
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability and potential degradation of the α′,β′-epoxyketone pharmacophore on ZnO nanocarriers: insights from reactive molecular dynamics and density functional theory calculations†
Cheherazade
Trouki
 ac,
Susanna
Monti
b and
Giovanni
Barcaro
*a
ac,
Susanna
Monti
b and
Giovanni
Barcaro
*a
aCNR-IPCF, Institute for Chemical and Physical Processes, Via G. Moruzzi 1, Pisa (PI) 56124, Italy. E-mail: giovanni.barcaro@cnr.it
bCNR-ICCOM, Institute of Chemistry of Organometallic Compounds, Via G. Moruzzi 1, Pisa (PI) 56124, Italy
cDepartment of Pharmacy, University of Pisa, Via Bonanno 6, Pisa (PI) 56126, Italy
First published on 8th June 2023
Abstract
We investigate the structure and dynamics of a zinc oxide nanocarrier loaded with Carfilzomib, an epoxyketone proteasome inhibitor developed for treating multiple myeloma. We demonstrate that, even though both bare and functionalized zinc oxide supports have been used for drug delivery, their interactions with the reactive functional groups of the ligands could be detrimental. This is because pharmacophores like α′,β′-epoxyketones should preserve the groups required for the drug activity and be capable of leaving the vehicle at the target site. Earlier studies showed that even when ZnO is functionalized with oleic acid surfactants, the drug could reach parts of the surface and remain stably adsorbed. Herein, we have used reactive molecular dynamics simulations and quantum chemistry calculations to explore the potential interactions of the Carfilzomib functional groups with the typical surfaces of ZnO supports. We have found that Carfilzomib can adsorb on the (0001)Zn-terminated polar surface through the carbonyl oxygens and the epoxyketone moiety. These strong connections could prevent the drug release and induce the epoxy ring opening with its consequential inactivation. Therefore, regulating the dosage to maintain the desired level of drug bioavailability is paramount. These findings emphasize the need for appropriate carrier functionalizations to efficiently entrap, transport, and release the cargo at the target sites and the crucial role played by predictive/descriptive computational techniques to complement and drive experiments to the most appropriate selections of the materials to optimize drug delivery.
Introduction
Designing efficient drug delivery systems is essential for enhancing intrinsic drug properties and overcoming resistance.1 Nanotechnology has provided new approaches to overcome conventional therapy limitations, specifically preparing tuned nanovehicles. Indeed, engineered nanoparticles have shown superior performance in undertaking their tasks due to their unique properties, including a high surface area-to-volume ratio that enables a high drug loading and controlled release. These delivery methods improved the stability and solubility of the drugs targeting selected cells/tissues, thus reducing systemic toxicity.2 Practically, drug-loaded nanocarriers outperformed free drugs because they could reduce structural degradation and prevent diffusion and accumulation in undesirable sites.3,4 Typical nanocarriers include liposomes, micelles, polymers, inorganic nanoparticles, and innovative platforms such as nanosheets and quantum dots.5,6 The inorganic ones have gained considerable interest due to their tunable physicochemical properties.7 Traditional cancer treatments often involve drugs acting directly on the target through physicochemical interactions or reactions. Therefore, it is crucial to elucidate the phenomena affecting a therapeutic molecule and its carrier, primarily if the therapeutic effect is based on the reaction with the specific target (i.e., one or more amino acids of a protein).8Reactive drugs with three-atom cyclic ethers, such as epoxides or oxiranes, which possess high ring strain, have gained significant attention. Although using epoxide-containing molecules has been controversial due to potential reactivity with biological functional groups, they have become a viable option for treating challenging diseases such as cancer and bacterial infections. This is testified by the epoxy molecules available on the market, with proper pharmacodynamics monitoring and targeted drug delivery controls.9 One of the classes of epoxy-containing molecules used in clinical practice is α′,β′-epoxyketones. This small molecular motif is found in several natural products and synthetic compounds. It consists of a ketone group and an epoxide moiety adjacent to each other in the molecule. The two carbons of the epoxide are located in position α–β to the ketone. The presence of this motif imparts specific biological activities to the molecule.10 The α′,β′-epoxyketone pharmacophore is valuable for developing new drugs, particularly those targeting cancer cells. Its ability to selectively bind to specific proteins and inhibit their activity has made it a promising target for drug discovery efforts. This class of molecules is widely used in treating multiple myeloma, a type of cancer affecting plasma cells in the bone marrow. An example of a compound that contains the α′,β′-epoxyketone pharmacophore is the natural product Epoxomicin. This pharmacophore is responsible for inhibitory activity against the proteasome, a cellular complex involved in protein degradation.11 Specifically, the α′,β′-epoxyketone group forms a covalent bond with the active site of the proteasome, thereby blocking its enzymatic activity. Another example of a compound containing this pharmacophore is Carfilzomib (CFZ), a proteasome inhibitor for treating multiple myeloma.12 Like Epoxomicin, CFZ binds to the proteasome through its α′,β′-epoxyketone group, inhibiting protein degradation and accumulation of damaged proteins in cancer cells, ultimately resulting in cell death.13 Unfortunately, drug resistance remains a significant concern even for very effective molecules when misused or used for prolonged periods. A promising strategy to overcome drug resistance is to combine the drug with a delivery system that can circumvent the mechanisms employed by the resistant cells. This approach could be advantageous for drugs like CFZ, where the resistance is mediated by a protein (P-glycoprotein) that recognizes the structure of the drug and expels it from the cancer cell, preventing it from reaching its target site.14,15 Therefore, it is essential to optimize their therapeutic efficacy to investigate the drug stability and potential degradation on different non-inert materials, like metal oxide nanocarriers (NCs).16–18 When dealing with nanoparticle-based drug delivery, surfactants are very often mandatory to prevent coalescence and improve colloidal stability.19,20 However, it was found that this does not preclude the possibility of drug interactions with uncovered regions of the NC surface.21
Given the complexity of the whole delivery scenario, an advanced experimental characterization to disclose the atomistic mechanisms of all the system components is impossible. Thus, experimental scientists often flank their studies with computational techniques for predicting/describing at the molecular/atomic level all the stages of the delivery process, including possible perturbing phenomena and abnormal behaviors. The most appropriate computational methods for such a task are molecular dynamics simulations based on force fields, which can explore the structure and dynamics of large-size systems for a long time, combined with quantum chemistry calculations to refine the classical pictures. We focus on ZnO NCs, which were employed in drug delivery against various types of cancer.22 The literature reports on several computational studies concerning the interactions between drugs, amino acids, or sugars with such inorganic substrates.23–27 The typical surfaces (or low-dimensional structural motifs) exposed by ZnO, which could be possibly doped or functionalized to tune drug-carrier interaction, were considered for designing and optimizing delivery or sensor devices.27–30 It is known that ZnO surfaces are highly reactive, able to induce dissociation of surfactants like carboxylic acids31 and small molecules used in drug loading and delivery process, such as ethanol32 and water.33 The effects of water–ZnO surface interactions on the oxide surface properties can impact the adsorption of cargo species.
