
Encapsulin cargo loading: progress and potential
Jesse A.
Jones
 a,
Robert
Benisch
b and
Tobias W.
Giessen
*ab
a,
Robert
Benisch
b and
Tobias W.
Giessen
*ab
aDepartment of Biological Chemistry, University of Michigan Medical School, Ann Arbor, MI, USA. E-mail: tgiessen@umich.edu
bProgram in Chemical Biology, University of Michigan, Ann Arbor, MI, USA
First published on 2nd May 2023
Abstract
Encapsulins are a recently discovered class of prokaryotic self-assembling icosahedral protein nanocompartments measuring between 24 and 42 nm in diameter, capable of selectively encapsulating dedicated cargo proteins in vivo. They have been classified into four families based on sequence identity and operon structure, and thousands of encapsulin systems have recently been computationally identified across a wide range of bacterial and archaeal phyla. Cargo encapsulation is mediated by the presence of specific targeting motifs found in all native cargo proteins that interact with the interior surface of the encapsulin shell during self-assembly. Short C-terminal targeting peptides (TPs) are well documented in Family 1 encapsulins, while more recently, larger N-terminal targeting domains (TDs) have been discovered in Family 2. The modular nature of TPs and their facile genetic fusion to non-native cargo proteins of interest has made cargo encapsulation, both in vivo and in vitro, readily exploitable and has therefore resulted in a range of rationally engineered nano-compartmentalization systems. This review summarizes current knowledge on cargo protein encapsulation within encapsulins and highlights select studies that utilize TP fusions to non-native cargo in creative and useful ways.
Introduction
All cells employ different methods of regulating their metabolism in time and space.1 Doing so allows compartmentalization and dynamic spatial control of otherwise incompatible chemical reactions or metabolic pathways.1–3 As opposed to the lipid-based organelles used by eukaryotes, prokaryotes mainly employ various protein-based modalities to establish and maintain discrete subcellular compartmental-ization.1–5 These strategies range from small homo-multimeric protein cages natively lacking sequestered protein cargo, to more complex, multi-component shell systems capable of housing multiple functional enzymes. A unifying theme for all prokaryotic protein compartments and cages is their ability to self-assemble and create sequestered spaces within cells. Through various biological and chemical engineering strategies, diverse nano-sized protein cages have been repurposed as programmable molecular containers for use in biocatalysis, biomedicine, and bionanotechnology.6,7Examples of prokaryotic protein cages include the small 8–12 nm ferritins, hollow shells comprised of identical protein subunits with the shell serving as a diffusion barrier and ferroxidase at the same time, allowing effective iron storage within, without the need to sequester proteinaceous cargo.8,9 In comparison, the much larger 40–200 nm bacterial microcompartments (BMCs) consist of multi-component cages sequestering multiple enzymes that act together to yield a complex metabolic organelle-like compartment.4,10 The more recently discovered encapsulin nanocompartments occupy the space between these two examples with respect to size, complexity, and ability to encapsulate cargo proteins, and will be the focus of this review.11–13
Encapsulins are icosahedral protein nanocages that range from 24 to 42 nm in size with varying triangulation numbers (T1, T3, or T4) formed via self-assembly of 60–240 subunits of the same shell protein exhibiting the HK97 (Hong Kong 97) phage-like fold (Fig. 1A–C).13–15 Notably, the eponymous feature of encapsulins is their ability to encapsulate specific cargo proteins during shell self-assembly using selective cargo loading mechanisms based on targeting domains (TDs) or targeting peptides (TPs) present at the N- or C-terminus of each cargo protein. This native, efficient, and modular cargo loading modality makes encapsulins excellent protein cargo carriers with potential broad applications as targeted drug delivery vehicles, vaccine platforms, and bionanoreactors, among others.14,16–22 Recent genome datamining studies have led to the grouping of encapsulins into four separate families that vary in sequence, operon configuration, overall structure, and encapsulation mechanism.23–25 Family 1 encapsulins are the most extensively studied, with experimental information available for multiple systems based on their shell structures, associated cargo function, and respective cargo loading process. Similar studies pertaining to Family 2 encapsulins have recently begun to emerge, though these studies remain nascent in comparison to the data available for Family 1 encapsulins. Family 3 and Family 4 encapsulins remain putative and currently lack experimental validation.
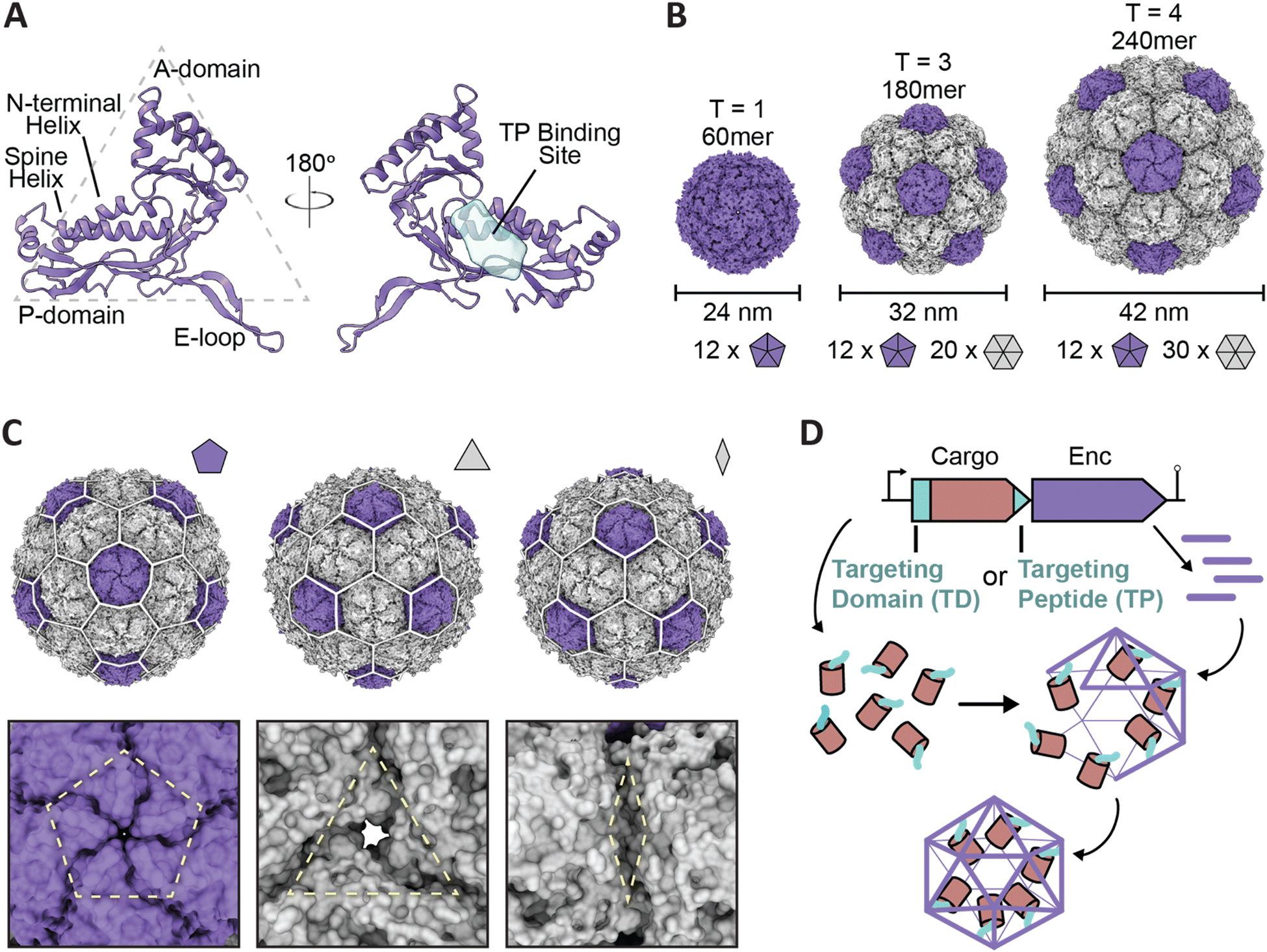 | ||
| Fig. 1 Overview of encapsulin nanocompartment structure and assembly. (A) An encapsulin shell monomer from the Thermotoga maritima encapsulin system in ribbon representation (purple; PDB: 3DKT). Left: exterior view. Right: interior view (180° rotated). The interior binding site of the Family 1 encapsulin targeting peptide outlined in light blue. (B) Exterior views of the T = 1 encapsulin from T. maritima (left; PDB: 3DKT), the T = 3 encapsulin from Myxococcus xanthus (center; PDB: 4PT2), and the T = 4 encapsulin from Quasibacillus thermotolerans (right; PDB: 6NJ8) highlighting the different sizes and assembly states of encapsulins. The number of pentameric and hexameric facets that make up the shell are shown at the bottom. (C) Schematic of the Q. thermotolerans encapsulin with a T = 4 icosahedral cage overlay highlighting the respective five-fold (left), three-fold (center), and two-fold (right) symmetry axes and pores, with respective magnified views below. (D) Schematic diagram of Family 1 and Family 2 core operon layouts (top) featuring the cargo (pink), respective targeting moieties (turquoise), and encapsulin shell (purple); note Family 1 and 2 cargo genes are found up- and downstream of the encapsulin gene, respectively. For simplicity, only the upstream operon organization is shown. Figures created using ChimeraX (Goddard et al., 2018). PDB, protein data bank; TD, Family 2 targeting domain; TP, Family 1 targeting peptide. | ||
As experimental details only exist for Family 1 and Family 2 encapsulins, this review will focus on the current understanding and use of encapsulin cargo loading of these two families. The native in vivo mechanisms involved in cargo loading as well as efforts undertaken to date to manipulate those mechanisms will be discussed. This review will also detail the practical application of encapsulin cargo loading as it pertains to recent bioengineering efforts as well as recent studies dissecting the mechanism of cargo encapsulation. Lastly, this review will discuss potential future challenges and directions, including prospective studies that may help further elucidate or manipulate encapsulin cargo loading, as well as the future potential that such rational manipulation of encapsulin cargo loading may hold for biocatalysis, biomedicine, and biomaterials research.
