEt-TBA, with a DF of ca. 1.4 for both M1bIm and M2bIm (Fig. 6a, b and Fig. S22, ESI†). The difference may be attributed to the absence of the Fmoc group in HPPA-TBA. However, when the bulkier tetrahexylammonium salt of HPPA− (HPPA-THA) was employed, more pronounced discrimination factors of 1.6 and 1.8 for M1bIm and M2bIm, respectively, were observed (Fig. 6a and b). This stresses the conclusion that additional π–π stacking interactions with the Fmoc group enhance the binding of protected amino acid and peptide species and that the presence of longer hydrophobic alkyl chains of counterions further prevents such interactions.37
From the cross-selectivity studies, it can be concluded that a stronger response of the imprinted material can be achieved if the analyte has a high number of aromatic moieties. Complementary π–π interactions together with hydrogen bonds and electrostatic interactions may be responsible for the binding of the analyte to the polymer network.46 These results for the M1 and M2 systems also showed that the behaviour is virtually independent of the synthetic method used, which underlines the robustness of the approach.
To further investigate the impact of the synthesised bis-imidazolium cross-linker in the polymer network on the behaviour of imprinted materials, two additional batches of imprinted polymers were prepared with slight modifications to the M1bIm and N1bIm recipes (Table S1, ESI†). The first batch (M1no, N1no) was synthesised without bIm-PF6 to evaluate the effect of a cooperative imprinting strategy on material performance. The second batch (M1D, N1D) replaced bIm-PF6 with an equivalent amount of divinylbenzene (DVB-80) cross-linker to assess the impact of increased cross-linking and more sites for π–π stacking on the MIP's affinity towards Fmoc-Y-pY-G-OMe-TBA, without the presence of synergistic interactions between functional monomers. Compared to M1bIm, both batches showed significantly lower imprinting factors (IF) (1.4 for M1no, 2.2 for M1D, see Fig. S26, ESI† for a comparison). This suggests that the higher affinity of the target molecule to imprinted materials, as compared to NIPs, cannot be solely attributed to the formation of tighter binding cavities in the dense polymer network. Our findings highlight the advantages of employing complementary imprinting using fCL and bIm-PF6 (for more details, see Section S12, ESI†).
Analytical performance of MIP and NIP particles in aqueous buffer
For the detection of oxoanions using organic molecular probes, fluorescence signalling relies on hydrogen bonding interaction typically occurring in a classical supramolecular chemistry sense via two hydrogen bonding partners in a low-dielectric microenvironment. However, for more challenging analytes such as sugar acids, glycosylated amino acids or peptides, which are present in a multicomponent network along with the indicator molecule, alternative signalling modes can become active, as shown above, and in ref. 28,29, making such approaches potentially useful for aqueous media where hydrogen bond-driven fluorescence signalling is not feasible.To evaluate the capability of the MIPs to capture the imprinted template in aqueous buffers, resembling real-life conditions for detecting phosphoproteins in cell lysates, we performed a four-fold scale-up synthesis of M1bIm and N1bIm, resulting in M3bIm and N3bIm, respectively (Table S2, ESI†). Characterisation confirmed the reproducibility of the synthesis, see, e.g., Table S4 and Fig. S10, ESI.† Furthermore, we obtained three decapeptides representing the ZAP70 kinase regulatory motif (Scheme S5, ESI†) and dissolved them in aqueous buffers of various pH values (from 1.0–9.2), corresponding to the operating range of fCL in its neutral form (Section S14, ESI†). Equilibrium binding tests were conducted by incubating M3bIm and N3bIm with the peptide mixtures, followed by quantification of unbound peptides using reverse phase HPLC with UV detection.
Fig. 7 illustrates the percentage binding of the different peptides to the MIPs and NIPs. In the low pH region (pH 1–5), it is evident that peptide uptake does not strongly correlate with the desired selectivity (YpY). Instead, binding appears to be influenced by the ionisation degree, as observed by the significant increase in binding of the multiply phosphorylated target when the buffer pH is increased from 3 to 5. This can be explained by examining the net charges of the peptides, where the charge of pYpY changes from −1 to −3 within this range (Table 2).