The present study builds upon an earlier work21 where we defined an atomistic multi-scale computational protocol for simulating the delivery of CFZ by ZnO NCs functionalized with oleic acid in ethanol or water. We found that, although the NC was initially uniformly covered with oleic acid, the relocation of the chains produced bare ZnO regions which could host CFZ molecules. These were directly connected with the metal oxide surfaces and remained stably adsorbed. Considering the high reactivity of the α′,β′-epoxyketone pharmacophore of CFZ, we have extended and refined the previous scenario focusing on the electronic interactions of CFZ with the ZnO interface considering various types of ZnO facets in the gas phase and water solution. We found that CFZ could remain stably adsorbed on the NC and react with the substrate, irreversibly damaging its pharmacophoric action.
Methods
Given the prohibitive computational cost of investigating the adsorption properties of CFZ on ZnO surfaces at the quantum chemistry (QC) level, we extracted small, low-weight molecular species (fragments), mimicking the action of the different parts of the drug. CFZ is a modified peptide that consists of four α-amino acids, α′,β′-epoxyketone, and morpholine. Considering that the adsorption of simple amino acids on ZnO has already been studied at the DFT level,34,35 we used, instead, ethyl-benzene (F02) to reproduce the aromatic side chains, N-methyl-acetamide (F04) for the interacting parts of the backbone, and acetyl-oxirane (F01) and morpholine (F03) to simulate the terminal moieties of the molecule. Methyl groups were added to these last two fragments to create steric hindrance and reproduce the tertiary amine of (F03). The structures of these molecules are shown in Fig. 1(a). For the interacting oxide substrates, we selected surfaces among the most relevant facets characterizing ZnO powders.36,37 The selected surfaces include two non-polar facets, (1010) and (1120), and two polar facets, (0001) zinc-terminated and oxygen-terminated, as shown in Fig. 1(b). Non-polar surfaces are stable and could be modeled by small bulk-terminated unit cells. In contrast, polar surfaces are unstable due to the divergence of the electric field. Previous studies have allowed surface reconstructions or used charged species that could be adsorbed on the surface to remove this instability.38 We chose the first approach to eliminate possible interferences with the CFZ fragments, following the protocol described here and relaxing the last layers of the surface. As expected, the polar topmost layers flattened, thus reducing the polarity (as shown in Fig. 1(b)).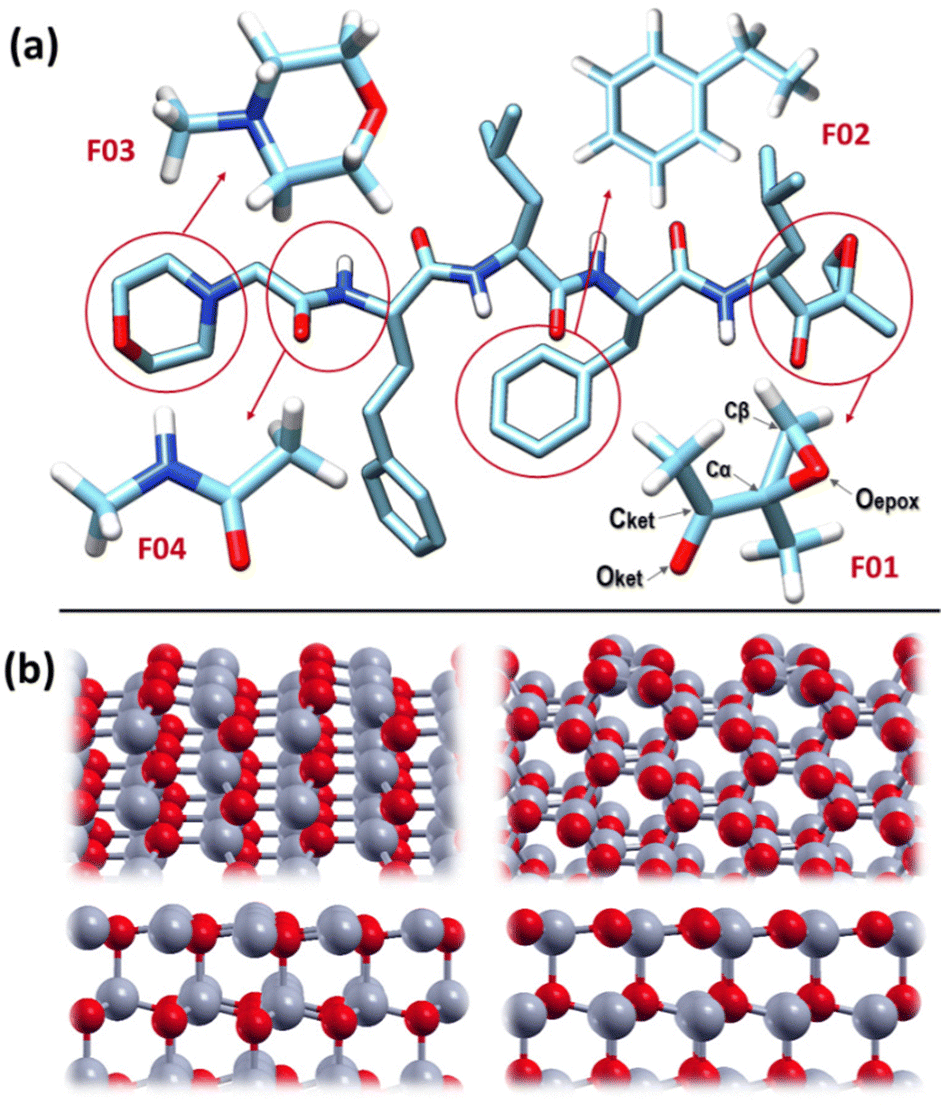 | ||
| Fig. 1 (a) Structure of CFZ and the selected fragments (F01–F04). (b) ZnO surfaces considered in this work, optimized at the DFT level: top left = (1010), top right = (1120), bottom left = (0001)Zn-terminated, bottom right = (0001)O-terminated. Color code: Zn = grey; O = red; C = light blue; N = blue; H = white. | ||
All QC calculations were carried out at the DFT level by using the Quantum Espresso software,39 which employs periodic boundary conditions, plane waves as basis sets, and PAW (Plane Augmented Waves) Pseudopotentials to derive Kohn–Sham single-particle wavefunctions to build up Bloch states. Values of 40 and 400 Ry have been employed as cut-offs on the kinetic energy/electronic density, respectively, and PBE/GGA has been used as xc-functional to describe exchange and correlation effects. Spin-restricted formalism, in conjunction with a Gaussian smearing technique with a value of 0.002 Ry, has been employed, verifying that all the investigated configurations (bare surfaces and surfaces with adsorbed species) adopt zero magnetization. We estimated the reaction barriers with the NEB (Nudged Elastic Band) algorithm implemented in the QE software by considering as initial and final structures the optimized conformations of reactants and products. Preliminary tests have been conducted to identify the optimal number of intermediate images (between reactants and products) that turned out to be seven. In building the ZnO surfaces, we have employed DFT-optimized values of the lattice constants of the wurtzite structure, corresponding to 3.29 Å and to 5.29 Å for the a and c unit cell parameters, respectively, in agreement with experimental values.36 In the case of the non-polar (1010) surface, a rectangular (2 × 3) cell has been built, extending 10.58 × 9.87 Å2 in the xy plane, with a thickness of 6 layers and a total of 72 atoms; the first Brillouin zone has been sampled by adopting a (2,2,1) uniform k-points mesh. In the case of the non-polar (1120) surface, a rectangular (2 × 2) cell has been built, extending 11.25 × 10.41 Å2 in the xy plane, with a thickness of 6 layers and a total of 96 atoms; the first Brillouin zone has been sampled by adopting a (2,2,1) uniform k-points mesh. Finally, in the case of the polar (0001) surface (both O- and Zn-terminated), an hexagonal (3 × 3) cell has been built, extending 9.87 × 9.87 Å2 in the xy plane, with a thickness of 6 layers and a total of 108 atoms; the first Brillouin zone has been sampled at the gamma point only, having verified that the results obtained by adopting a denser (2,2,1) k-points mesh do not differ quantitatively. For all four surfaces considered, a minimal space of about 12 Å has been inserted along the z-direction, and the bottom three layers have been frozen in their bulk positions. In contrast, the top three layers have been fully relaxed.
