
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrids of manganese oxide and lipid liquid crystalline nanoparticles (LLCNPs@MnO) as potential magnetic resonance imaging (MRI) contrast agents†
Dorota
Flak
 a,
Tomasz
Zalewski
a,
Katarzyna
Fiedorowicz
a,
Łucja
Przysiecka
a,
Marcin
Jarek
a,
Adam
Klimaszyk
a,
Marek
Kempka
ab,
Agnieszka
Zimna
c,
Natalia
Rozwadowska
c,
Jonathan
Avaro
d,
Marianne
Liebi
e and
Grzegorz
Nowaczyk
*a
a,
Tomasz
Zalewski
a,
Katarzyna
Fiedorowicz
a,
Łucja
Przysiecka
a,
Marcin
Jarek
a,
Adam
Klimaszyk
a,
Marek
Kempka
ab,
Agnieszka
Zimna
c,
Natalia
Rozwadowska
c,
Jonathan
Avaro
d,
Marianne
Liebi
e and
Grzegorz
Nowaczyk
*a
aNanoBioMedical Centre, Adam Mickiewicz University Poznań, Wszechnicy Piastowskiej 3, 61-614 Poznań, Poland. E-mail: nowag@amu.edu.pl
bDepartment of Biomedical Physics, Faculty of Physics, Adam Mickiewicz University Poznań, Uniwersytetu Poznańskiego 2, 61-614 Poznań, Poland
cInstitute of Human Genetics Polish Academy of Sciences, Strzeszyńska 32, 60-479 Poznań, Poland
dEmpa - Swiss Federal Laboratories for Materials Science and Technology, Center for X-ray Analytics and Biomimetic Membranes and Textiles, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland
eEmpa - Swiss Federal Laboratories for Materials Science and Technology, Center for X-ray Analytics, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland
First published on 1st September 2023
Abstract
Due to the health risks associated with the use of Gd-chelates and the promising effects of using nanoparticles as T1 contrast agents (CAs) for MRI, Mn-based nanoparticles are considered a highly competitive alternative. The use of hybrid constructs with paramagnetic functionality of Mn-based nanoparticles is an effective approach, in particular, the use of biocompatible lipid liquid crystalline nanoparticles (LLCNPs) as a carrier of MnO nanoparticles. LLCNPs possess a unique internal structure ensuring a payload of different polarity MnO nanoparticles. In view of MRI application, the surface properties including the polarity of MnO are crucial factors determining their relaxation rate and thus the MRI efficiency. Two novel hybrid constructs consisting of LLCNPs loaded with hydrophobic MnO-oleate and hydrophilic MnO-DMSA NPs were prepared. These nanosystems were studied in terms of their physico-chemical properties, positive T1 contrast enhancement properties (in vitro and in vivo) and biological safety. LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA hybrids exhibited a heterogeneous phase composition, however with differences in the inner periodic arrangement and structural parameters, as well as in the preferable localization of MnO NPs within the LLCNPs. Also, these hybrids differed in terms of particle size-related parameters and colloidal stability, which was found to be strongly dependent on the addition of differently functionalized MnO NPs. Embedding both types of MnO NPs into LLCNPs resulted in high relaxivity parameters, in comparison to bare MnO-DMSA NPs and also commercially developed CAs (e.g. Dotarem and Teslascan). Further biosafety studies revealed that cell internalization pathways were dependent on the prepared hybrid type, while viability, effects on the mitochondria membrane potential and cytoskeletal networks were rather related to the susceptibility of the particular cell line. The high relaxation rates achieved with the developed hybrid LLCNPs@MnO enable them to be possibly used as novel and biologically safe MRI T1-enhancing CAs in in vivo imaging.
1. Introduction
Magnetic resonance imaging (MRI) is a non-invasive technique supporting disease diagnosis and pathological characterization with ease and in a comparatively safe manner. Nowadays, it is routinely used in clinics due to its associated intrinsic spatial resolution, deep tissue penetration and three-dimensional anatomical information it gathers.1 The MRI signal is dependent on the density of protons and their relaxations, which are largely dependent on the chemical environment. The rates of relaxation are characterized by a longitudinal relaxation time (T1), describing the energy exchange between spins and lattices, and by a transverse relaxation time (T2), describing the energy exchange between spins. Both the proton density and the T1 and T2 relaxation times are used to contrast MRI images. However, MRI suffers from inherently low sensitivity; hence, exogenous contrast agents (CAs) are often introduced in order to affect the relaxation times of water protons, and therefore brighten (positive CAs) or darken (negative CAs) their immediate surrounding areas in the image, respectively.2 To date, numerous design approaches have been used to develop CAs with improved molar relativities, either longitudinal (r1) or transverse (r2), beginning with the coordination of metallic complexes including lanthanide complexes and finally nanoparticles.2–5 Most of these are based on highly paramagnetic ions such as Gd3+, Fe3+ and Mn2+. Gold-standard CAs used in clinical MRI are Gd-based positive contrast agents. These are mainly macrocyclic- (Dotarem, Gadovist, and Prohance) and linear- (Magnevist, Multihance, Omniscan, Optimark, and Primovist) type Gd-chelate formulations. These Gd-based chelate CAs, although the most frequently used agents in MRI body scans worldwide, suffer from possible toxicity and long-term accumulation in brain tissues, and may also induce nephrogenic systemic fibrosis (NSF) and resultant renal impairment.6–8 For these reasons, in 2017, the European Medicines Agency (EMA) concluded its review of gadolinium-based CAs, with recommendations to restrict the use of some linear gadolinium agents used in MRI body scans and to suspend the authorization of others. In the same recommendation, the EMA permitted the use of macrocyclic Gd-based chelate formulations.9 A fairly good commercial Mn-based alternative to Gd-based CAs is Teslascan (the active substance is mangafodipir trisodium). It was initially approved by the Food and Drug Administration (FDA) and the EMA for the detection of lesions in the liver that might be caused by liver cancer or cancer that has spread to the liver from other parts of the body.10 However, in 2012, based on the decision of the marketing authorization holder, the EMA made a decision to withdraw the marketing authorization for Teslascan for commercial reasons.11 Due to the reduced number of available T1 CAs and possible health risks of some Gd-chelates, alternative CAs are being searched and developed. A promising approach is the use of nanoparticles in two ways: (1) formation of NPs with paramagnetic ions incorporated into a nanostructured framework (e.g. metal oxides, sodium gadolinium fluoride, and metal ferrites); (2) formation of hybrid constructs consisting of inorganic and organic components, with paramagnetic functionality either in an inorganic part or an organic part (e.g. SiO2, Au NPs or polymeric NPs, functionalized with either DOTA, DTPA–lanthanide complexes or Gd2O3, MnO NPs or Mn2+, Gd3+ ions incorporated in the organic matrix, e.g. functional polymeric NPs).12–15Taking into account the health risks associated with the use of Gd-chelates and promising effects of using nanoparticles as CAs, manganese-based NPs are seen in the light of recent research as a highly competitive alternative for T1 CAs.16,17 Mn has been used as a T1 CA since the very beginning of MRI development, however, applied in the form of MnCl2 or Mn2+ complexes.18–21 A Mn2+ ion has five unpaired electrons in a 3d orbital and can therefore produce a large magnetic moment and cause an efficient nearby water proton relaxation. Moreover, due to the biophysical similarity of Mn2+ to Ca2+ ions, it has been exploited to map the brain activity in vivo in animals in so called manganese enhanced magnetic resonance imaging (MEMRI), recognized as an important tool in all sorts of neurological studies.22 It is also an essential element for living organisms, crucial for normal body growth and functioning, and involved in several cellular processes; therefore, it can be considered in vivo tolerable. However, manganese deregulation can result in a disease state; for example, excess of Mn is known to induce cytotoxicity manifested as the abnormal redox potential and changes in the central nervous system.23,24
Manipulation of NPs’ properties such as particle morphology and surface functionalization, realized by the rational design and synthesis of NPs, creates an opportunity for the development of new and efficient CAs. MnO NPs have been proposed as efficient T1 CAs, particularly ultrasmall NPs and coated/functionalized with biocompatible molecules.16,17,25,26 The signal contrast enhancement in MRI is expected via an improved contact between the Mn2+ ions of the MnO core and the surrounding water molecules. Na et al. reported on the MnO NPs (7, 15, 20, and 25 nm) coated with a mPEG-2000 PE (1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N [methoxy(polyethylene glycol)-2000]) shell.27 This study showed that the smaller the NP size the brighter the signal in T1-weighted MRI and proved that the paramagnetic Mn2+ ions on the surface of NPs are responsible for the shortening of the T1 relaxation times, and also T2 shortening effects were observed. Létourneau et al. used ultrasmall (8.4 nm) and nanoaggregates of MnO NPs coated with thiolated molecules (DMSA, meso-2,3-dimercaptosuccinic acid) and aminated- and thiolated-polyethylene glycol (NH2-PEG(5000)-SH) for enhanced cell labelling and colloidal stability, respectively.28 A higher signal was obtained for cells labelled with aggregated MnO NPs due to the higher relaxometric properties of the agglomerates, suggesting that the expected agglomeration of NPs in endosomes does not adversely affect further cell tracking procedures with MRI. Gianolo et al. reported on the relaxivity and physico-chemical data for a high relaxivity of a previously developed Mn(II)-containing polymeric nanomaterial (SN132D Spago Nanomedical AB) with a NP size of 5.6 nm.12 The per Mn relaxivity of this system is among the highest reported for Mn-containing systems and also significantly higher in comparison to that of Gd-based CAs, currently used in clinics. This was ascribed to several complementary characteristics such as slow rotational dynamics, due to the nanosize of used particles, the fast exchange rate of the coordinated water molecules and the presence of a large contribution from second coordination sphere water molecules. Moreover, a further relaxivity increase was observed in human serum due to the increased medium viscosity and competitive chelation of Mn2+. Another rational design approach for MnO-based CAs was provided by Mauri et al., who proposed the embedding of MnO NPs in functional polymers, via either individual NPs coated with PMA (poly(isobutylene-alt-maleic anhydride) or multiple NPs embedded in large PLGA envelopes (poly(lactic-co-glycolic acid) in order to enhance the MRI contrast.14 The PLGA envelope was likely to reduce the MnO NP interaction with water molecules by decreasing both r1 and r2 more pronouncedly than in the case of PMA-coated MnO NPs. However, the PLGA-based system exhibited significant dose- and time-dependent r1 increases in plasma, due to the release of active MnO NPs from the polymer matrix in plasma.