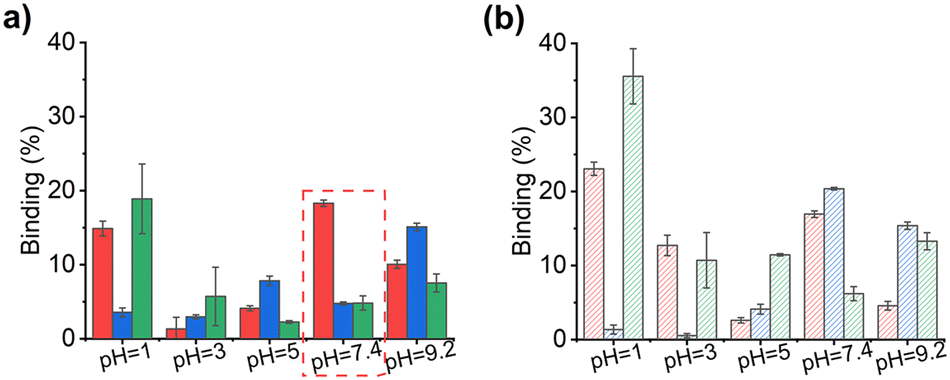 | ||
| Fig. 7 Binding of M3bIm (a) and N3bIm (b) core–shell polymer@SiO2 particles (10 mg mL−1) after incubation in decapeptides mixture of GADDSYpYTAR, GADDSpYpYTAR and GADDSYYTAR (20 μM) in aqueous buffers; M3bIm binding – solid bar, N3bIm binding – shaded bar, GADDSYpYTAR percentage bound – red, GADDSpYpYTAR percentage bound – blue and GADDSYYTAR percentage bound – green. | ||
| pH | GADDSYYTAR | GADDSYpYTAR | GADDSpYpYTAR |
|---|---|---|---|
| 1 | 2 | 2 | 2 |
| 3 | 1 | 0 | −1 |
| 5 | −1 | −2 | −3 |
| 7.4 | −1 | −3 | −5 |
| 9.2 | −1 | −3 | −5 |
Of particular interest is the binding behaviour in neutral buffer conditions. MIPs exhibit preferential uptake of the phosphopeptide complement, with YpY binding more strongly than pYpY and YY (Fig. 7a). In contrast, NIPs show no such selectivity (Fig. 7b). The higher non-specific binding of the NIPs could be mitigated by employing “dummy NIPs”, as demonstrated in previous studies, which yield more realistic control polymers for highly hydrophilic biomolecule targets.28 In this case, the increased non-specific binding may result from hydrophobic interactions and the presence of bIm-PF6 cross-linkers that are more randomly distributed throughout the network, offering cationic sites for non-specific interactions.
To interpret these findings in aqueous media at varying pH, it is important to understand the relationship between fluorescence assays in organic media and the capture data obtained by HPLC in aqueous media. Weaker (and different) or no fluorescence signalling would be expected in highly polar or protic environments, but binding within the cavities can still occur, similar to conventional non-fluorescent MIPs. The intention of testing the system in aqueous media was not to replicate the fluorescence response but rather to assess whether the discrimination observed in the polymerisation solvent is maintained in an analytically important pH range in water, particularly near-neutral pH. The step from chloroform to buffered aqueous media involves different swelling behaviour, electrolyte content, ionic components of the buffer, and charge states of the analytes. As the charge state of the analyte significantly influences its conformational structure, our goal was to minimise the factors influencing the system and determine if the binding pattern remains comparable to the fluorescence assay in near-neutral pH conditions relevant to sensing in diluted biological fluids. A comparison of the data in Fig. 6 and 7 confirms that this is indeed the case.
Contrary to intuition, we did not expect the polymers to maintain the binding pattern over a wide pH range (pH 1.0–9.2), where the charge states of the species vary between +1 and −5 (Table 2). These charge states affect peptide conformation and the presence of counterions in close proximity to the charged groups. It would be unlikely for a MIP with selective binding pockets to accommodate a wide range of species with different conformations, charges, and counterions. Therefore, it is expected that discriminative binding would be observed within a narrow but relevant pH window, as observed in the present case.
Fluorescence measurements performed on this system in differently buffered solutions did not reveal clear fluorescence changes, which was anticipated because, as explained in the beginning of this section, the present signalling mode is solely based on hydrogen bond interactions which are too inefficient for the present system. However, the favourable binding behaviour at neutral pH is encouraging for the development of fluorescent indicator monomers and cross-linkers. Once a more suitable fluorescent monomer or cross-linker capable of responding to the target phosphate group in water is identified, possibly also involving alternative signalling modes, its incorporation into the present MIP system holds promise for creating an fMIP system capable of binding and sensing in water.