Through reactive molecular dynamics simulations40,41 based on the force field already employed in the previous study,21 we tried to disclose the behavior of the drug close to the various types of ZnO surfaces mentioned above. We designed and optimized four extended interfaces, namely (0001)Zn-terminated, (0001)O-terminated, (1010), and (1120), where the xy cells sizes were 19.7 × 17.1 Å2, 20.2 × 29.1 Å2, 39.0 × 20.4 Å2, and 20.8 × 22.5 Å2, respectively, considering six layers in each case. Periodic boundary conditions were applied in all directions. The z-direction was extended to 400 Å to avoid interactions with the periodic image. Then, we placed an energy-minimized conformation of CFZ near the interface and carried out RMD simulations in the gas phase. The simulations were run in the NVT ensemble, slowly raising the temperature from 10 K to 300 K (50 ps) and equilibrating the system for nearly 100 ps. The production dynamics was approximately 500 ps, and system structures were collected every 0.002 ps. The temperature was controlled through Berendsen's thermostat with a relaxation constant of 0.1 ps, and the time step was set to 0.2 fs. The final 100 ps of the trajectories were analyzed to identify potential binding sites of CFZ, the most interacting groups, and possible reactions. MD simulations in water solutions were used to unveil the effect of water on the reaction mechanisms determined in the gas phase. The starting structure was the initial configuration of the in vacuo MD. Water molecules were included in the simulation box (around 300 water molecules) and equilibrated in steps. First, the solute was frozen, and the waters were randomized and heated to 300 K (about 100 ps); then, the constraints were released, and the system was equilibrated for about 50 ps. The simulation protocol of the production stage was similar to the one in the gas phase. The total simulation time was about 500 ps. All the RMD simulations were carried out with the Amsterdam Density Functional (ADF) modeling suite.42
Results and discussion
Reactive molecular dynamics
The analysis of the trajectories revealed that the molecule could not bind stably either to the polar (0001)O-terminated or the non-polar (1120) surfaces. In the first case, the migration to farther regions was swift (a few picoseconds). In the second case, the molecule spent approximately 400 ps near the interface exploring various portions of the surface without establishing any stable connection and then moved to a longer distance. On the contrary, the adsorption of CFZ onto the (0001)Zn-terminated polar and (1010) non-polar surfaces was persistent and mainly dominated by the interaction of the oxygen atoms of CFZ with Zn. This scenario is evident in the atom–atom radial distribution functions trend shown in Fig. 2 and in the image of the final configurations adopted by CFZ on the (0001)Zn-terminated and (1010) interfaces (Fig. 3). The sharp peak at about 2.2 Å (Fig. 2 – top) indicates that almost all CFZ carbonyl oxygens were coordinated to a Zn atom. The smaller broader peak at about 3.2 Å suggests a weaker interaction between the oxygens in the rings and the surface. This type of binding, however, seems slightly more robust in the case of (1010), where only the morpholine oxygen was in close contact with the slab (peak at about 2.6 Å) together with nearby carbons (peak at about 2.8 Å). Indeed, as apparent in the plots, the other portion of CFZ, namely backbone oxygens and –NH groups, remained relatively far. On the polar (0001)Zn-terminated surface, instead, CFZ was strongly adsorbed through multidentate coordination of the oxygens (Fig. 4) with a dominant contribution due to the Oket (population density map: around 29%) followed by the four central carbonyl O where the maximum population density is around 22%. In contrast, morpholine O was more mobile and explored a larger area (population density map: around 4%). The firm anchoring of the adsorbed CFZ molecule translated in a reduced flexibility of all the backbone torsional angles, as shown in the Ramachandran plots of Fig. S1 (ESI†). Indeed, the regions explored by the ϕ–Ψ angles are less extended than those reported in ref. 21 corresponding to the in vacuo scenario. This behavior is reflected in the geometrical descriptors shown in Fig. S2 (ESI†). Closer inspection of the trajectory indicated that after only 120 ps, this sort of immobilization of the molecule on the surface involving both carbon and nitrogen atoms caused the opening of the epoxide ring. The various steps of the mechanism are displayed in Fig. 5. The interactions of Oepox with Zn sites, which were attracted from CFZ, favored the elongation of the Oepox–Cα bond and the direct coordination of the abandoned Cα to the Zn nearby. This was not the case for the (1010) surface, where the epoxy ketone moiety remained permanently far. To verify if this phenomenon could be related to the lack of solvent around the solute, we examined the behavior of this type of CFZ arrangement when water molecules were included in the simulations. | ||
| Fig. 2 Atom–atom radial distribution functions of Zn and C, N, and O of CFZ. (0001)Zn-terminated (top), (1010) (middle), (1120) (bottom) surfaces. | ||
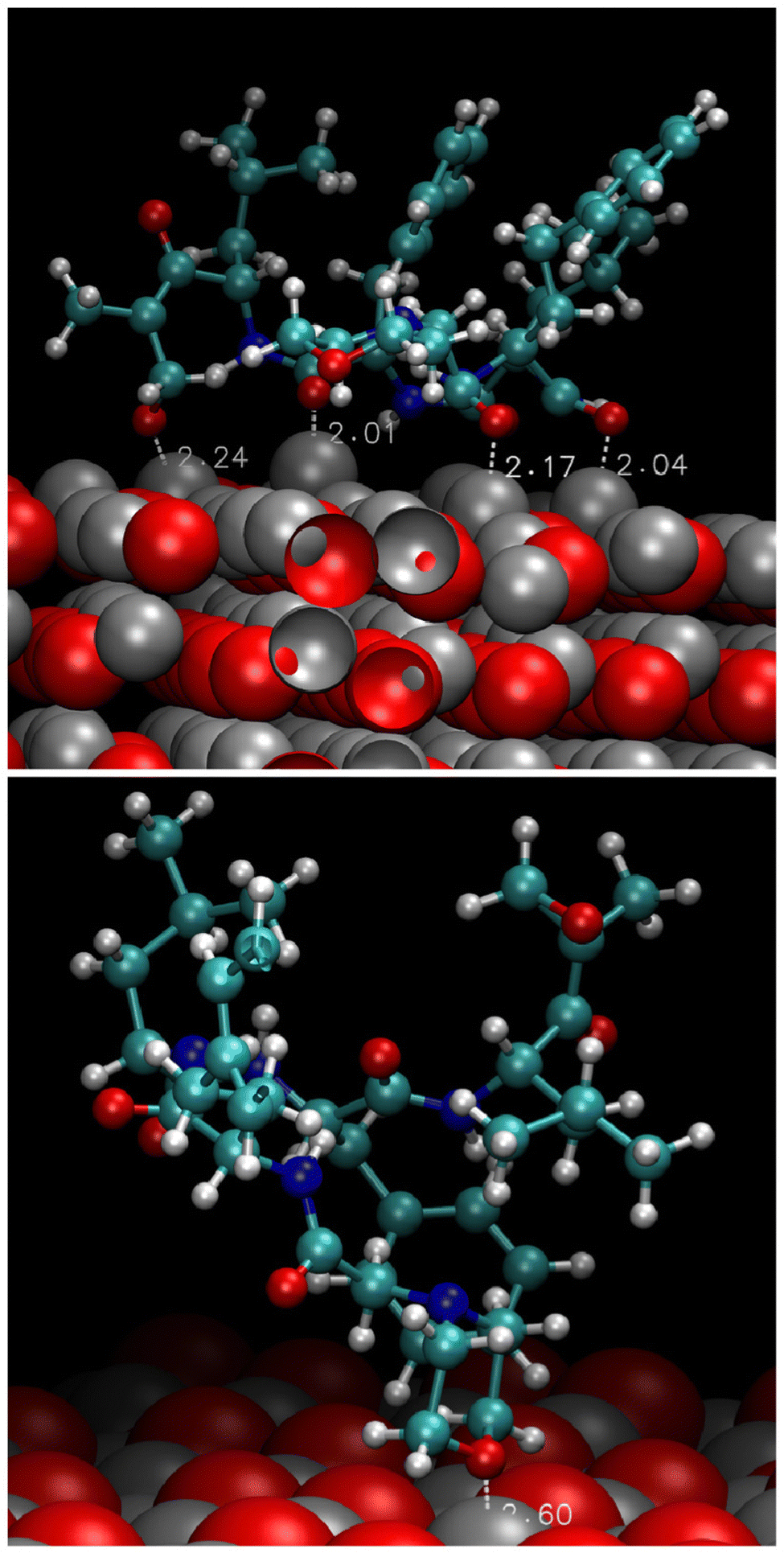 | ||
| Fig. 3 Final arrangements of CFZ on the (0001)Zn-terminated (top) and (1010) (bottom) surfaces. The surface is rendered through vdW spheres, whereas CFZ by balls and sticks. Color code: Zn = grey; O = red; C = light blue; N = blue; H = white. | ||
 | ||
