
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired mechanically stable all-polysaccharide based scaffold for photosynthetic production†
Tuuli
Virkkala
 a,
Sergey
Kosourov
b,
Ville
Rissanen
a,
Vilja
Siitonen
b,
Suvi
Arola
a,
Yagut
Allahverdiyeva
*b and
Tekla
Tammelin
*a
a,
Sergey
Kosourov
b,
Ville
Rissanen
a,
Vilja
Siitonen
b,
Suvi
Arola
a,
Yagut
Allahverdiyeva
*b and
Tekla
Tammelin
*a
aVTT Technical Research Centre of Finland Ltd, VTT, PO Box 1000, FI-02044 Espoo, Finland. E-mail: tekla.tammelin@vtt.fi
bMolecular Plant Biology, Department of Life Technologies, University of Turku, FI-20014 Turku, Finland. E-mail: allahve@utu.fi
First published on 21st August 2023
Abstract
We demonstrate the construction of water-stable, biocompatible and self-standing hydrogels as scaffolds for the photosynthetic production of ethylene using a bioinspired all-polysaccharidic design combining TEMPO-oxidised cellulose nanofibers (TCNF) and a cereal plant hemicellulose called mixed-linkage glucan (MLG). We compared three different molecular weight MLGs from barley to increase the wet strength of TCNF hydrogels, and to reveal the mechanisms defining the favourable interactions between the scaffold components. The interactions between MLGs and TCNF were revealed via adsorption studies and interfacial rheology investigations using quartz crystal microbalance with dissipation monitoring (QCM-D). Our results show that both the MLG solution stability and adsorption behaviour did not exactly follow the well-known polymer adsorption and solubility theories especially in the presence of co-solute ions, in this case nitrates. We prepared hydrogel scaffolds for microalgal immobilisation, and high wet strength hydrogels were achieved with very low dosages of MLG (0.05 wt%) to the TCNF matrix. The all-polysaccharic biocatalytic architectures remained stable and produced ethylene for 120 h with yields comparable to the state-of-the-art scaffolds. Due to its natural origin and biodegradability, MLG offers a clear advantage in comparison to synthetic scaffold components, allowing the mechanical properties and water interactions to be tailored.
1. Introduction
Plant cell walls are strong natural composites formed by a fibrous backbone and an amorphous matrix.1,2 The backbone consists of cellulose fibrils that interact with diverse and structurally complex matrix polysaccharides, such as hemicelluloses, to form highly durable and load-bearing networks with efficient water and nutrient transport properties.3,4 In addition to the interactions with cellulose, hemicelluloses interact strongly with water, which is a crucial constituent of the plant cell wall influencing its structural and mechanical properties.5,6 Inspired by the cell wall architecture, hemicelluloses have been studied for their interactions with cellulosic materials in the development of polysaccharide-based structures for various applications.7–9 Recently, mixed-linkage glucans (MLGs) i.e., hemicelluloses found in cell walls of grassy plants such as cereals,10 have gained attention due to their intrinsic interactions with cellulose,11 unique ability to efficiently gel nanocellulosic materials even at ultra-low solids contents,12 and hydrate cellulosic networks due to their strong water interactions.13Cell factories from photosynthetic microorganisms, i.e. eukaryotic microalgae and procaryotic cyanobacteria, have been widely researched as an alternative to petroleum-based fossil resources in the production of sustainable chemicals and fuels.14,15 Despite the promise of photosynthetic microbial cell factories, technical hurdles associated with suspension cultures, such as the self-shading effect leading to low light utilisation efficiency as well as high water and energy consumption, have limited harnessing their theoretical potential in chemicals and fuels production.16–18 Thin-layer immobilisation, i.e. binding the cells within a gel-like matrix, has been suggested to help address these difficulties.17,19–21 Previously, nanocellulose-based materials have been demonstrated as a viable alternative to conventionally used alginate-based matrices,16,22,23 since alginate-based matrices have their shortcomings in terms of mechanical stability especially in media with high content of chelating agents.
In addition to unique mechanical and colloidal properties24,25 and beyond their biocompatibility, nanocelluloses are interesting for biological and biomimicking applications due to their life-sustaining property.26 Fundamentally, cellulose has a hierarchical structure with individual molecules packed into microfibrils, further into microfibril bundles, and ultimately cellulose fibers.27
Due to the mechanical properties, optical transparency, and highly hygroscopic nature, TEMPO-oxidised cellulose nanofibers (TCNFs)28 are an attractive choice for immobilisation of photosynthetic microorganisms that require both an aqueous environment and sufficient light for growth.16,22 The challenge with TCNF gels is in their poor aqueous stability, which can be increased by various cross-linking strategies.29 Di- or multivalent metal cations, for example Ca2+, have been shown to efficiently improve the wet strength of TCNF-based structures,30 but the ionic cross-linking is reversible by nature, limiting the feasibility of this strategy when submerged in nutrient-rich growth medium containing chelating agents.22 Addition of synthetic polyvinyl alcohol (PVA) has also been suggested to improve the wet strength of TCNF.31 However, this strategy has significance mainly in dry films since formation of the cross-linked covalent ester bond requires efficient drying.32 Furthermore, PVA constitutes a possible source of microplastics in the environment due to its limited biodegradability33 and finding bio-based alternatives is favourable. Therefore, mixed-linkage glucans are interesting as an alternative bioinspired approach, mimicking the natural architecture and function of the grass cell wall.
The linear MLG homopolysaccharides consist of consecutive glucose units connected by two types of linkages: β-(1,4)-linked glucoses form cellotriosyl and cellotetraosyl units that are joined together by single interspersed β-(1,3)-linkages.34 The cellotriosyl and cellotetraosyl units constitute approximately 58–72% and 21–34% of the polymer, respectively.35 Additionally, MLGs also contain larger segments (5–11 glucose units), comprising around 10–20% of the polymer.36 This type of linking results in an irregular shape that renders the polymer flexible and soluble in water and prevents intermolecular alignment of MLGs into structured microfibrils.37,38 Together with a high degree of polymerisation, however, the asymmetric conformation allows MLGs to form a gel-like matrix between cellulose fibrils in the cell wall.39 Besides providing additional strength coupled with a certain extent of flexibility and elasticity, the gel-like matrix is thought to provide the cell wall with porosity, which is essential for water and nutrient exchange.40 These features make MLGs an interesting component for leaf-inspired nanocellulose-based scaffolds for biocatalytic solid-state photosynthetic cell factories (SSPCFs).
This paper demonstrates a bioinspired, all-polysaccharidic scaffold for SSPCFs obtained from bio-based components, cellulose nanofibers (TCNF) and hemicellulose from barley (MLG), in a design that mimics the composition and architecture of the plant cell wall (Fig. 1). The aim is to reveal the mechanism of how MLGs contribute to TCNF gel wet strength and stability. For this, we conduct a rigorous interfacial and bulk investigation to clarify MLG binding and stability of interactions as well as gel strength in physiological conditions. Quartz crystal microbalance with dissipation monitoring (QCM-D) is applied to study adsorption of three commercially available MLGs with different molecular weights on TCNF surfaces. We model the viscoelastic properties of the MLG adlayers by fitting the Voigt model41 to the adsorption and energy dissipation data. The swelling and water binding of the adsorbed TCNF–MLG layers are considered due to the key role of water interactions at the hemicellulose–cellulose interface.5,6 Furthermore, we attempt to relate the interfacial rheology results to bulk rheological properties to gain in-depth understanding of the materials’ macroscale properties. Finally, we highlight the performance of TCNF–MLG scaffolds with embedded ethylene-producing cyanobacteria in challenging submerged production conditions for a prolonged time span of 120 h. The research paves the way towards a bioinspired, fully polysaccharide-based, and tailored scaffold for entrapment of engineered cyanobacteria as biocatalytic architectures for photosynthetic chemicals production.
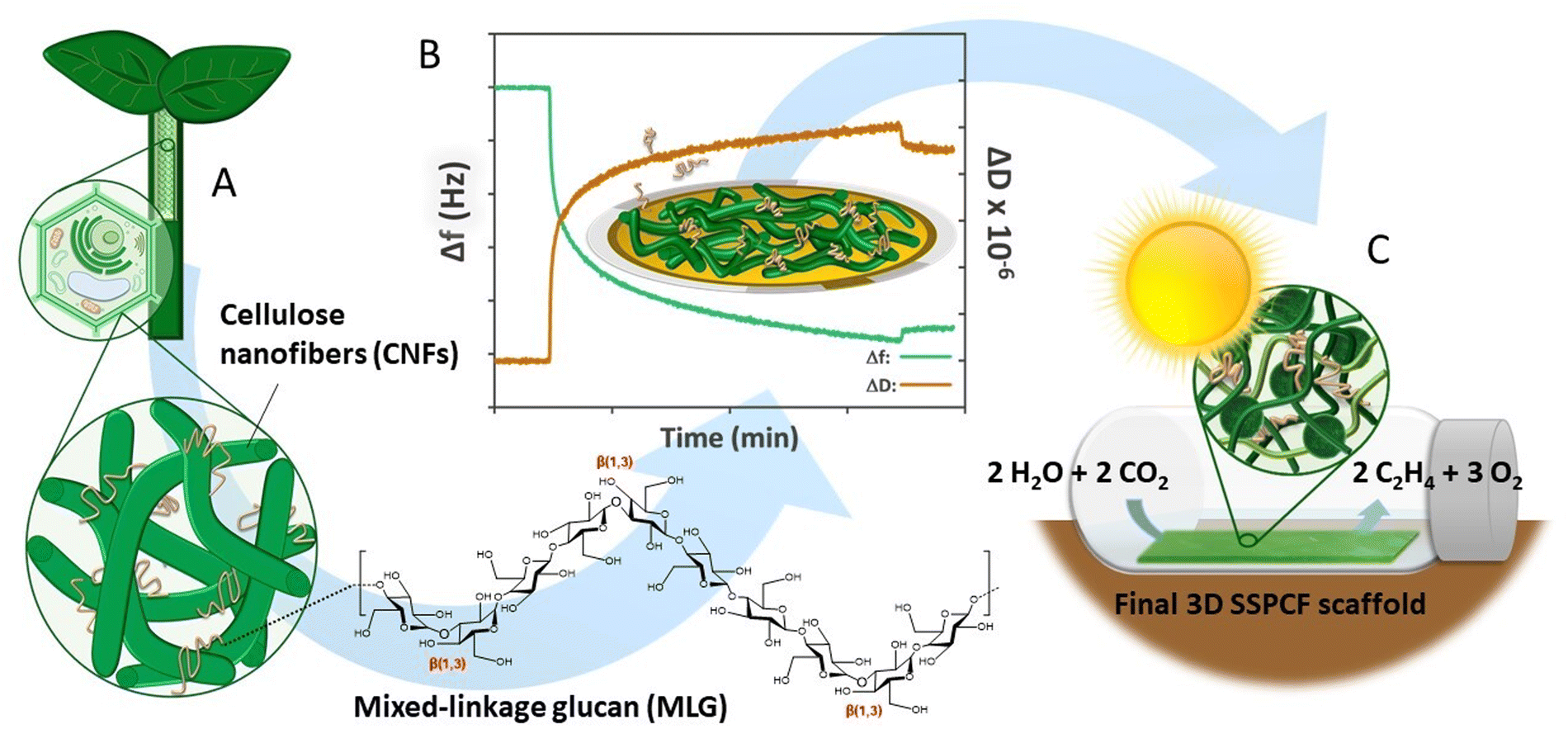 | ||
| Fig. 1 Conceptual view of the design steps of a bioinspired scaffold for photosynthetic production from mixed-linkage glucan (MLG) and nanocellulose: (A) The native cell wall structure of cereal plants with MLGs interlaced in the cellulose network was used as inspiration. (B) An interfacial study using quartz crystal microbalance with dissipation monitoring (QCM-D) and nanocellulose ultra-thin films was conducted to understand the component interactions in an aqueous environment. (C) Nanocellulose and MLGs were combined to fabricate the final 3D scaffold architecture and its performance was demonstrated with an ethylene bioproduction assay. | ||
2. Experimental
2.1. Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 min. The cell pellets were resuspended in fresh BG-11 medium to obtain culture with an optical density (OD720) of approximately 1.0 as measured with AquaPen-C AP-C 100 handheld fluorometer (Photon Systems Instruments, Czech Republic).