Conclusions
We have developed a fluorescent molecularly imprinted polymer (fMIP) capable of distinguishing phosphorylated and non-phosphorylated epitopes of ZAP70. Our study demonstrated that cooperative imprinting using a colourless and non-fluorescent bis-imidazolium cross-linker improves the generation of imprinted cavities compared to using a fluorescent urea cross-linker alone. Including counterions in the imprinting models enhances the evaluation and prediction of MIP performance. By utilising complementary and cooperative interactions, bulky polar anions can be effectively imprinted into the organic polymer, as observed in fluorescence rebinding studies. However, imprinting protected epitopes may decrease selectivity towards target analytes due to the co-imprinting of protective groups. Nevertheless, when assessing peptide recognition in aqueous systems at neutral pH, we obtained favourable results with remarkable peptide specificity that aligned with the intended selectivity. Despite the challenges involved in transferring the MIP system from an organic to an aqueous environment, these findings suggest that our imprinting strategy is robust and holds great promise for developing fluorescent indicator-containing MIPs capable of binding and indicating target analytes under realistic conditions. The synthesised particles can potentially be integrated into microfluidic devices for the rapid detection of cancer biomarkers,19 such as small-number amino acid peptides, by transferring them from an aqueous sample to an organic signalling phase. Ongoing research in our laboratories focuses on both the development of indicator monomers/cross-linkers and the advancement of microfluidic assay platforms.Experimental
Synthesis of M1bIm, N1bIm, M2bIm and N2bIm core–shell polymer@SiO2 particles
MIP and NIP layers were grafted from the RAFT-SiO2 particles using two different recipes (Table 1). EGDMA was purified by passing through a Pasteur pipette containing inhibitor remover for hydroquinone and monomethyl ether hydroquinone. MAAm was used without additional purification. A 2 mM stock solution of freshly prepared Fmoc-Y-pY-G-OMe-TBA (for more details, see Section S1, ESI†) was prepared by dissolving in anhydrous chloroform and briefly sonicated. bIm-PF6 (0.31 mg, 0.53 μmol for M1bIm and N1bIm or 0.85 mg, 1.46 μmol for M2bIm and N2bIm), MAAm (1.83 mg, 21.07 μmol for M1bIm and N1bIm or 5.06 mg, 58.32 μmol for M2bIm and N2bIm), EGDMA (20.6 μL, 105.37 μmol for M1bIm and N1bIm or 56.12 μL, 291.62 μmol for M2bIm and N2bIm), and fluorescent cross-linker fCL (0.55 mg, 1.05 μmol for M1bIm and N1bIm or 1.52 mg, 2.92 μmol for M2bIm and N2bIm) were combined in specific amounts according to the recipes. In the MIP vial, the template solution (0.53 mL for M1bIm or 1.46 mL for M2bIm) and chloroform (1.47 mL for M1bIm or 0.54 mL for M2bIm) were added, while only chloroform (2 mL) was added to the NIP vials. The solutions were sonicated for 10 min. A small portion of the reaction solution (30 μL) was used for control purposes in prepolymerisation studies. RAFT-SiO2 particles (20 mg) were then added, and the suspensions were sonicated for another 10 min. Under argon, the initiator ABDV (2 mg for M1bIm and N1bIm or 0.79 mg for M2bIm and N2bIm) was added to the reaction mixture. The mixture was cooled, degassed with argon for 5 min, and then mixed at moderate speed in a hybridisation oven at 50 °C. After 16 h, the temperature was increased to 70 °C. After an additional 2 h, the particles were precipitated using n-hexane (4 mL), washed with chloroform (2 mL) and acetonitrile (2× 4 mL and 2× 1.8 mL) with centrifugation at 6500 ×g for 5 min and 4800 ×g for 10 min in between the washing steps. The particles were incubated for 1 h in methanol/acetic acid 9:1 solution (3× 1.8 mL) using a rotator (40 rpm), followed by sonication (10 min) and centrifugation (4800 ×g, 10 min). The particles were additionally washed with methanol (2× 1.8 mL), sonicated, and centrifuged (4800 ×g, 10 min) before drying overnight in a vacuum oven.