Family 1 encapsulins – targeting peptides and cargo loading
As Family 1 encapsulins (Pfam ID: PF04454) were the first discovered family, most of the published encapsulin-based research has been focused on them. Originally identified in the 1990s as high-molecular-weight aggregates found in the supernatant of Brevibacterium linens M18, the first encapsulin was initially misidentified as a possible antibacterial bacteriocin dubbed linocin M18.26 Additional homologs were soon discovered in Mycobacterium species and Thermotoga maritima, some of which were thought to display proteolytic activity.27,28 However, additional studies in the 2000s resulted in researchers being unable to replicate the previously perceived proteolytic effects while concurrent structural studies proved encapsulins to be homomultimeric, self-assembling capsid-like nanocompartments sequestering enzymes in their interior.13,29–31Family 1 encapsulins are mainly found in the bacterial phyla Actinobacteria, Proteobacteria, and Firmicutes, and are sorted into several operon types according to their native enzyme cargo.23 The operon arrangement for Family 1 encapsulins generally follows a layout comprised of an upstream gene encoding for the respective cargo protein followed by the gene encoding for the encapsulin shell protein, with or without flanking co-regulated accessory proteins (Fig. 1D).23,32 In order for cargo encapsulation to occur, a targeting peptide (TP), sometimes also referred to as a cargo-loading peptide (CLP), is strictly necessary and is found at the C-terminus of all cargo proteins (Fig. 1D). TPs are usually separated from the catalytically active folded domain of the cargo by a flexible linker with high glycine and proline content of ca. 10–50 residues in length (Fig. 2A). This arrangement likely minimizes steric clashes between adjacent cargo proteins within the shell, thus maximizing cargo loading capacity. A corollary of this feature is that cargo proteins are generally poorly resolved in encapsulin structures due to their high mobility caused by being flexibly tethered to the shell interior. Cargo protein loading is not necessary for shell assembly. Encapsulin shells generally self-assemble very efficiently even in the absence of any cargo. This implies a cargo loading mechanism where co-expression of cargo and shell – as insured by a tight operon structure – allows efficient TP–shell interactions during shell self-assembly.
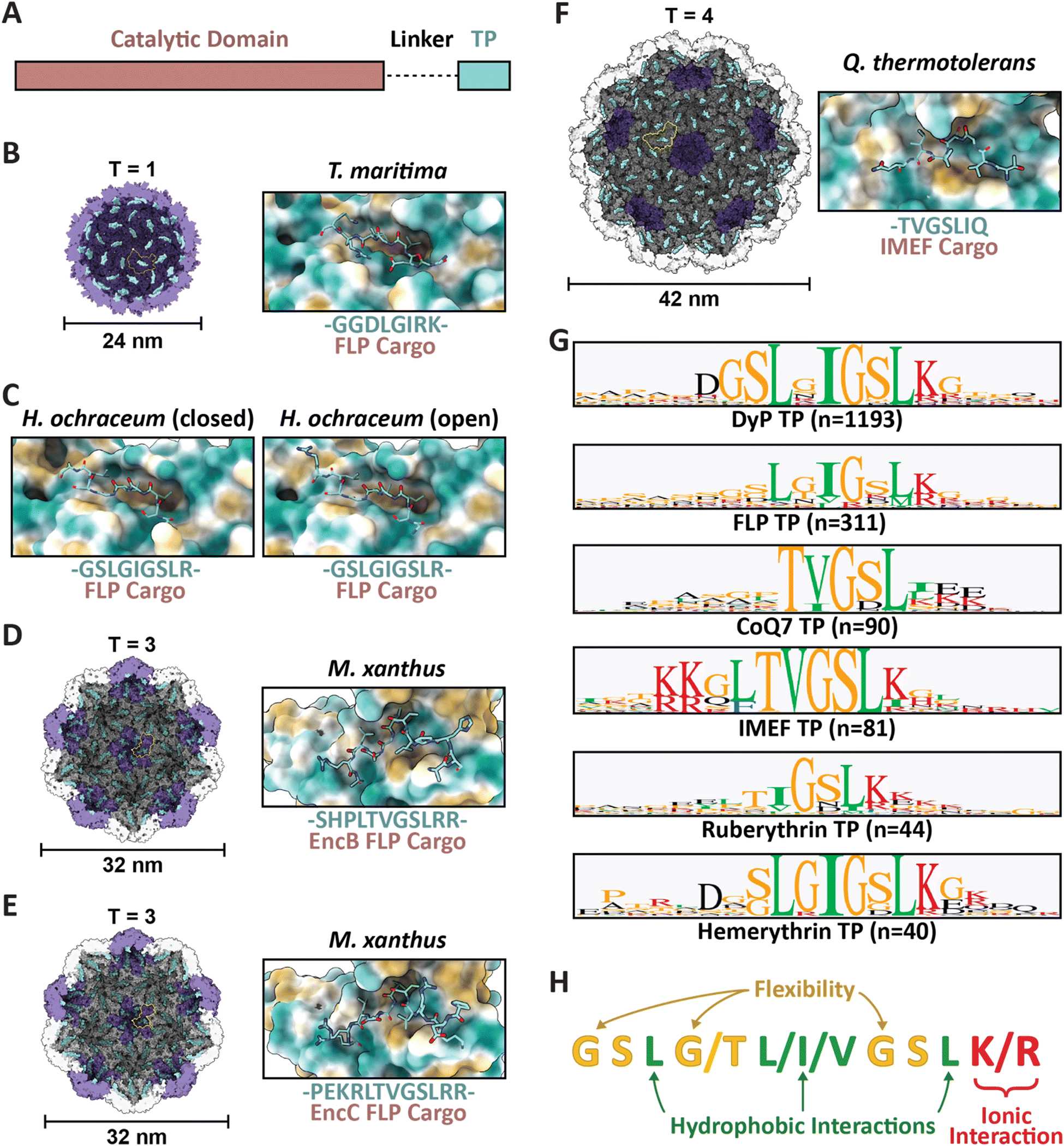 | ||
| Fig. 2 Family 1 cargo loading is mediated by specific TP–shell interactions. (A) Schematic representation of Family 1 cargo components, including the catalytic domain (pink), proline- and glycine-rich flexible linker (dash), and targeting peptide (turquoise). (B) Cutaway view of the T. maritima T1 encapsulin shell (PDB: 3DKT) with one shell protein subunit highlighted (yellow) and the encapsulin (purple) and GGDLGIRK TP of the FLP cargo (turquoise) shown in surface representation (left) and zoomed-in view of the conserved binding pocket (hydrophobic representation) with the resolved residues of the bound TP shown in stick representation (turquoise; right). (C) Zoomed-in view of the H. ochraceum T1 encapsulin TP–shell interaction (PDB: 7OE2) highlighting the binding pocket and the GSLGIGSLR TP sequence of the FLP cargo as found in the closed (left) and open (right) pentamer conformations of the shell. (D) Cutaway view of the M. xanthus T3 encapsulin shell (PDB: 7S2T) with one shell protein subunit highlighted (yellow) and the SHPLTVGSLRR TP (turquoise) of the EncB FLP cargo shown in surface representation (left). A zoomed-in view of the TP–shell interaction is shown on the right. (E) Similar overview of the M. xanthus T3 shell interaction with the PEKRLTVGSLRR TP of the EncC FLP cargo (PDB: 7S4Q). (F) Analogous overview of the Q. thermotolerans T4 shell interaction with the TVGSLIQ TP of the IMEF cargo (PDB: 6NJ8). (G) Consensus sequences for TPs from each of the major Family 1 cargo classes after alignment via Clustal Omega 1.2.3 with 20 residues centred on the consensus peak or, when limited by sequence length, using the last 20 C-terminal residues; visualized using GraphPad Prism v9.0.2; n, number of cargo sequences used. (H) Schematic of general binding mode for Family 1 TPs. Figures created using ChimeraX (Goddard et al., 2018). TP, targeting peptide; PDB, protein data bank; FLP, ferritin-like protein; IMEF, iron-mineralizing encapsulin-associated firmicute. | ||
Several examples now exist in the literature providing reliable structural data illustrating TP–shell interactions (Fig. 2). Of these examples, the TPs of two ferritin-like protein (Flp) cargos bound to the interior surface of their respective T1 shells have been resolved – GGDLGIRK in the T. maritima system (Fig. 2B), and GSLGIGSLR in the Haliangium ochraceum system determined in both the “closed” and “open” pentameric conformations based on a shift in the encapsulin A-domain (Fig. 2C).13,33 Further, two Flp TP–shell interactions were resolved for the T3 Myxococcus xanthus system showing the TPs to be SHPLTVGSLRR for the EncB cargo (Fig. 2D) and PEKRLTVGSLRR for the EncC cargo, both found to bind to all available binding sites in pentameric and hexameric shell facets (Fig. 2E).34 Lastly, the TP of an iron-mineralizing encapsulin-associated firmicute (IMEF) cargo protein bound to the hexamers of its native T4 shell from Quasibacillus thermotolerans was determined to be TVGSLIQ (Fig. 2F).14 Based on the cumulative structural data, the TP binding site has been determined to reside on the luminal surface of each Family 1 encapsulin shell protein subunit in a conserved cleft between the N-terminal helix and the P-domain (Fig. 1A and 2). TP lengths range from 7 to 12 residues that rigidly interact with the binding site. In many cargo proteins, additional C-terminal residues, usually less than 10, can be found after the rigidly interacting binding motif. However, they do not seem to be important for TP–shell interaction based on their absence in the structural data available at this time, though further research is warranted. Because the TP binding pocket completely resides within a single shell protein subunit and does not cross subunit boundaries, the maximal cargo loading is set by the total number of shell protein subunits – 60 for T1, 180 for T3, and 240 for T4 shells. However, cargo loading capacity is likely further determined by cargo protein size and oligomerization state. Bioinformatic analyses have provided further evidence that Family 1 TPs are often comprised of 10–20 C-terminal residues containing GSL or double GSL motifs – with exceptions as exemplified by the T. maritima system – often with an immediately subsequent positively charged residue.23,35 Based on structural information and the consensus TP sequences of the main cargo types (Fig. 2G), a general TP binding mode can be derived where two or three hydrophobic residues (isoleucine, leucine, or valine) spaced one or two residues apart – the spacers often containing glycines for flexibility – specifically interact with hydrophobic patches within the binding pocket. In many instances, positively charged residues (lysines or arginines) follow this motif and seem to interact less specifically with negatively charged surface patches of the shell protein (Fig. 2H).