000g for 15 min. The cell pellets were resuspended in fresh BG-11 medium to obtain culture with an optical density (OD720) of approximately 1.0 as measured with AquaPen-C AP-C 100 handheld fluorometer (Photon Systems Instruments, Czech Republic).
For ethylene production experiments, an ethylene-producing cyanobacterium clone (S5) by heterologous expression of ethylene forming enzyme (Efe) from Pseudomonas syringae in Synechocystis sp. PCC 6803 (shortly, Synechocystis efe)45,46 was applied. The cells were maintained in BG-11 medium containing 20 mM HEPES-NaOH (pH 7.5) at 30 °C supplemented with 25 μg mL−1 spectinomycin and 10 μg mL−1 chloramphenicol. The cultures were agitated on a rotary shaker (120 rpm) in a growth chamber (MLR-351, versatile environmental test chamber Sanyo, Japan) with 1% CO2 in the atmosphere. The cells were illuminated continuously with fluorescent light of about 35 μmol photons m−2 s−1 (Philips Master TL-D 36W/865). Experimental cultures were grown without antibiotics and without agitation in 1 L flat round neck Roux type flask (DURAN®, Schott, DWK Life Sciences GmbH) in BG-11 medium. These cultures were continuously bubbled using sterile filtered air supplemented with 1% CO2 (0.2 μm, Acro 37 TF, Gelman Sciences, USA). The cells were collected at OD750 1.4–1.9 by centrifugation at 5000 g for 10 min (Avanti JXN-26, JA10, Beckman Coulter). The cells were resuspended in fresh BG-11 and the OD750 was adjusted to 1.0 prior to immobilisation using the UV-1800 spectrophotometer (Shimadzu, Japan).
000–100000, 06090-100) was purchased from Polysciences Inc. (Warrington, PA, USA). AT-cut gold-coated quartz crystal sensors (QSX 301) with a fundamental resonance frequency f0 = 5 MHz and a sensitivity constant C = 0.177 mg m−2 Hz−1 were obtained from Biolin Scientific AB, Sweden.
2.2. Methods
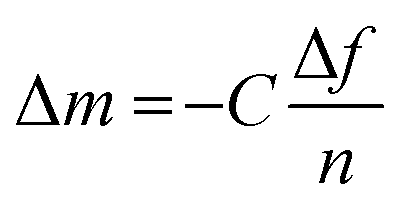 | (1) |
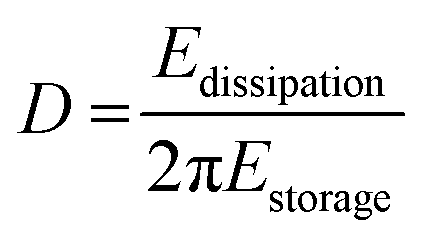 | (2) |
The spincoated TCNF thin films were allowed to stabilise in Milli-Q water overnight prior to the measurement to allow only the effect of MLG adsorption on frequency and dissipation to be investigated. During the experiment, water was first introduced to the flow cells at a rate of 0.1 mL min−1 at 23 °C for ≥30 min to obtain a steady baseline. Then, water was replaced with 0.01 wt% MLG solutions. The change in frequency (Δf) and change in dissipation (ΔD) were followed for approximately 5 h (300 min) until the equilibrium was reached. Finally, the flow cells were rinsed with water for ≥30 min. For LVMLG, which possessed the most promising mechanical properties based on the preliminary studies, the same experiment was repeated by using BG-11 as a buffer to investigate adsorption behaviour also in nutrient-rich growth medium. Similarly, the TCNF model films were first allowed to stabilize in BG-11 overnight, followed by the adsorption of LVMLG solution prepared in BG-11. The data represented was acquired with the third overtone (15 MHz, f0 = 5 MHz, n = 3).
| G = G′ + iG′ = μf + 2πifηf = μf(1 + 2πifτf) | (3) |
:1 ratio with TCNF. The mixtures were homogenized with IKA T 18 digital Ultra-Turrax® at >10000 rpm for at least 2 minutes until homogeneous. To get rid of air bubbles, the homogenized mixtures were centrifuged at 4500 G for 3 min. A handheld coating applicator with 2 mm gap was used to spread the gels into thin hydrogel films on coronated PTFE. An excess amount of 50 mM CaCl2 was sprayed onto the gel films to further cross-link them.16,22,30,51 To enhance cross-linking, the gel films were dried at +23 °C and 50% relative humidity (RH) until the original dry weight content of TCNF and MLG was reached, i.e., until 50% of water from the diluted gels had evaporated. After the removal of the excess of water, the self-standing gel films were cut into circles with approximately 5 cm diameters and immersed in 50 mM CaCl2 for 30 min. After this, CaCl2 was removed, and the samples were submerged in Milli-Q water and left to swell overnight prior to the rheological measurements to obtain the aqueous conditions for wet strength characterisation.
Immobilisation of Synechocystis within MLG-Ca2+-TCNF matrices for the rheology experiments and photosynthetic activity monitoring followed the same methodology as described above but using cell suspension instead of water. Cells were suspended in BG-11 growth medium with OD720 of 1.0 ± 0.1 (determined via AquaPen, Photon Systems Instruments, Czech Republic) and added to mixtures of 1 wt% TCNF and 0.01 wt% MV- and HVMLG or 0.05 wt% LVMLG in 1:1 ratio with TCNF. For rheology, control samples without the cells were prepared by replacing the cell suspension with BG-11 growth medium. The hydrogel films with the cells were then prepared like the MLG-Ca2+-TCNF hydrogel films. The samples were cut as described above, immersed in CaCl2 for 30 min, and left to swell in BG-11 overnight. For photosynthetic activity monitoring, the hydrogels were cut into 3 × 1 cm pieces, CaCl2 treated, and stored on plates with BG-11.
For ethylene production studies, the MLG-Ca2+-TCNF hydrogel films with Synechocystis efe were prepared as described above, except OD750 was adjusted to 1.0 using the UV-1800 spectrophotometer (Shimadzu, Japan). LVMLG in a concentration of 0.05 wt% was used in the final formulation. The hydrogel films were coated on the surface of a coronated PTFE support using the automatic film applicator (TQC Sheen) and 56 μm-, 100 μm- and 200 μm-graded Mayer rods to produce matrices with different thicknesses. 2 mm films were also prepared using the handheld applicator with 2 mm gap on the same support. Finally, the hydrogel matrices were stabilised by spraying 50 mM CaCl2 over the surface of hydrogel layers, washed once in BG-11 medium, and the excess water was removed by evaporation yielding hydrogels with 50% water content. The films were placed in Petri dishes containing BG-11 and cultivated in the growth chamber at 30 °C under around 35 μmol photons m−2 s−1 fluorescent light and 1% CO2 in air atmosphere. After 3 days of cultivation, the films were used for ethylene production experiments.
δ) is given by the ratio of G′′ to G′ (tanδ = G′′/G′). G′, G′′, and tanδ denoted the rest behaviour of the materials in the linear viscoelastic (LVE) region. Critical stress denoting the onset of non-linear, irreversible deformation was determined from the stress/strain curve as the point in stress where a non-linear relationship between stress and strain was observed.
The rheological measurements were performed with an AR-G2 rheometer (TA Instruments, New Castle, DE, USA) at 22 °C. To avoid slip, a 40 mm cross-hatched plate-plate geometry was used. The measuring gap was adjusted to yield a normal force of 0.3–0.8 N between the sample and the rheometer, and the samples were let to relax under the measuring head for 2 min prior to all measurements. Stress sweeps were conducted between 0.1–600 Pa oscillation range under a constant 0.1 Hz frequency, taking 10 points per measuring decade. At least five technical replicates with uniform thicknesses of 1000 ± 500 μm were measured for each sample and included in further analysis. TRIOS software (TA Instruments, New Castle, DE, USA) was used for operating the rheometer and data collection.