| Fig. 4 Top: Final arrangement of CFZ on the (0001)Zn-terminated surface. The surface is rendered through vdW spheres, whereas CFZ by balls and sticks. Color code: Zn = grey; O = red; C = light blue; N = blue; H = white. Bottom: Contour plot of CFZ oxygen population density (%) on the surface. The red-yellow areas are the most populated. The strongest connection (top contours) corresponds to the open epoxy oxygen. The four contours in the middle of the surface identify the positions of the backbone carbonyl O. The elongated blue contour on the left corresponds to the morpholine oxygen. | ||
 | ||
| Fig. 5 Snapshots extracted from the RMD simulations in the gas phase depicting the epoxy ring opening on the (0001)Zn-terminated surface. The surface is rendered through vdW spheres, whereas CFZ by balls and sticks. Color code: Zn = grey; O = red; C = light blue; N = blue; H = white. | ||
The reactive scenario in the solution is shown in Fig. 6; the corresponding radial distribution functions are reported in Fig. S3 (ESI†). After the equilibration of the solvent, a compact water layer was deposited on the (0001) surface. However, CFZ remained close to the interface kept there by intermolecular hydrogen bonds with the adsorbed water molecules. As is evident in the pictures, also these indirect bindings induced the breaking of the Oepox–Cβ bond. The oxygen received an H+ from the closest water, whereas the Cβ an OH−. After a while, the molecule detached from the surface. This is apparent in the Zn–O radial distribution function trend in water (Fig. S3, ESI†), with a broad peak at about 4.8 Å, and in the other RDF CFZ-water plots, which confirm the complete solvation of the drug. Considering that the (1010) surface could also host adsorbed CFZ molecules, we solvated the other modeled configuration to disclose possible opening effects induced by adsorbed waters (Fig. 7). Examination of the snapshots of Fig. 7 and the trend of the RDF for the solvated (1010) slab (not shown), we found that the epoxy ketone fragment remained intact, a water layer solvated the surface, and water shells surrounded all the groups of the molecules. In the final configuration, after 500 ps, CFZ was moving in the solution.
 | ||
| Fig. 6 Snapshots extracted from the RMD simulations in water solution depicting the epoxy ring opening on the (0001)Zn-terminated surface. The surface is rendered through vdW spheres, whereas CFZ and water molecules close to the surface are rendered by balls and sticks. Color code: Zn = grey; O = red; C = light blue; N = blue; H = white. | ||
 | ||
| Fig. 7 Snapshots extracted from the RMD simulations in water solution depicting the adsorbed and desorbed structures of CFZ on the (1010) surface. The surface is rendered through vdW spheres, whereas CFZ and water molecules close to the surface are rendered by balls and sticks. Color code: Zn = grey; O = red; C = light blue; N = blue; H = white. | ||
Density functional theory calculations
We show, in Fig. 8, the QC adsorption energies of the four fragments representing the different parts of CFZ (F01–F04) interacting with bare ZnO surfaces considered in RMD simulations. The adsorption energy (Eads) was calculated according to the following expression:| Eads = Etot − EZnO,opt − EF0x,opt |
 | ||
| Fig. 8 Adsorption energies of different orientations (F0Xa, F0Xb) of the CFZ fragments on the four selected surfaces. (a) = (0001)Zn-terminated; (b) = (0001)O-terminated; (c) = (1010); (d) = (1120). Polar surfaces = red bars. Non-polar surfaces = blue bars. On the left, the distances between the center of mass of each fragment and the average height of the topmost layer is reported. For each panel, the configuration of the fragment with the greater adsorption energy is shown. Ball and stick atomic models; color coding: Zn = grey; O = red; C = cyan; N = blue; H = white. | ||
We can observe a remarkable difference between the polar (panels (a) and (b) of Fig. 8), and non-polar surfaces (panels (c) and (d) of Fig. 8), with the polar (0001)Zn-terminated surface exhibiting dominant adsorption. The strong reactivity of the (0001)Zn-terminated surface is well-known in the literature and has been exhaustively investigated at the QC level. Carboxyl groups can dissociatively adsorb with the formation of negatively charged carboxylate (firmly bound to two zinc atoms of the surface) and positively charged H+ tightly bound to a positive zinc ion of the surface.43 Gao et al.44 observed a similar reactivity of ethanol and Iachella et al.45 of water on a similar polar surface. Water dissociation was accompanied by the stabilization of the OH group at the bridge position between two Zn ions of the surface and by adsorption of the other H atom on a further vicinal Zn surface ion. Bridge stabilization of adsorbed OH group on Zn-terminated surfaces is also compatible with recent measurements by Manzano et al.46 As observed in RMD simulations in line with previous literature, the adsorbed fragments preferred to establish favorable interactions between the Zn atoms of the topmost layers and the nucleophilic oxygen atoms of CFZ. This agrees with previous literature in which drug/ZnO interactions were mainly established via the coordination of carbonyl oxygens to surface zinc atoms.47–49 No reactivity was observed between all the fragments and the non-polar surfaces or the polar (0001)O-terminated surface. In this case, the adsorption energies were mainly between −0.9 and −4.1 kcal mol−1, where the weaker interaction corresponds to structures in which an oxygen atom pointed toward the surface, creating a repulsive effect with the exposed surface O. The only exception was given by N-methyl-acetamide (fragment F04), with adsorption energy between −6.3 and −8.5 kcal mol−1 due to a hydrogen bond (2.19–2.75 Å) between the –NH and surface O. This interaction was so favorable that, even when the –NH was placed on the opposite side, the molecule rotated completely during optimization to expose the –NH toward the surface at hydrogen bonding distance.