In sum, cargo loading in Family 1 encapsulin systems results from a combination of mass action based on the relative expression levels of cargo and shell proteins, and specific TP-mediated protein–protein interactions with the final number of encapsulated cargo proteins being additionally determined by the relative size of the shell and cargo as well as the cargo oligomerization state.
Family 2 encapsulins – targeting domains and cargo loading
Family 2 encapsulins (Pfam ID: PF19307) can be distinguished from other encapsulin families by sequence similarity, distinct insertion domains within the shell protein, as well as their operon structure.23,36 In Family 2 encapsulin systems, most putative cargo proteins are encoded by genes found immediately downstream of the encapsulin gene. This family is further subdivided into Family 2A and Family 2B, differentiated by the absence (2A) or presence (2B) of an insertion domain, annotated as a cyclic nucleoside monophosphate (cNMP)-binding domain, within the E-loop of the encapsulin shell protein. In contrast to the short C-terminal TPs found in Family 1 encapsulin systems, Family 2 cargo proteins contain long (20–260 residues), unannotated, intrinsically disordered targeting domains (TDs) generally located at their N-terminus (Fig. 3A).23,36,37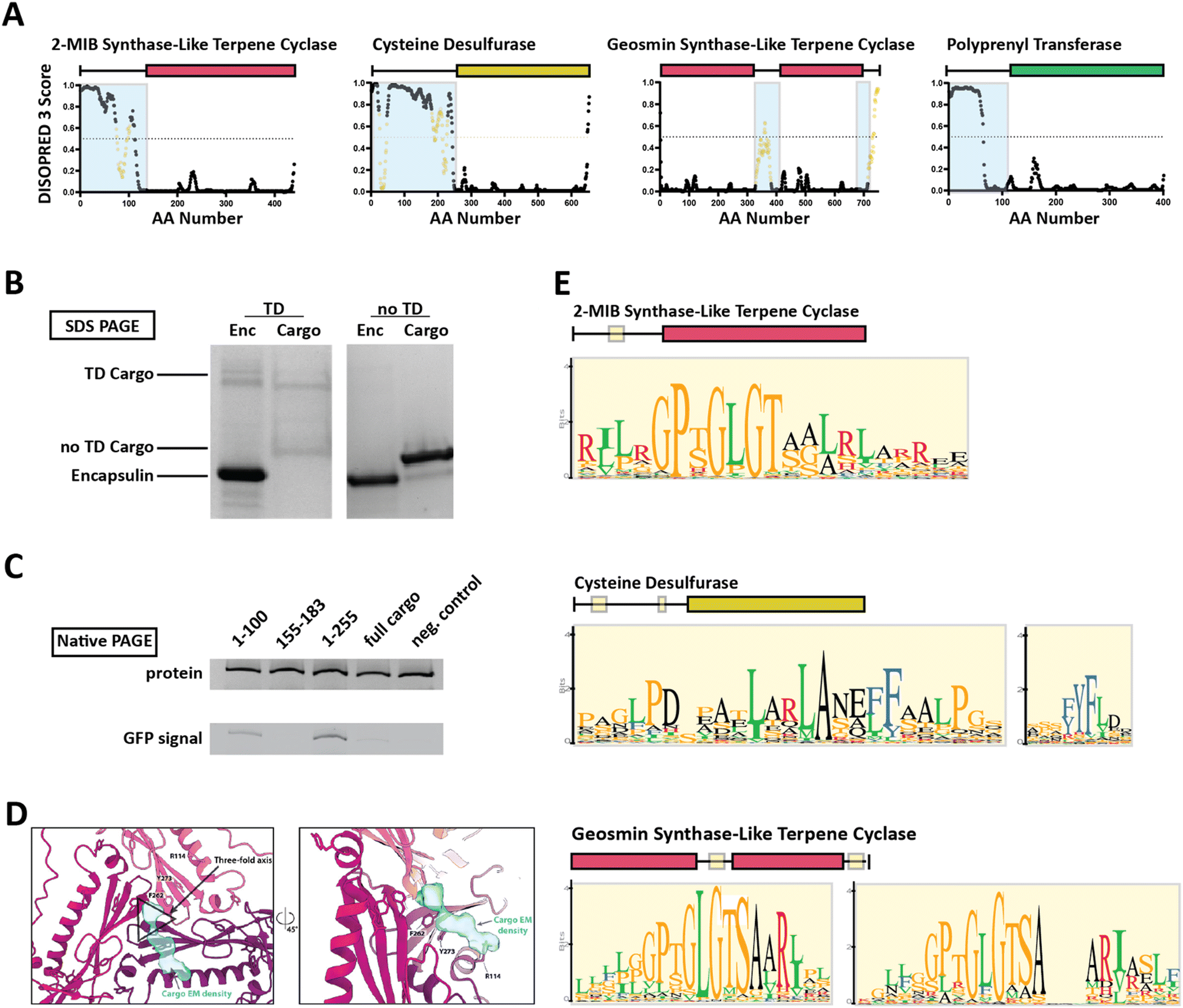 | ||
| Fig. 3 Family 2 encapsulin systems utilize N-terminal targeting domains (TDs) to direct cargo to the interior of the shell. (A) Intrinsic disorder statistics plots generated using DISOPRED3 for four different Family 2 cargo types. Light blue background highlights the disordered regions while positions with relatively high sequence similarity, potentially representing conserved interaction motifs, are shown in yellow. Adapted with changes with open access permission from ref. 23via a creative common license (https://creativecommons.org/licenses/by/4.0/). (B) SDS-PAGE gel of gel-filtration chromatography fractions containing the S. elongatus T1 encapsulin refolded in the presence of desulfurase cargo with and without its native TD. Adapted with changes with open access permission from ref. 36via a creative common license. (C) Native PAGE gel showing Coomassie stain (top) and GFP signal (bottom) of purified S. elongatus encapsulin loaded with GFP reporter fused to different truncations of the N-terminal native TD of the system. Adapted with changes with open access permission from ref. 36via a creative common license. (D) View from the shell interior along the 3-fold symmetry axis (black triangle) of the S. elongatus Family 2 encapsulin (pinks and purple) highlighting additional non-shell density attributed to the native TD (turquoise). Adapted with changes with open access permission from ref. 36via a creative common license. (E) Sequence logos of conserved motifs found within different Family 2 cargo types. Adapted with changes with open access permission from ref. 23via a creative common license. DISOPRED3 outputs were visualized using GraphPad Prism v9.0.2. | ||
So far, little experimental evidence for Family 2 encapsulin systems has been published. However, the N-terminal targeting domain hypothesis has recently been confirmed for one cysteine desulfurase-encapsulating Family 2A system found in Synechococcus elongatus.36 Using in vitro assays, it was shown that the N-terminal 255 residue long domain found in the desulfurase cargo is necessary and sufficient for cargo encapsulation (Fig. 3B and C). Furthermore, structural analysis of the cargo-loaded shell highlighted a resolvable, low-resolution density close to the 3-fold symmetry axis of the shell, suggesting a potential binding region for TDs on the shell interior (Fig. 3D). One caveat of this analysis is the fact that cargo-loading was carried out in vitro using a protein refolding procedure which could have resulted in a non-native mode of cargo encapsulation. Computational analysis of the N-terminal TD within the desulfurase cargo identified 20–30 residue long conserved motifs of high sequence identity (Fig. 3E), separated by long stretches of divergent, mostly hydrophobic residues.23,36 To explore the contributions of each of the conserved motifs to cargo loading, different parts of the TD, containing different combinations of motifs, were N-terminally fused to a fluorescent reporter (GFP) followed by co-expression and purification. The results did not clearly identify a single motif or sub-region sufficient for maximal cargo loading. Instead, it seems that the full-length TD is needed to mediate optimal cargo encapsulation (Fig. 3C). This may have important mechanistic implications for the Family 2 cargo loading process which seems to be quite different compared to Family 1, relying on potentially multiple specific discontiguous interactions based on conserved sequence motifs separated by long flexible and hydrophobic linker regions which themselves might possess affinity for the interior of the encapsulin shell.