3. Results and discussion
3.1. Basic properties of MLGs – Polymers in aqueous solution
Table 1 lists the molecular weights of the three MLGs as given by the manufacturer, as well as ζ-potential values of 0.01 wt% MLG solutions in mV. To investigate the overall charge of MLGs under physiological conditions, the ζ-potentials were measured in water at pH ranges of 7.4–7.6 and 8.4–8.6, and in BG-11 growth medium with pH of 7.4–7.5. BG-11 medium contains a large number of dissolved salts used as a nutrient by photosynthetic cells.44 Conductivities of ultrapure water and the growth medium are approximately 0.60 and 2400 μS cm−1, respectively, indicating a high ionic strength of the nutrient rich medium. All aqueous solutions of MLGs displayed only very low negative ζ-potentials, and therefore MLG systems can be regarded as neutral polymers or at most weak anionic polyelectrolytes when dispersed in aqueous solution.| MLG type | M w (kg mol−1) | DPwb | ζ-Potentialc (mV) | ||
|---|---|---|---|---|---|
| a M w as informed by the manufacturer (Megazyme Ltd, Bray, Ireland). b DP is calculated from the equation DPw = Mw/M0, where M0 = 0.162 kg mol−1, molecular weight of MLGs monomer unit (glucose). c ζ-Potentials values should not be taken as absolute values but as estimates on the overall charge of the MLG solution since the measurement of ζ-potentials is not trivial due to the fast aggregation of MLG polymers. LVMLG, MVMLG, and HVMLG – barley low, medium, and high viscosity MLG, respectively. | |||||
| pH | |||||
| 7.4–7.6 | 8.4–8.6 | 7.4–7.5 (BG-11) | |||
| LVMLG | 179 | 1105 | −5.41 ± 2.0 | −2.48 ± 2.1 | −0.06 ± 0.1 |
| MVMLG | 238 | 1469 | −1.33 ± 0.3 | −1.51 ± 1.7 | −0.28 ± 0.4 |
| HVMLG | 495 | 3056 | −0.89 ± 0.7 | −10.14 ± 6.3 | −0.91 ± 0.8 |
Fig. 2 represents size distributions of the MLGs as a function of light intensity percentage measured using DLS. The lower molecular weight LV- and MVMLG displayed a clear bimodal size distribution with two distinct peaks due to the strong tendency of the MLG polymers, and hemicelluloses more generally, to aggregate in solution.53 The peaks around 35 and 43 nm for LV- and MVMLG, respectively, represent the individual polymer chains still present in the solution, whereas the peaks around 220 nm correspond to the formed polymer agglomerates. Aggregation seems to be reduced with increasing MLG molecular weight, and HVMLG displayed unimodal size distribution seen as a peak around 75 nm. Moreover, the number of individual chains in relation to agglomerated particles was higher in BG-11 regardless of MLG molecular weight, seen as a higher intensity of the smaller nm peaks.
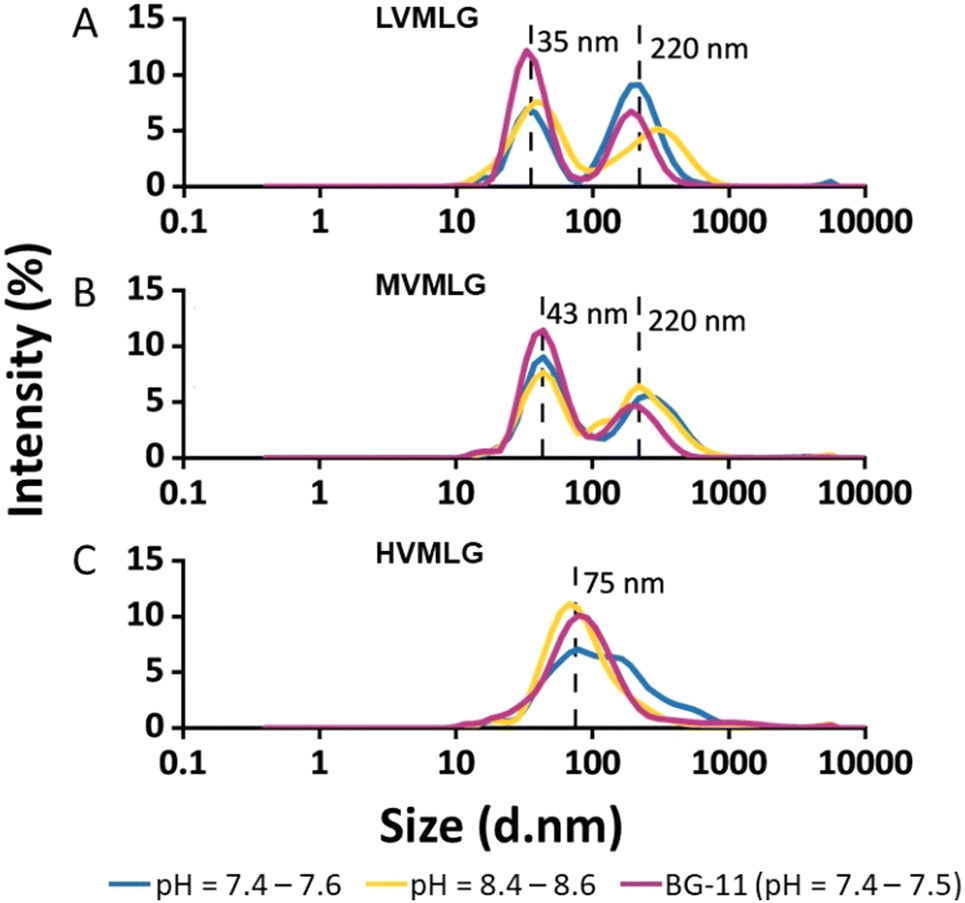 | ||
| Fig. 2 Hydrodynamic diameters of MLGs. Size distributions of (A) LV-, (B) MV-, and (C) HVMLG determined by dynamic light scattering (DLS) measurements. The analyses were conducted for 0.01 wt% solutions in water at pH ranges of 7.4–7.6 (blue line) and 8.4–8.6 (yellow line), and in BG-11 growth medium (pink line) with neutral pH and approximately 20 mM ionic strength. Peak values in nm are indicated with dashed lines. | ||
As shown in Fig. 2, neutral MLG polymers have poor long-term stability. Indeed, polymer aggregates are detected even in freshly prepared MLG solutions, which supports the notion that the solubility of MLG polymers is limited indicating that polymer–polymer interactions are favoured over polymer–solvent interactions. The solubility of MLGs is therefore determined by a favourable free energy change when polymer segments are exchanged to solvent molecules during dissolution.54 Given that MLG dissolution requires elevated temperatures i.e., the process is endothermic, the solubility must be attributed to a gain in entropy during mixing of polymer and solvent.
MLGs, having relatively high molecular weights when considering hemicelluloses, behave as random coil polysaccharides in solution. Coupled with the asymmetric conformations, these features are likely to drive the aggregation and gel formation processes of MLGs.11,43 It has been proposed that the interchain aggregates probably develop due to hydrogen bond formation between adjacent β-(1,4)-linked cellulose-like regions.53,55,56 According to Stokes–Einstein diffusion theory, in diffusion-controlled transport where no shear flow is present, the rate of diffusion is lower the larger the polymer coil. As a result, coils formed by shorter MLG chains have higher mobility and they experience less spatial hindrance than the larger MLG coils, which increases possibilities for both translational (collision) and rotational diffusion (sticking).53 Furthermore, the higher friction between polymer and solvent molecules retards the movement of larger molecules due to the entry of the viscosity effect as predicted in Smoluchowski coagulation model. In other words, the lower Mw MLG coils are more mobile and are more probable to associate with each other, which increases the chances for aggregate formation. The longer HVMLG chains remain as individual coils in solution as suggested in Fig. 2C. Similar observations were also made by Lazaridou et al.43
Additionally, the data suggests that increasing salt concentration has a positive effect on the solubility and stability of MLGs, which is seen as an increase in the amount of individual and non-aggregated MLG chains (Fig. 2). Based on ζ-potential measurements (Table 1), the overall charge of MLG system is even lower in higher salt concentration as compared with the systems in pure water at similar pH. The circumspect explanation could be that the long-range attractive interactions between the polymer chains are screened in the presence of high concentration of ionic species weakening the mobility of the MLG molecules in the electric field. Therefore, the polymer chains are not moving towards each other as efficiently, which increases MLG solution stability and less aggregates are formed. Another possible explanation is that the ionic species in the growth medium, such as nitrates, stabilise the electric double layer of MLGs, which otherwise is very weak. Furthermore, short-range repulsive hydration forces along with probable specific ionic interactions in the presence of nitrates may also contribute to the enhanced MLG dispersion stability.57,58 Despite the efforts to understand solution properties of MLGs, the effect of salt on polymer solvency is not fully understood and further research is needed.
ζ-Potential and particle size measurements are by no means trivial. The poor long-term stability and low charge of MLGs hamper the polymer particle size and charge determinations. Despite preparing the solutions directly prior to running the samples and filtering them into the cuvettes, sometimes large standard deviations were detected. These findings express the importance on working with freshly prepared MLG solutions, as aggregation is strongly time-dependent phenomenon.