Ethyl-benzene (fragment F02) exhibited weak van der Waals interactions on all selected surfaces in horizontal and vertical orientations, with adsorption energies ranging from −0.9 to −3.5 kcal mol−1. The Zn-terminated surface showed greater adsorption energies, ranging from −6.5 to −9.2 kcal mol−1, attributed to the electron density of the aromatic ring drawn towards the exposed electrophilic Zn atoms, favored by the electron-donating ethyl group. For N-methyl-morpholine (fragment F03), the adsorption energies varied depending on the orientation to the surface, except for the non-polar surface, where they remained around −10 kcal mol−1. The most significant difference in adsorption energies was on the non-polar (1120) surface (ΔE = 13.3 kcal mol−1) and on the polar (0001)Zn-terminated surface (ΔE = 13.4 kcal mol−1). The greater adsorption energy corresponded to the structures where the ring's oxygen and nitrogen atoms were oriented towards the partially positive surface, adopting a horizontal orientation. In this orientation, the nitrogen atom attracted the surface Zn directly underneath, moving it by approximately 0.5 Å from the surface plane formed by the other Zn atoms at a Zn–N distance of 2.25 Å. Similarly, the oxygen atom of the ring attracted the corresponding surface Zn atom, but the displacement of the Zn atom was less pronounced (0.2 Å), reaching a Zn–O distance of 2.28 Å after complete optimization. This orientation's stronger adsorption is in line with the DFT studies by Balachandran and coworkers50 on N-methyl-morpholine, which showed charge transfer interactions within the molecule and highlighted the electronegative charge regions at both N and O atoms of the ring through molecular electrostatic potential analysis.
Increased interaction between the exposed surface Zn atoms and the nucleophilic O of the fragments F01, F03, and F04 justifies the larger values found for the (0001)Zn-terminated surface. For this surface, we can distinguish between configurations in which the structure of all the considered fragments was preserved, qualitatively analogous to what was observed on the two non-polar and on the (0001)O-terminated surfaces, and a configuration corresponding to a robust structural modification, given by a ring-opening of the epoxide moiety in the acetyl-oxirane fragment F01, resulting in the formation of a strong complex, characterized by a center of mass of the fragment at only 2.3 Å from the surface and with an adsorption energy of −79.6 kcal mol−1 (see Fig. 9(b)); this latter configuration lies 58.6 kcal mol−1 lower than the configuration of F01 with the epoxide ring closed and adsorbed via the interaction of Oket with an underneath Zn atom (see Fig. 9(a)). Such a modified structure is very similar to that observed in the terminal epoxyketone moiety of CFZ in RMD dynamics, as, also in this case, the Cα atom of the epoxide ring underwent a transition from sp3 to sp2, adsorbing its trigonal plane parallel to the surface. This arrangement, whose characteristic distances are reported in the inset of Fig. 10(a), was favored by a charge donation process from the p density of the Cα–Cket bond and from Oepox to four Zn atoms of the surface, which moved outward by about 0.5 Å to the flatter Zn atoms within the unit cell. This analysis is confirmed by the density of states (DOS) of this configuration, shown in Fig. 10, which allowed us to locate a state