For other Family 2 cargo types besides desulfurases, recent bioinformatic analyses have shown similar motif-containing N-terminal domains annotated as mostly disordered.23 So far, one other putative cargo type, a 2-methylisoborneol (2-MIB) synthase with a similarly long, disordered N-terminal domain has been confirmed as a Family 2 cargo protein.38 However, no detailed structural or mechanistic analysis of this system is currently available in the literature.36,38
Of additional note regarding Family 2 encapsulins is the putative existence of two-component shells, so far only bioinformatically predicted, where the Family 2 operon encodes two distinct encapsulin shell genes.23 As experimental data for these systems is currently lacking, it is not yet known how these encapsulins might assemble. However, if the gene products do assemble into functional encapsulin shells with two different types of subunits, there is a possibility that these systems can natively encapsulate defined stoichiometries of two distinct cargo proteins into the same two-component nanocompartment based on specific interactions of two distinct TDs with two distinct shell protein binding sites. If confirmed, such systems may hold significant potential for novel more complex bioengineering applications, beyond what is currently possible with engineered Family 1 systems.
Encapsulin engineering and applications
Protein-based nanocages have gained significant popularity for various engineering applications including for the delivery of therapeutics, as diagnostics, as small molecule and materials nanoreactors, and more.19,20,39–43 As opposed to lipid-based compartments, protein-based cages can be genetically engineered, generally self-assemble into defined 3D architectures, and can be easily chemically functionalized. Therefore, they represent excellent platforms for rational bioengineering. This has inspired a wide array of protein cage engineering using viral capsids, BMCs, ferritins, and de novo designed protein cages.4,21,42–50 Similar efforts at engineering Family 1 encapsulins have recently been undertaken making use of the key advantage of encapsulins, namely, their modular native in vivo cargo loading mechanism. With respect to non-native cargo encapsulation, a protein of interest can be easily genetically functionalized with the respective TP leading to efficient in vivo cargo encapsulation upon co-expression with the cognate encapsulin shell protein.48,51 In addition, in vitro assembly of enzyme-loaded encapsulin nanoreactors is also possible based on in vitro disassembly of the encapsulin shell via exposure to extreme pH or denaturants followed by co-assembly of both shell and separately purified cargo after exchange into physiological buffer, yielding cargo-loaded protein cages.52Potential benefits of encapsulating non-native cargo proteins are abundant and include improving the stability of cargo proteins under harsh conditions like elevated temperature, extreme pH, or exposure to proteases; controlling or improving catalysis; delivering a therapeutic or diagnostic payload; or a combination of the above. Below, we will first highlight efforts towards engineering TPs and modulating their targeting strength, followed by a discussion of select recent studies showcasing the progress made in employing encapsulins as bioengineering tools. Particular emphasis is placed on examples that improve cargo stability, add control over chemical reactions, or show therapeutic or diagnostic application potential (Table 1).
| Achievement | Ref. |
|---|---|
| Use of targeting peptides to encapsulate non-native cargo | 19,20,22,35,36,41,48,51–63 |
| Improved cargo stability | 57–59 |
| Control of chemical reactions | 22,53,59,60,62,64 |
| Therapeutic or diagnostic development | 19,20,53,58,60,65 |
Targeting peptide engineering
So far, the prevailing strategy for encapsulating non-native cargo proteins inside encapsulins has been to genetically fuse the known native TP of a given system to the C-terminus of the cargo of interest. Relatively few reports have tried to change or optimize TP sequences for modulating cargo loading efficiency. To rationally and reliably mutate TPs for controlling cargo loading extent, or relative stoichiometry when encapsulating multiple cargos at once, TP–shell interactions need to be understood in detail. So far, only six TP–shell interactions have been structurally resolved, always of native TPs and their cognate encapsulin shell (Fig. 2). However, rational changes to TP sequences for altering TP–shell binding affinity will likely require further structural analyses of systematically mutated TPs. So far, only a few studies have systematically probed TP–shell interactions using experimental and computational approaches (Fig. 4).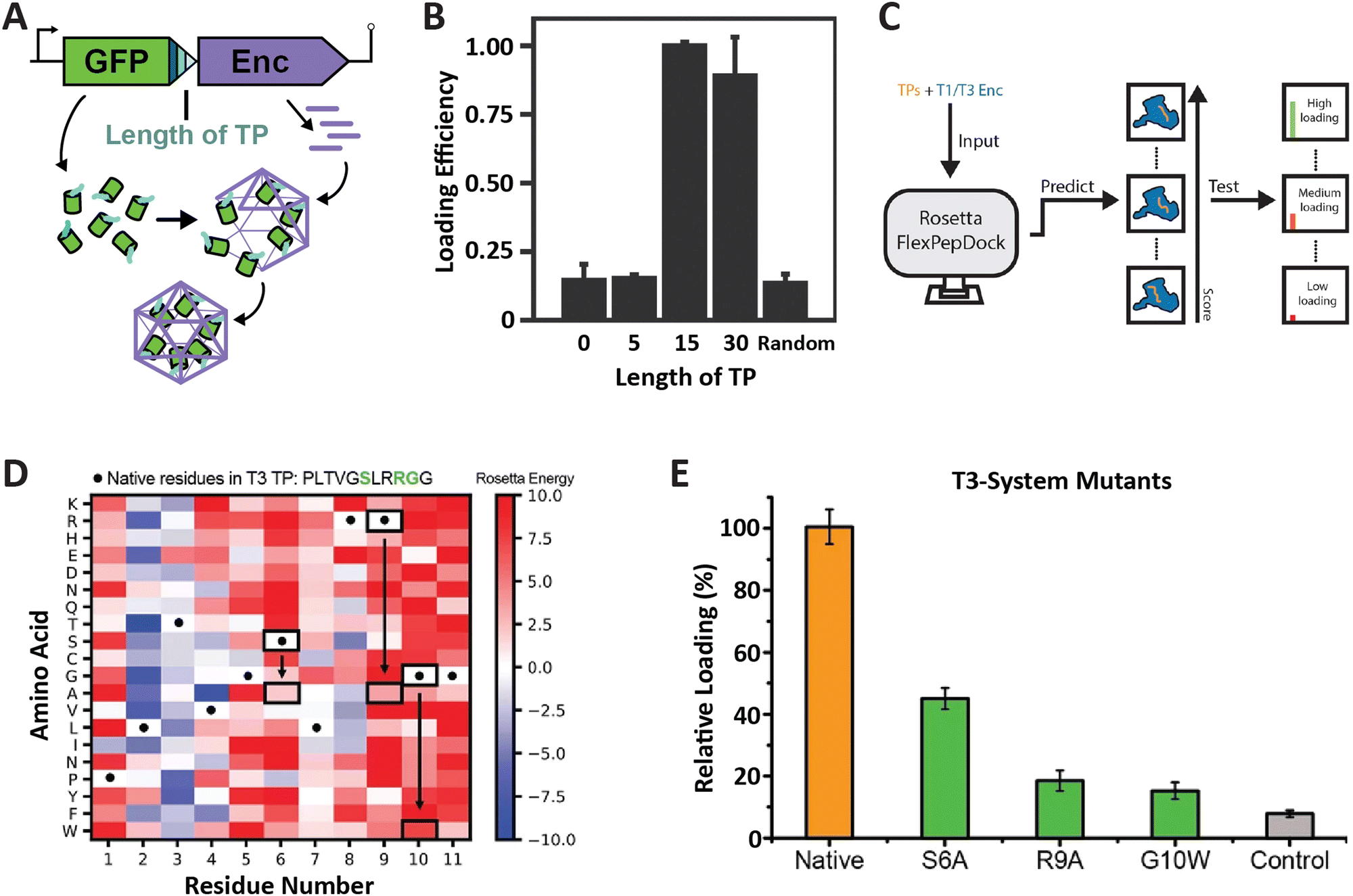 | ||
| Fig. 4 Characterization of T1 and T3 encapsulin targeting peptides. (A) Operon design of TP-fused sfGFP and the corresponding T. maritima encapsulin used for heterologous co-expression and downstream cargo loading analysis. Different TP truncations are highlighted. (B) Comparison of normalized sfGFP fluorescence in purified encapsulins to investigate the influence of TP truncation on cargo loading highlighting that the 15 C-terminal residues are sufficient for maximal cargo encapsulation. (A) and (B) adapted with permission from ref. 35 Copyright 2016 American Chemical Society (ACS). (C) Schematic of computational flexible docking and experimental workflow used to predict and analyze the relative strength of TP–shell binding in single residue TP mutants. (D) Heat map of computational point mutations with the color gradient representing the Rosetta Energy score (blue, improved binding; red, worse binding). (E) Experimental analysis of cargo loading for the three TP mutants highlighted in green in panel (D) highlighting that most single residue substitutions lead to decreased cargo encapsulation. (C)–(E) adapted with changes with open access permission from ref. 54via a creative common license (https://creativecommons.org/licenses/by/4.0/). sfGFP, super folder green fluorescent protein. | ||
One study utilized the T1 encapsulin from T. maritima and its native TP fused to a fluorescent reporter (sfGFP) in order to assess the minimal TP needed to attain maximal cargo loading (Fig. 4A and B).35 Different truncations of the C-terminal 30 residues found in the native T. maritima cargo protein were appended to the reporter. Bulk fluorescence after purification was used as a readout of cargo loading extent. Results showed that the 15 C-terminal residues of the native cargo were sufficient for optimal cargo loading. These include the 8 rigidly bound residues that could be structurally resolved (Fig. 2). It is likely that only these residues are needed for binding. In a similar study focused on the T1 encapsulin from Mycobacterium smegmatis, different truncations of the 19 C-terminal residues found in the native cargo were appended to an eGFP reporter.53 The results indicated that the 12 C-terminal residues were required to attain maximal cargo loading levels. Even though no structural information for the M. smegmatis TP–shell interaction is available, the 12 C-terminal residues contain a double GSL motif confirming its importance for cargo encapsulation.