Titrations of particles
The fluorescence response of MIP and NIP core–shell polymer@SiO2 particles to analyte addition was evaluated in 10 × 10 mm quartz cuvettes. The particles were suspended in chloroform (0.5 mg mL−1) and sonicated for 15 min prior to measurement. A freshly prepared analyte stock solution (1 mM) was used. Before each measurement, the suspension was equilibrated for 2 min using a magnetic stirring bar and a motorised cuvette holder. Fluorescence measurements were conducted with an excitation wavelength of 385 nm, and fluorescence excitation spectra were recorded at an emission wavelength of 490 nm. Slits were adjusted to keep the signal intensity below 106 counts per second.Imprinting factor (IF) and discrimination factor (DF)
IF, the imprinting factor, can be determined by calculating the ratio of fluorescence intensity change between MIP and NIP core–shell polymer@SiO2 particles. This is expressed in eqn (1), where Fx represents the emission intensity at λ = 503 nm of a particle suspension upon reaching signal saturation with the template/analyte, and Fmin represents the emission intensity at λ = 503 nm of a particle suspension in the absence of a template: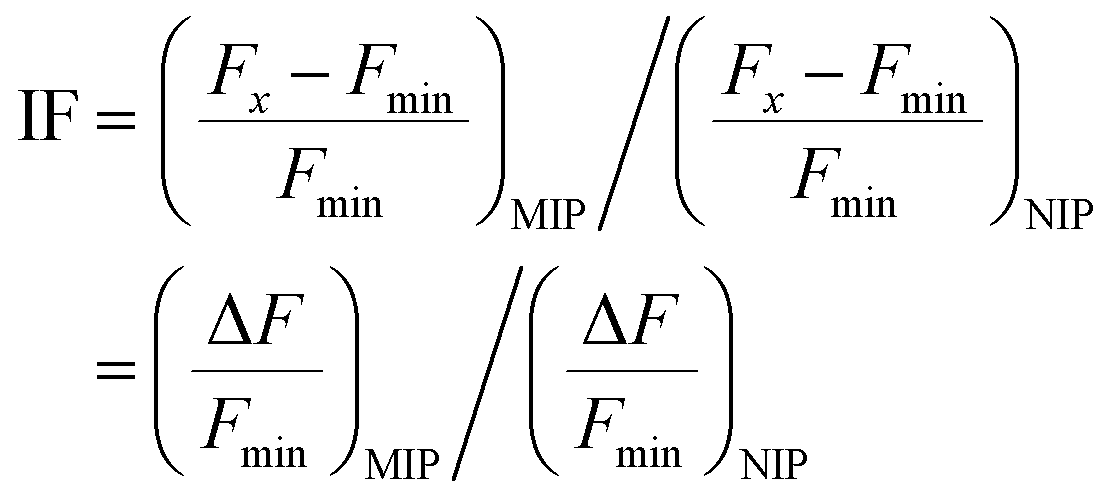 | (1) |
DF, the discrimination factor, can be calculated as the ratio of intensity change in MIP core–shell polymer@SiO2 particles upon the addition of the template and a competitor molecule. This is shown in eqn (2), where ΔF represents Fx–Fmin of Eqn 1 after achieving signal saturation with the analyte as well as the competitor, and Fmin represents the emission intensity at λ = 503 nm of a particle suspension in the absence of a guest:
 | (2) |
Capture studies in aqueous buffers
To assess the binding of MIP and NIP core–shell polymer@SiO2 particles, a 20 μM solution (C0) of three decapeptides (GADDSYpYTAR (YpY) + GADDSpYpYTAR (pYpY) + GADDSYYTAR (YY)) in aqueous buffers with pH values of 1.0, 3.0, 5.0, 7.4, 9.2, were prepared. The particles were suspended in a peptide mixture (10 mg mL−1) and sonicated for 15 min. Following vigorous shaking during a 2 h incubation, centrifugation at 14000 × g for 15 min was performed. The resulting supernatant was collected, concentrated using a Genevac EZ-2 evaporator, and reconstituted in a water/acetonitrile 95:5 v/v solution containing 0.1% trifluoroacetic acid. The peptide concentrations were determined using HPLC (n = 3). The binding of the polymer was calculated using eqn (3), where Cfree represents the concentration of peptide in the supernatant, and C0 represents the concentration of peptide in the peptide mixture: | (3) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank M. Grüneberg (BAM) for TGA measurements and synthetic support, K. Keil (BAM) for SiO2 particle synthesis, zeta potential and pKa measurements, V. Perez Padilla (BAM) for experimental support, B. Kobin (Humboldt University Berlin, HUB) for MS analysis, J. Krone (TU Berlin) and A. Meckelburg (BAM) for performing elemental analyses, and A. Zimathies (BAM) for N2 adsorption/desorption measurements. This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 722171.Notes and references
- R. Vaidyanathan, R. H. Soon, P. Zhang, K. Jiang and C. T. Lim, Lab Chip, 2019, 19, 11–34 CAS.
- P. Pinzani, V. D’Argenio, M. D. Re, C. Pellegrini, F. Cucchiara, F. Salvianti and S. Galbiati, Clin. Chem. Lab. Med., 2021, 59, 1181–1200 CrossRef CAS PubMed.
- M. Ignatiadis, G. W. Sledge and S. S. Jeffrey, Nat. Rev. Clin. Oncol., 2021, 18, 297–312 CrossRef PubMed.
- V. Singh, M. Ram, R. Kumar, R. Prasad, B. K. Roy and K. K. Singh, Protein J., 2017, 36, 1–6 CrossRef CAS PubMed.
- Y. P. Lim, Clin. Cancer Res., 2005, 11, 3163–3169 CrossRef CAS PubMed.
- K. Rikova, A. Guo, Q. Zeng, A. Possemato, J. Yu, H. Haack, J. Nardone, K. Lee, C. Reeves, Y. Li, Y. Hu, Z. Tan, M. Stokes, L. Sullivan, J. Mitchell, R. Wetzel, J. MacNeill, J. M. Ren, J. Yuan, C. E. Bakalarski, J. Villen, J. M. Kornhauser, B. Smith, D. Li, X. Zhou, S. P. Gygi, T.-L. Gu, R. D. Polakiewicz, J. Rush and M. J. Comb, Cell, 2007, 131, 1190–1203 CrossRef CAS PubMed.
- J. A. Orchard, R. E. Ibbotson, Z. Davis, A. Wiestner, A. Rosenwald, P. W. Thomas, T. J. Hamblin, L. M. Staudt and D. G. Oscier, Lancet, 2004, 363, 105–111 CrossRef CAS PubMed.
- P. Zhang, L.-Y. Jiang, J.-T. Ma and Q. Jia, Chin. J. Anal. Chem., 2021, 49, 24–33 CAS.
- J. Chen, S. Shinde, P. Subedi, C. Wierzbicka, B. Sellergren, S. Helling and K. Marcus, J. Chromatogr. A, 2016, 1471, 45–50 CrossRef CAS PubMed.
- A. İncel, S. Shinde, I. A. Diez, M. M. Stollenwerk, O. N. Jensen and B. Sellergren, ChemRxiv, Cambridge: Cambridge Open Engage, 2022, DOI:10.26434/chemrxiv-2022-bzkrq.
- Q. S. Li, F. Shen, X. Zhang, Y. F. Hu, Q. X. Zhang, L. Xu and X. Q. Ren, Anal. Chim. Acta, 2013, 795, 82–87 CrossRef CAS PubMed.
- L. Xu, Y. F. Hu, F. Shen, Q. S. Li and X. Q. Ren, J. Chromatogr. A, 2013, 1293, 85–91 CrossRef CAS PubMed.
- X. Q. Yang and Y. Xia, J. Sep. Sci., 2016, 39, 419–426 CrossRef CAS PubMed.
- M. Emgenbroich, C. Borrelli, S. Shinde, I. Lazraq, F. Vilela, A. J. Hall, J. Oxelbark, E. De Lorenzi, J. Courtois, A. Simanova, J. Verhage, K. Irgum, K. Karim and B. Sellergren, Chem. – Eur. J., 2008, 14, 9516–9529 CrossRef CAS PubMed.
- A. Incel, I. A. Diez, C. Wierzbicka, K. Gajoch, O. N. Jensen and B. Sellergren, Anal. Chem., 2021, 93, 3857–3866 CrossRef CAS PubMed.
- C. Wierzbicka, M. Liu, D. Bauer, K. Irgum and B. Sellergren, J. Mater. Chem. B, 2017, 5, 953–960 RSC.
- R. Gui and H. Jin, J. Photochem. Photobiol., C, 2019, 41, 100315 CrossRef CAS.
- D. Y. Li, Y. P. Qin, H. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2015, 66, 224–230 CrossRef CAS PubMed.
- S. C. Burnage, J. Bell, W. Wan, E. Kislenko and K. Rurack, Lab Chip, 2023, 23, 466–474 RSC.
- W. Wan, A. B. Descalzo, S. Shinde, H. Weißhoff, G. Orellana, B. Sellergren and K. Rurack, Chem. – Eur. J., 2017, 23, 15974–15983 CrossRef CAS PubMed.
- P. Narayanaswamy, S. Shinde, R. Sulc, R. Kraut, G. Staples, C. H. Thiam, R. Grimm, B. Sellergren, F. Torta and M. R. Wenk, Anal. Chem., 2014, 86, 3043–3047 CrossRef CAS PubMed.
- V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- S. Pal, T. K. Ghosh, R. Ghosh, S. Mondal and P. Ghosh, Coord. Chem. Rev., 2020, 405, 213128 CrossRef CAS.
- Y. Hu, S. Long, H. Fu, Y. She, Z. Xu and J. Yoon, Chem. Soc. Rev., 2020, 50, 589–618 RSC.
- K. Dong, S. Zhang and J. Wang, Chem. Commun., 2016, 52, 6744–6764 RSC.
- S. Farshbaf and P. Anzenbacher, Chem. Commun., 2019, 55, 1770–1773 RSC.
- S. Shinde, Z. El-Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. Gjoerloff Wingren and B. Sellergren, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef CAS PubMed.
- M. Kimani, S. Beyer, Z. El-Schich, K. Gawlitza, A. Gjörloff-Wingren and K. Rurack, ACS Appl. Polym. Mater., 2021, 3, 2363–2373 CrossRef CAS.
- S. Jiang, T. Wang, S. Behren, U. Westerlind, K. Gawlitza, J. L. Persson and K. Rurack, ACS Appl. Nano Mater., 2022, 5, 17592–17605 CrossRef CAS PubMed.
- A. C. Chan, M. Iwashima, C. W. Turck and A. Weiss, Cell, 1992, 71, 649–662 CrossRef CAS PubMed.
- Q. Li, T. Wang, Y. Jin, C. Wierzbicka, F. Wang, J. Li and B. Sellergren, Sens. Actuators, B, 2022, 368, 132193 CrossRef CAS.
- O. Bakulina, F. K. Merkt, T. O. Knedel, C. Janiak and T. J. J. Muller, Angew. Chem., Int. Ed., 2018, 57, 17240–17244 CrossRef CAS PubMed.
- S. J. Pike, J. J. Hutchinson and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6700–6706 CrossRef CAS PubMed.
- V. Amendola, D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS PubMed.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53–R88 CrossRef CAS.
- S. Shinde, M. Mansour, L. Mavliutova, A. Incel, C. Wierzbicka, H. I. Abdel-Shafy and B. Sellergren, ACS Omega, 2022, 7, 587–598 CrossRef CAS PubMed.
- S. Wagner, C. Zapata, W. Wan, K. Gawlitza, M. Weber and K. Rurack, Langmuir, 2018, 34, 6963–6975 CrossRef CAS PubMed.
- Bindfit, https://app.supramolecular.org/bindfit, (accessed November 2022).
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- G. Wulff and K. Knorr, Bioseparation, 2001, 10, 257–276 CrossRef CAS PubMed.
- R. S. Fernandes, I. M. Raimundo and M. F. Pimentel, Colloids Surf., A, 2019, 577, 1–7 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- J. E. S. Schier, D. Cohen-Sacal, O. R. Larsen and R. A. Hutchinson, Polymers, 2017, 9, 368 CrossRef PubMed.
- S. Banerjee, ARKIVOC (Gainesville, FL, U. S.), 2016, 82–110.
- M. Divya, T. Benny, P. Christy, E. P. Aparna and K. S. Devaky, Bioorg. Chem., 2017, 74, 91–103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: synthetic and experimental details, quantum chemical calculations, binding constant calculations, calculations of pKa values and molar absorption coefficient of fCL, functionalised SiO2 particles characterisation, spectroscopic properties and response behaviour of fCL and MIP and NIP core–shell polymer@SiO2 particles, TEM images analysis for SiO2 core particles and MIP and NIP core–shell polymer@SiO2 particles, calculation of fCL amount in polymer shell of MIP and NIP core–shell polymer@SiO2 particles, LOB, LOD and LOQ calculations and calculations of relative measurement uncertainties. See DOI: https://doi.org/10.1039/d3tb01474f |
| This journal is © The Royal Society of Chemistry 2023 |