3.2. TCNF–MLG interactions and viscoelastic properties of adsorbed MLG layers – Polymers at interfaces
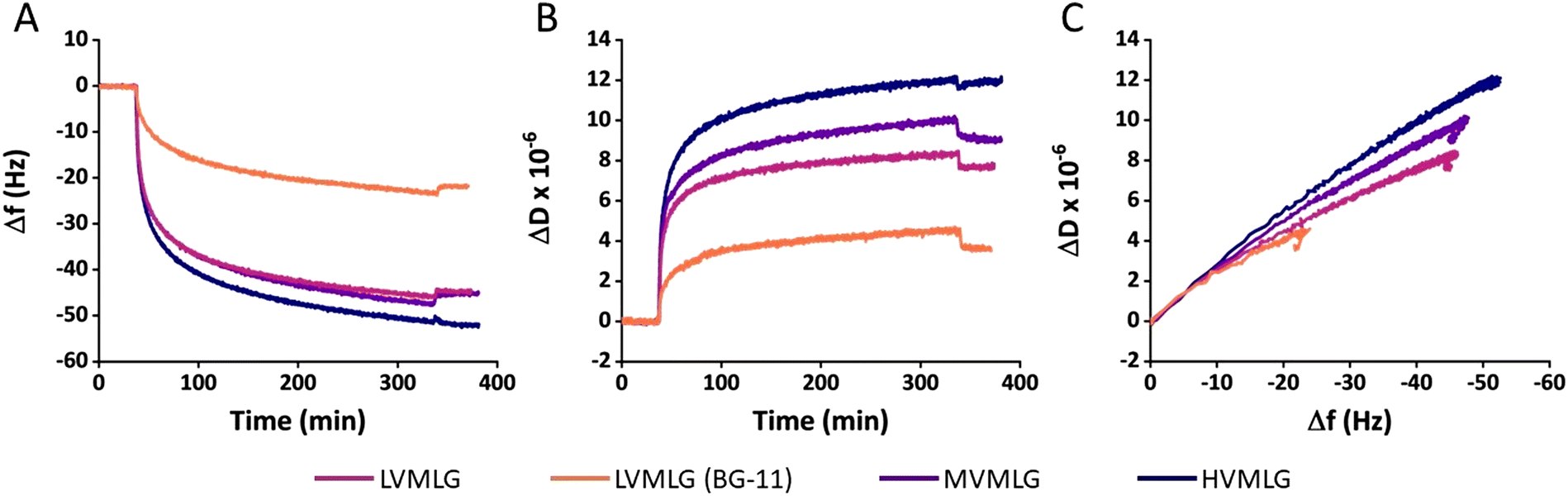 | ||
| Fig. 3 Adsorption of MLGs on TCNF. Changes is (A) frequency, (B) dissipation factor, and (C) dissipation factor as a function of frequency during adsorption of 0.01 wt% MLG solutions on TCNF surfaces. A baseline for TCNF surfaces was first obtained, after which MLG solutions were introduced at around 30 min. Lastly, a rinsing step was applied at 350 min. Adsorption of LVMLG in both water and BG-11 growth medium is shown. 15 MHz, f0 = 5 MHz, n = 3. | ||
However, the different adsorbed MLG layers display different viscoelastic properties (Fig. 3B). Higher ΔD values were obtained with increasing MLG molecular weight as final levels of approximately 11, 9 and 7 × 10−6 were reached for HV-, MV-, and LVMLG, respectively. By simplified interrelation of the dissipation changes to viscoelastic properties of thin films, the results denote that layer softness increases in the order HVMLG > MVMLG > LVMLG as the amount of bound water increases. A more profound effect was again seen when the adsorption was conducted nutrient-rich growth medium. Change in dissipation of LVMLG in BG-11 attained 4 × 10−6, which is only slightly above 50% of that in water. Moreover, plotting ΔD as a function of Δf allows concomitant comparison of how much energy the MLG layers bind during adsorption, i.e. how much energy the frequency changes correspond to. The slope of a ΔD/Δf curve is indicative of how dissipative a layer is and softening or packing of the layer is seen as a change in the slope. From Fig. 3C we can make an interpretation that MLG layers adsorbed from water are softer and more mobile than MLG layers adsorbed from BG-11, which are more densely packed. All in all, each MLG layer remained stable after rinsing i.e., Δf and ΔD did not change significantly once pure solvent (either water or BG-11) was pumped through the cells after the adsorption on TCNF (Fig. 3A and B). These results are in line with the previously reported findings stating that adsorption of MLG on cellulose surfaces is irreversible.11 We have established that MLGs behave as neutral polymers in aqueous environments. For TCNF with a similar carboxylate content to what was used here, a negative ζ-potential around −75 mV in water has been reported.59 Regarding TCNF–MLG interactions, these findings indicate that the attribution of electrostatic interactions to adsorption of neutral MLG chains on the negatively charged TCNF surface is negligible i.e., the driving force for adsorption must be non-electrostatic in nature. The unspecific binding suggests the mechanism of adsorption to be entropy-driven.60 Therefore, binding is likely promoted by an increase in entropy that is gained by the loss of preferential polar interactions when water molecules at the TCNF–liquid interface are released into solution during MLG binding to the surface to minimise the change in Gibbs free energy.60,61
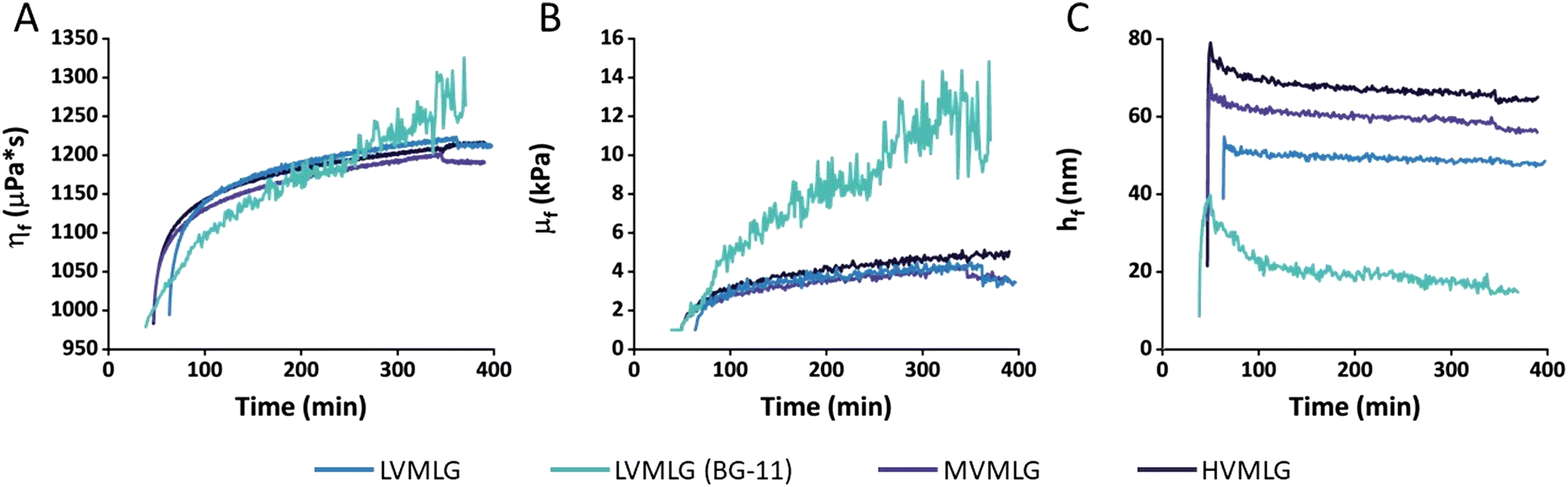 | ||
| Fig. 4 Viscoelastic modelling of MLG adsorption data. (A) Shear viscosities (ηf), (B) shear elastic moduli (μf), and (C) hydrodynamic thicknesses (hf) of the different MLG layers as a function of time obtained via fitting the the Voigt model to the adsorption data. Values for LVMLG in both water and BG-11 growth medium are shown. 15 MHz, f0 = 5 MHz, n = 3. Assumed layer density 1.05 g cm−3. | ||
Viscoelastic modelling of the QCM-D data provides a tool for interpretation of adsorbed mass and structural changes in the adsorbed layers. In the context of this paper, it enables comparison of thin film rheology to rheological properties of the materials in bulk. As all ΔD values are >1 × 10−6 suggesting that TCNF–MLG layers are viscoelastic, it is reasonable to assume that the exploitation of Voigt model is warranted. Despite of its simplicity, the model fits the experimental adsorption data very well with the difference between measured data and calculated values being almost negligible for several overtones (Fig. S2, ESI†). It should be noted that even though the model is a useful estimate on the viscoelastic properties of the TCNF–MLG layers, the final calculated values are not to be taken as absolute values but as a tool enabling comparison of layer properties to each other.
Fig. 5 shows changes in areal mass (Δm) of spincoated TCNF surfaces and subsequent MLG layers adsorbed on the TCNF surfaces. For LVMLG, two different layers are shown: one adsorbed in water and one in BG-11 growth medium. Areal mass changes due to MLG adsorption on TCNF reached approximately 400 ng cm−2 for both LVMLG and MVMLG, which is as expected due to their moderately similar Mw and DP values (Table 1). HVMLG being twice as large as MVMLG yields two times higher dry areal mass change of ∼800 ng cm−2. Interestingly, significantly higher dry mass of LVMLG was generated from nutrient rich medium being as high as 1200 ng cm−2, which is approximately three times higher than the areal mass adsorbed from pure water. Furthermore, AFM imaging, which was used to probe the coverage, topography, and surface roughness of the dry TCNF–MLG layers, reveals that all different surfaces appeared to be even in coverage and the height images show only minor topological differences (AFM height images in Fig. 5). No MLG agglomerates were detected pointing towards the uniform thickness and density of the adsorbed layers that is a prerequisite for the viscoelastic treatment of the adsorbed layers by the Voigt model.