In another recent study, a combined computational and experimental approach was taken to probe the influence of single residue substitutions within the TPs of the T1 T. maritima and T3 M. xanthus encapsulin systems.54 Rosetta-based force-field modelling was employed to predict the influence of mutations within the two TPs (Fig. 4C). Select TP mutants were then experimentally characterized. Computational prediction and experiment were found to generally agree. This approach led to further interesting insights, including the fact that most mutations were computationally predicted to have a negative effect on binding strength, in particular, the mostly conserved GSL-like motifs (Fig. 4D and E). The cumulative results of these studies highlight the fact that TPs vary with respect to native specificity and binding strength, and that TP–shell binding is significantly influenced by specific hydrophobic and ionic interactions, as well as TP flexibility.
Improved cargo stability under harsh conditions
Improved stability of cargo proteins upon encapsulation is an often-observed phenomenon in encapsulin systems and encompasses thermal stability, increased catalytic lifetime of sequestered enzymes, and protease resistance (Fig. 5A). Increased thermal stability is generally found in encapsulins originating from thermotolerant or thermophilic organisms. For example, encapsulation of IMEF cargo inside its native Q. thermotolerans T4 shell increased its melting temperature by 10 °C.14 This improved thermal stability is not only seen for native cargo proteins, but also when non-native cargo is encapsulated. Multiple non-native enzymes showed prolonged catalytic activity at elevated temperatures when encapsulated inside the T1 protein shell of the thermophile M. hassiacum. This included an encapsulated dye-decolorizing peroxidase (DyP) cargo which was active at 40 °C for 25 hours while the free DyP lost activity at that temperature after 30 minutes.57 Additionally, the carbohydrate oxidase mChitO lost activity after 90 minutes at 50 °C whereas when encapsulated, mChitO still retained 50% activity after four hours. The same enzymes in their encapsulated forms also exhibited prolonged resistance against digestion with proteinase K. Similarly, another recent study showed that protein cargo encapsulated within a T1 encapsulin from the acidophile Acidipropionibacterium acidipropionici was protected from pepsin digest at pH 3 as well as trypsin and chymotrypsin degradation at pH 7.5. In general, the encapsulin shell is able to protect cargo from proteolytic degradation due to the physical sequestration of the cargo within a stable protein shell which is itself often highly protease resistant. The often observed increased thermal stability of encapsulated cargo might be a result of high local cargo molarity, tethering, and confinement within the tight encapsulin shell that leads to a favorable molecular crowding effect, where protein–protein interactions are optimized to prevent complete cargo unfolding, multimer dissociation, or aggregation and encourage rapid protein re-folding or re-association when partial denaturation or dissociation occurs.57,66,67 A similar mechanism might be responsible for the increased catalytic lifetime reported for many encapsulated enzymes.30,36 Increased stability of proteins of interest upon encapsulation could be exploited for numerous applications, such as producing encapsulated biocatalysts that exhibit long-term stability under harsh conditions or acting as robust delivery vehicles for biological therapeutic or diagnostic proteins.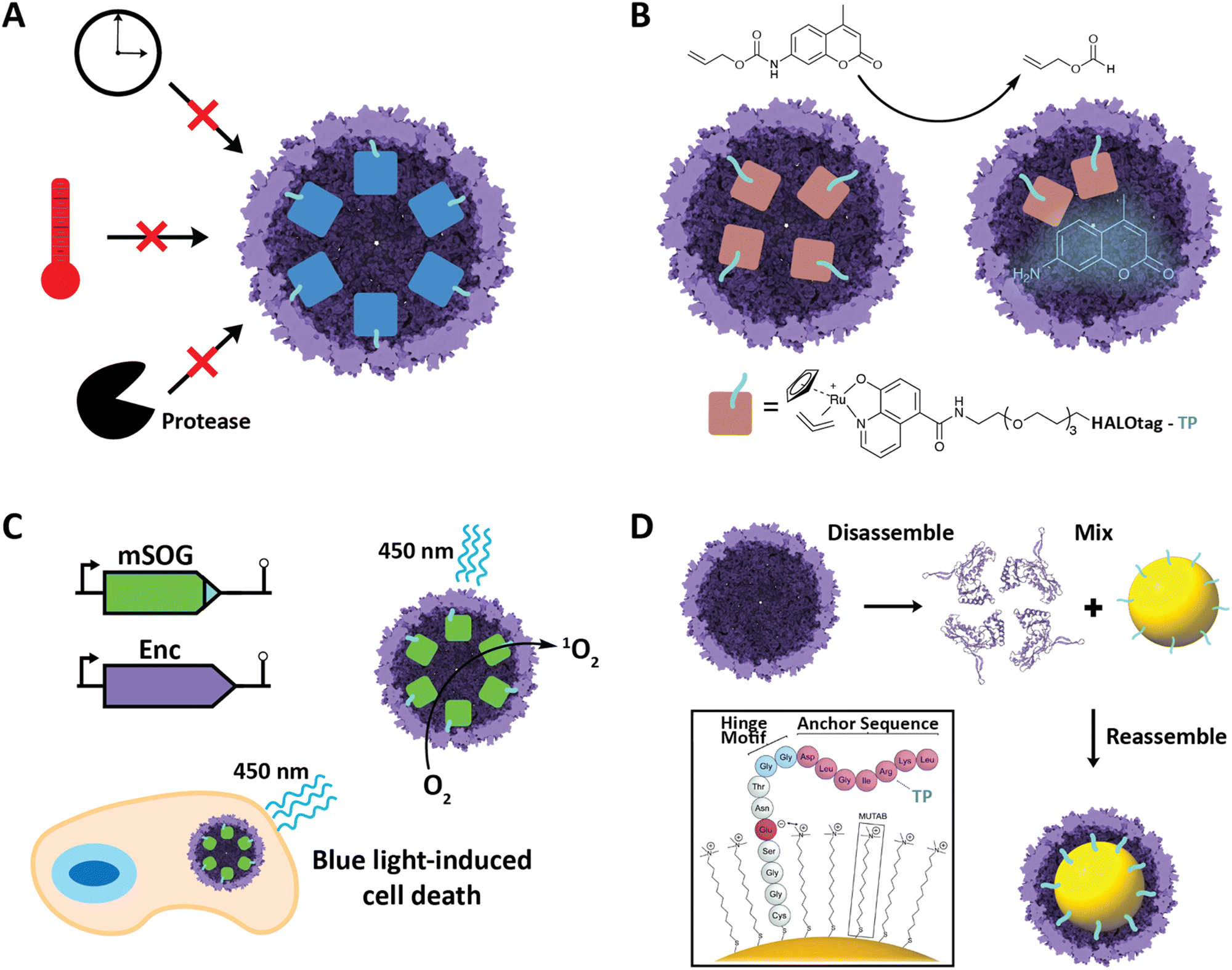 | ||
| Fig. 5 Select engineering applications of encapsulins. (A) Cargo encapsulation within encapsulin protein shells can have a variety of beneficial effects, including increased cargo stability over time, increased thermal stability, and increased resistance against proteases. (B) Encapsulation of a ruthenium-based metal organocatalyst based on a covalently modifiable HaloTag yielding a system able to catalyze de-N-allylation of the shown pro-fluorophore both in vitro and in vivo. Adapted with changes with open access permission from ref. 53via a creative common license (https://creativecommons.org/licenses/by/4.0/). (C) In vivo encapsulation of a light-controllable minimal singlet oxygen generator (mSOG) able to generate large amounts of toxic ROS via singlet oxygen species inside mammalian cancer cells resulting in cell death. Adapted with permission from ref. 58 Copyright 2021 American Chemical Society (ACS). (D) In vitro assembly of gold nanoparticles encapsulated within an encapsulin protein shell using a synthetically modified TP. Adapted from ref. 52 with permission from the Royal Society of Chemistry. Copyright 2018 The Royal Society of Chemistry. | ||
Encapsulation of metal catalysts and control of chemical reactions
Encapsulins have recently been employed to create metal catalyst-loaded nanocages for biocatalysis applications. An encapsulin-based nanoreactor containing the organometallic ruthenium catalyst [CpRu(HQ)(allyl)]PF6 was assembled by encapsulating a proteinaceous TP-fused HaloTag in vivo which was then purified and covalently modified with the ruthenium catalyst in vitro via a specific chloroalkane spacer.53 The resulting encapsulated catalyst was capable of de-N-allylation of a coumarin pro-fluorophore with a yield and kinetics similar to that of free PEGylated catalyst (Fig. 5B). The nanoreactor was also shown to be active in cultured mammalian cells. This successfully demonstrates the use of engineered encapsulins to conduct bio-orthogonal transition-metal catalysis in live cells. Furthermore, it demonstrates the possibility of using encapsulins for the delivery of catalysts that convert pro-drugs to their bioactive forms in situ. This is especially noteworthy as it opens up the possibility of delivering catalysts with low biocompatibility in an encapsulated, biocompatible, and active form. Other secondary tags besides HaloTag have recently been employed for similar purposes, including simple avidin tags. In general, this approach is promising for creating modular nanoreactors containing potentially multiple co-localized transition metal organocatalysts and enzymes in the same compartment leading to novel chemoenzymatic nanoreactor capabilities.Engineered encapsulins as therapeutics and diagnostics
Nanoscale encapsulation systems – including protein nanocages – have long been used as engineering platforms for nano-medicine applications.43,68–72 Different platform types, e.g., lipid- vs. protein-based compartments, offer different advantages and disadvantages, including accessible size range, stability, and ease of functionalization.73 As any therapeutics or diagnostics delivery system is required to be biocompatible, large enough to carry a cargo of interest, and targetable to a specific biological site, encapsulins in particular have recently shown significant promise.20,39,74,75A recent innovative study involved the use of non-UV light to control the production of toxic reactive oxygen species (ROS) inside cancer cells.58 A TP was genetically fused to a minimal singlet oxygen generator (mSOG), a flavoprotein that produces mainly singlet oxygen ROS upon exposure to blue light. The TP-fused mSOG was then encapsulated inside the T. maritima T1 encapsulin shell via co-expression yielding a nanocage-based platform for the delivery of photodynamic therapeutics (Fig. 5C). Encapsulated mSOG was shown to generate higher ROS levels than either the free mSOG or the encapsulin control due to the encapsulin's additive O2 generative effect when combined with the free mSOG, which in turn is likely due to the observed non-specific adsorption of endogenous flavin molecules such as flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and riboflavin to the encapsulin from T. maritima.52,56 Furthermore, the use of encapsulated mSOG led to increased cell death in a lung cancer cell culture model, being attributed to the fact that encapsulated mSOG was shown to be taken up by cells while free mSOG was not internalized, which was consistent with previous reports that show free mSOG to be incapable of penetrating tumor cells.76 This system provides a novel, highly controllable method for the light-triggered generation of toxic ROS without the use of potentially harmful UV light or the need for additional small-molecule substrates. As ROS generation can be used as both a therapeutic modality and effective bioimaging signal, the platform represents a theranostic encapsulin-based delivery system.