Another approach for the development of a new class of nanoparticle-based CAs exploits the use of a hybrid construct consisting of lipid liquid crystalline nanoparticles (LLCNPs) as a carrier of nanoparticles with specific magnetic properties. LLCNPs are highly curved lipid-based NPs formed by more exotic highly ordered membrane-like phases, such as cubosomes and hexosomes or less ordered spongosomes, in comparison to simple vesicle-like liposomes.29,30 LLCNPs are colloidal NPs, formed mainly by the steric or electrostatic stabilization process modulated by the self-assembly of surfactant-like lipids forming non-lamellar phases in excess water and in the presence of a stabilizer. Cubosomes are NPs with their internal structure consisting of a single membrane bilayer forming a lattice structure type network with two continuous intertwining, but non-intersecting aqueous channels – a 3D well-ordered bicontinuous cubic phase (V2). However, in the case of hexosomes, the internal structure comprises the 2D columnar hexagonal (H2) phases. Spongosomes are another type of LLCNPs, and consist of a randomly ordered bicontinuous cubic phase.31,32 The unique internal structure and morphology of LLCNPs result in a higher membrane surface area in LLCNPs, in comparison to simple vesicle-like NPs. This translates to a significantly higher number of lipid molecules per particle, which also enables a higher payload of drug molecules and/or diagnostic agents. Moreover, LLCNPs can entrap hydrophilic, hydrophobic and amphiphilic molecules in the aqueous channels, hydrophobic region or at their interphases, respectively.29 Likewise, considering the high biocompatibility, biodegradability, loading/release efficiencies, and facile fabrication strategies, LLCNPs have potential to be applied further in clinics next to liposomes, niosomes, transfersomes, and solid lipid nanoparticles.33,34
So far, LLCNPs have been used as carriers of nanoparticulated inorganic CAs such as SPIONs,35 mixed ferrite,36 and also formed nanoassemblies with Gd-containing amphiphiles such as Gd-DTPA-BSA and PE-DTPA-Gd,37 and with higher molecular weight Gd-DTPA-monophytanyl or Gd-DTPA-monooleyl amphiphiles forming supramolecular assemblies.38,39 These two approaches exhibit a certain collection of advantages and disadvantages; however, both can be considered to have high potential to be used as combined diagnostic and therapeutic nanomedicine. However, before any nanoparticles can be applied to clinical practice, their biosafety needs to be adequately evaluated, using in vitro cell culture models first. Such tests usually include an assessment of nanoparticle uptake (efficiency and route) and their impact on cell viability and cytotoxicity. Prolonged incubation of nanosystems with cells may lead to apoptosis (programmed cell death) via different pathways; therefore, the possible DNA, membrane and cytoskeletal damage should be considered.
In this work, we studied a new class of safe hybrid MRI positive contrast agents based on combined constructs consisting of LLCNPs loaded with two types of inorganic MnO nanoparticles. MnO-oleate capped NPs possessed a hydrophobic character, while MnO-DMSA NPs showed a hydrophilic character due to functionalization with carboxylic acid ligands. These surface properties of MnO NPs are relevant first for their interaction with lipid molecules and finally lipid NPs, and also in view of MRI, for their interaction with water molecules affecting the relaxation rate from the NPs to the surrounding medium.
The positive T1 contrast enhancement phenomena of the prepared hybrid lipid@MnO systems were first studied by in vitro quantification of their relaxometric performance and hydration state (q) of the Mn(II) ions. Finally, it was followed by in vivo MRI using a mouse model. The research was completed with complex characterization of the physico-chemical properties of the obtained hybrid systems as MRI contrast agents, which nevertheless qualify these systems for advanced development research. In order to prove their biosafety, we tested the effects of different formulations on viability and genotoxicity as well as on changes in the mitochondrial membrane potential and cytoskeletal network in three cell lines. Here, we used HeLa cells as a standard model system of cancer cells, and LN229 and U251MG cells as glioblastoma cell lines.
2. Experimental
2.1. Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.15 at 60 °C until the viscous transparent melt was obtained. Then, the pre-warmed up to ∼60 °C dH2O was added to gain a concentration of 20 mg mL−1 (2% w/w) of GMO considering the temperature-composition phase diagram48 and left overnight at room temperature for hydration to form a bulk (gel) lipid phase. LLCNPs were formed from the bulk lipid phase by homogenization (Branson Sonifier 250, Emerson Electric Company) at an output power of 60 W until a milky homogenous dispersion was formed.
:0.15 at 60 °C until the viscous transparent melt was obtained. Then, the pre-warmed up to ∼60 °C dH2O was added to gain a concentration of 20 mg mL−1 (2% w/w) of GMO considering the temperature-composition phase diagram48 and left overnight at room temperature for hydration to form a bulk (gel) lipid phase. LLCNPs were formed from the bulk lipid phase by homogenization (Branson Sonifier 250, Emerson Electric Company) at an output power of 60 W until a milky homogenous dispersion was formed.
For the formulation of LLCNPs loaded with the hydrophobic oleate-capped MnO (LLCNPs@MnO-oleate), these MnO nanoparticles were diluted in chloroform and added to the mixture of GMO and Pluronic F127 at 10% w/w to the lipid. The resulting solution was then vacuum evaporated at 60 °C in order to remove the organic solvent and then added to the pre-warmed up to ∼60 °C dH2O to gain a concentration of 2% w/w of GMO in a final mixture. The further steps in the preparation of LLCNPs@MnO-oleate were performed as for unloaded LLCNPs.
For the formulation of LLCNPs loaded with the hydrophilic MnO-DMSA (LLCNPs@MnO-DMSA), functionalized MnO-DMSA nanoparticles were added as a pre-warmed (∼60 °C) aqueous suspension to the melt of GMO and Pluronic F127 at 10% w/w to the lipid, and the resulting mixture was added to the pre-warmed (∼60 °C) dH2O to gain a concentration of 2% w/w of GMO in a final mixture. The further steps in the preparation of LLCNPs@MnO-DMSA were performed as for unloaded LLCNPs. To sum up the composition, the weight ratio of all components, i.e. GMO:F-127:MnO, was kept as 1:0.15:0.1. Both the obtained MnO-loaded LLCNP dispersions appeared as milky light-brown. Before further analyses, as-prepared LLCNP dispersions were subjected to stabilization at room temperature for 24 h.
For uptake studies, LLCNPs were stained with the fluorescent dye Nile Red (NR, Merck). Briefly, for NR-stained LLCNPs@MnO-oleate, the NR solution in chloroform was added to the GMO and Pluronic F127 mixture in an amount of 0.1% w/w with respect to the GMO, prior to the further addition of MnO-oleate NPs. Further synthesis steps were similar as above for LLCNPs@MnO-oleate. In the case of NR-stained LLCNPs@MnO-DMSA, NR solution in ethanol was added to the GMO and Pluronic F127 mixture in an amount of 0.1% w/w with respect to the GMO. Then, the solution was vacuum evaporated at 60 °C to remove the organic solvent, prior the further addition of the MnO-DMSA NPs. Further synthesis steps were similar to the above for LLCNPs@MnO-DMSA. Finally, for both NR-stained LLCNPs@MnO-oleate and NR-stained LLCNPs@MnO-DMSA, unattached NR molecules were removed by ultrafiltration and centrifugation using Amicon® Ultra-4 Centrifugal Filter Devices (10000 MWCO, Merck Millipore) according to manufacturer's instructions.
2.2. Characterization of physicochemical properties
:1 ratio with dH2O and deposited on previously treated plasma lacey/carbon grids (Ted Pella Inc.) and freeze-dried at −173 °C using the Cryoplunge 3 System (Gatan Inc.).
| MnO-DMSA | LLCNPs | LLCNPs@MnO-oleate | LLCNPs@MnO-DMSA |
|---|---|---|---|
| [Mn ion]/mM | μg mL−1 | [Mn ion]/mM | [Mn ion]/mM |
| 0.08 | 50 | 0.01 | 0.01 |
| 0.19 | 100 | 0.02 | 0.02 |
| 0.38 | 150 | 0.03 | 0.03 |
| 0.56 | 200 | 0.04 | 0.05 |
| 0.75 | 300 | 0.06 | 0.07 |
| 1.00 | 400 | 0.08 | 0.09 |
To acquire information about the transverse relaxation time T2 (spin–spin interactions), the Carr–Purcell–Meiboom–Gill (CPMG) sequence was used. Since all collected data exhibited a single-exponential decay of echo's train (behavior), they were fitted using the mono-function M(t) = M0*exp(−t/T2).
Finally, the dependence of relaxation times T1, and T2vs. Mn(II) concentration (relaxation rates 1/T1, 1/T2) was then used to calculate the relaxivity parameters r1 and r2, respectively.
000 ms, Te – 10 ms, numbers of echoes – 64, average – 1, FOV – 25 × 25 mm, matrix – 256 × 256, and slice thickness – 2 mm.
2.3. Biological characterization
The influence of different endocytic inhibitors on the cellular uptake of LLCNPs was also assessed. Briefly, the seeded cells were incubated separately with methyl-β-cyclodextrin (2.5 mg mL−1), as an inhibitor of caveolae/lipid raft-dependent endocytosis, chlorpromazine hydrochloride (5 μg mL−1), as an inhibitor of clathrin-mediated endocytosis, and cytochalasin D (5 μg mL−1), as an inhibitor of F-actin polymerization (for 1 h at 37 °C). Subsequently, cells were incubated with 10 μg mL−1 of Nile Red stained LLCNPs for 1 h and imaged as described above.
To determine the intracellular localization of MnO-loaded LLCNPs, the samples were analyzed using a transmission electron microscope (TEM) according to our previous procedure.50 Briefly, the HeLa cells after 30 min or 3 h incubation with nanoparticles were fixed with 4% formaldehyde and 1% glutaraldehyde in PBS for 2 h. After fixation, the samples were postfixed in 1% osmium tetroxide for 1 h at room temperature, dehydrated through a graded series of ethanol concentrations (50, 70, 80, 90, 96, and 100%) and embedded in epoxy resin. Ultrathin sections were prepared using an ultramicrotome (RMC PowerTome PT-XL), collected onto TEM grids, stained with 2% uranyl acetate and analyzed using a transmission electron microscope (Jeol JEM-1400 TEM).
All cells were cultured in Dulbecco's modified Eagle's medium (DMEM, ThermoFisher Scientific) supplemented with 10% fetal bovine serum (FBS, Gibco® Qualified, ThermoFisher Scientific) and 100 units per mL penicillin–streptomycin (ThermoFisher Scientific). Cells were maintained under standard cell culture conditions at 37 °C in a humidified atmosphere containing 5% CO2. Cell cultures were monitored every day until they reached 90% of confluence and were passaged using the trypsin/EDTA protocol in a 1:9 ratio. In vitro cell culture was routinely checked for Mycoplasma contamination by the polymerase chain reaction (PCR).