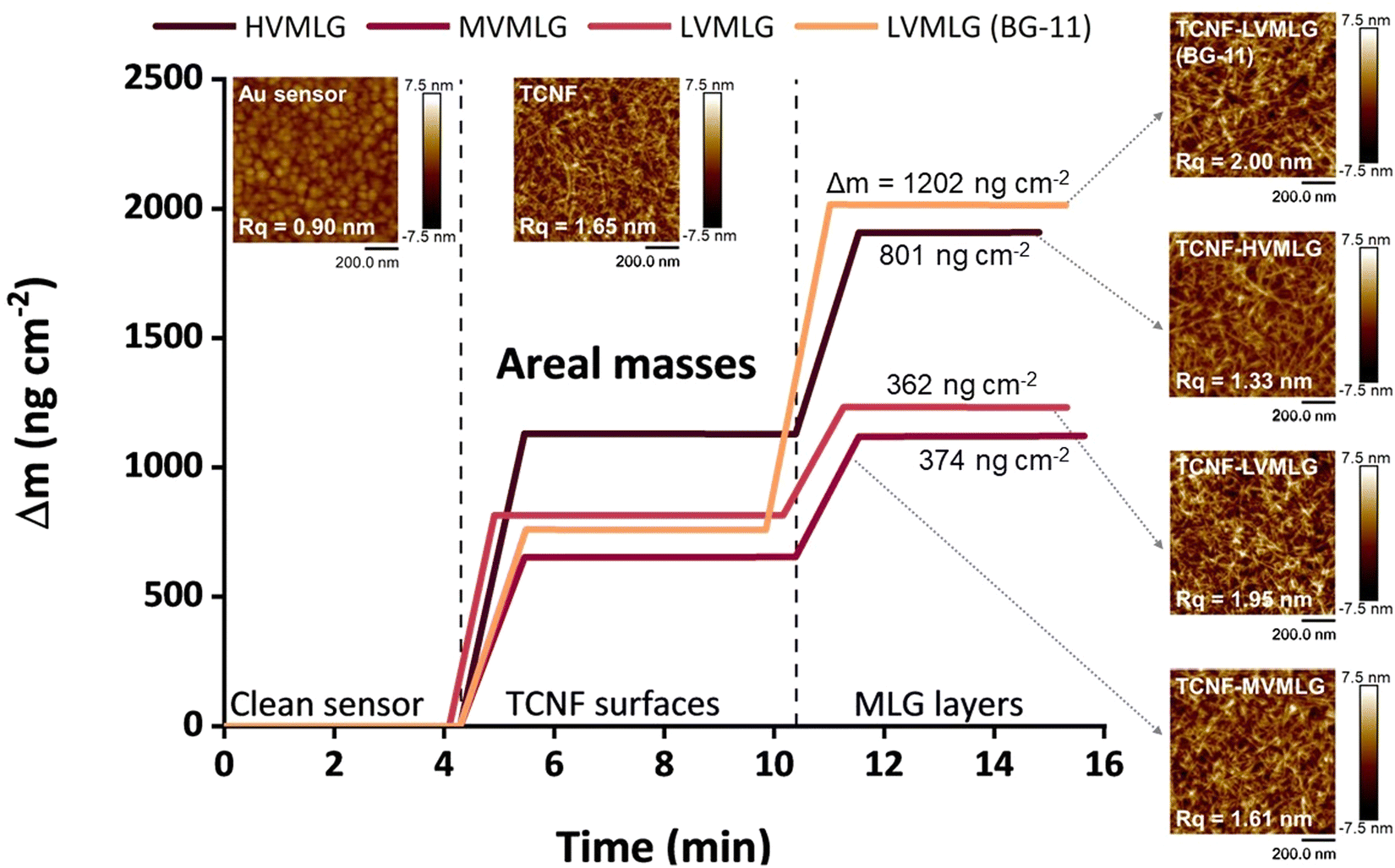 | ||
| Fig. 5 Adsorbed MLG layers at the solid–air interface. Areal masses of clean sensors, added TCNF layers, as well as different MLG layers on TCNF surfaces. 15 MHz, f0 = 5 MHz, n = 3. Layer density assumed as 1.05 g cm−3. Final Δm between TCNF and TCNF–MLG surfaces are given. 1 × 1 μm AFM height images of the corresponding surfaces are included for visual representation and the clean Au sensor is shown as a reference. The images were flattened and analysed with NanoScope Analysis software (Bruker), and the root-mean-square roughness (Rq) values of each surface are embedded in the images. | ||
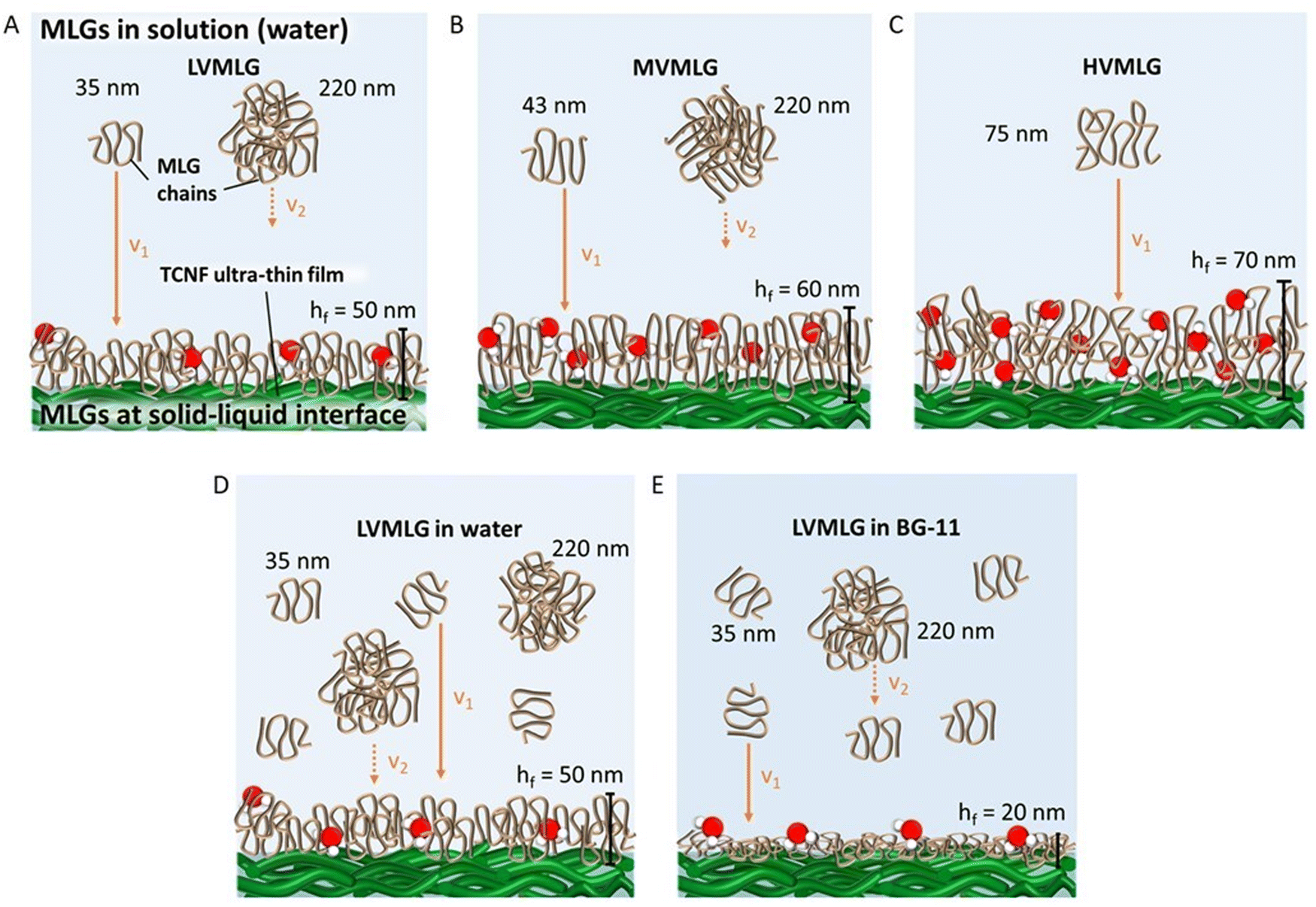 | ||
| Fig. 6 Schematic representation of MLG polymers in solution and at the solid–liquid nanocellulose (TCNF) interface. (A) LV- and (B) MVMLG solutions contain both individual chains and agglomerated particles. In a diffusion-controlled system, the smaller particles migrate faster to the surface and the larger aggregates stay in solution. v1 and v2 along with the orange arrows represent the diffusion rates of individual and agglomerated MLG chains (v1 > v2). Red-and-white objects represent water molecules trapped inside the layers. For the sake of clarity, free water molecules are excluded. (C) Longer HVMLG chains with internal loops and coils form even thicker layers with more bound water. In comparison to LVMLG in water (D), in BG-11 (E) the polymer chains adsorb in a flatter conformation allowing a denser layer to be formed. Water molecules cannot penetrate into the structure, even though still associated with the layer. As a result, the obtained layers are much thinner. | ||
Especially the longer HVMLGs contain segments forming internal loops and coils protruding out into the solution phase. In aqueous environments, water molecules are likely to strongly interact with the loops and coils keeping water molecules trapped inside the layer structure. Consequently, the amount of water in the MLG layer increases with the Mw of the MLG polymer, which is reflected as larger dissipation values and increased thickness corresponding to softer layers. The shorter LV- and MVMLGs, in contrast, form more stable and rigid layers with less incorporated water molecules.
The growth media had a highly pronounced effect on the LVMLG solution properties, adsorption properties, and, especially, the rheological properties of the thin films (Fig. 6D and E). The ions in BG-11 stabilised the LVMLG solutions, seen as an increase in the amount of individual polymer chains compared to agglomerated polymers (Fig. 2). Demonstrated by the accumulation of a higher relative areal mass on the sensor surface (Fig. 5), adsorption of LVMLG in BG-11 is promoted. Arumughan et al.63 recently showed that this is likely caused by the anion specific interactions resulting mainly from the nitrate ions present in the medium: chaotropic ions, such as nitrate,58 accumulate near the chaotropic cellulose surface64 altering the orientation of water molecules at the interface. The release of these unfavourably organised water molecules from the interface during adsorption generates a higher entropy gain that further promotes adsorption. MLGs also express self-affinity as shown by the aggregation behaviour present in aqueous solutions (Fig. 2), which combined with possible conformational changes could lead to formation of internal connections and exclusion of water molecules from the structure. In other words, the TCNF surface can accommodate more MLG molecules, allowing a higher dry mass to be built on top of the nanocellulose layer. Overall, the increased electrolyte concentration due to the presence of ionic species in the growth medium enables MLG chains to pack more densely in BG-11 (Fig. 6E) than in water (Fig. 6D), attributing to the formation of stronger, more stable, rigid, and flatter layers from BG-11.
In terms of the overall viscoelastic nature of the TCNF–MLG thin films, the μf values calculated using the Voigt model in kPa (103 × Pa) range are substantially higher in scale than the ηf values in μPa s (10−6 × Pa s), which indicates that the MLG layers all exhibit predominantly elastic character. As a reference, μf values in 105 Pa range have been obtained for cationic starches.62 In comparison, the elastic moduli of MLG layers are lower. As the hydrodynamic thicknesses are overall rather high, all adsorbed MLG layers still show notable softness and viscoelastic character associated with strong water binding ability. The notable association of the adsorbed layers with water is in line with previous research reporting the significant role of water at hemicellulose–cellulose interfaces.5
To the authors’ knowledge, there is little existing literature regarding MLG modelling that these results could be compared to. Kiemle et al.11 reported estimated layer thicknesses of 15 nm when investigating adsorption of 0.01 wt% MVMLG solutions onto regenerated cellulose surfaces at approximately 20 mM ionic strength, however, in sodium acetate instead of BG-11. Our results showed a thickness of approximately 20 nm for LVMLG in the growth medium. The experimental condition used by Kiemle et al. (T = 48 °C, pH 5.5) differ from our conditions. In addition, the molecular weight of MLG in their studies was different, making direct comparison challenging. Nevertheless, the thickness obtained for LVMLG here seems realistic despite being a little higher than what was obtained for MVMLG at similar ionic strength.
3.3. Effect of MLG polymer size and concentration on matrix wet strength
Rheological measurements enable analysis of material behaviour under shear stress, which several papers list as a substantial challenge in photobioreactors.65–67 To optimise the grade and amount of MLG in the TCNF-based hydrogel scaffold for mechanical stability and durability in submerged production conditions, small deformation oscillatory rheology was used. The aqueous conditions for wet strength determination were obtained by letting the samples swell overnight in water/BG-11 prior to the measurements. Fig. 7 displays critical stress values of self-standing MLG-Ca2+-TCNF hydrogel films in Pa as a function of MLG concentration. Due to the nature of the samples and the measurement,51 high variance in both was seen even though several replicates were measured, making it difficult to find considerable differences between the yield behaviour of the matrices with different molecular weight MLGs. Generally, however, it seemed that increasing the amount of MLG decreased the wet strength i.e., 0.01–0.05 wt% MLG additions yielded better results than the higher concentrations. Also, it seemed that with LVMLG a higher amount was required to reach similar critical stresses that were reached already at 0.01 wt% with HV- and MVMLG. Overall, the wet strengths of all MLG-Ca2+-TCNF hydrogel films were comparable to the previously reported TCNF-based matrix formulation with synthetic PVA.22 Matrices with 0.1–0.2 wt% HVMLG were omitted from the rheological investigation due to lower biological compatibility based on preliminary visual assessment (Fig. S3, ESI†) and because the higher MLG concentrations were not expected to benefit the mechanical properties. The rest properties of the MLG-Ca2+-TCNF matrices are additionally included in the ESI† (Fig. S4, ESI†).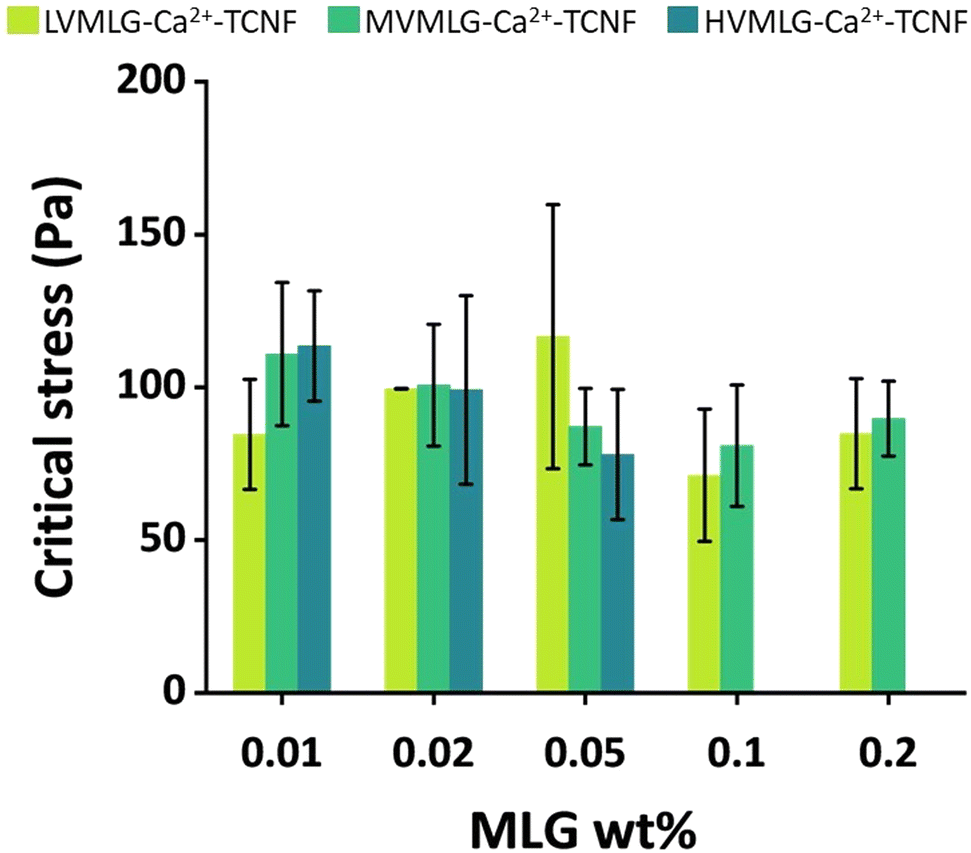 | ||
| Fig. 7 Wet mechanical properties of self-standing MLG-Ca2+-TCNF hydrogel films. Critical stress values of the matrices with LV-, MV-, and HVMLG in 0.01–0.2 wt% concentrations. Standard deviations from at least five replicate measurements are shown. | ||
As seen in the QCM-D experiments, the irreversible adsorption of MLGs onto TCNF proposedly leads to so-called polymer entanglement, meaning that MLGs physically bridge the cellulosic network.11 These same interactions are assumed to be present also in bulk, meaning that the viscoelastic properties of the thin films are reflected to the bulk rheological properties of MLG-Ca2+-TCNF matrices. However, a few important considerations need to be addressed.
First, the mechanical properties of the colloidal hydrogel matrix are attributed to properties of both TCNFs and the interlaced MLGs, which intrinsically have very different network structures.12 Pure TCNFs are rather rigid structures in comparison to the more flexible MLG chains having an irregular shape due to the β-(1,3)-linkages in their backbone. When extended, the length of a (HV)MLG chain in comparison to a cellulose fiber can be a few times higher. Consequently, a network containing both TCNF and MLG is generally likely to be softer in nature than a TCNF network by itself, despite being more stable in an aqueous environment, and the softness increases the longer the interlaced MLG chains are and the more the system contains MLG.12 Secondly, the ratio of TCNF to MLG is substantially higher in the hydrogels in comparison to the thin films investigated by the surface sensitive techniques. The matrix network mainly consists of the more rigid TCNFs with small amounts of interlaced MLGs. Therefore, the softening effect of MLGs is not as prominent as it could be if the amount of MLG in the matrix was increased significantly. Moreover, the MLG-Ca2+-TCNF hydrogel films are dried during the preparation procedure. With areal masses, the total removal of water was seen to have a profound effect on the interactions in the TCNF–MLG network. Even though the hydrogel matrices are dried to a much lesser extent, the rheological properties may be affected by the process. Lastly, only molecular weight effects were investigated with the surface sensitive techniques. As shown by the rheology results, concentration is another factor influencing the viscoelastic properties and wet strength of the MLG-Ca2+-TCNF matrices.
It is also important to note that all the hydrogel films studied in this paper also contained divalent Ca2+ cations, as di- and multivalent ions have been shown to have a strong effect on wet mechanical stability of TCNF-based networks.30,51,68 In fact, the TCNF–MLG formulations discussed here did not form self-standing hydrogels without the Ca2+ ions (Fig. S5, ESI†). Differences in wet strength and yielding behaviour of the matrices, however, likely arise from the MLG component as all hydrogels were allowed to saturate with Ca2+ ions and it can be assumed that MLGs do not interact with Ca2+ ions due to their neutral charge.
The dissipation monitoring and modelling of TCNF–MLG thin films discussed in Section 3.2 indicated that larger MLG polymer sizes contribute to formation of softer layers than the shorter ones. Thus, one could expect HVMLG-Ca2+-TCNF hydrogel films to have softer mechanical properties than the matrices with the other two MLGs. However, drying likely allows interactions with water to be replaced with particle–particle interactions. This effect is more pronounced with longer MLG chains containing internal loops and coils with multiple interaction sites that can attach onto several TCNF fibrils once bound water molecules are expelled from the structure. As a result, mechanical properties of the matrices with the higher molecular weight MLGs appear to be slightly better in comparison to LVMLG-Ca2+-TCNF when the MLG concentration is low. When the concentration is increased, the presence of the irregular, interlaced MLG chains seems to result in softening of the network. Again, this effect is more pronounced the longer the MLGs are. Additionally, increasing the MLG concentration may cause entanglement and coiling of the long polymer chains with each other resulting in larger local MLG aggregates that soften the overall matrix network structure.
On the other hand, at a constant concentration, the number of individual polymer chains per mass unit is higher for MLGs with lower molecular weight. In other words, the number of chains interacting with TCNF fibrils likely increases even though there are fewer interaction sites per chain. In addition, surface interaction studies showed adsorption of LVMLG on TCNF to result in more rigid layers. At very low MLG concentrations, these features are probably less prominent and the longer polymer chains may yield better matrix wet mechanical properties. However, when the concentration is raised a little (to 0.02–0.05 wt%), LVMLG-Ca2+-TCNF matrices display slightly better wet strength. This increase in concentration may bring a sufficient amount of LVMLGs in contact with TCNFs allowing a mechanically stable network to be established. Further increase of MLG concentration seems to have the same effect as with the higher molecular weight MLGs, i.e., the TCNF-based network appears to lose some of its rigidity as the amount of the softer polymeric component in the matrix increases.
The effect of medium composition and incorporation of cells on matrix wet strength was also investigated, and the rheological properties MLG-Ca2+-TCNF hydrogel films prepared in BG-11 with and without Synechocystis cells are included in the ESI† (Table S2 and Fig. S6). Unlike with the thin films, interpretation of the results at bulk level was more challenging and clear trends were difficult to observe due to heterogeneity of the samples and possible artefacts resulting from sensitivity of the measurement. As for the long-term stability of the hydrogel scaffolds with entrapped cells, while all MLG-Ca2+-TCNF matrices remained stable in BG-11 for almost two weeks, LVMLG-Ca2+-TCNF showcased superior performance in comparison to the other two matrices and retained its mechanical integrity for over 50 days in submerged conditions (Fig. S7, ESI†). Additionally, the strong interactions with TCNF observed for LVMLG in the nutrient rich growth medium as shown by the interfacial studies (Fig. 4–6) also support the choice of LVMLG to be used in the scaffold formulation. Hence, we chose to test the performance and productivity of LVMLG-Ca2+-TCNF in ethylene production. The 0.05 wt% concentration was used as it showed slightly more promising results than the other concentrations.