This mSOG encapsulin platform was further engineered to display the Designed Ankyrin repeat protein (DARPin), an antibody mimic, on the exterior of the shell through genetic fusion of DARPin to the surface-exposed C-terminus of the encapsulin shell protein.19 This enabled the targeted delivery of mSOG to Human Epidermal growth factor Receptor 2 (HER2)-positive breast cancer cells and the subsequent light-triggered induction of toxic ROS leading to apoptosis. This platform self-assembles in a single step when expressed in Escherichia coli and offers specific targeting, photodynamic therapeutic ROS generation, and potential modular functionalization with different DARPin molecules selected to specifically bind to other targets.
Another innovative example of using encapsulins as reporters or diagnostic platforms was based on the in vivo encapsulation of the enzyme tyrosinase, able to polymerize melanin inside the encapsulin protein shell.60 The sequestered and concentrated melanin could then be used for imaging and tracking purposes due to its strong near-infrared absorption. Further, cells expressing this system did not show any growth defect based on melanin toxicity which is usually observed for non-encapsulated melanin.
Production of inorganic materials using encapsulins
Encapsulins have recently been used to create inorganic–organic biomaterials platforms. To introduce non-organic cargo into encapsulin shells, a recent study showed that TP-functionalized inorganic nanoparticles could be encapsulated by the T. maritima T1 encapsulin by following an in vitro co-assembly protocol.52 Gold nanoparticles (AuNPs) were surface-modified with TP-containing synthetic peptides consisting of an N-terminal cysteine residue for direct binding to the gold surface, followed by four residues ending in a glutamate able to electrostatically interact with the positive charge of an (11-mercaptoundecyl)-N,N,N-trimethylammonium bromide (MUTAB) shell around the AuNP, a short four-residue hinge motif ending in a flexible double glycine, and culminating in the seven residue native T. maritima TP. This allowed AuNPs to be encapsulated following denaturation of the encapsulin shell protein at pH 2, mixing of the functionalized TP-containing AuNPs, and refolding and reassembly of the encapsulin shell at pH 7 (Fig. 5D). The process was proven to be extremely efficient with 99% AuNP encapsulation as confirmed by TEM. This approach may be useful in the creation of protein-coated inorganic nanoparticles for use in antimicrobials and anti-cancer applications.Engineered encapsulins as catalytic enzyme nanoreactors
Engineered encapsulin-based nanoreactors aim to emulate the advantages observed for naturally-occurring protein organelles and compartments.77–79 These include the ability to co-localize multiple enzymes which can improve intermediate channeling and pathway flux; increasing the local molarities of enzymes, substrates, and intermediates; preventing unwanted side reactions and toxic or reactive intermediate leakage; and generally improving the stability and performance of a sequestered enzymatic process.24,80 Recent progress in utilizing encapsulins as enzyme nanoreactors include the encapsulation of the pyruvate dehydrogenase Aro10p in yeast to produce and protect an intermediate of the high-value opioid precursor norcoclaurine.59 When TP-fused Aro10p was encapsulated inside the Myxococcus xanthus T3 encapsulin shell, the Aro10p-generated reactive intermediate 4-hydroxyphenylacetaldehyde (4-HPPA) was protected from endogenous detoxification enzymes and could undergo spontaneous reaction with dopamine yielding norcoclaurine, whereas overexpression of free Aro10p did not lead to detectable levels of the opioid precursor.Another recent example of employing encapsulins as biocatalytic enzyme nanoreactors is the use of the DyP-peroxidase-loaded M. hassiacum T1 encapsulin together with free eugenol oxidase in an enzyme cascade, yielding lignin-like crosslinked reaction products.57 The main challenge for all protein-based nanoreactors encapsulating non-native enzymes is to overcome the often observed decrease in catalytic activity upon enzyme encapsulation.57 This is generally due to the protein shell not being optimized for the influx of the particular substrates and cofactors needed by a specific non-native sequestered enzyme. Future efforts aimed at pore engineering to improve molecular flux across encapsulin shells will likely be able to address this problem and result in fully catalytically active nanoreactors.22,63,81,82
Conclusions and future challenges
Encapsulins offer advantages relative to other nanocages used in bioengineering, including their exclusively proteinaceous nature, biophysical robustness, genetic engineerability, and facile in vivo cargo loading that negates the need for additional methods like cargo-scaffold or cargo-capsid genetic fusions, covalent conjugation, or harsh refolding procedures.7,40,83,84 Encapsulin research has made substantial progress over the past decade, generating novel insights into shell structure and dynamics, cargo encapsulation mechanisms, biological function, and engineering applications.24 This progress has been almost exclusively confined to Family 1 systems. However, recent bioinformatic analyses have unveiled thousands of novel encapsulin systems across numerous bacterial and archaeal phyla.23 Many of these systems likely contain novel useful features for future bioengineering and synthetic biology applications including dynamic and controllable pores, shell insertion domains, and two-component protein shells.38 Recent projects aimed at engineering encapsulins have highlighted a range of promising application areas, many of which have been highlighted in this review. However, some of these studies have also revealed a number of outstanding challenges to be overcome, including limitations associated with the various techniques used for determining cargo loading, such as the potential imprecision of gel densitometry, the difficulty of deconvoluting shell and cargo UV/Vis signals, and the cost and nascence of mass photometry.85,86 Notably, recent studies have also highlighted the current insufficient understanding of the cargo encapsulation mechanism which at the moment prevents the rational design of stoichiometrically defined cargo-loaded encapsulins. Therefore, systematic studies probing the effects of TP mutations on cargo loading and TP binding mode should be prioritized in future studies, along with elucidating the mechanistic details of pore dynamics and molecular flux across the protein shell, and investigating potential two-component shell assembly, all potentially highly useful features for various engineering applications. We envision encapsulins as a modular and robust platform technology that, once their basic biophysical and biochemical characteristics are thoroughly understood, will find many applications in biomedicine, biocatalysis, biomaterials research, and bionanotechnology.Author contributions
Jesse A. Jones, Robert Benisch, and Tobias W. Giessen conceived and wrote the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the NIH (R35GM133325). Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.References
- E. Cornejo, N. Abreu and A. Komeili, Curr. Opin. Cell Biol., 2014, 26, 132–138 CrossRef CAS PubMed.