of intermixing at about 1.05 eV below the Fermi level (see PDOS in the inset), conveying contributions from the π density of the Cα–Cket bond donated on the states of an underneath Zn atom of the surface. As the epoxide is a highly reactive functional group,51 it often undergoes a ring-opening reaction to relieve the ring's tension, so we sought to determine whether the three-membered ring of the epoxyketone was the primary trigger of the reaction on polar Zn-terminated surfaces. Our analysis showed that the carbonyl group, rather than the epoxide ring, initiated the ring-opening reaction after adsorbing on an underneath Zn atom of the surface. The reason behind this preference is the presence of the ketone group, which withdraws electrons and destabilizes the charge that develops during the opening process at the α-position.52
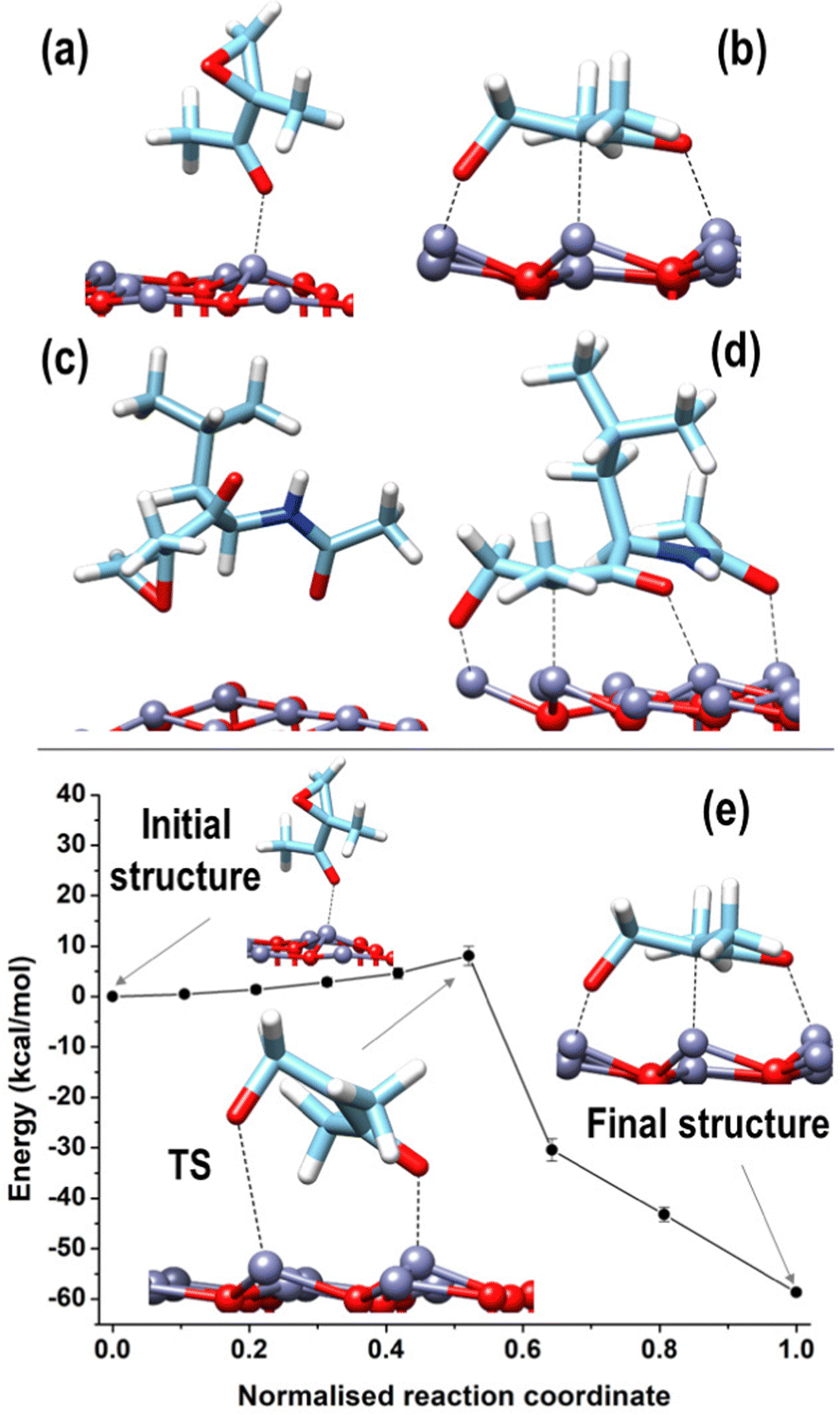 | ||
| Fig. 9 F01 (a), (b) and F01enl (c), (d) adsorbed on the bare (in vacuo) (0001)Zn-terminated surface in configurations where the epoxide ring is closed (a)–(c) or open (b)–(d). (e) Energy profile calculated by NEB and corresponding to a ring opening of the epoxide ring in F01 fragment. In the Initial Structure, F01 is adsorbed according to configuration (a), whereas in the final structure, according to configuration (b); TS corresponds to the NEB transition state. Ball and stick atomic models; color coding: Zn = grey; O = red; C = cyan; N = blue; H = white. | ||
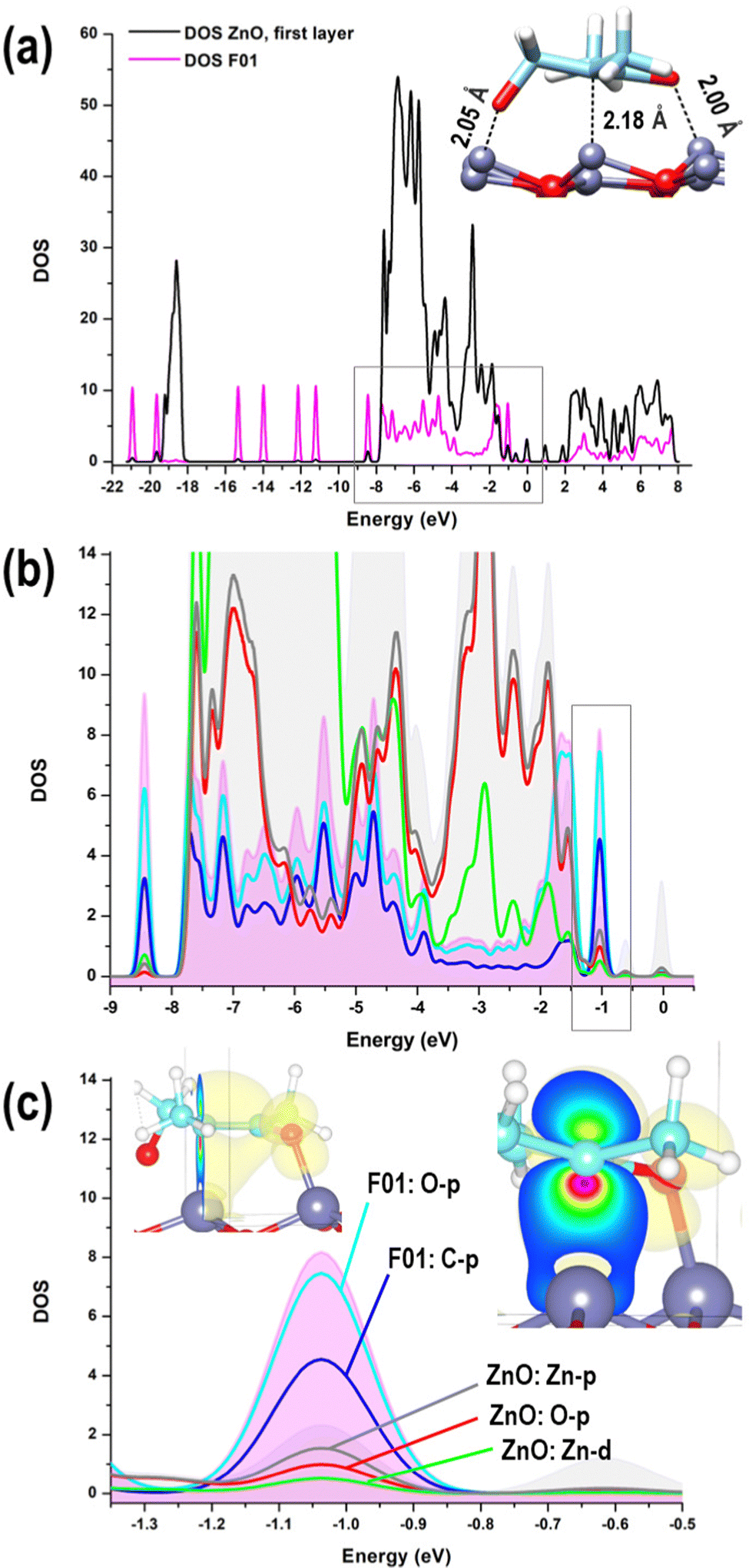 | ||
| Fig. 10 (a) Contribution deriving from the topmost layer of the (0001)Zn-terminated surface (in black) and from F01 (in pink) to the total Density Of States of the configuration shown in Fig. 9(b); (b) the area highlighted in grey in panel (a) is reported together with the main contributions describing the interaction between the oxide surface and F01; (c) a further zoom on the PDOS contributions in the peak located at about 1.05 eV below the Fermi level shows the charge donation process from the p density of the Cα–Cket bond to an underneath Zn atom of the surface. Ball and stick atomic models; color coding: Zn = grey; O = red; C = cyan; N = blue; H = white. | ||
The epoxide ring opening is a relevant process for the stability of the CFZ structure and its activity as an anti-cancer drug. For this purpose, we enlarged the F01 fragment to create a model of the epoxyketone moiety closer to the reference structure of the CFZ molecule by substituting the terminal methyl group linked to Cket in F01 with a fragment of the peptide backbone carrying the lateral group of the leucine amino acid. Using this modified version of the F01 component (from now on called F01enl), we could confirm the stabilization accompanying the epoxide ring opening by sampling the two configurations reported in Fig. 9(c) and (d). In Fig. 9(c), F01 was stabilized with its epoxide ring closed and absorbed on a Zn atom of the underneath polar surface through the carbonyl oxygen of the backbone; the Oepox pointed towards the surface, in trans position (dihedral angle of about 143°) relative to Oket, which pointed far from the surface. This configuration was less stable (by about 59.3 kcal mol−1) than that of Fig. 9(d), which was characterized by the opening of the epoxide ring very similar to that of Fig. 9(b), with the addition of a further bonding established between the carbonyl O of the backbone and a Zn of the surface.
Given the similarity between the behavior (and relative energetics) of F01 and F01enl, we decided to estimate the barrier associated with the ring-opening process by using NEB connecting the two minima shown in Fig. 9(b) and (a). Moving along the potential energy profile shown in Fig. 9(e), we observed an initial region of flat energy, corresponding to epoxyketone rotation approaching the surface around its pivotal Oket bound to the underneath surface Zn atom. The NEB transition state, located at an energy higher by 8.1 ± 1.9 kcal mol−1 with respect to the initial state, is characterized by a highly distorted geometry, with a distance of 2.11 Å between the Cα and Oepox atoms and a distance of 3.08 Å between Oepox and the nearest Zn atom of the surface.
To establish a connection with the results of RMD in water, we performed some specific calculations to investigate the epoxide ring opening in F01 on the (0001)Zn-terminated surface with one water molecule (low-density regime) and a few water molecules corresponding to the first-hydration shell, extracted from RMD upon opening of the epoxide ring (high-density regime). The most significant configurations corresponding to the low-density regime are reported in Fig. 11(a)–(c): a water molecule can be firmly anchored via its O atom on any Zn of the surface, although previous literature has shown a preference towards dissociation;44 we observed that, in almost all sampled cases, F01 did react by inducing water dissociation via the extraction of a hydrogen atom that could be adsorbed either on Oepox, accompanied by epoxide ring opening, see configurations in Fig. 11(a) and (b), or on Cket, preserving the epoxide ring closed. In the former case, Cα passed from sp3 to sp2, analogously to what was observed in the configuration of Fig. 9(b). However, in both Fig. 11(a) and (b) cases, Cα did not show direct contact with any underneath Zn since the OH group (resulting from the Oepox and extracted H atom) was stabilized at a larger distance from the surface to the case of Fig. 9(b). In the latter case, a new mechanism was singled out. Being the epoxide ring initially on the opposite side of the fragment with respect to the water molecule, hydrogen adsorbed directly on Cket by inducing a transition from sp2 to sp3, accompanied by an enhancement of the Cket–Oket distance and a decrease of the Oket–Zn separation. Among the three configurations reported, Fig. 11(a) was the lowest-energy one, followed by Fig. 11(b), higher in energy by 6.2 kcal mol−1, and Fig. 11(c), higher by 32.4 kcal mol−1, respectively. The minor energy differences between configurations Fig. 11(a) and (b) demonstrate that they belong to the same reactive scenario. Interestingly, in all cases, as discussed for the bare surfaces, the reaction of F01 was promoted by the interaction of Oket with a Zn atom of the surface, triggering the opening of the ring in Fig. 11(a) and (b) or Cket hybridization in Fig. 11(c). In conclusion, in the low-density case, epoxide ring opening was foreseen, analogously to the bare case, triggered by the interaction between the fragment and the surface.
 | ||
| Fig. 11 F01 configurations on (0001)Zn-terminated in (a)–(c) low density regime (1 water molecule) and (d), (e) high density regime (5 water molecules). Ball and stick atomic models; color coding: Zn = grey; O = red; C = cyan; N = blue; H = white. Hydrogen bonds: green dashed lines. Possible bonds: black dashed lines. | ||
In the high-density regime (Fig. 11(d)and (e)), we could observe a configuration in which the native structure of F01 was preserved, namely that of Fig. 11(d), whereas in that of Fig. 11(e) the same reaction mechanism of Fig. 11(a) and (b) was noticed. Despite the higher surface coverage, F01 interacted with the surface in Fig. 11(e) configuration via Oket, promoting the ring-opening pathway found for the bare surface at the low-density regime. On the contrary, due to the steric hindrance of the water molecules underneath F01 in Fig. 11(d), the fragment could not reach the surface, preventing ring opening. This latter configuration had a higher energy, by about 84.5 kcal mol−1, than that of Fig. 11(d), suggesting that the reactive pathway is preferred. In sum, in the high-density case, we could observe a reduced reactivity of the epoxide ring due to water passivation. Nevertheless, when in contact with some uncovered Zn, F01 could react with an irreversible alteration of its structure.
Conclusions
In conclusion, based on a joint RMD/DFT multi-scale computational investigation, our study provided valuable insights into the adsorption behavior and the electronic properties of the α′,β′-epoxyketones pharmacophore contained in CFZ with ZnO-based nanocarriers. Our main finding demonstrated that, in case the epoxide moiety of CFZ entered in contact with uncovered Zn atoms of the polar (0001)Zn-terminated oxide substrate, an interaction between the carbonyl group next to the epoxide could trigger a structural change to the drug. More specifically, an opening of the epoxide ring could take place, determining a detrimental effect on the activity of CFZ, mainly for two reasons: (a) the enhancement of the drug-substrate interaction decreases the probability of release of the drug from the NC; (b) although released, the irreversible alteration of the drug structure (due to the epoxide ring opening) prevents its efficacy when interacting with the proteasome in cancer target cells. We have shown that, in a water solution, if CFZ is in contact with uncovered Zn atoms, also water molecules could trigger possible reactions between the drug and the surface, involving H atoms exchange between the solvent and the drug. This latter evidence strongly indicates that, although typical NC surfaces are passivated by a surfactant or solvent species in operating conditions, this does not exclude the possibility of drug structure alteration due to the interaction with the NC. Possible reduced availability of the active drug is consequently envisaged, a factor that should be carefully considered when regulating the drug dose in the preparation of this kind of therapeutic device.The present investigation aims at highlighting the pivotal role that computational modeling could play in synergy with experiments in all the phases of the development of therapeutic devices. Given an active drug, computational modeling can predict the optimal nanocarrier in terms of the nature of the material, dimension, and morphology and the best strategy for efficient entrap, transport, and release to the target sites.
For all these reasons, the present investigation, declined according to a rigorous multi-scale protocol, provides a foundation for future studies in the field of drug delivery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
All the authors acknowledge the MITHoS project (2020MHL8S9) funded by MUR (Ministero Italiano dell'Università e della Ricerca) within the PRIN 2020 program.References
- K. Park, J. Controlled Release, 2014, 190, 3–8 CrossRef CAS PubMed.
- J. Wang, Q. Ni, Y. Wang, Y. Zhang, H. He, D. Gao, X. Ma and X. J. Liang, J. Controlled Release, 2021, 331, 282–295 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- D. Mandal, K. Kushwaha and J. Gupta, OpenNano, 2023, 9, 100112 CrossRef.
- M. X. Zhao and B. J. Zhu, Nanoscale Res. Lett., 2016, 11, 1–9 CrossRef PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- V. Chandrakala, V. Aruna and G. Angajala, Emergent Mater., 2022, 5, 1593–1615 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- A. R. Gomes, C. L. Varela, E. J. Tavares-da-Silva and F. M. F. Roleira, Eur. J. Med. Chem., 2020, 201, 112327 CrossRef CAS PubMed.
- G. Ernouf, I. K. Wilt, S. Zahim and W. M. Wuest, ChemBioChem, 2018, 19, 2448–2452 CrossRef CAS PubMed.
- I. M. Konstantinova, A. S. Tsimokha and A. G. Mittenberg, Int. Rev. Cell. Mol. Biol., 2008, 267, 59–124 Search PubMed.
- G. Perel, J. Bliss and C. M. Thomas, P & T: a Peer-rev. J. Formul. Manage., 2016, 41, 303–307 Search PubMed.
- K. B. Kim and C. M. Crews, Nat. Prod. Rep., 2013, 30, 600–604 RSC.
- G. Kozalak, İ. Bütün, E. Toyran and A. Koşar, Pharmaceuticals, 2023, 16, 111 CrossRef CAS PubMed.
- P. Robak, I. Drozdz, J. Szemraj and T. Robak, Cancer Treat. Rev., 2018, 70, 199–208 CrossRef CAS PubMed.
- P. Makvandi, C. Yu Wang, E. N. Zare, A. Borzacchiello, L. Na Niu and F. R. Tay, Adv. Funct. Mater., 2020, 30, 1910021 CrossRef CAS.
- A. Mishra, D. Pradhan, J. Halder, P. Biswasroy, V. K. Rai, D. Dubey, B. Kar, G. Ghosh and G. Rath, J. Inorg. Biochem., 2022, 237, 111938 CrossRef CAS PubMed.
- A. I. Ribeiro, A. M. Dias and A. Zille, ACS Appl. Nano Mater., 2022, 5, 3030–3064 CrossRef CAS PubMed.
- N. M. Kovalchuk and M. J. H. Simmons, Adv. Colloid Interface Sci., 2023, 312, 102844 CrossRef CAS PubMed.
- H. Cortés, H. Hernández-Parra, S. A. Bernal-Chávez, M. L. Del Prado-Audelo, I. H. Caballero-Florán, F. V. Borbolla-Jiménez, M. González-Torres, J. J. Magaña and G. Leyva-Gómez, Materials, 2021, 14, 3197 CrossRef PubMed.
- C. Trouki, G. Barcaro and S. Monti, Nanoscale, 2022, 14, 13123–13131 RSC.
- S. Anjum, M. Hashim, S. A. Malik, M. Khan, J. M. Lorenzo, B. H. Abbasi and C. Hano, Cancers, 2021, 13, 4570 CrossRef CAS PubMed.
- S. Große Holthaus, S. Köppen, T. Frauenheim and L. C. Ciacchi, J. Chem. Phys., 2014, 140, 234707 CrossRef PubMed.
- I. Schäfer, G. Lasko, T. A. Do, J. Pleiss, U. Weber and S. Schmauder, Comput. Mater. Sci., 2014, 95, 320–327 CrossRef.
- F. Buonocore, C. Arcangeli, F. Gala, G. Zollo and M. Celino, J. Phys. Chem. B, 2015, 119, 11791–11797 CrossRef CAS PubMed.
- S. Singh, B. Singh, P. Sharma, A. Mittal, S. Kumar, G. S. S. Saini, S. K. Tripathi, G. Singh and A. Kaura, Mater. Des., 2017, 134, 10–22 CrossRef CAS.
- H. Hematian, E. Ukraintsev and B. Rezek, ChemPhysChem, 2022, 23, e202100639 CAS.
- M. H. Mohammed and F. H. Hanoon, Comput. Theor. Chem., 2021, 1194, 113079 CrossRef CAS.
- S. C. Mandal and J. Chakrabarti, Phys. Chem. Chem. Phys., 2023, 25, 7805–7814 RSC.
- E. Ukraintsev, H. Hematian and B. Rezek, Langmuir, 2023, 39, 1764–1774 CrossRef CAS PubMed.
- M. Y. Sengul, C. A. Randall and A. C. T. van Duin, ACS Appl. Mater. Interfaces, 2018, 10, 37717–37724 CrossRef CAS PubMed.
- J. Gao and A. V. Teplyakov, J. Catal., 2013, 300, 163–173 CrossRef CAS.
- D. Raymand, A. C. T. van Duin, W. A. Goddard, III, K. Hermansson and D. Spångberg, J. Phys. Chem. C, 2011, 115, 8573–8579 CrossRef CAS.
- Y. K. Gao, F. Traeger, O. Shekhah, H. Idriss and C. Wöll, J. Colloid Interface Sci., 2009, 338, 16–21 CrossRef CAS PubMed.
- S. Irrera, D. Costa and P. Marcus, J. Mol. Struct., 2009, 903, 49–58 CrossRef CAS.
- C. Wöll, Prog. Surf. Sci., 2007, 82, 55–120 CrossRef.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951 RSC.
- J. Goniakowski, F. Finocchi and C. Noguera, Rep. Prog. Phys., 2008, 71, 016501 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.-Condes. Matter, 2009, 21, 395502 CrossRef PubMed.
- A. C. T. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- K. Chenoweth, A. C. T. Van Duin and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS PubMed.
- ReaxFF 2019.102, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com Search PubMed.
- M. M. Islam, B. Diawara, P. Marcus and D. Costa, Catal. Today, 2011, 177, 39–49 CrossRef CAS.
- J. Gao and A. V. Teplyakov, J. Catal., 2013, 300, 163–173 CrossRef CAS.
- M. Iachella, J. Cure, M. Djafari Rouhani, Y. Chabal, C. Rossi and A. Estève, J. Phys. Chem. C, 2018, 122, 21861–21873 CrossRef CAS.
- A. M. Lucero Manzano, J. D. Fuhr, E. D. Cantero, M. Famá, E. A. Sánchez, V. E. Esaulov and O. Grizzi, Appl. Surf. Sci., 2022, 572, 151271 CrossRef CAS.
- H. Li, R. P. H. Mohammad, G. E. Abdol, S. Ahmadi and A. Sarkar, Braz. J. Phys., 2022, 52, 53 CrossRef CAS.
- W. Xu, H. Cao, H. Chen, Y. Yang and A. Sarkar, Struct. Chem., 2021, 32, 63–68 CrossRef CAS.
- M. M. Kadhim, T. Z. Taban, S. A. Abdullaha, N. Alnasoud, S. K. Hachim and S. Alomar, J. Mol. Model., 2023, 29, 47 CrossRef CAS PubMed.
- V. Balachandran, G. Mahalakshmi, A. Lakshmi and A. Janaki, Spectrochim. Acta, Part A, 2012, 97, 1101–1110 CrossRef CAS PubMed.
- F. Moschona, I. Savvopoulou, M. Tsitopoulou, D. Tataraki and G. Rassias, Catalysts, 2020, 10, 1117 CrossRef CAS.
- C. Lauret, Tetrahedron: Asymmetry, 2001, 12, 2359–2383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: details of the geometric parameters considered in the analysis of the MD simulations. See DOI: https://doi.org/10.1039/d3tb00902e |
| This journal is © The Royal Society of Chemistry 2023 |