3. Results and discussion
3.1. Synthesis and physico-chemical characterization
The schematic representation of the preparation route of hybrid constructs consisting of lipid-based LLCNPs loaded with inorganic MnO nanoparticles is given Fig. S1 (ESI†). The GMO-based LLCNPs matrix was used to embed and carry MnO NPs in a hydrophobic form synthesized in an organic solvent and further functionalized to a hydrophilic form. In brief, thermal decomposition of the Mn(II) oleate complex in the presence of strongly coordinating stearic acid as a surfactant resulted in quasi-spherical hydrophobic MnO-oleate capped NPs exhibiting a medium range of polydispersity, as revealed with TEM and DLS analyses. Excess of stearic acid inhibits shape-controlled growth and hence results in spherical or octahedral nanocrystals (Fig. 1A). The particle size calculated from the TEM analysis was around 8.3 nm (Fig. 1B) and the related hydrodynamic particle size (dNumber) was 15.9 nm, with a PDI (polydispersity index) of 0.308, measured in hexane. The XRD pattern (Fig. 1D) with broad and low intensity reflexes indicates that small nanocrystallines of MnO were prepared, crystallizing in the cubic structure, with a relatively insignificant contribution of the tetragonal structure, which occurred as a result of the possible surface oxidation towards Mn3O4.38 Additionally, sharp XRD reflexes assigned to stearic acid are also observed.51 To confirm the possible existence of the tetragonal phase characteristic for Mn3O4 nanoparticles detailed, the fast Fourier transformation (FFT) analysis of HRTEM images was performed (Fig. 1C). It was found that besides evident spots corresponding to the cubic MnO (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) structure some additional spots are also present on FFT diffractograms, which can be found as originating from tetragonal Mn3O4.52 The analogous results were also obtained for MnO-DMSA based on FFT difractogram analysis showing additional reflection rings not derived from the cubic phase (Fig. S2, ESI†). Additionally, TEM, morphological studies led to the conclusion that DMSA-functionalization had an important impact on the agglomeration of MnO-DMSA NPs, which is visible in TEM results, and is supported by particle size measurements (DLS), where the obtained dNumber was 120.4 nm, with a PDI of 0.148.
m) structure some additional spots are also present on FFT diffractograms, which can be found as originating from tetragonal Mn3O4.52 The analogous results were also obtained for MnO-DMSA based on FFT difractogram analysis showing additional reflection rings not derived from the cubic phase (Fig. S2, ESI†). Additionally, TEM, morphological studies led to the conclusion that DMSA-functionalization had an important impact on the agglomeration of MnO-DMSA NPs, which is visible in TEM results, and is supported by particle size measurements (DLS), where the obtained dNumber was 120.4 nm, with a PDI of 0.148.
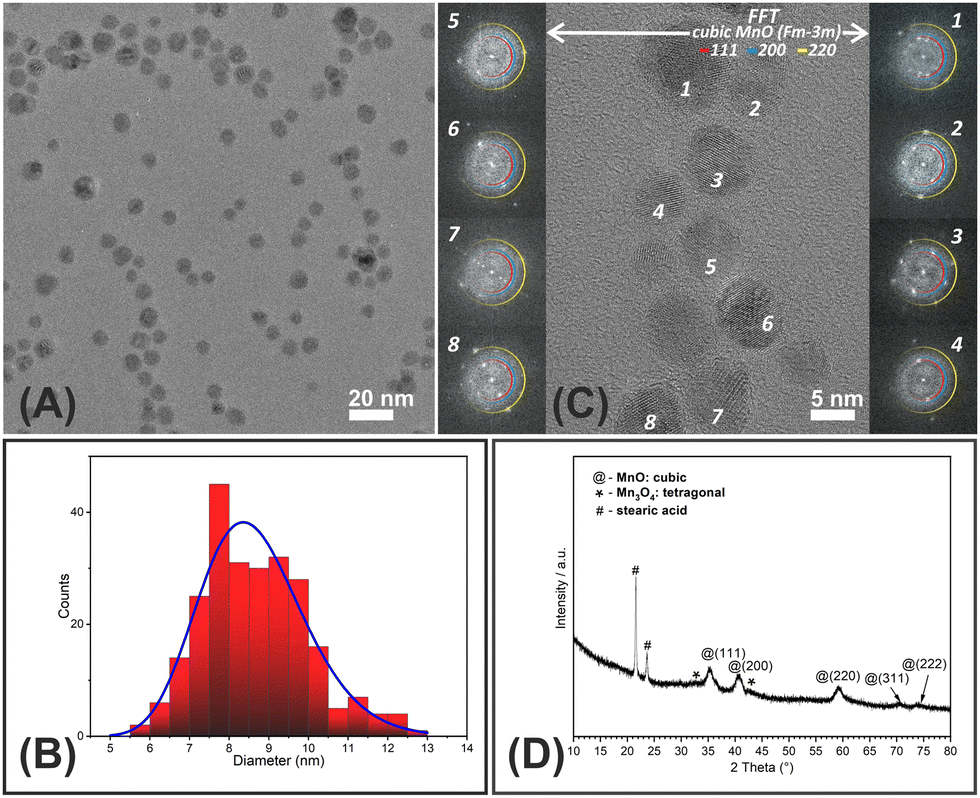 | ||
| Fig. 1 (A) HRTEM image of MnO NP population considered for the size distribution evaluation (B). (C) HRTEM image of MnO NPs with corresponding FFT diffractograms. Insets in columns are FFTs corresponding to numbered nanoparticles. The coloured rings on FFT diffractograms indicate the position of expected and existing reflections from (111 – red, 200 – blue and 220 – yellow) atomic planes of the cubic MnO monocrystal. The most noticeable discrepancy between the MnO diffraction pattern and the obtained results refers to particle 7. Since the diffraction patterns of cubic MnO and tetragonal Mn3O4 are overlapping in a reciprocal space, some reflections look like blurred. (D) XRD pattern of MnO NPs with denoted reflection peaks of the cubic phase (@), tetragonal one (*) and stearic acid (#). | ||
The strategy for an LLCNP hybrid construct preparation assumed a loading of MnO NPs into a lipid-based carrier as efficient T1 CAs. This is expected to deliver a sufficient amount of Mn ions to the area of interest to cause an increase in the T1 relaxivity, and also simultaneously decrease the metal-associated toxicity and enhanced bioavailability, particularly improved via the DMSA-functionalization of MnO NPs. In the first preparation approach of hybrid constructs, the hydrophobic MnO-oleate capped NPs were directly loaded into a GMO-based matrix, resulting in the LLCNPs@MnO-oleate. In the second approach, DMSA-functionalized MnO NPs (MnO-DMSA) were loaded into a GMO-based matrix. DMSA-functionalization imparted the hydrophilicity to MnO NPs, owing to which a water-based solution was obtained for further preparation of MnO-loaded LLCNPs (LLCNPs@MnO-DMSA). DMSA ligands bind to the surface of metal oxide providing disulfide bridges between particle cores, and the resulting MnO-DMSA NPs were anionic with a zeta potential of −14.2 mV. DMSA ligands additionally provided biocompatibility to the NPs, as required for biomedical applications, and also, as it will be further presented, imposed the different cell uptake mechanism with all associated consequences (such as biological effects and final MRI efficiency).
Both types of hybrid MnO-loaded LLCNPs appear as milky-brown homogenous dispersions in comparison to milky-white unloaded LLCNPs. Cryo-TEM images (Fig. 2A, B and C) revealed that the obtained LLCNPs were of the shape and structure of cubosomes, spongosomes and liposomes. For comparison, the cryo-TEM images of pure unmodified GMO-based LLCNPs are given elsewhere.47 The inner periodic arrangement depended strongly on the addition of differently functionalized MnO NPs. Although cryo-TEM images do not provide certain information that obtained colloidal dispersions containing very well, long-distance arranged liquid crystals, the SAXS data confirm clearly that at least for the LLCNPs@MnO-DMSA system, such ordering reflecting the Pn-3m phase is present (Fig. 2D). Considering the peak positions (110 and 111) from SAXS scattering curves, the calculated lattice constant is of values of 8.6 nm and 8.8 nm for LLCNPs and LLCNPs@MnO-DMSA, respectively. This result suggests that the system with MnO-DMSA NPs can swell more efficiently. Moreover, the lack of clear diffraction peaks besides the broad background component in SAXS data and cryo-TEM images of lamellar, but not ordered structures proved that for LLCNPs@MnO-oleate, the existence of the cubic phase within LLCNPs is not dominating.53,54 It is anticipated that this system exhibits mainly a sponge-like phase, less hydrated with only a short-range arrangement. Based on cryo-TEM images, significant differences are also found in the preferential localization of MnO NPs depending on their surface functionalization. In contrast to the MnO-oleate NPs, which are preferably incorporated into the interior of LLCNP forming clusters (Fig. 2A), the MnO-DMSA NPs were located on the LLCNPs’ surface as well as in their core (Fig. 2B and C).
 | ||
| Fig. 2 Morphological (cryo-TEM) and structural (SAXS) analyses of LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA systems. (A) Cryo-TEM image of LLCNPs@MnO-oleate shows the inner localization of MnO nanoparticles and the less ordered sponge-like structure of lipid nanoparticles. (B) Cryo-TEM image of LLCNPs surrounded efficiently by MnO-DMSA NPs. (C) LLCNPs@MnO-DMSA systems of observed alternative structures with MnO-DMSA in the core of LLCNPs. (D) SAXS scattering curves of pure LLCNPs based on GMO and with embedded MnO NPs (oleate- and DMSA-functionalized). In the SAXS spectra, the reflexes 110 and 111 characteristic for the Pn-3m cubic structure are present within the regions marked by dotted lines. | ||
The particle size and zeta potential (ζ) results of prepared LLCNP dispersions are reported in Table 2, which summarizes the mean particle size from the distribution of the particle size (hydrodynamic diameter) measurements weighted by the intensity (dIntensity) and the number (dNumber), the polydispersity index (PdI), and the ζ potential. The results also include the stability of prepared NPs and hybrid constructs assessed after one-month storage at room temperature (_1 m), and in the presence of the human serum albumin (_HSA) reflecting the plasma environment and to estimate better their potential in biomedical applications.
| Sample | d Intensity/nm | SD | d Number/nm | SD | PDI | SD | Zeta/mV | SD |
|---|---|---|---|---|---|---|---|---|
| a Bimodal PSD, values in brackets are the area under PSD peaks. b Measurements in hexane. | ||||||||
| MnO-oleate cappedb | 83.6 | 1.10 | 15.9 | 3.00 | 0.308 | 0.035 | −31.1 | 14.20 |
| MnO-DMSA | 209.8 | 10.80 | 120.4 | 18.15 | 0.148 | 0.010 | −14.2 | 0.51 |
| MnO-DMSA_1 m | 239.7 | 15.42 | 132.6 | 16.03 | 0.255 | 0.076 | −11.2 | 0.93 |
| MnO-DMSA_HSA | 4.6 (53.4)a | 0.13 | 3.6 | — | — | — | — | — |
| 313.0 (38.3)a | 106.40 | — | 0.16 | 0.553 | 0.006 | −3.6 | 0.17 | |
| LLCNPs | 175.9 | 9.94 | 82.9 | 13.10 | 0.172 | 0.016 | −19.8 | 0.60 |
| LLCNPs _1m | 193.8 | 7.80 | 64.6 | 38.00 | 0.162 | 0.032 | −17.8 | 0.56 |
| LLCNPs _HSA | 4.6 (62.9)a | 0.10 | 3.4 | — | — | — | — | — |
| 234.5 (35.9)a | 11.75 | — | 0.10 | 0.440 | 0.009 | −1.6 | 0.86 | |
| LLCNPs@MnO-oleate | 246.0 | 6.33 | 167.8 | 6.63 | 0.138 | 0.023 | −20.2 | 1.21 |
| LLCNPs@MnO-oleate _1m | 500.3 | 27.69 | 242.4 | 6.65 | 0.297 | 0.032 | −28.5 | 0.61 |
| LLCNPs@MnO-oleate _HSA | 4.5 (48.1)a | 0.05 | 3.5 | — | — | — | — | — |
| 447.5 (51.9)a | 47.10 | — | 0.09 | 0.656 | 0.012 | −0.8 | 0.02 | |
| LLCNPs@MnO-DMSA | 170.6 | 5.75 | 78.1 | 12.63 | 0.162 | 0.014 | −16.2 | 1.17 |
| LLCNPs@MnO-DMSA _1m | 202.7 | 4.90 | 143.1 | 3.30 | 0.102 | 0.012 | −21.2 | 2.08 |
| LLCNPs@MnO-DMSA _HSA | 4.6 (59.5)a | 0.06 | 3.4 | — | — | — | — | — |
| 240.2 (36.7)a | 7.44 | — | 0.01 | 0.484 | 0.006 | −3.0 | 0.85 | |
| HSA | 4.8 (51.9)a | 0.04 | 3.5 | 0.10 | 0.501 | 0.004 | −9.0 | 0.52 |
| 88.3 (46.7)a | 1.76 | — | — | — | — | — | — | |
The as-prepared unloaded LLCNPs have a size (dIntensity) of 175.9 nm and loading with MnO significantly affected the size only in the case of LLCNPs@MnO-oleate, having a size of 246 nm, while LLCNPs@MnO-DMSA preserved a similar size of 170.6 nm. PdI in all cases was similar below 0.2, indicating monomodal to a medium range of the polydispersity of prepared dispersions. Regarding colloidal stability, all samples exhibited medium-range stability expressed with a zeta potential negative value of around −20 mV. Therefore, it can be concluded that the MnO-loading did not affect the colloidal stability in dH2O. The summary of the full DLS results of the prepared nanoparticle size (Z-ave – cumulant mean, dIntensity – intensity distribution, dNumber – number distribution, PdI – polydispersity index) and zeta potential including standard deviation (SD, n = 3) is given in Table S1 (ESI†). Furthermore, the particle size parameters and zeta potential of prepared nanoparticles in the culture DMEM medium (Dulbecco's modified Eagle's medium) after 24 h incubation are given in Table S2 (ESI†).
Results further indicate that the size (dIntensity) of particles in prepared dispersions in general increased after 1 month of storage. For LLCNPs and LLCNPs@MnO-DMSA, the size increase was rather small, about 20–30 nm, with an almost no change in the PdI. While in the case of LLCNPs@MnO-oleate the size substantially increased by about 250 nm, which was accompanied by an almost double increase of PdI to 0.297, indicating increased polydispersity. However, still this value stands for the medium range of polydispersity. The ζ potential after 1 month of storage slightly increased in the case of both MnO-loaded LLCNPs, while for MnO-DMSA and unloaded LLCNPs the change was negligible.
In the presence of HSA, the size of particles increased in the case of all samples. The highest increase by 200 nm was again observed for LLCNPs@MnO-oleate. This fraction is due to the contribution of the HSA protein (see the measurement for HSA in Table 2). The size increase was also accompanied by the PdI increase (double, and even triple). Again, the highest value of 0.656 was obtained for LLCNPs@MnO-oleate. The high PdI is also due to the HSA protein contribution to the PSD (particle size distribution). As expected, a large decrease of ζ was observed for all samples incubated with HSA, due to a strong interaction of prepared particles with protein molecules. LLCNPs@MnO-oleate can be therefore considered less stable than LLCNPs@MnO-DMSA in terms of the size and size-related parameters and therefore more prone to interaction with the surrounding protein-containing environment. Similarly, the PDI increased for nanoparticles incubated for 24 h in the DMEM, while the ζ changes were negligible, except for LLCNPs@MnO-oleate (Table S2, ESI†).
3.2. NMR relaxometric determination of the contrast efficiency of prepared NPs
The ability of prepared NPs to reduce the longitudinal relaxation rate (R1) of water protons in a field value of 0.4 T at room temperature was evaluated through the longitudinal relaxivity assigning (r1). Relaxation rates calculated as 1/T1 for MnO-DMSA, LLCNPS@MnO-oleate, and LLCNPs@MnO-DMSA, were plotted as a function of Mn ion concentration (C [Mn ion]) (Fig. 3A and B), and these values depend linearly on the nanoparticles, and hence on Mn ion concentration (Table 1). The longitudinal relaxivity (r1) was determined as a slope of the plotted dependence R1vs. [Mn ion] using the equation: R1 = r1*[Mn ion] + 1/T1_water. r1 was calculated for all prepared NPs, and it was shown to be 2.8 mM−1 s−1 for MnO-DMSA, 12.4 mM−1 s−1 and 9.1 mM−1 s−1 for LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA, respectively. These results confirm that the obtained lipid-based NPs can serve as T1 CAs in MR imaging. Embedding both MnO-oleate capped and MnO-DMSA into LLCNPs leads to the increase of r1, and the highest was obtained for LLCNPs@MnO-oleate when compared with MnO-DMSA. The lower r1 value for MnO-DMSA is probably due to the presence of DMSA-ligands, which gain the hydrophilicity to the NPs, but at the same time also may reduce their interaction and energy transfer with water molecules. In the case of LLCNPs@MnO-DMSA, the r1 value is definitely increased in comparison to MnO-DMSA, which may result from the MnO-DMSA better distribution over the LLCNPs surface (as observed in Fig. 2B), and hence improved spin–lattice relaxation achieved from the NPs to the surrounding water molecules. Yet, the r1 value of LLCNPs@MnO-DMSA was lower than in case of LLCNPs@MnO-oleate, which can be due to a smaller size of the overall hybrid LLCNPs@MnO-DMSA (∼170 nm), when compared to LLCNPs@MnO-oleate (∼246 nm). In addition, much higher r1 values for LLCNPs@MnO-DMSA in comparison to MnO-DMSA can be related to the higher rotational effect assigned to loaded LLCNPs, since the MnO NPs are, as mentioned above, better distributed mainly over the surface of LLCNPs, while for MnO-DMSA NPs most of them remain in the middle of the aggregates covered with the DMSA ligands. Moreover, the ordered liquid crystalline structure (cubic phases next to the lamellar ones) of the prepared hybrid LLCNPs@MnO-DMSA, possessing extensive continuous water channels, allows an efficient water exchange through the swollen matrix, resulting in higher relaxivity. The similar effect can also explain the higher r1 value for LLCNPs@MnO-oleate than that for MnO-DMSA, and the remaining in the less ordered sponge-like phase have better contact with the water molecules in the structure of hybrid nanoparticles resulting in more efficient molecular energy transfer and improved relaxivity.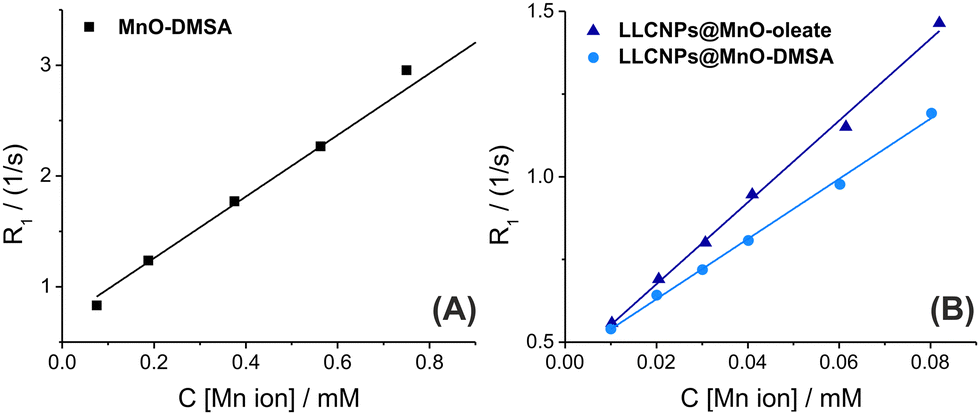 | ||
| Fig. 3 Comparison of the relaxation rate (R1) of (A) MnO-DMSA NPs and (B) LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA at different concentrations of Mn ions, in water and room temperature at 0.4 T. | ||
In this work, the prepared lipid@MnO hybrid NPs are characterized by much higher values of r1 than those of commercially developed CAs (2.2 mM−1 s−1 for Teslascan – Mn-based CAs, or 3.6 mM−1 s−1 for DOTAREM – Gd-based CAs) measured at the similar magnetic field (Table 3 in Rohrer et al.4). This can be attributed to their larger sizes, resulting in longer rotational correlation times.
| s | r 1/(mM−1 s−1) | r 2/(mM−1 s−1) | Field/solvent | r 2 O [mM−1 s−1] | q |
|---|---|---|---|---|---|
| a Results presented by Rohrer et al.4 | |||||
| MnO-DMSA | 2.8 | 8.4 | 0.4 T, H2O | 965 | 1.89 |
| LLCNPs@MnO-oleate | 12.4 | — | 0.4 T, H2O | 2089 | 4.09 |
| LLCNPs@MnO-DMSA | 9.1 | — | 0.4 T, H2O | 1734 | 3.4 |
| Teslascana | 2.2 | 2.0 | 0.47 T, H2O | — | — |
| Dotarema | 3.6 | 4.3 | 0.47 T, H2O | — | — |
| [(Mn(H2O)6]2+ | — | — | — | 2873 | 5.6 |
However, in order to fully understand the effect of different NPs on the relaxivity, it is imperative to determine the hydration state (q) of the Mn(II) ion and the transverse relaxivities r2O. This can be accomplished through 17O T2 measurements. As it was demonstrated for different Mn(II) complexes,49 the hydration state of Mn(II) can be accurately estimated directly from H217O line widths for micromolar Mn ion concentration and can be extended to other types of Mn-containing systems. Such an experimental NMR approach was therefore used in this study, and the results indicate that the method works well also in the case of NPs, including lipid-based ones. The highest number of inner-sphere water ligands (q) was obtained for LLCNPs@MnO-oleate, slightly lower for LLCNPs@MnO-DMSA, and the lowest for MnO-DMSA (Table 3), with respective values of transverse relaxivities (r2O). These results are in accordance with the longitudinal relaxivity measurements (r1), and confirm that NP-based CAs investigated in this work exhibit a solid NP-water proton interaction-related base.
3.3. Studies on enhancing magnetic resonance imaging (MRI) by prepared NPs (in vitro MRI)
To directly observe the contrast enhancement, in vitro MRI imaging was performed on prepared NP dispersions. For this, the longitudinal relaxation times were measured with an MRI scanner at 9.4 T and room temperature, and both T1- and T2-weighted images were obtained, as a reference.As it is visible in Fig. 4, T1-weighted MRI images confirmed the effectiveness of the prepared NPs as positive CAs. As the molar concentration of Mn ions increases (Fig. 4A to C), the obtained images are brighter, which indicates a clear signal intensity dependence on the concentration of Mn ions. These results are in agreement with the one obtained for the dependence of the longitudinal relaxation rate R1vs. [Mn ions]. However, in the case of MnO-DMSA, the signal does not change continuously, as in the case of LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA, which show similar efficiency. In the case of MnO-DMSA, the higher Mn ion concentration (sub-mM up to 1 mM range) has to be used in order to obtain a similar signal to DMSA dispersions, for which the Mn ion concentration was lower (in μM range). Although, CAs based on paramagnetic Mn ions are mainly used as positive CAs, bringing the brightening the T1-depending image through shortening the longitudinal relaxation time T1, the effectiveness of the prepared NPs as T2 negative contrast agents was also investigated. The calculated r2 values obtained from T2 maps are 84 mM−1 s−1 for MnO-DMSA, 208 mM−1 s−1 for LLCNPs@MnO-oleate and 172 mM−1 s−1 for LLCNPs@MnO-DMSA. Obtained T2-weighted MR images also show the dose-dependence of the signal intensity, however, also indicate that prepared lipid-based NPs are not as efficient as for T1 contrasting.
 | ||
| Fig. 4 T 1-weighted and T2-weighted MR images of (A) MnO-DMSA, (B) LLCNPs@MnO-oleate and (C) LLCNPs@MnO-DMSA at different concentrations of Mn ions, in water and room temperature and under the same MRI experimental conditions carried out at 9.4 T. | ||
The positive contrast enhancement using prepared lipid@MnO hybrid NPs can be further considered as competitive, since the obtained values of r1, as mentioned before, exceed those of commercially developed like Teslascan. These systems can also conquer other under-development hybrid systems based on manganese oxide nanoparticles. For example, Mauri et al. reported on MnO NPs embedded in functional polymers, PLGA and PMA, exhibiting r1 values of 0.24 and 7.7 mM−1 s−1 (measured at 0.5 T in PBS), respectively.14 In turn, Na et al. used mPEG-2000 PE to coat MnO NPs with different sizes (7–25 nm), and resultant r1 values were below 0.37 mM−1 s−1 (for 7 nm NPs, measured at 3 T).25 Much the same, MnO-DMSA NPs were investigated by Létourneau et al.26 They investigated non-aggregated and aggregated MnO NPs coated with DMSA and aminated- and thiolated-polyethylene glycol (MnO-DMSA-PEG) exhibiting r1 values of 3.4 mM−1 s−1 and surprisingly 17 mM−1 s−1 (measured at 1.41 T, 20 °C), respectively. Furthermore, they showed in in vitro studies that the expected high positive signal enhancement was not significantly affected upon further agglomeration in endosomes.
3.4. Biocompatibility studies in vitro
 | ||
| Fig. 5 CLSM images of HeLa cells incubated with 10 μg mL−1 of Nile Red stained LLCNPs, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA together with selected transport inhibitors. Red signals are related to the Nile Red dye, and blue signals are related to Hoechst nuclei staining. Scale bar: 10 μm. | ||
To determine whether MnO-loaded LLCNPs uptake was an energy-dependent or –independent mechanism, cell cultures were incubated at 4 °C to inhibit active processes by inactivation of the temperature-fragile enzymes. Interestingly, the differences between samples could be observed. In the case of cells treated with LLCNPs@MnO-DMSA, similarly, as for unloaded LLCNPs, the lowered temperature did not result in an inhibition of endocytosis (energy-independent), whereas in the case of treatment with LLCNPs@MnO-oleate the internalization was inhibited completely, suggesting a different energy-dependent uptake mechanism.
Furthermore, to investigate in detail different types of endocytosis, suitable pharmacological inhibitors were used. The differences are clearly visible when different transport inhibitors are used, and are shown in Fig. 5. Treatment with chlorpromazine resulted in a strong inhibition (almost 100%) of LLCNPs@MnO-oleate uptake in the HeLa cell line, while in the case of LLCNPs@MnO-DMSA only a slight decrease in signal was visible. Taking into account that chlorpromazine is an inhibitor of clathrin-mediated endocytosis, these results together with energy-dependence results indicated that this is the main route of LLCNPs@MnO-oleate uptake. The uptake inhibition by methyl-β-cyclodextrin (MβCD), an inhibitor of lipid raft formation, indicates the role of cholesterol in LLCNP internalization. As can be seen in the corresponding image panel in Fig. 5, the depletion of cellular cholesterol resulted in the inhibition of LLCNPs@MnO-DMSA uptake in HeLa cells, but not of LLCNPs@MnO-oleate. On the other hand, the cellular internalization of LLCNPs@MnO-oleate by cells treated with a cytochalasin D inhibitor showed a fluorescence signal decreasing in comparison to the control. In the case of LLCNPs@MnO-DMSA treated cells, such a decrease was only slight. Therefore, cytochalasin D is responsible for F-actin depolymerization, and as such, it indicates the role of F-actin in cellular uptake, particularly for LLCNPs@MnO-oleate and less in the case of LLCNPs@MnO-DMSA.
Based on the results obtained from internalization studies, it can be concluded that the main mechanism of LLCNPs@MnO-oleate uptake is energy-dependent, clathrin-mediated endocytosis, whereas in the case of LLCNPs@MnO-DMSA it is energy-independent and cholesterol-dependent internalization.
In our former research, the internalization studies performed on the GMO-based LLCNPs on normal and cancer cell lines indicated efficient uptake and localized the nanoparticles in the cytoplasm, close to the perinuclear membrane.47 Such intracellular localization was also observed in other research concerning LLCNPs.55,56 Results presented in this study for MnO-labeled LLCNPs also indicate similar localization. However, the additional analysis of the uptake mechanism indicated significant differences between the two investigated hybrid constructs. The one consisting of hydrophilic LLCNPs@MnO-DMSA exhibited behavior similar to bare LLCNPs, as it was internalized by cells mainly in an energy-independent and cholesterol-dependent manner. This kind of LLCNP–cell membrane interaction could be related to membrane fusion as proposed by Dyett et al.57 However, different uptake mechanisms could also be considered, like the endocytosis–independent mechanism of internalization, previously proposed for phytantriol-based hexosomes,58 where the crucial role of interaction between the membrane-lipids and nanoparticles, then membrane destabilization and direct nanoparticle translocation was indicated.
On the other hand, the intracellular transport of LLCNPs@MnO-oleate was inhibited by chlorpromazine, an inhibitor that causes clathrin to accumulate in late endosomes and thereby inhibits coated pit-mediated endocytosis,59 showing as the main uptake route energy-dependent and clathrin-mediated endocytosis. This kind of internalization is typical for inorganic nanoparticles; however, it was also observed for bare LLCNPs by Zeng et al.60 The obtained results are in agreement with the work of Abdel-Bar and el Basset Sanad, where distinct endocytic routes were observed, and these were also dependent on the LLCNP size.61
To investigate the MnO-loaded LLCNP intracellular localization at the ultrastructural level, the TEM imaging of HeLa cells treated with prepared NPs was performed. In Fig. 6A, the TEM micrograph of non-treated control HeLa cells is shown. As presented in Fig. 6B, after incubation with LLCNPs@MnO-oleate, numerous endosomes’ vesicles are visible inside cells’ cytoplasm, which confirms the clathrin-dependent endocytosis as a main route of hydrophobic MnO-loaded LLCNP uptake. Meanwhile, also freely occurring lipid aggregates can be observed (Fig. 6C). On the other hand, in the case of hydrophilic MnO-DMSA loaded into LLCNPs, they are mainly internalized without the endocytosis pathway and are observed as lipidic aggregates inside the cell cytoplasm, with the visible MnO nanoparticles at the edge of the lipidic structure (Fig. 6D). These results are in agreement with our confocal imaging observations of the internalization pathways. The differences in the uptake are evidently related to the type of prepared hybrid constructs, loaded either with hydrophobic MnO-oleate or hydrophilic MnO-DMSA, which is also related to the difference in their physico-chemical properties, e.g., size, structure, and colloidal stability in different media.
 | ||
| Fig. 6 TEM micrographs of HeLa cells treated with MnO-loaded LLCNPs showing the uptake and intracellular localization: (A) HeLa cells without any treatment, (B) and (C) cells incubated for 30 min with LLCNPs@MnO-oleate, and (D) cells incubated for 30 min with LLCNPs@MnO-DMSA. The second-row images are magnified images marked with square regions; e denotes endosomes. | ||
Summarizing, it is not possible to unequivocally indicate one single mechanism of cell uptake for each prepared type of MnO-loaded LLCNPs. Such behavior could be related to sample heterogeneity, which is composed of lipid-based nanostructures of various sizes and internal phase assembly.
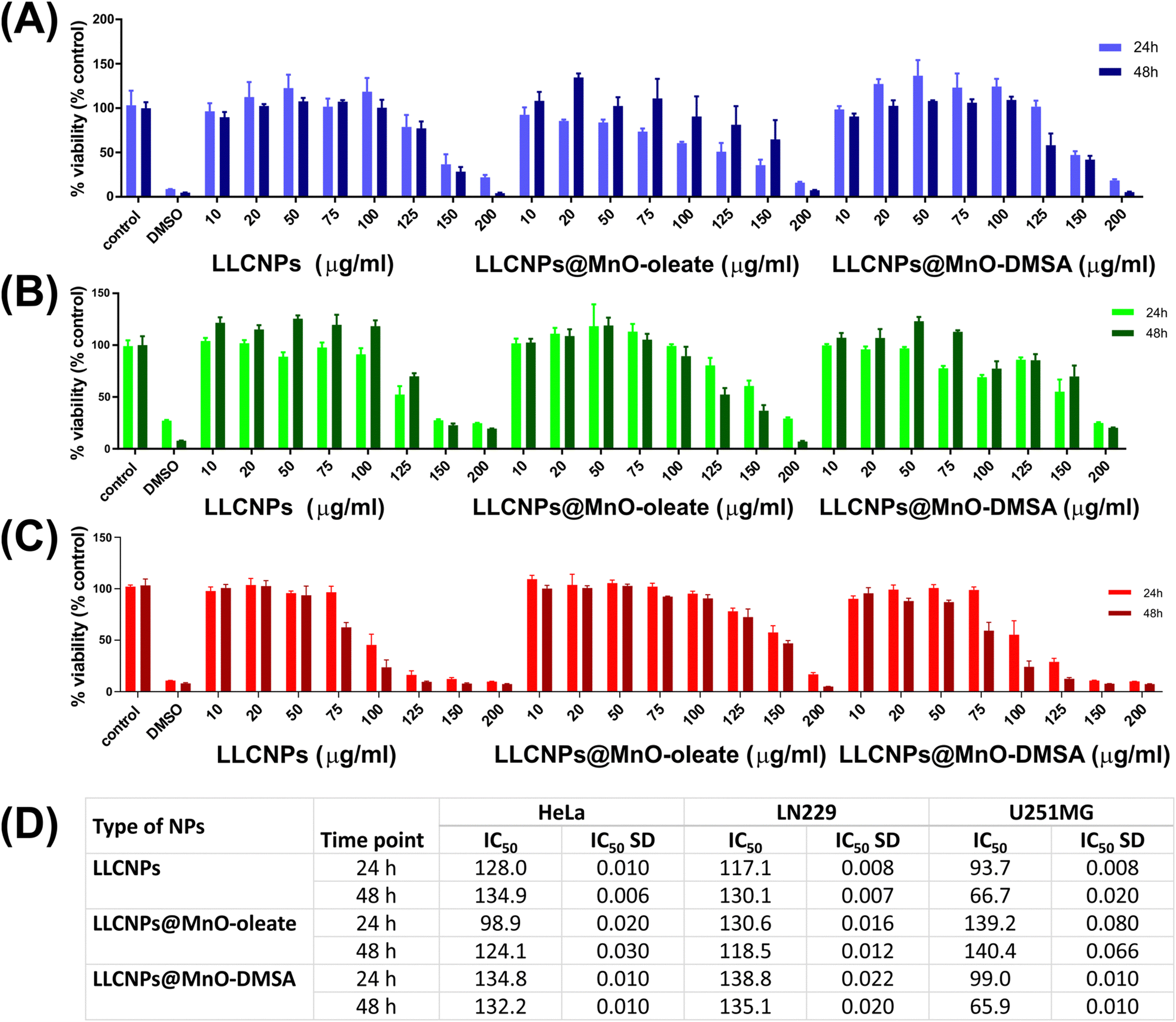 | ||
| Fig. 7 Cell viability of (A) HeLa, (B) LN229, and (C) U251MG, after 24 h and 48 h of incubation with different concentrations (10, 20, 50, 75, 100, 125, 150, and 200 μg mL−1) of the prepared LLCNPs, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA, investigated with WST-1. Controls are cells non-treated, and DMSO is cells treated with 50% v/v DMSO. Data are given as mean ± SD for four replicates. (D) Table with the calculated half maximal inhibitory concentration (IC50). | ||
In the case of LN229 cells after 24 h incubation, the decrease in viability was observed at 125 μg mL−1 for LLCNPs (****p < 0.0001) and LLCNPs@MnO-oleate (*p = 0.0418) and at 75 μg mL−1 (**p = 0.0066) for LLCNPs@MnO-DMSA, with respective values of IC50 = 117.1 μg mL−1, IC50 = 130.6 μg mL−1 and IC50 = 138.8 μg mL−1. After an additional 24 h of incubation, the recovery of LN229 cells treated was observed in the case of LLCNPs and LLCNPs@MnO-DMSA exposure, except for the highest doses.
U251MG cells were more sensitive to LLCNPs and LLCNPs@MnO-DMSA because reduced viability was noted after 24h incubation already at 100 μg mL−1 dose exposure (****p < 0.0001). IC50 values were 93.7 μg mL−1 and 99 μg mL−1, respectively. An additional 24 h of incubation resulted in similar or slightly lower viability values for all nanoparticle types of exposures.
Interestingly, IC50 showed that HeLa cells were the most susceptible for LLCNPs@MnO-oleate amongst investigated cell lines, while U251MG cells were the most susceptible for LLCNPs and LLCNPs@MnO-DMSA, but the most resistant for LLCNPs@MnO-oleate. There are no earlier studies on MnO-loaded LLCNPs for direct comparison. Investigated glioblastoma cell lines (LN229 or U251MG) have been used as an in vitro models of nanoparticle based anti-cancer treatment so far for cubosome nanoparticles,46 but also for inorganic iron oxide or silver nanoparticles,63,64 confirming different viability profiles vs. nanoparticle concentration, depending on the nanoparticle types. Studies performed in the other glioblastoma cell line (T98G) revealed that cubosomes themselves were not toxic up to a 90 μg mL−1 dose exposure, even after 72 h of incubation.65 Therefore, it could be concluded that the cytotoxic effect of lipid-based LLCNPs is strongly cell type-specific, which is in accordance with the recent review work of Almoshari et al.66 It is important to note that the other latest review by Sobańska et al. highlights the significance of investigating the cytotoxicity of manganese oxide nanoparticles, especially in neural cells (neurotoxicity), since Mn itself is a crucial cofactor in neurotransmitters (like glutamine synthetase), and its unbalanced level may lead to neurological disorders.67 Therefore, in our work, we also intended to show how MnO-loaded LLCNPs affected the viability of glioblastoma cells (LN229 and U251MG).
Additionally, the viability of MSU1.1 cells treated with the same concentrations of LLCNP formulation is shown in Fig. S4 (ESI†). The obtained results indicated that the level of cytotoxicity performed on normal human fibroblasts was similar to cancer cells.
 | ||
| Fig. 8 The alkaline comet assay performed for (A) HeLa, (B) LN229, and (C) U251MG cell lines. Cells were treated for 24 h with three concentrations (10, 75 and 125 μg mL−1) of LLCNPs, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA. Controls (negative) are the non-treated cells, and H2O2 (3%, 20 min) treated cells are the positive control. A green spot in images represents the DNA of an individual cell. Graphs shows the calculated tail moment, and the horizontal line shows the mean value. | ||
 | ||
| Fig. 9 Effects of LLCNPs, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA (24 h incubation with 10, 75 and 100 μg mL−1) on mitochondrial membrane potential in (A) HeLa, (B) LN229, and (C) U251MG cells. Representative images of the control (untreated cells) and cells treated with uncoupler-FCCP (positive control) are also given. | ||
 | ||
| Fig. 10 CLSM images of the actin cytoskeleton staining of (A) HeLa, (B) LN229, and (C) U251MG cells after the treatment with the unloaded LLCNPs, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA in concentrations of 10, 75 and 100 μg mL−1 for 24 h and with no treatment (control). F-actin and nuclear DNA were stained with Oregon Green 488 Phalloidin (green) and Hoechst (blue), respectively. Scale bar: 10 μm. | ||
In order to gain deeper insight into the cytoskeletal rearrangement after lipid-based and MnO-loaded NP treatment, additional analysis of gene expression selected based on their function in cytoskeleton network formation was performed. As the cytoskeleton is composed of a network of various proteins that include microfilaments (actin), proteins involved in the actin turnover (e.g., profilin) and microtubules (tubulin), and lamins, genes encoding these proteins were selected, and their expression profile was analyzed by quantitative real-time PCR in all three cell lines (Fig. 11).
 | ||
| Fig. 11 4 Expression of ACTB, ACTG, TUBA4a, PFN1, LMNA, and LMNB genes (normalized to GPADH) in (A) HeLa, (B) LN229 and (C) U251MG cells upon incubation with 10 and 75 μg mL−1 of prepared LLCNPs, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA. Data are given as mean ± SD, n = 3, *p ≤ 0.05, p** ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. | ||
Beta (ACTB) and gamma (ACTG1) actins are nearly identical in the amino acid sequence but play different roles in cells. Beta actin is essential for cell migration and proliferation, while gamma actin is involved in cell motility.71 The results on their expression regulation after incubation with LLCNPs show that, in HeLa cells, only the incubation with 75 μg mL−1 LLCNPs@MnO-oleate led to the downregulation of both genes (Fig. 11A). In the U251MG cell line, the expression of ACTB was downregulated nearly at all tested concentrations of all three types of NPs, when compared to the control (Fig. 10C). In contrast, ACTG1 expression remained at the same level. Interestingly, in the LN229 cell line, the most robust upregulation of ACTB was observed at 75 μg mL−1 of LLCNPs@MnO-oleate and that of ACTG1 at 75 μg mL−1 of LLCNPs (Fig. 11B).
Tubulin expression (TUBA4a), in turn, increased in a dose-dependent manner in LN229, irrespective of LLCNP formulations, when compared to the control (non-treated cells) (Fig. 10B). In the HeLa cell line, the only significant increase in tubulin expression was noted for cells treated with the highest dose of 75 μg mL−1 unloaded LLCNPs (Fig. 11A), while in U251MG cells between controls, 10 and 75 μg mL−1 of LLCNPs@MnO-DMSA (Fig. 11C).
Profilin is an actin-binding protein involved in the regulation of actin and microtubule assembly into the cytoskeleton.72 Interestingly, in our studies, the expression of the profilin 1 (PFN1) was upregulated in the case of incubation with LLCNPs in HeLa (Fig. 11A) and LN229 (Fig. 11B) cell lines treated with 75 μg mL−1 of NPs. In LN229, the significant increase was also noted for treatment with 75 μg mL−1 of LLCNPs@MnO-oleate. Surprisingly, in the U251MG cells, a decrease in PFN1 expression between controls and cells treated with 10 μg mL−1 LLCNPs@MnO-oleate was observed (Fig. 11C).
Laminin is a member of the family of membrane glycoproteins that play a role in cell adhesion, migration, growth and differentiation.73 It is known that the cytoskeletal reorganization is dependent on laminin polymerization.74 Three non-identical chains, laminin alpha, beta, and gamma, constitute the whole family.75 Here, the expression of laminins alfa and beta (LMNA and LMNB) were analyzed. Unlike in previous genes, the expression of LMN in turn varied between different types of cells. For example, in the HeLa cells, the decrease in LMNA and LMNB was observed between controls and cells treated with 10 μg mL−1 of LLCNPs. Interestingly, treatment with 75 μg mL−1 of LLCNPs@MnO-DMSA had a stronger downregulating effect than 10 μg mL−1 (Fig. 10A). In the LN229 cells, the only increase in LMNA regulation was noted for the cells treated with 10 μg mL−1 LLCNPs@MnO-DMSA, while the LMNB expression responded to different formulations of NPs, and the upregulation was noted for those treated with 75 μg mL−1 LLCNPs@MnO-oleate and 10 μg mL−1 LLCNPs@MnO-DMSA, with the highest increase at 75 μg mL−1 after the incubation with LLCNPs (Fig. 10B). In the U251MG cell line, the upregulation of LMNA and LMNB was noted in the cell treated with 10 μg mL−1 LLCNPs@MnO-oleate, but only the LMNB expression remained elevated after incubation with the 75 μg mL−1 concentration. LMNA and LMNB were also downregulated after the cells were treated with 75 μg mL−1 LLCNPs@MnO-DMSA, when compared to the control cells (Fig. 10C). In other studies, the downregulation of LMN was also observed after nanoparticle treatment, but only in cancer tissues, whereas in healthy tissues it remained at a similar level as compared to control.76
Till now, in the literature, there is no analysis concerning the cytoskeleton network gene expression after LLCNP treatment, so we cannot compare our results with others; however, we definitely can conclude that their expression profile is no concentration-dependent, except for tubulin in LN229 cells. The differences which occurred between cells treated with different NPs could be related rather to the characteristics of the selected cell lines, and how they are prone to metastasis. The additional analysis in different time-points, along with the protein expression, should be further evaluated. It is also important to note that the expression of ACTB incubated with different types and concentrations of LLCNPs was unstable in each investigated cell line, which strongly restricts its usage as a reference gene for the real-time PCR.
 | ||
| Fig. 12 In vivo T 1-weighted MR images before (left column) and after administration (right column) of (A) MnO-DMSA, (B) LLCNPs@MnO-oleate, and (C) LLCNPs@MnO-DMSA. The images were collected using a 9.4 T horizontal MRI scanner, at 37 °C. Regions of hind limbs are marked with a white square with an enhanced contrast by administered NPs as contrast agents. | ||
The LLCNPs loaded with both hydrophobic and hydrophilic MnO NPs (LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA) manifested a remarkable T1 contrast effect (brightening) in vivo compared to MnO-DMSA NPs themselves. However, stronger T1-contrasting efficiency was observed for LLCNPs@MnO-oleate, which is consistent with relaxivity data. The contrast enhancement was not observed after 3 days after contrast administration (Fig. S5, ESI†). This indicates that contrasting NPs were cleared from the area of injection, ensuring their biosafety. Further studies of these NPs in vivo bio-distribution will be necessary to unequivocally determine their biosafety. So far most of the in vivo studies on MnO-based CAs focused mainly on inorganic MnO NPs, without the use of any additional nanocarrier. Therefore, it is difficult to make any comparison and justification of the developed lipid@MnO hybrid system here. However, the results presented in this work indicate that prepared novel hybrid based on LLCNP and MnO systems may be a good choice for the currently intensively searched new positive T1 MRI contrast agent.
4. Conclusions
In summary, it was proven that two distinct systems were successfully fabricated, LLCNPs@MnO-oleate and LLCNPs@MnO-DMSA, which significantly differ morphologically; nevertheless, both can serve as novel efficient T1 CAs for MRI. Moreover, these hybrid nanosystems are a suitable alternative to Gd-based or withdrawn from the market Mn-based Teslascan CAs.As reported, LLCNPs were prepared using an efficient and simple top-down approach from the GMO lipid, Pluronic F127 surfactant, and loaded either with hydrophobic MnO-oleate or hydrophilic MnO-DMSA. Due to the amphiphilic character of LLCNPs, loading with hydrophobic and hydrophilic NPs was feasible. In detail, the obtained hybrids exhibited a heterogeneous phase composition as they were of the shape and structure of cubosomes, spongosomes and liposomes. LLCNPs@MnO-oleate exhibited a rather internal short-range arrangement in the dominating sponge-like phase, with the MnO-oleate NPs preferably embedded into the interior of LLCNPs. In turn, LLCNPs@MnO-DMSA exhibited an internal long-range arrangement in the dominating cubic phase, with MnO-DMSA NPs embedded on the surface, as well as in the core of LLCNPs. These two hybrids also exhibited different particle size distributions, with a larger average size for LLCNPs@MnO-oleate than that for LLCNPs@MnO-DMSA. Taking structural and morphological observations into account, it could be assumed that the surface modification of MnO NPs with DMSA does not affect GMO based LLCNPs causing their significant phase rearrangement as compared to MnO-oleate NPs. For future possible biomedical applications, it was important to examine their colloidal stability in the water environment, which was assessed as of medium range. Studies in the protein-rich medium on the other hand showed that LLCNPs@MnO-oleate is less stable and hence more prone to interact with proteins.
Regarding the relaxivity parameters, embedding both types of MnO NPs into LLCNPs resulted in a high relaxivity r1, particularly in the case of LLCNPs@MnO-oleate, in comparison to bare MnO-DMSA NPs and also commercially developed so far CAs, e.g., Gd-based DOTAREM or already withdrawn from the use Mn-based Teslascan. Results show that LLCNPs@MnO hybrids with larger sizes than chelate molecules or bare MnO-DMSA, and with extensive water channels within their internal structure, provide an efficient water molecule exchange. These, in turn, affect the higher number of inner-sphere water ligands, and hence, a higher value of relaxivity r1, particularly in the case of LLCNPs@MnO-oleate. The comprehensive analysis of morphology and related ability to modify molecular NMR relaxation processes led to the conclusion that not necessarily perfect liquid crystalline structure CA hybrids can guarantee the highest, desired impact on molecular relaxation parameters and in addition, the multicomponent composition of the system is found to be even an advantage. These results are well reflected in the in vitro MRI results showing the ability of particular novel CAs to influence the contrast in the recorded images.
Furthermore, the biosafety of prepared novel LLCNPs@MnO CAs was investigated in terms of uptake and intracellular localization as well as viability, genotoxicity, effects on mitochondria membrane potential, and cytoskeletal rearrangements. Both studied hybrids and bare LLCNPs were efficiently internalized by cells; however, the structure of the nanoparticles affected the mechanism of their uptake. In the case of bare LLCNPs and hydrophilic MnO-loaded LLCNPs, they internalized mainly via energy-independent and cholesterol-dependent pathways, whereas spongosomes of hydrophobic-loaded MnO LLCNPs internalized mainly via clathrin-dependent endocytosis. The cell viability studies showed that lower concentrations of LLCNPs in all three cell lines remained at the same high level. However, in the highest doses (above 100 μg ml−1), the viability decreased. Despite the cytotoxic effect, there were no observed genotoxic effects at the studied concentrations. The analysis of cytoskeleton rearrangements indicated that prepared hybrid nanosystems did not disturb the structure of the cytoskeleton at low concentrations, although gene expression changed dynamically. At the highest concentrations, however, disturbances in the actin cytoskeleton structure appeared, which, in turn, may be responsible for the decrease in cell viability. Furthermore, the results show that the mitochondria in glioblastoma cell lines were less susceptible to the harmful effects of NPs than in HeLa cells, which may be a premise for the use of these systems in neurological diseases.
Author contributions
Dorota K. Flak: conceptualization, resources, investigation, formal analysis, visualization, methodology, writing – original draft, review and editing, and supervision. Tomasz Zalewski: conceptualization, investigation, formal analysis, visualization, methodology, and writing – original draft, review and editing. Katarzyna Fiedorowicz: conceptualization, investigation, formal analysis, visualization, methodology, and writing – original draft, review and editing. Łucja Przysiecka: conceptualization, investigation, formal analysis, visualization, methodology, and writing – original draft, review and editing. Marcin Jarek: investigation, formal analysis, visualization, and writing – review and editing. Adam Klimaszyk: investigation, formal analysis, and visualization. Marek Kempka: investigation, formal analysis, and writing – review and editing. Agnieszka Zimna: resources, investigation, and writing – review and editing. Natalia Rozwadowska: conceptualization, resources, investigation, and writing – review and editing. Jonathan Avaro: investigation, formal analysis, visualization, and writing – review and editing. Marianne Liebi: resources and formal analysis. Grzegorz Nowaczyk: conceptualization, resources, investigation, formal analysis, visualization, writing – original draft, review and editing, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the research grant Sonata Bis 6 2016/22/E/ST3/00458 from the National Science Centre, Poland. We are grateful to Jacek Jenczyk and Maciej Nowaczyk for their assistance in creating the graphical abstract, and Karolina Gębicka for her help in preparing the samples.Notes and references
- R. Botár, E. Molnár, G. Trencsényi, J. Kiss, F. K. Kálmán and G. Tircsó, J. Am. Chem. Soc., 2020, 142, 1662–1666 CrossRef PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- J. Pellico, C. M. Ellis and J. J. Davis, Contrast Media Mol. Imaging, 2019, 2019, 1845637 Search PubMed.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H.-J. Weinmann, Invest. Radiol., 2005, 40, 715–724 CrossRef.
- Y.-D. Xiao, R. Paudel, J. Liu, C. Ma, Z.-S. Zhang and S.-K. Zhou, Int. J. Mol. Med., 2016, 38, 1319–1326 CrossRef CAS PubMed.
- 6FDA, FDA Drug Safety Communication (2015) FDA evaluating the risk of brain deposits with repeated use of gadolinium-based contrast agents for magnetic resonance imaging (MRI). http://www.fda.gov/Drugs/DrugSafety/ucm455386.html.
- M. Rogosnitzky and S. Branch, BioMetals, 2016, 29, 365–376 CrossRef CAS PubMed.
- A. Weller, J. L. Barber and Ø. E. Olsen, Pediatr. Nephrol., 2014, 29, 1927–1937 CrossRef PubMed.
- EMA, European Medicines Agency, PRAC confirms restrictions on the use of linear gadolinium agents, https://www.ema.europa.eu/en/documents/referral/gadolinium-article-31-referral-prac-confirms-restrictions-use-linear-gadolinium-agents_en.pdf, 2017.
- EMA, https://www.ema.europa.eu/en/documents/overview/teslascan-epar-summary-public_en.pdf, 2007.
- EMA, https://www.ema.europa.eu/en/documents/public-statement/public-statement-teslascan-withdrawal-marketing-authorisation-european-union_en.pdf, 2012.
- E. Gianolio, S. Bäckström, R. M. Petoral Jr, A. Olsson, S. Aime and O. Axelsson, Eur. J. Inorg. Chem., 2019, 1759–1766 CrossRef CAS.
- D. S. Karaman, M. P. Sarparanta, J. M. Rosenholm and A. J. Airaksinen, Adv. Mater., 2018, 30, 1703651 CrossRef PubMed.
- M. Mauri, V. Collico, L. Morelli, P. Das, I. García, J. Penaranda Avila, M. Bellini, R. Rotem, M. Truffi, F. Corsi, R. Simonutti, L. M. Liz-Marzán, M. Colombo and D. Prosperi, ACS Appl. Nano Mater., 2020, 3, 3787–3797 CrossRef CAS.
- L. Moriggi, C. Cannizzo, E. Dumas, C. R. Mayer, A. Ulianov and L. Helm, J. Am. Chem. Soc., 2009, 131, 10828–10829 CrossRef CAS PubMed.
- L. García-Hevia, M. Bañobre-López and J. Gallo, Chem. - Eur. J., 2019, 25, 431–441 CrossRef PubMed.
- Z. Zhen and J. Xie, Theranostics, 2012, 2, 45–54 CrossRef CAS PubMed.
- M. H. Mendonça-Dias, E. Gaggelli and P. C. Lauterbur, Semin. Nucl. Med., 1983, 13, 364–376 CrossRef PubMed.
- C. F. G. C. Geraldes, A. D. Sherry, R. D. Brown and S. H. Koenig, Magn. Reson. Med., 1986, 3, 242–250 CrossRef CAS.
- C. A. Massaad and R. G. Pautler, Methods in molecular biology, Clifton, N.J., 2012, 711, 145–174 Search PubMed.
- A. C. Silva, J. H. Lee, I. Aoki and A. P. Koretsky, NMR Biomed., 2004, 17, 532–543 CrossRef CAS.
- J. Yang and Q. Li, Front. Neurol., 2020, 11, 143 CrossRef PubMed.
- D. S. Harischandra, S. Ghaisas, G. Zenitsky, H. Jin, A. Kanthasamy, V. Anantharam and A. G. Kanthasamy, Front. Neurosci., 2019, 13, 654 CrossRef PubMed.
- T. V. Peres, M. R. C. Schettinger, P. Chen, F. Carvalho, D. S. Avila, A. B. Bowman and M. Aschner, BMC Pharmacol. Toxicol., 2016, 17, 57 CrossRef.
- X. Cai, Q. Zhu, Y. Zeng, Q. Zeng, X. Chen and Y. Zhan, Int. J. Nanomed., 2019, 14, 8321–8344 CrossRef CAS.
- X. Qian, X. Han, L. Yu, T. Xu and Y. Chen, Adv. Funct. Mater., 2020, 30, 1907066 CrossRef CAS.
- H. B. Na, J. H. Lee, K. An, Y. I. Park, M. Park, I. S. Lee, D.-H. Nam, S. T. Kim, S.-H. Kim, S.-W. Kim, K.-H. Lim, K.-S. Kim, S.-O. Kim and T. Hyeon, Angew. Chem., 2007, 119, 5493–5497 CrossRef.
- M. Létourneau, M. Tremblay, L. Faucher, D. Rojas, P. Chevallier, Y. Gossuin, J. Lagueux and M.-A. Fortin, J. Phys. Chem. B, 2012, 116, 13228–13238 CrossRef PubMed.
- I. D. M. Azmi, P. P. Wibroe, L.-P. Wu, A. I. Kazem, H. Amenitsch, S. M. Moghimi and A. Yaghmur, J. Controlled Release, 2016, 239, 1–9 CrossRef CAS PubMed.
- H. M. G. Barriga, M. N. Holme and M. M. Stevens, Angew. Chem., Int. Ed., 2018, 58, 2958–2978 CrossRef.
- D. Zhuang, H. Zhang, G. Hu and B. Guo, J. Nanobiotechnol., 2022, 20, 284 CrossRef CAS PubMed.
- X. Mulet, B. J. Boyd and C. J. Drummond, J. Colloid Interface Sci., 2013, 393, 1–20 CrossRef CAS.
- T. Madheswaran, M. Kandasamy, R. J. C. Bose and V. Karuppagounder, Drug Discovery Today, 2019, 24, 1405–1412 CrossRef CAS PubMed.
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Vaccines, 2021, 9, 359 CrossRef CAS PubMed.
- D. P. Acharya, B. A. Moffat, A. Polyzos, L. Waddington, G. Coia, D. K. Wright, H. X. Wang, G. F. Egan, B. W. Muir and P. G. Hartley, RSC Adv., 2012, 2, 6655–6662 RSC.
- M. Szlezak, D. Nieciecka, A. Joniec, M. Pękała, E. Gorecka, M. Emo, M. J. Stébé, P. Krysiński and R. Bilewicz, ACS Appl. Mater. Interfaces, 2017, 9, 2796–2805 CrossRef CAS PubMed.
- N. Tran, N. Bye, B. A. Moffat, D. K. Wright, A. Cuddihy, T. M. Hinton, A. M. Hawley, N. P. Reynolds, L. J. Waddington, X. Mulet, A. M. Turnley, M. C. Morganti-Kossmann and B. W. Muir, Mater. Sci. Eng., C, 2017, 71, 584–593 CrossRef CAS PubMed.
- A. Gupta, L. de Campo, B. Rehmanjan, S. A. Willis, L. J. Waddington, T. Stait-Gardner, N. Kirby, W. S. Price and M. J. Moghaddam, Langmuir, 2015, 31, 1556–1563 CrossRef CAS PubMed.
- A. Gupta, T. Stait-Gardner, L. de Campo, L. J. Waddington, N. Kirby, W. S. Price and M. J. Moghaddam, J. Mater. Chem. B, 2013, 2, 1225–1233 RSC.
- K. An, M. Park, J. H. Yu, H. B. Na, N. Lee, J. Park, S. H. Choi, I. C. Song, W. K. Moon and T. Hyeon, Eur. J. Inorg. Chem., 2012, 2148–2155 CrossRef CAS.
- S. I. C. J. Palma, M. Marciello, A. Carvalho, S. Veintemillas-Verdaguer, M. D. P. Morales and A. C. A. Roque, J. Colloid Interface Sci., 2015, 437, 147–155 CrossRef CAS PubMed.
- K. R. Wierzbinski, T. Szymanski, N. Rozwadowska, J. D. Rybka, A. Zimna, T. Zalewski, K. Nowicka-Bauer, A. Malcher, M. Nowaczyk, M. Krupinski, M. Fiedorowicz, P. Bogorodzki, P. Grieb, M. Giersig and M. K. Kurpisz, Sci. Rep., 2018, 8, 3682 CrossRef PubMed.
- H. Ljusberg-Wahren, L. Nyberg and K. V. Larsson, Chim. Oggi, 1996, 14, 40–43 CAS.
- L. de Campo, A. Yaghmur, L. Sagalowicz, M. E. Leser, H. Watzke and O. Glatter, Langmuir, 2004, 20, 5254–5261 CrossRef CAS.
- D. Demurtas, P. Guichard, I. Martiel, R. Mezzenga, C. Hebert and L. Sagalowicz, Nat. Commun., 2015, 6, 8915 CrossRef CAS PubMed.
- D. K. Flak, V. Adamski, G. Nowaczyk, K. Szutkowski, M. Synowitz, S. Jurga and J. Held-Feindt, Int. J. Nanomed., 2020, 15, 7415–7431 CrossRef CAS PubMed.
- J. Jagielski, L. Przysiecka, D. Flak, M. Diak, Z. Pietralik-Molińska, M. Kozak, S. Jurga and G. Nowaczyk, J. Nanobiotechnol., 2021, 19, 168 CrossRef CAS PubMed.
- H. Qiu and M. Caffrey, Biomaterials, 2000, 21, 223–234 CrossRef CAS PubMed.
- E. M. Gale, J. Zhu and P. Caravan, J. Am. Chem. Soc., 2013, 135, 18600–18608 CrossRef CAS PubMed.
- D. Flak, L. Przysiecka, G. Nowaczyk, B. Scheibe, M. Kościński, T. Jesionowski and S. Jurga, J. Nanopart. Res., 2018, 20, 306 CrossRef.
- Y. Chen, X. Zhang, B. Wang, M. Lv, Y. Zhu and J. Gao, RSC Adv., 2017, 7, 15625–15631 RSC.
- A. López-Ortega, D. Tobia, E. Winkler, A. López-Ortega, D. Tobia, E. Winkler, I. V. Golosovsky, G. Salazar-Alvarez, S. Estradé, M. Estrader, J. Sort, M. Angel González, S. Suriñach, J. Arbiol, F. Peiró, R. D. Zysler, M. Dolors Baró and J. Nogués, J. Am. Chem. Soc., 2010, 132(27), 9398–9407 CrossRef.
- B. Angelov, A. Angelova, R. Mutafchieva, S. Lesieur, U. Vainio, V. M. Garamus, G. V. Jensen and J. S. Pedersen, Phys. Chem. Chem. Phys., 2011, 13, 3073–3081 RSC.
- J. Borné, T. Nylander and A. Khan, Langmuir, 2001, 17, 7742–7751 CrossRef.
- A. R. Faria, O. F. Silvestre, C. Maibohm, R. M. R. Adco, B. F. B. Silva and J. B. Nieder, Nano Res., 2018, 12, 991–998 CrossRef.
- M. M. Mehanna, R. Sarieddine, J. K. Alwattar, R. Chouaib and H. Gali-Muhtasib, Int. J. Nanomed., 2020, 15, 9557–9570 CrossRef CAS.
- B. P. Dyett, H. Yu, J. Strachan, C. J. Drummond and C. E. Conn, Nat. Commun., 2019, 10, 4492 CrossRef PubMed.
- L. Rodrigues, F. Schneider, X. Zhang, E. Larsson, L. W. K. Moodie, H. Dietz, C. M. Papadakis, G. Winter, R. Lundmark and M. Hubert, J. Colloid Interface Sci., 2019, 553, 820–833 CrossRef CAS PubMed.
- K. Jacobson, O. G. Mouritsen and R. G. W. Anderson, Nat. Cell Biol., 2007, 9, 7–14 CrossRef CAS PubMed.
- N. Zeng, X. Gao, Q. Hu, Q. Song, H. Xia, Z. Liu, G. Gu, M. Jiang, Z. Pang, H. Chen, J. Chen and L. Fang, Int. J. Nanomed., 2012, 7, 3703–3718 CAS.
- H. M. Abdel-Bar and R. A. el Basset Sanad, Biomed. Pharmacother., 2017, 93, 561–569 CrossRef CAS PubMed.
- V. Meli, C. Caltagirone, A. M. Falchi, S. T. Hyde, V. Lippolis, M. Monduzzi, M. Obiols-Rabasa, A. Rosa, J. Schmidt, Y. Talmon and S. Murgia, Langmuir, 2015, 31, 9566–9575 CrossRef CAS PubMed.
- P. V. AshaRani, M. P. Hande and S. Valiyaveettil, BMC Cell Biol., 2009, 10, 65 CrossRef CAS.
- M. Norouzi, V. Yathindranath, J. A. Thliveris, B. M. Kopec, T. J. Siahaan and D. W. Miller, Sci. Rep., 2020, 10, 11292 CrossRef CAS PubMed.
- E. Nazaruk, A. Majkowska-Pilip and R. Bilewicz, ChemPlusChem, 2017, 82, 570–575 CrossRef CAS.
- Y. Almoshari, Pharmaceutics, 2022, 14, 600 CrossRef CAS PubMed.
- Z. Sobańska, J. Roszak, K. Kowalczyk and M. Stępnik, Nanomaterials, 2021, 11, 1084 CrossRef PubMed.
- M. Freag, Y. Elnaggar, D. Abdelmonsif and O. Y. Abdallah, Int. J. Nanomed., 2016, 11, 4799–4818 CrossRef CAS PubMed.
- A. Azqueta and M. Dusinska, Front. Genet., 2015, 6, 239 Search PubMed.
- A. M. Falchi, A. Rosa, A. Atzeri, A. Incani, S. Lampis, V. Meli, C. Caltagirone and S. Murgia, Toxicol. Res., 2015, 4, 1025–1036 CrossRef CAS.
- P. Vedula and A. Kashina, J. Cell Sci., 2018, 131, jcs215509 CrossRef PubMed.
- M. L. Pimm, J. Hotaling and J. L. Henty-Ridilla, in International Review of Cell and Molecular Biology, ed. C. Thomas and L. Galluzzi, Academic Press, 2020, vol. 355, pp. 155–204 Search PubMed.
- Y. Sun, T. L. Wang, W. S. Toh and M. Pei, Eur. Cells Mater., 2017, 34, 40–54 CrossRef CAS PubMed.
- H. Colognato, D. A. Winkelmann and P. D. Yurchenco, J. Cell Biol., 1999, 145, 619–631 CrossRef CAS PubMed.
- A. Goddi, L. Schroedl, E. M. Brey and R. N. Cohen, Metabolism, 2021, 120, 154775 CrossRef CAS PubMed.
- M. Rocha, R. Arcanjo, C. Lopes, M. Carneiro, A. Souza and S. Báo, Sci. Rep., 2017, 7, 17904 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01110k |
| This journal is © The Royal Society of Chemistry 2023 |