- Y. Diekmann and J. B. Pereira-Leal, Biochem. J., 2013, 449, 319–331 CrossRef CAS PubMed.
- T. Gabaldón and A. A. Pittis, Biochimie, 2015, 119, 262–268 CrossRef PubMed.
- C. Aussignargues, M. E. Pandelia, M. Sutter, J. S. Plegaria, J. Zarzycki, A. Turmo, J. Huang, D. C. Ducat, E. L. Hegg, B. R. Gibney and C. A. Kerfeld, J. Am. Chem. Soc., 2016, 138, 5262–5270 CrossRef CAS PubMed.
- C. A. Kerfeld, S. Heinhorst and G. C. Cannon, Annu. Rev. Microbiol., 2010, 64, 391–408 CrossRef CAS PubMed.
- T. G. W. Edwardson, M. D. Levasseur, S. Tetter, A. Steinauer, M. Hori and D. Hilvert, Chem. Rev., 2022, 122, 9145–9197 CrossRef CAS PubMed.
- J. A. Jones and T. W. Giessen, Biotechnol. Bioeng., 2020, 118, 491–505 CrossRef PubMed.
- M. Rivera, Acc. Chem. Res., 2017, 50, 331–340 CrossRef CAS PubMed.
- K. Honarmand Ebrahimi, P. L. Hagedoorn and W. R. Hagen, Chem. Rev., 2015, 115, 295–326 CrossRef CAS PubMed.
- C. A. Kerfeld, M. R. Sawaya, S. Tanaka, C. V. Nguyen, M. Phillips, M. Beeby and T. O. Yeates, Science, 2005, 309, 936–938 CrossRef CAS PubMed.
- T. W. Giessen, Curr. Opin. Chem. Biol., 2016, 34, 1–10 CrossRef CAS PubMed.
- C. A. McHugh, J. Fontana, D. Nemecek, N. Cheng, A. A. Aksyuk, J. B. Heymann, D. C. Winkler, A. S. Lam, J. S. Wall, A. C. Steven and E. Hoiczyk, EMBO J., 2014, 33, 1896–1911 CrossRef CAS PubMed.
- M. Sutter, D. Boehringer, S. Gutmann, S. Günther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban and N. Ban, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CrossRef CAS PubMed.
- T. W. Giessen, B. J. Orlando, A. A. Verdegaal, M. G. Chambers, J. Gardener, D. C. Bell, G. Birrane, M. Liao and P. A. Silver, eLife, 2019, 8, e46070 CrossRef PubMed.
- D. J. Tso, R. W. Hendrix and R. L. Duda, J. Mol. Biol., 2014, 426, 2112–2129 CrossRef CAS PubMed.
- T. W. Giessen and P. A. Silver, Nat. Microbiol., 2017, 2, 17029 CrossRef PubMed.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS.
- A. N. Gabashvili, N. S. Chmelyuk, V. A. Sarkisova, P. A. Melnikov, A. S. Semkina, A. A. Nikitin and M. A. Abakumov, Int. J. Mol. Sci., 2022, 23, 15591 CrossRef CAS PubMed.
- A. Van de Steen, R. Khalife, N. Colant, H. Mustafa Khan, M. Deveikis, S. Charalambous, C. M. Robinson, R. Dabas, S. Esteban Serna, D. A. Catana, K. Pildish, V. Kalinovskiy, K. Gustafsson and S. Frank, Synth. Syst. Biotechnol., 2021, 6, 231–241 CrossRef CAS.
- P. Lagoutte, C. Mignon, G. Stadthagen, S. Potisopon, S. Donnat, J. Mast, A. Lugari and B. Werle, Vaccine, 2018, 36, 3622–3628 CrossRef CAS PubMed.
- L. H. Lua, N. K. Connors, F. Sainsbury, Y. P. Chuan, N. Wibowo and A. P. Middelberg, Biotechnol. Bioeng., 2014, 111, 425–440 CrossRef CAS PubMed.
- M. C. Jenkins and S. Lutz, ACS Synth. Biol., 2021, 10, 857–869 CrossRef CAS PubMed.
- M. P. Andreas and T. W. Giessen, Nat. Commun., 2021, 12, 4748 CrossRef CAS PubMed.
- T. W. Giessen, Annu. Rev. Biochem., 2022, 91, 353–380 CrossRef PubMed.
- J. C. Tracey, M. Coronado, T. W. Giessen, M. C. Y. Lau, P. A. Silver and B. B. Ward, Sci. Rep., 2019, 9, 20122 CrossRef CAS PubMed.
- N. Valdés-Stauber and S. Scherer, Appl. Environ. Microbiol., 1994, 60, 3809–3814 CrossRef PubMed.
- I. Rosenkrands, P. B. Rasmussen, M. Carnio, S. Jacobsen, M. Theisen and P. Andersen, Infect. Immun., 1998, 66, 2728–2735 CrossRef CAS PubMed.
- P. M. Hicks, K. D. Rinker, J. R. Baker and R. M. Kelly, FEBS Lett., 1998, 440, 393–398 CrossRef CAS.
- F. Akita, K. T. Chong, H. Tanaka, E. Yamashita, N. Miyazaki, Y. Nakaishi, M. Suzuki, K. Namba, Y. Ono, T. Tsukihara and A. Nakagawa, J. Mol. Biol., 2007, 368, 1469–1483 CrossRef CAS PubMed.
- R. Rahmanpour and T. D. Bugg, FEBS J., 2013, 280, 2097–2104 CrossRef CAS PubMed.
- H. Contreras, M. S. Joens, L. M. McMath, V. P. Le, M. V. Tullius, J. M. Kimmey, N. Bionghi, M. A. Horwitz, J. A. Fitzpatrick and C. W. Goulding, J. Biol. Chem., 2014, 289, 18279–18289 CrossRef CAS PubMed.
- T. D. Goddard, C. C. Huang, E. C. Meng, E. F. Pettersen, G. S. Couch, J. H. Morris and T. E. Ferrin, Protein Sci., 2018, 27, 14–25 CrossRef CAS PubMed.
- J. Ross, Z. McIver, T. Lambert, C. Piergentili, J. E. Bird, K. J. Gallagher, F. L. Cruickshank, P. James, E. Zarazúa-Arvizu, L. E. Horsfall, K. J. Waldron, M. D. Wilson, C. L. Mackay, A. Baslé, D. J. Clarke and J. Marles-Wright, Sci. Adv., 2022, 8, eabj4461 CrossRef CAS PubMed.
- E. Eren, B. Wang, D. C. Winkler, N. R. Watts, A. C. Steven and P. T. Wingfield, Structure, 2022, 30(551–563), e554 Search PubMed.
- C. Cassidy-Amstutz, L. Oltrogge, C. C. Going, A. Lee, P. Teng, D. Quintanilla, A. East-Seletsky, E. R. Williams and D. F. Savage, Biochemistry, 2016, 55, 3461–3468 CrossRef CAS PubMed.
- R. J. Nichols, B. LaFrance, N. R. Phillips, D. R. Radford, L. M. Oltrogge, L. E. Valentin-Alvarado, A. J. Bischoff, E. Nogales and D. F. Savage, eLife, 2021, 10, e59288 CrossRef CAS PubMed.
- D. T. Jones and D. Cozzetto, Bioinformatics, 2015, 31, 857–863 CrossRef CAS PubMed.
- M. P. Andreas and T. W. Giessen, MethodsX, 2022, 9, 101787 CrossRef CAS PubMed.