3.4. Biological compatibility of scaffold and ethylene production by cyanobacteria entrapped in MLG-Ca2+-TCNF matrices
The long-term biocompatibility of MLG-Ca2+-TCNF matrices was assessed by entrapping wild-type Synechocystis cells in the hydrogel thin films and monitoring the cells’ photosynthetic activity periodically during cultivation under normal room temperature and light conditions. The samples contained HV-/MVMLG and LVMLG in 0.01 wt% and 0.05 wt% concentrations, respectively. Fig. 8A shows that the cells retained their photosynthetic activity in the LVMLG-Ca2+-TCNF hydrogel for over 50 days. In fact, cells inside the MV- and HVMLG-Ca2+-TCNF matrices also showcased good photosynthetic activities after almost two weeks. However, the monitoring was stopped as the samples started to deteriorate. It has been shown that the cells lose their photosynthetic activity if deprived of nutrients, such as N, S, P, and Ci amongst others, and that their photosynthetic apparatus is susceptible to light, requiring constant recovery.69–74 Overall, these results indicate that nutrients are efficiently distributed inside the matrix and that the material supports long-term cell viability, i.e., the hydrogels are biocompatible.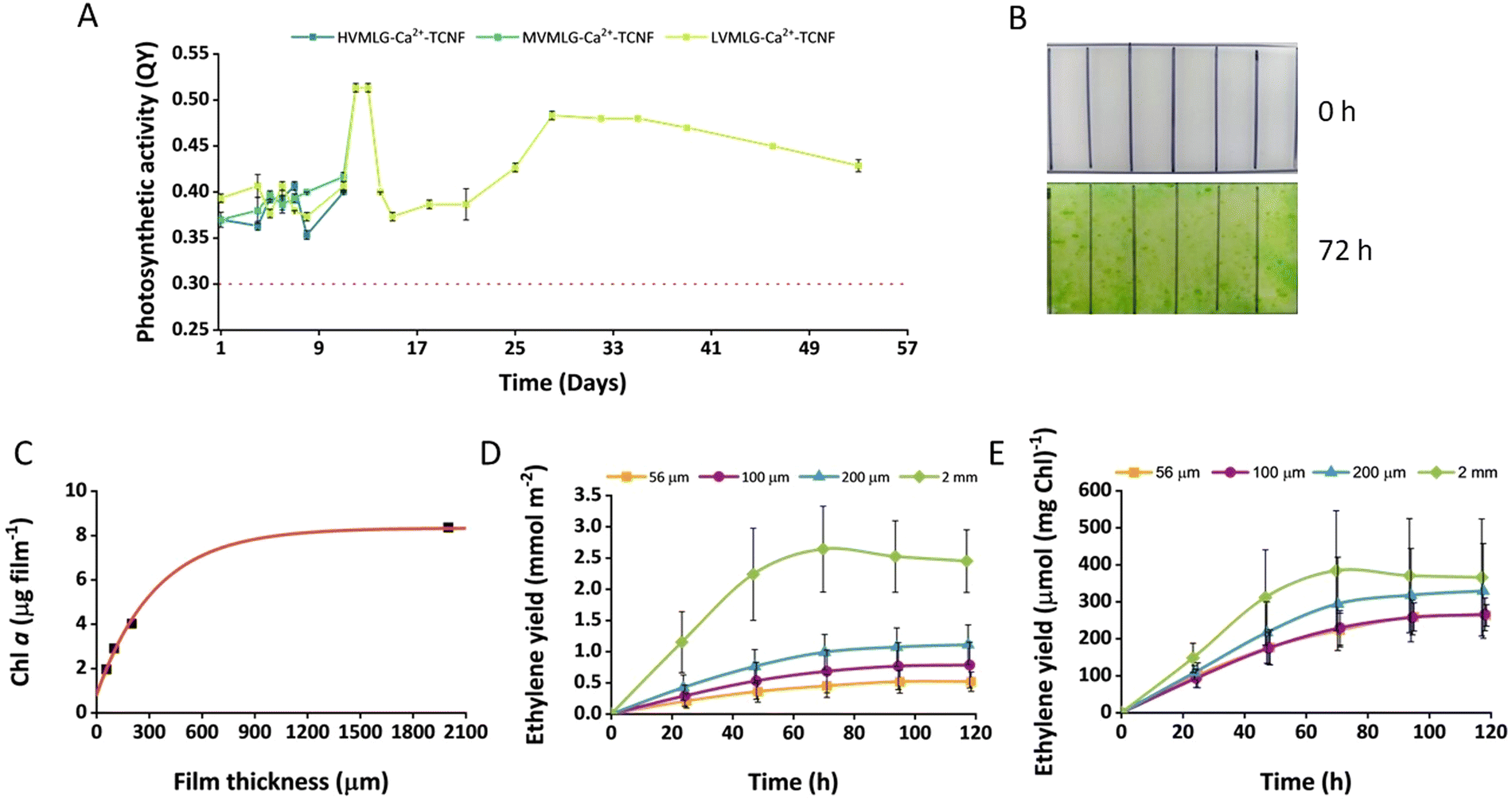 | ||
| Fig. 8 Cell fitness of wild-type Synechocystis and biocatalytic performance of ethylene-producing Synechocystis efe cells entrapped within thin-layer MLG-Ca2+-TCNF matrices under submerged cultivation. (A) The long-term viability of the wild-type cells in the nanocellulose-based scaffolds was monitored via photosynthetic activity. The given values are averages measured from three replicates and standard deviations are shown. The dashed red line shows the approximate value (0.3) below which the cells’ fitness was considered as significantly affected. (B) For ethylene production experiments, the thin LVMLG-Ca2+-TCNF films with Synechocystis efe (efe films) were adapted in BG-11 medium under 1% CO2 in air atmosphere for 3 days. The change in the cell density of efe films after 3 d adaptation. 100 μm-thick film is shown as an example. (C) Dependence of Chl content of the efe film on the film thickness after 3 d adaptation. (D) Ethylene production yields by the efe films with different thicknesses. (E) Specific ethylene production yields by the efe films normalised to the total Chl content of the film. Values are the mean of 6 independent replicates and standard deviations are included. | ||
Moreover, the performance of the MLG-Ca2+-TCNF scaffolds in bioproduction was evaluated using Synechocystis efe strain, which was specifically engineered for the photosynthetic production of ethylene.45,46 The cells were entrapped in MLG-Ca2+-TCNF films with LVMLG in 0.05 wt% concentration. The cells were entrapped at very low cell density and the fabricated films were allowed to adapt for 3 days in BG-11 medium under 1% CO2 in an air atmosphere. As shown in Fig. 8B, MLG-Ca2+-TCNF films demonstrated a dramatic increase in the cell density (determined by the change in Chl content) after 3 days of submerged cultivation. In addition to supporting photosynthetic activity, the increased cell density also supports the notion that the matrix is biocompatible with cyanobacteria. Interestingly, the Chl content of the films determined after 3 days of cultivation depended exponentially on the film thickness (Fig. 8C). As demonstrated in Fig. 8D, the immobilised cells were capable of producing ethylene for 5 days under submerged cultivation conditions. The overall ethylene production yield increased with the increase of the film thickness and was highest in 2 mm-thick films due to a high initial biomass content (Fig. 8C). At the same time, the specific ethylene production yield (calculated per mg Chl) showed almost similar dependence, but the differences were not statistically significant (Fig. 8E). Moreover, the specific activity showed a tendency to increase with increasing film thickness.
The Synechocystis efe strain synthesises ethylene thanks to the over-expressed ethylene-forming enzyme (Efe) from Pseudomonas syringae.75 The process depends on photosynthetic CO2 fixation and thus, requires inorganic carbon supplementation in the form of CO2 or bicarbonate and moderate (10–100 μmol photons m−2 s−1) illumination.21 It has been shown that the entrapment of Synechocystis efe cells within thin Ca2+–alginate films improves the ethylene production yield and the long-term catalytic performance of the entrapped cells for up to one month under semi-wet cultivation compared to suspension cultures.21 However, Ca2+–alginate films showed low mechanical stability under high (above 200 mM) bicarbonate loadings and thus, were unsuitable for submerged production.22 On the other hand, entrapment of cyanobacterial cells in the self-standing PVA-Ca2+-TCNF films resolved the problem.22 We expected a similar result from MLG-Ca2+-TCNF formulations.
The all-polysaccharidic MLG-Ca2+-TCNF films were mechanically stable, and the cell density was sufficient for checking ethylene production after the three-day adaptation period. The exponential dependency between film Chl content and film thickness indicates that cell growth is limited by the distribution of nutrients (and/or CO2) within the thicker films. We exclude a significant light limitation since the immobilised cultures can reach much higher cell densities under the same illumination conditions (but during a longer growth period). Additionally, the tendency of the specific activity to increase in the thicker films suggests that the process is not light-limited under these cultivation conditions, while the limitation of biomass formation results in slightly better ethylene productivity. In general, the ethylene production capacity of the cells during submerged cultivation conditions for 5 days is comparable to the data obtained previously.22 The specific ethylene production activity by the MLG-Ca2+-TCNF films was higher than in Ca2+–alginate films under semi-wet cultivation,21 as well as Ca2+–alginate hydrogel and PVA-Ca2+-TCNF films under submerged cultivation.22 Thus, MLG-Ca2+-TCNF matrices have the potential for engineering SSPCFs for photosynthetic production of chemicals and biofuels.
4. Conclusions
This work aimed to fabricate strong, biocompatible hydrogels from plant cell wall components. Obtaining all polysaccharide-based materials with sufficient mechanical performance in challenging aqueous and physiological conditions is not a trivial task though. In this research, we have shown that the interactions between the polysaccharidic components, nanocellulose and MLG, are strong enough to generate a scaffold for solid-state photosynthetic cell factories with sufficient wet strength in addition to being biocompatible and enhancing the viability of the cyanobacterial cells. Moreover, the ethylene production experiments showcase that the scaffold has the potential for bioproduction in submerged conditions. As a plant cell wall component and natural polymer, MLG offers a clear advantage in terms of biodegradability and end-of-life in comparison to fossil-based alternatives, such as PVA, as a structural component of the 3D SSPCF platform, allowing tailoring of the mechanical properties and water interactions of the matrix architecture.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 899576 and the Academy of Finland project #322752/#322754. S. A. was funded by the Academy of Finland grant #326262/311608. The authors acknowledge the support from the Academy of Finland funded flagship programme FinnCERES Materials Bioeconomy Ecosystem. This work made use of the Bioeconomy Facilities at Aalto University and VTT. Photosynthetic measurements and ethylene production experiments were performed in the University of Turku PhotoSYN infrastructure. We thank Teemu Suutari and Juan José Valle-Delgado for assistance with QCM-D and AFM measurements.Notes and references
- J. U. Fangel, P. Ulvskov, J. P. Knox, M. D. Mikkelsen, J. Harholt, Z. A. Popper and W. G. T. Willats, Front. Plant Sci., 2012, 3 DOI:10.3389/fpls.2012.00152.
- Y. Zhang, J. Yu, X. Wang, D. M. Durachko, S. Zhang and D. J. Cosgrove, Science, 2021, 372, 706–711 CrossRef CAS PubMed.
- D. J. Cosgrove and M. C. Jarvis, Front. Plant Sci., 2012, 3 DOI:10.3389/fpls.2012.00204.
- J. P. Moore, M. Vicré-Gibouin, J. M. Farrant and A. Driouich, Physiol. Plant., 2008, 134, 237–245 CrossRef CAS PubMed.
- R. Cresswell, R. Dupree, S. P. Brown, C. S. Pereira, M. S. Skaf, M. Sorieul, P. Dupree and S. Hill, Biomacromolecules, 2021, 22, 4669–4680 CrossRef CAS PubMed.
- A. Khodayari, W. Thielemans, U. Hirn, A. W. van Vuure and D. Seveno, Carbohydr. Polym., 2021, 270, 118364 CrossRef CAS PubMed.
- P. Eronen, M. Österberg, S. Heikkinen, M. Tenkanen and J. Laine, Carbohydr. Polym., 2011, 86, 1281–1290 CrossRef CAS.
- J. Lucenius, J. J. Valle-Delgado, K. Parikka and M. Österberg, J. Colloid Interface Sci., 2019, 555, 104–114 CrossRef CAS PubMed.
- T. M. Tenhunen, M. S. Peresin, P. A. Penttilä, J. Pere, R. Serimaa and T. Tammelin, React. Funct. Polym., 2014, 85, 157–166 CrossRef CAS.
- M. E. Vega-Sánchez, Y. Verhertbruggen, H. V. Scheller and P. C. Ronald, Plant Signaling Behav., 2013, 8 DOI:10.4161/psb.23143.
- S. N. Kiemle, X. Zhang, A. R. Esker, G. Toriz, P. Gatenholm and D. J. Cosgrove, Biomacromolecules, 2014, 15, 1727–1736 CrossRef CAS PubMed.
- S. Arola, M. Ansari, A. Oksanen, E. Retulainen, S. G. Hatzikiriakos and H. Brumer, Soft Matter, 2018, 14, 9393–9401 RSC.
- J. C. Muñoz-García, K. R. Corbin, H. Hussain, V. Gabrielli, T. Koev, D. Iuga, A. N. Round, D. Mikkelsen, P. A. Gunning, F. J. Warren and Y. Z. Khimyak, Biomacromolecules, 2019, 20, 4180–4190 CrossRef PubMed.
- M. I. Khan, J. H. Shin and J. D. Kim, Microb. Cell Fact., 2018, 17 DOI:10.1186/s12934-018-0879-x.
- R. H. Wijffels, O. Kruse and K. J. Hellingwerf, Curr. Opin. Biotechnol, 2013, 24, 405–413 CrossRef CAS PubMed.
- M. Jämsä, S. Kosourov, V. Rissanen, M. Hakalahti, J. Pere, J. A. Ketoja, T. Tammelin and Y. Allahverdiyeva, J. Mater. Chem. A, 2018, 6, 5825–5835 RSC.
- H. Leino, S. N. Kosourov, L. Saari, K. Sivonen, A. A. Tsygankov, E. M. Aro and Y. Allahverdiyeva, Int. J. Hydrogen Energy, 2012, 37, 151–161 CrossRef CAS.
- G. Peers, Trends Biotechnol., 2014, 32, 551–555 CrossRef CAS PubMed.
- S. Kosourov, G. Murukesan, M. Seibert and Y. Allahverdiyeva, Algal Res., 2017, 28, 253–263 CrossRef.
- S. N. Kosourov and M. Seibert, Biotechnol. Bioeng., 2009, 102, 50–58 CrossRef CAS PubMed.
- S. Vajravel, S. Sirin, S. Kosourov and Y. Allahverdiyeva, Green Chem., 2020, 22, 6404–6414 RSC.
- V. Rissanen, S. Vajravel, S. Kosourov, S. Arola, E. Kontturi, Y. Allahverdiyeva and T. Tammelin, Green Chem., 2021, 23, 3715–3724 RSC.
- H. Lee, D. Shin, J. Choi, C. S. Ki and J. Hyun, Carbohydr. Polym., 2022, 290, 758–767 CrossRef PubMed.
- E. Kontturi, P. Laaksonen, M. B. Linder, Nonappa, A. H. Gröschel, O. J. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779 CrossRef PubMed.
- M. C. Li, Q. Wu, R. J. Moon, M. A. Hubbe and M. J. Bortner, Adv. Mater., 2021, 33, 2006052 CrossRef CAS PubMed.
- K. Heise, E. Kontturi, Y. Allahverdiyeva, T. Tammelin, M. B. Linder, Nonappa and O. Ikkala, Adv. Mater., 2021, 33, 2004349 CrossRef CAS PubMed.
- M. C. Jarvis, Philos Trans. R. Soc., A, 2018, 376 DOI:10.1098/rsta.2017.0045.
- T. Saito, Y. Nishiyama, J. L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS PubMed.
- A. Walther, F. Lossada, T. Benselfelt, K. Kriechbaum, L. Berglund, O. Ikkala, T. Saito, L. Wågberg and L. Bergström, Biomacromolecules, 2020, 21, 2536–2540 CrossRef CAS PubMed.
- H. Dong, J. F. Snyder, K. S. Williams and J. W. Andzelm, Biomacromolecules, 2013, 14, 3338–3345 CrossRef CAS PubMed.
- M. Hakalahti, A. Salminen, J. Seppälä, T. Tammelin and T. Hänninen, Carbohydr. Polym., 2015, 126, 78–82 CrossRef CAS PubMed.
- M. Hakalahti, A. Mautner, L. S. Johansson, T. Hänninen, H. Setälä, E. Kontturi, A. Bismarck and T. Tammelin, ACS Appl. Mater. Interfaces, 2016, 8, 2923–2927 CrossRef CAS PubMed.
- E. Chiellini, A. Corti, S. D’Antone and R. Solaro, Prog. Polym. Sci., 2003, 28, 963–1014 CrossRef CAS.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- W. Cui, P. J. Wood, B. Blackwell and J. Nikiforuk, Carbohydr. Polym., 2000, 41, 249–258 CrossRef CAS.
- J. R. Woodward, G. B. Fincher and B. A. Stone, Carbohydr. Polym., 1983, 3, 207–225 CrossRef CAS.
- R. A. Burton and G. B. Fincher, Mol. Plant, 2009, 2, 873–882 CrossRef CAS PubMed.
- N. Böhm and W.-M. Kulicke, Carbohydr. Res., 1999, 315, 302–311 CrossRef.
- S.-J. Kim and F. Brandizzi, Plant Cell Physiol., 2021, 62, 1839–1846 CrossRef CAS PubMed.
- P. J. Harris and G. B. Fincher, in Chemistry, Biochemistry, and Biology of 1-3 Beta Glucans and Related Polysaccharides, ed. A. Bacic, G. B. Fincher and B. A. Stone, Elsevier, 2009, ch. 4.6, pp. 621–654 Search PubMed.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391–396 CrossRef CAS.
- Scandinavian Pulp, Paper and Board Testing Committee, SCAN-CM 65:02 Total acidic group content conductometric titration method, Stockholm, Sweden, 2002.
- A. Lazaridou, C. G. Biliaderis and M. S. Izydorczyk, Food Hydrocolloids, 2003, 17, 693–712 CrossRef CAS.
- R. Rippka, J. Deruelles, J. B. Waterbury, M. Herdman and R. Y. Stanier, J. Gen. Microbiol., 1979, 111, 1–61 Search PubMed.
- K. Thiel, E. Mulaku, H. Dandapani, C. Nagy, E. M. Aro and P. Kallio, Microb. Cell Fact., 2018, 17 DOI:10.1186/s12934-018-0882-2.
- V. Carbonell, E. Vuorio, E. M. Aro and P. Kallio, World J. Microbiol. Biotechnol., 2019, 35 DOI:10.1007/s11274-019-2652-7.
- M. Hakalahti, M. Faustini, C. Boissière, E. Kontturi and T. Tammelin, Biomacromolecules, 2017, 18, 2951–2958 CrossRef CAS PubMed.
- P. Eronen, M. Österberg, S. Heikkinen, M. Tenkanen and J. Laine, Carbohydr. Polym., 2011, 86, 1281–1290 CrossRef CAS.
- M. S. Peresin, K. Kammiovirta, H. Setälä and T. Tammelin, J. Polym. Environ., 2012, 20, 895–904 CrossRef CAS.
- G. S. Tóth, V. Siitonen, L. Nikkanen, L. Sovic, P. Kallio, R. Kourist, S. Kosourov and Y. Allahverdiyeva, Biotechnol. Biofuels Bioprod., 2022, 15 DOI:10.1186/s13068-022-02248-1.
- T. Levä, V. Rissanen, L. Nikkanen, V. Siitonen, M. Heilala, J. Phiri, T. C. Maloney, S. Kosourov, Y. Allahverdiyeva, M. Mäkelä and T. Tammelin, Biomacromolecules, 2023 DOI:10.1021/acs.biomac.3c00261.
- H. K. Lichtenthaler, Methods Enzymol., 1987, 148, 350–382 CAS.
- W. Li, S. W. Cui, Q. Wang and R. Y. Yada, Food Hydrocolloids, 2011, 25, 189–195 CrossRef CAS.
- G. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Chapman & Hall, London, 1993 Search PubMed.
- J.-L. Doublier and P. J. Wood, Cereal Chem., 1995, 72, 335–340 CAS.
- J. R. Woodward, D. R. Phillips and G. B. Fincher, Carbohydr. Polym., 1988, 8, 85–97 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, 3rd edn, 2011, ch. 15, pp. 341–380 Search PubMed.
- K. P. Gregory, G. R. Elliott, H. Robertson, A. Kumar, E. J. Wanless, G. B. Webber, V. S. J. Craig, G. G. Andersson and A. J. Page, Phys. Chem. Chem. Phys., 2022, 24, 12682–12718 RSC.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS PubMed.
- S. Lombardo and W. Thielemans, Cellulose, 2019, 26, 249–279 CrossRef CAS.
- T. Benselfelt, E. D. Cranston, S. Ondaral, E. Johansson, H. Brumer, M. W. Rutland and L. Wågberg, Biomacromolecules, 2016, 17, 2801–2811 CrossRef CAS.
- T. Tammelin, J. Merta, L. S. Johansson and P. Stenius, Langmuir, 2004, 20, 10900–10909 CrossRef CAS PubMed.
- V. Arumughan, H. D. Özeren, M. Hedenqvist, M. Skepö, T. Nypelö, M. Hasani and A. Larsson, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-c02j8.
- N. Mittal, T. Benselfelt, F. Ansari, K. Gordeyeva, S. V. Roth, L. Wågberg and L. D. Söderberg, Angew. Chem., 2019, 131, 18735–18742 CrossRef.
- M. J. Barbosa, Hadiyanto and R. H. Wijffels, Biotechnol. Bioeng., 2004, 85, 78–85 CrossRef CAS PubMed.
- C. Gudin and D. Chaumont, Bioresour. Technol., 1991, 38, 145–151 CrossRef.
- C. Posten, Eng. Life Sci., 2009, 9, 165–177 CrossRef CAS.
- T. Benselfelt, M. Nordenström, M. M. Hamedi and L. Wågberg, Nanoscale, 2019, 11, 3514–3520 RSC.
- V. Kumaresan, F. Nizam, G. Ravichandran, K. Viswanathan, R. Palanisamy, P. Bhatt, M. V. Arasu, N. A. Al-Dhabi, K. Mala and J. Arockiaraj, Algal Res., 2017, 23, 96–103 CrossRef.
- S. Kosourov, H. Leino, G. Murukesan, F. Lynch, K. Sivonen, A. A. Tsygankov, E. M. Aro and Y. Allahverdiyeva, Appl. Environ. Microbiol., 2014, 80, 5807–5817 CrossRef CAS PubMed.
- R. Schwarz and K. Forchhammer, Microbiology, 2005, 151, 2503–2514 CrossRef CAS PubMed.
- D. Kirilovsky, Photosynth. Res., 2015, 126, 3–17 CrossRef CAS.
- A. Latifi, M. Ruiz and C. C. Zhang, FEMS Microbiol. Rev., 2009, 33, 258–278 CrossRef CAS PubMed.
- A. Derks, K. Schaven and D. Bruce, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 468–485 CrossRef CAS PubMed.
- K. Nagahama, T. Ogawa, T. Fujii, M. Tazaki, S. Tanase, Y. Morino and H. Fukuda, J. Gen. Microbiol., 1991, 137, 2281–2286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00919j |
| This journal is © The Royal Society of Chemistry 2023 |