- H. Moon, J. Lee, J. Min and S. Kang, Biomacromolecules, 2014, 15, 3794–3801 CrossRef CAS PubMed.
- T. W. Giessen and P. A. Silver, ChemBioChem, 2016, 17, 1931–1935 CrossRef CAS PubMed.
- R. M. Putri, C. Allende-Ballestero, D. Luque, R. Klem, K. A. Rousou, A. Liu, C. H. Traulsen, W. F. Rurup, M. S. T. Koay, J. R. Castón and J. J. L. M. Cornelissen, ACS Nano, 2017, 11, 12796–12804 CrossRef CAS PubMed.
- R. M. Putri, J. W. Fredy, J. J. Cornelissen, M. S. Koay and N. Katsonis, Chem. Phys. Chem., 2016, 17, 1815–1818 CrossRef CAS PubMed.
- Y. Ma, R. J. Nolte and J. J. Cornelissen, Adv. Drug Delivery Rev., 2012, 64, 811–825 CrossRef CAS PubMed.
- L. Schoonen, R. J. Nolte and J. C. van Hest, Nanoscale, 2016, 8, 14467–14472 RSC.
- L. Schoonen and J. C. van Hest, Adv. Mater., 2016, 28, 1109–1128 CrossRef CAS PubMed.
- S. Michel-Souzy, N. M. Hamelmann, S. Zarzuela-Pura, J. M. J. Paulusse and J. J. L. M. Cornelissen, Biomacromolecules, 2021, 22, 5234–5242 CrossRef CAS PubMed.
- J. Snijder, O. Kononova, I. M. Barbu, C. Uetrecht, W. F. Rurup, R. J. Burnley, M. S. Koay, J. J. Cornelissen, W. H. Roos, V. Barsegov, G. J. Wuite and A. J. Heck, Biomacromolecules, 2016, 17, 2522–2529 CrossRef CAS PubMed.
- W. F. Rurup, J. Snijder, M. S. Koay, A. J. Heck and J. J. Cornelissen, J. Am. Chem. Soc., 2014, 136, 3828–3832 CrossRef CAS PubMed.
- F. Sainsbury, P. Saxena, A. A. Aljabali, K. Saunders, D. J. Evans and G. P. Lomonossoff, Methods Mol. Biol., 2014, 1108, 139–153 CrossRef CAS PubMed.
- S. A. McConnell, K. A. Cannon, C. Morgan, R. McAllister, B. R. Amer, R. T. Clubb and T. O. Yeates, ACS Synth. Biol., 2020, 9, 381–391 CrossRef CAS PubMed.
- A. Tamura, Y. Fukutani, T. Takami, M. Fujii, Y. Nakaguchi, Y. Murakami, K. Noguchi, M. Yohda and M. Odaka, Biotechnol. Bioeng., 2015, 112, 13–20 CrossRef CAS PubMed.
- M. Künzle, J. Mangler, M. Lach and T. Beck, Nanoscale, 2018, 10, 22917–22926 RSC.
- P. Lohner, M. Zmyslia, J. Thurn, J. K. Pape, R. Gerasimaitė, J. Keller-Findeisen, S. Groeer, B. Deuringer, R. Süss, A. Walther, S. W. Hell, G. Lukinavičius, T. Hugel and C. Jessen-Trefzer, Angew. Chem., Int. Ed., 2021, 60, 23835–23841 CrossRef CAS PubMed.
- W. J. Altenburg, N. Rollins, P. A. Silver and T. W. Giessen, Sci. Rep., 2021, 11, 4951 CrossRef CAS PubMed.
- X. Xiong, C. Sun, F. S. Vago, T. Klose, J. Zhu and W. Jiang, Biomolecules, 2020, 10, 1342 CrossRef PubMed.
- M. Künzle, J. Mangler, M. Lach and T. Beck, Nanoscale, 2019, 11, 6497 RSC.
- N. Lončar, H. J. Rozeboom, L. E. Franken, M. C. A. Stuart and M. W. Fraaije, Biochem. Biophys. Res. Commun., 2020, 529, 548–553 CrossRef PubMed.
- D. Diaz, X. Vidal, A. Sunna and A. Care, ACS Appl. Mater. Interfaces, 2021, 13, 7977–7986 CrossRef CAS PubMed.
- Y. H. Lau, T. W. Giessen, W. J. Altenburg and P. A. Silver, Nat. Commun., 2018, 9, 1311 CrossRef PubMed.
- F. Sigmund, C. Massner, P. Erdmann, A. Stelzl, H. Rolbieski, M. Desai, S. Bricault, T. P. Wörner, J. Snijder, A. Geerlof, H. Fuchs, M. Hrabe de Angelis, A. J. R. Heck, A. Jasanoff, V. Ntziachristos, J. Plitzko and G. G. Westmeyer, Nat. Commun., 2018, 9, 1990 CrossRef PubMed.
- J. Snijder, M. van de Waterbeemd, E. Damoc, E. Denisov, D. Grinfeld, A. Bennett, M. Agbandje-McKenna, A. Makarov and A. J. Heck, J. Am. Chem. Soc., 2014, 136, 7295–7299 CrossRef CAS PubMed.
- J. A. Jones, A. S. Cristie-David, M. P. Andreas and T. W. Giessen, Angew. Chem., Int. Ed., 2021, 60, 25034–25041 CrossRef CAS PubMed.
- E. M. Williams, S. M. Jung, J. L. Coffman and S. Lutz, ACS Synth. Biol., 2018, 7, 2514–2517 CrossRef CAS PubMed.
- P. Ebensperger, M. Zmyslia, P. Lohner, J. Braunreuther, B. Deuringer, A. Becherer, R. Süss, A. Fischer, H. J. Jessen and C. Jessen-Trefzer, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-1xxjh-v2.
- R. Klem, M. V. de Ruiter and J. J. L. M. Cornelissen, Mol. Pharmacol., 2018, 15, 2991–2996 CrossRef CAS PubMed.
- J. M. Rodríguez, C. Allende-Ballestero, J. J. L. M. Cornelissen and J. R. Castón, Nanomaterials, 2021, 11, 1467 CrossRef PubMed.
- H. X. Zhou, G. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine, 2010, 6, 714–729 CrossRef CAS PubMed.
- A. Maham, Z. Tang, H. Wu, J. Wang and Y. Lin, Small, 2009, 5, 1706–1721 CrossRef CAS PubMed.
- M. Khoshnejad, H. Parhiz, V. V. Shuvaev, I. J. Dmochowski and V. R. Muzykantov, J. Controlled Release, 2018, 282, 13–24 CrossRef CAS PubMed.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS PubMed.
- T. H. Lee, T. S. Carpenter, P. D’haeseleer, D. F. Savage and M. C. Yung, Biotechnol. Bioeng., 2020, 117, 603–613 CrossRef CAS PubMed.
- B. Choi, H. Moon, S. J. Hong, C. Shin, Y. Do, S. Ryu and S. Kang, ACS Nano, 2016, 10, 7339–7350 CrossRef CAS PubMed.
- K. E. Mironova, G. M. Proshkina, A. V. Ryabova, O. A. Stremovskiy, S. A. Lukyanov, R. V. Petrov and S. M. Deyev, Theranostics, 2013, 3, 831–840 CrossRef PubMed.
- N. W. Kennedy, C. E. Mills, T. M. Nichols, C. H. Abrahamson and D. Tullman-Ercek, Curr. Opin. Microbiol., 2021, 63, 36–42 CrossRef CAS PubMed.
- T. W. Giessen and P. A. Silver, J. Mol. Biol., 2016, 428, 916–927 CrossRef CAS PubMed.
- H. Kirst and C. A. Kerfeld, BMC Biol., 2019, 17, 79 CrossRef PubMed.
- A. M. Stewart, K. L. Stewart, T. O. Yeates and T. A. Bobik, Trends Biochem. Sci., 2021, 46, 406–416 CrossRef CAS PubMed.
- L. S. R. Adamson, N. Tasneem, M. P. Andreas, W. Close, E. N. Jenner, T. N. Szyszka, R. Young, L. C. Cheah, A. Norman, H. I. MacDermott-Opeskin, M. L. O'Mara, F. Sainsbury, T. W. Giessen and Y. H. Lau, Sci. Adv., 2022, 8, eabl7346 CrossRef CAS PubMed.
- N. Tasneem, T. N. Szyszka, E. N. Jenner and Y. H. Lau, ACS Nano, 2022, 16, 8540–8556 CrossRef CAS PubMed.
- M. Uchida, E. Manzo, D. Echeveria, S. Jiménez and L. Lovell, Curr. Opin. Virol., 2022, 52, 250–257 CrossRef CAS PubMed.
- D. P. Patterson, B. LaFrance and T. Douglas, Chem. Commun., 2013, 49, 10412–10414 RSC.
- D. Wu, P. Hwang, T. Li and G. Piszczek, Gene Ther., 2022, 29, 691–697 CrossRef CAS PubMed.
- M. Gassmann, B. Grenacher, B. Rohde and J. Vogel, Electrophoresis, 2009, 30, 1845–1855 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |